

Abstract
Background
Patients with Duchenne muscular dystrophy exhibit progressive cardiac and skeletal muscle dysfunction. Based on prior data, cardiac dysfunction in Duchenne muscular dystrophy patients may be influenced by myocardial fibrosis and steroid therapy. We examined the longitudinal relationship of myocardial fibrosis and ventricular dysfunction using cardiac magnetic resonance in a large Duchenne muscular dystrophy cohort.
Methods and Results
We reviewed 465 serial cardiac magnetic resonance studies (98 Duchenne muscular dystrophy patients with ≥4 cardiac magnetic resonance studies) for left ventricular ejection fraction (LVEF) and presence of late gadolinium enhancement (LGE), a marker for myocardial fibrosis. LVEF was modeled by examining LGE status, myocardial fibrosis burden (as assessed by the number of LGE‐positive left ventricular segments), patient age, and steroid treatment duration. An age‐only model demonstrated that LVEF declined 0.58±0.10% per year. In patients with both LGE‐negative and LGE‐positive studies (n=51), LVEF did not decline significantly over time if LGE was absent but declined 2.2±0.31% per year when LGE was present. Univariate modeling showed significant associations between LVEF and steroid treatment duration, presence of LGE, and number of LGE‐positive left ventricular segments; multivariate modeling showed that LVEF declined by 0.93±0.09% for each LGE‐positive left ventricular segment, whereas age and steroid treatment duration were not significant. The number of LGE‐positive left ventricular segments increased with age, and longer steroid treatment duration was associated with lower age‐related increases.
Conclusion
Progressive myocardial fibrosis, as detected by LGE, was strongly correlated with the LVEF decline in Duchenne muscular dystrophy patients. Longer steroid treatment duration was associated with a lower age‐related increase in myocardial fibrosis burden.
Keywords: cardiomyopathy, magnetic resonance imaging, morbidity
Introduction
Duchenne muscular dystrophy (DMD) composes part of the clinical spectrum of the dystrophinopathies and is caused by mutations in the dystrophin gene.1 Because of improvements in respiratory care, cardiac dysfunction is now a leading cause of morbidity and mortality in DMD patients.2–5 Left ventricular (LV) dilation and the development of depressed LV ejection fraction (LVEF) are common findings in patients with DMD2,6 and often present in the second decade of life.7 The exact mechanisms of the development of cardiac dysfunction in DMD are unclear, but the presence of myocardial fibrosis may indicate progression of cardiac disease.8–11 In a cross‐sectional study, we showed that late gadolinium enhancement (LGE), a marker for myocardial fibrosis,12 is associated with LV systolic dysfunction and increases with age13; however, this finding has not been thoroughly investigated in a longitudinal study. The primary aims of this study were to evaluate the longitudinal relationship of LVEF and myocardial fibrosis burden in a large DMD patient cohort and to investigate the associations with age and duration of steroid treatment. Our secondary aim was to evaluate the relationship of LGE to all‐cause death, LV assist device implantation, heart transplant, and clinically significant arrhythmias.
Methods
Study Population
All male patients with DMD who underwent clinical cardiac magnetic resonance (CMR) studies at Cincinnati Children's Hospital Medical Center between January 2005 and January 2013 were identified by querying the CMR database, and those who had undergone ≥4 studies in which LGE status could be determined were selected. At our institution, we routinely perform annual CMR studies on every DMD patient who is able to undergo the scan without sedation; only patients who refused or who could not tolerate lying in the scanner did not undergo CMR. An annual CMR study was recommended regardless of previous refusal or inability to undergo CMR. Intravenous access with administration of gadolinium is attempted on every DMD CMR study. All selected patients were confirmed to have a pathological dystrophin mutation. The institutional review board approved the study.
Ventricular Function Imaging
Cardiac functional imaging was performed with a retrospectively, vectorcardiographically gated, segmented steady‐state free precession technique after localized shimming and/or frequency adjustment, as necessary.14–15 CMR studies were conducted on clinical 3‐T or 1.5‐T scanners. The scanner used for each study was based solely on clinical availability, independent of the patient's clinical status or the machine used for previous studies. No sedation or anesthesia was used for these studies. Patients were imaged with a breath‐held technique, as tolerated; for those patients who could not adequately breath hold, a free‐breathing technique with multiple‐signal averaging was used. Standard functional imaging included a short‐axis stack of segmented steady‐state free precession cine images from cardiac base to apex; the short axis was prescribed as the perpendicular plane to the LV long axis in 2‐ and 4‐chamber views, based on previously published protocols.14,16 Typical scan parameters used were slice thickness of 5 mm and in‐plane resolution of 1.5 mm. A minimum of 12 slices were performed with 30 phases per slice. The typical temporal resolution of the segmented steady‐state free precession cine images was 30 to 40 ms. LVEF was calculated using standard planimetry techniques (QMass MR, version 7.5; Medis Medical Imaging Systems) by an expert reader (R.F., K.N.H., J.J.S., M.D.T.) and then exported to a spreadsheet.
LGE Imaging
LGE imaging was performed with a standard inversion recovery sequence protocol 8 to 10 minutes after injection with 0.2 mmol/kg gadolinium diethylenetriamine penta‐acetic acid. A study was considered LGE positive (LGE+) if any LV segment showed myocardial hyperenhancement. We then identified which segments were LGE+ (LGE+ LV segments) based on the 16‐segment American Heart Association model.17
Patient Clinical Characteristics
The medical records for all patients in the study were reviewed. Start and stop dates for use of steroids (deflazacort and/or prednisone) were extracted; steroid treatment data were available for all patients in the study cohort. Steroid treatment duration was calculated from the steroid start date to the date of CMR or the date of stopping steroids, whichever was sooner; steroid‐naïve patients were assigned steroid treatment durations of 0 for each CMR. The duration of steroid treatment was a time‐varying variable and was calculated for each CMR for each patient. We also identified date of death, LV assist device implantation, and orthotopic heart transplant, if applicable. All Holter monitor data were reviewed, and episodes of nonsustained ventricular tachycardia, atrial fibrillation, and atrial tachycardia were considered to be clinically significant. Holter studies were ordered based solely on the preference of the individual patient's neurologist or cardiologist.
Statistical Analysis
We analyzed the association of depressed LVEF and LGE status using the chi‐square test and the relationship of LVEF to age using linear mixed‐model analysis, accounting for the correlated values within study patients. For patients who were initially LGE negative (LGE−) but then developed LGE over the course of the study (the LGE−/+ group), we assigned an estimated date of development of LGE (tLGE), defined as the date midway between the patient's last LGE− and first LGE+ study. We then performed a piecewise linear regression of LVEF versus time, divided into time before tLGE (negative) and time after tLGE (positive). To assess for independent determinants of LVEF, we performed multivariable regression with a model including patient age, number of LGE+ LV segments, duration of steroid treatment, and interaction of age and duration of steroid treatment (these are correlated), using linear mixed‐model analyses. Finally, we assessed the effects of age and steroid treatment duration on the percentage of LGE+ LV segments using fractional logit regression. All tests were 2‐sided, and a P value of <0.05 was considered statistically significant. All analyses were performed using SAS version 9.3 (SAS Institute).
Results
Characteristics of Study Cohort
We identified 335 DMD patients who had undergone at least 1 CMR in the study period, and 98 of these patients had ≥4 CMR studies (465 total studies) in which LGE status could be adequately determined (Table 1). Patient age at the time of CMR ranged from 6.6 to 29.4 years (median 12.2, mean 13.1±4.1 years), which was similar to the overall cohort. Forty‐five patients (46%) developed LGE before depressed LVEF, 11 (11%) patients developed depressed LVEF before LGE, and 3 patients (3%) developed both on the same study. In addition, 51 patients (52%) were initially LGE− and subsequently developed LGE during the course of the study (the LGE−/+ group). In terms of steroid treatment, 50 patients (51%) were treated with deflazacort only, 12 (12%) were treated with prednisone only, 33 (34%) were treated with both; only 3 (3%) had never been treated with steroids (Table 1). The mean age of initiating steroid therapy was 7.0±2.5 years, with a mean duration of use 7.6±3.4 years.
Table 1.
Patient Characteristics and Cardiac Magnetic Resonance Study Results
| Patient Characteristics (n=98) | Results |
|---|---|
| Age at CMR, y | 6.6 to 29.4 (median 12.2, mean 13.1±4.1) |
| Age at first CMR, y | 6.6 to 22.5 (median 9.9, mean 10.6±3.4) |
| Age at last CMR, y | 9.4 to 29.4 (median 14.2, mean 15.2±4.0) |
| Normal LVEF and LGE− on all CMR, n (%) | 39 (40) |
| LGE+ on ≥1 CMR, n (%) | 57 (58) |
| Depressed LVEF on ≥1 CMR, n (%) | 23 (23) |
| Had either depressed LVEF or LGE on ≥1, n (%) CMR | 59 (60) |
| Had both depressed LVEF and LGE on ≥1 CMR, n (%) | 21 (21) |
| Age of first LGE+ study, y | 8.4 to 27.5 (median 13.5, mean 14.0±3.6) |
| Age of first CMR with depressed LVEF, y | 6.9 to 22.4 (median 15.0, mean 14.7±4.5) |
| Developed LGE before depressed LVEF, n (%) | 45 (46) |
| Developed depressed LVEF before LGE, n (%) | 11 (11) |
| Developed LGE and depressed LVEF on same study, n (%) | 3 (3) |
| Had only LGE, n (%) | 36 (37) |
| Had only depressed LVEF, n (%) | 2 (2) |
| Treated with deflazacort only, n (%) | 50 (51) |
| Treated with prednisone only, n (%) | 12 (12) |
| Treated with both deflazacort and prednisone, n (%) | 33 (34) |
| No steroid treatment, n (%) | 3 (3) |
| Age at initiation of steroids, y | 3.2 to 19.4 (median 6.8, mean 7.0±2.5) |
| Duration of steroid treatment, y | 0.2 to 20.1 (median 7.2, mean 7.6±3.4) |
CMR indicates cardiac magnetic resonance; LGE, late gadolinium enhancement; LGE+, LGE positive; LGE−, LGE negative; LVEF, left ventricular ejection fraction.
Prevalence and Age of Onset of LGE and Depressed LVEF
There were 146 LGE+ studies (31.4%) and 57 studies (12.3%) that demonstrated depressed LVEF; these were similar to the cross‐section of the entire DMD cohort (23.9% LGE+, 5.7% LGE indeterminate, and 70.4% LGE−; 11.7% with depressed LVEF). The ages of onset of LGE and depressed LVEF are described in Table 1. The relative risk of having depressed LVEF given an LGE+ study was 7.6 (95% CI 1.8 to 30.4; P=0.0002).
Effect of Age and Time Since Development of LGE on LVEF
An age‐only model for the entire cohort demonstrated that LVEF declined by 0.58±0.10% per year (mean±SE; P<0.0001, r2=0.067) (Figure 1). To further delineate the relationships among age, LGE, and LVEF, we transformed age into time after tLGE for the LGE−/+ group, as described. LVEF did not decline significantly before the development of LGE (0.21±0.22% per year; P=0.34) (Figure 2) but declined significantly after the development of LGE (2.2±0.31% per year; P<0.0001). The rate of LVEF decline accelerated by 2.0±0.45% per year (P<0.0001) at tLGE.
Figure 1.
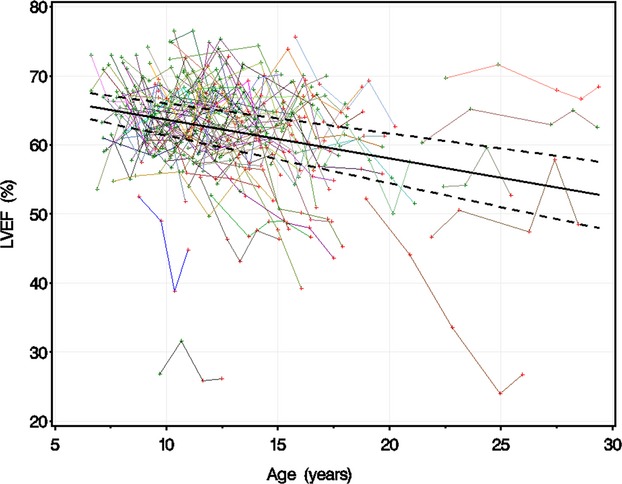
LVEF vs age. LGE‐negative studies are marked in green, and LGE‐positive studies are marked in red. The studies for each individual patient are linked with a colored line. Dashed lines are upper and lower 95th CIs of predicted mean LVEF. LVEF declined 0.58±0.10% per year in an age‐only model, accounting for correlated values within patients. LGE indicates late gadolinium enhancement; LVEF, left ventricular ejection fraction.
Figure 2.
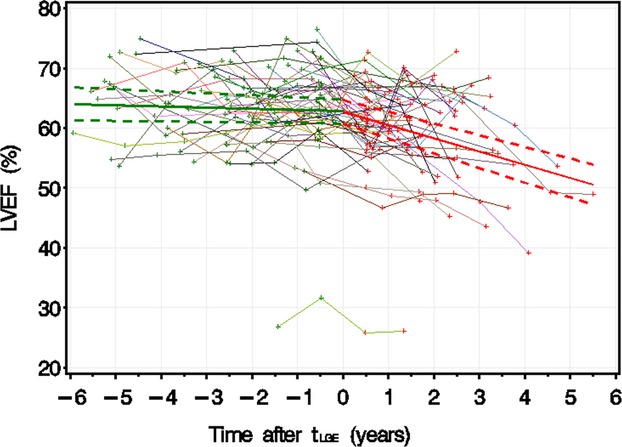
LVEF vs time after tLGE. This figure demonstrates LVEF vs tLGE for patients with at least 1 LGE‐negative study followed by at least 1 LGE‐positive study (LGE−/+ group). LGE‐negative studies are marked in green, and LGE‐positive studies are marked in red. The studies for each individual patient are linked with a colored line. Dashed lines are upper and lower 95th CIs of predicted mean LVEF. LVEF remained stable over time if patients were LGE negative but declined by 2.2±0.31% per year if patients were LGE positive. LGE indicates late gadolinium enhancement; LVEF, left ventricular ejection fraction; tLGE, time after development of LGE.
Predictors of LVEF in DMD Patients
Univariate analyses showed that an increased steroid treatment duration was associated with an LVEF decline of 0.43±0.11% per year of treatment (P<0.0001, r2=0.027). In addition, LVEF was lower if LGE was positive (−3.9±0.58%; P<0.0001, r2=0.094). For each additional LGE+ LV segment, there was an associated decline of LVEF by 0.93±0.09% (P<0.0001, r2=0.174) (Figure 3). Multivariate analyses showed that only the number of LGE+ LV segments was a significant predictor of LVEF (β=−0.93±0.09, P<0.0001).
Figure 3.
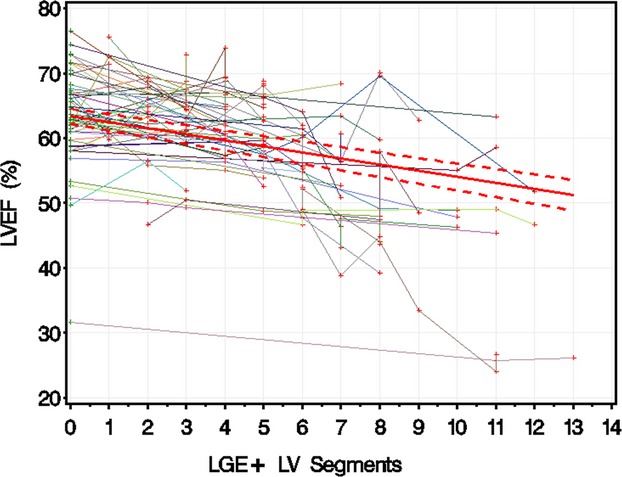
LVEF vs number of left ventricular segments positive for LGE. LGE‐negative studies are marked in green, and LGE‐positive studies are marked in red. The studies for each individual patient are linked with a colored line. Dashed lines are upper and lower 95th CIs of predicted mean LVEF. Each additional LGE‐positive LV segment was associated with an LVEF decline of 0.93±0.09%. LGE indicates late gadolinium enhancement; LV, left ventricular; LVEF, left ventricular ejection fraction.
Modifiers of the Myocardial Fibrosis Burden
Fractional logit regression showed that the odds of developing an LGE+ LV segment increased with age (Figure 4; Table 2), implying that the number of LGE+ LV segments increases slowly at younger ages, accelerates, and then levels off. A multivariate fractional logit regression model of the percentage of LGE+ LV segments was constructed for the entire cohort using age, duration of steroid treatment, and their interaction as predictors. There was a significant age–steroid treatment duration interaction, suggesting that a longer steroid treatment duration was associated with an attenuated age‐related increase in the number of LGE+ LV segments, although the effect was small (Table 2).
Figure 4.
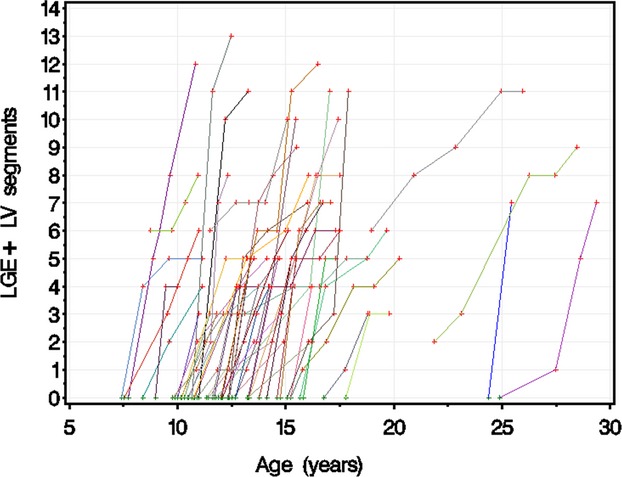
Number of LGE‐positive LV segments vs patient age. This figure demonstrates the number of LGE‐positive LV segments related to age. The points for each patient are connected with a colored line. LGE indicates late gadolinium enhancement; LV, left ventricular.
Table 2.
Parameter Estimates of Age and Steroid Therapy Duration on Late Gadolinium Enhancement–Positive Segment Percentage From Fractional Logit Regression
| Models | Effect | Estimate | Standard Error | P Value |
|---|---|---|---|---|
| Age only | Age | 0.133 | 0.017 | <0.0001 |
| Intercept | −4.09 | 0.270 | <0.0001 | |
| Age and steroid duration | Age | 0.292 | 0.0393 | <0.0001 |
| Steroid duration | 0.494 | 0.0773 | <0.0001 | |
| Age–steroid duration interaction | −0.020 | 0.00344 | <0.0001 | |
| Intercept | −7.31 | 0.681 | <0.0001 |
Cardiac Outcomes
Of the 98 total patients in the cohort, 4 died during the study period; of these 4 patients, 3 were LGE+ on their last CMR study and 3 had LVEF <55 (mean 48.0±15%). No patients in this cohort had undergone heart transplant or LV assist device implantation. Given the low rate of these events, statistical testing could not be performed. At least 1 Holter study was performed on 76 of the 98 patients. Nonsustained ventricular tachycardia (1 patient), atrial fibrillation (1 patient), and nonsustained atrial tachycardia (8 patients) were infrequently observed. There was no statistically significant difference in risk of arrhythmias based on having ≥1 CMR with LGE, and there was no relationship to LVEF.
Discussion
In our longitudinal model of LVEF in DMD, we found that the development of LGE was associated with a 2.2% decline in LVEF per year, whereas there was no statistically significant decline in LVEF over time in patients without LGE. In addition, the strongest correlate of LVEF was the number of LGE+ LV segments, a quantitative measure of myocardial fibrosis burden; age and steroid treatment duration (independent of fibrosis burden) were not statistically significant predictors of LVEF in multivariate modeling. Furthermore, a longer steroid treatment duration correlated with a smaller age‐related increase in myocardial fibrosis burden. In our cohort, the age of onset of depressed LVEF and LGE were quite variable, with some patients developing LGE or depressed LVEF at an early age and others being LGE− and maintaining normal LVEF into their late 20s.
Given low rates of death, LV assist device implantation, and heart transplant, it was not possible to draw conclusions about these events and their relationship to myocardial fibrosis burden. Significant arrhythmia was infrequent, and the risk did not appear to be related to LGE status or LVEF.
Comparison to Previous Studies
Our study is the largest longitudinal examination of CMR in the DMD population to date, but direct comparison with other studies is challenging because of variations in age range and disease severity. Our cohort demonstrated similar proportions of patients with normal LVEF (77%) compared with other studies (70%,2 76%,5 88%9). Previous studies reported a broad range of LGE positivity in DMD patients (from 32%9 to 70%10); comparing the 58% that we observed is difficult, given that the other studies had patients of different age ranges and provided limited data regarding steroid duration, which was related to the development of LGE in our study. Our data also corroborate previous reports that, in general, LGE appears to develop before depressed LVEF,10–11 but our results extend this observation to show that once LGE has developed, there is a decline in LVEF, on average, over time.
There are no large, recent studies on the arrhythmia burden in DMD patients, so the low arrhythmia rates we observed are difficult to compare. This area requires further, larger studies.
Potential Mechanisms of Cardiac Dysfunction in DMD Patients
Increasing histopathological evidence shows that the fibrofatty replacement of myocytes is a significant pathophysiological mechanism in the development of cardiac dysfunction in DMD mouse models.18–21 Combined with previous DMD patient imaging studies,9,11,13,22 our results support the hypothesis that fibrofatty replacement resulting in diffuse myocardial fibrosis is a key step in the development of cardiac dysfunction in this population. Furthermore, our results suggest that increased age correlates with a higher myocardial fibrosis burden, that a longer steroid treatment duration correlates with a smaller age‐related increase in myocardial fibrosis, and that the fibrosis burden correlates with the development LV dysfunction.
It is worth noting that the LGE technique requires a minimum threshold volume of myocardial fibrosis before becoming evident on CMR and, consequently, underestimates the total myocardial fibrosis burden. This may explain why some patients with moderately or severely depressed LVEF are LGE−. There are now quantitative CMR techniques that are more sensitive to diffuse myocardial fibrosis and that may further delineate the relationship of myocardial fibrosis to LVEF in DMD patients. In addition to the factors studied in this report, other modifiers that increase or decrease myocardial fibrosis development likely still need identification and examination.
Implications for DMD Patient Care
Angiotensin‐converting enzyme inhibitors,21 aldosterone antagonists,21 and angiotensin receptor blockers18,23 decrease myocardial fibrosis and improve circumferential strain in DMD mouse models. In humans with DMD, steroids5,24–27 and angiotensin‐converting enzyme inhibitors28–31 have shown a protective effect on cardiac function in some32 but not all studies.33–34 All of these classes of agents may have antifibrotic effects, but the critical mechanism of action in DMD patients is not known. If the antifibrotic properties are the primary mechanism of such agents, these studies also lend credence to the theory that myocardial fibrosis leads to LV systolic dysfunction. Our study supports the theory that steroids decrease myocardial fibrosis burden at a given age.
Studies in other models of progressive LV systolic dysfunction suggest that myocardial fibrosis may be preventable. In animal models of heart failure, aldosterone antagonists have been shown to prevent the development of myocardial fibrosis35; in a prospective trial in anthracycline‐induced cardiomyopathy in adults, enalapril and carvedilol led to preservation of LV systolic function,36 and this population shows signs of increased myocardial fibrosis on CMR before the development of identifiable LGE.37 Consequently, initiating antifibrotic therapies before the development of overt LV systolic dysfunction may be helpful in delaying DMD‐related cardiac dysfunction, and the results of a clinical trial evaluating the use of eplerenone for the prevention of LV dysfunction and myocardial fibrosis in DMD patients was recently published.38
The presence of LGE has been linked to increased rates of adverse outcomes such as cardiac death in nonischemic dilated,39–40 hypertrophic,41–43 and other nonischemic cardiomyopathies44; a similar examination of the relationship of LGE to outcomes in DMD‐related cardiac dysfunction would be of great benefit. Given that our data show that the presence of LGE and myocardial fibrosis burden correlate with LVEF, our study suggests that assessment of myocardial LGE should be part of the evaluation of DMD patients if it is clinically feasible and if it presents a low risk to the patient.
Limitations
The CMR data for our cohort were collected retrospectively, which limits some comparisons and does not allow for the determination of causal relationships. Based on the selection criteria for inclusion in this analysis, we necessarily chose a cohort that survived long enough to undergo ≥4 CMR studies; therefore, these patients may have had a milder cardiac phenotype. The specific reasons for some patients not tolerating a CMR study could not be identified and thus affected which patients met eligibility criteria. The relative tachycardia of these patients and their diffuse LGE patterns complicate quantitative myocardial fibrosis assessment. In addition, some techniques for myocardial fibrosis quantification were not available for clinical use until recently. Some patients with overt dysfunction are on β‐blockers and/or angiotensin‐converting enzyme inhibitors, which may confound the evaluation of LV function and LGE, although not all studies suggest that these medications affect the rate of LVEF decline.33 In addition, our study was not powered to take the effects of multiple medications into account. This area requires further study.
Conclusions
Our study demonstrates that the development of LGE is a strong marker for the progression of LV systolic dysfunction in DMD patients and that myocardial fibrosis burden correlates strongly with LVEF. A longer steroid treatment duration correlated with a smaller age‐related increase in myocardial fibrosis burden. This observation could not be attributed to the effect of steroids alone, given the inability to control for other medications and other potential modifiers of the fibrosis burden. These findings support the theory that progressive fibrofatty myocardial replacement is a substrate for myocardial dysfunction in DMD patients. Further investigations into the use of antifibrotic agents in DMD populations should be performed with myocardial fibrosis imaging and LVEF as therapeutic end points.
Sources of Funding
All or portions of this manuscript were submitted as a thesis in partial fulfillment of requirements for a Master of Science degree. This publication was supported by an Institutional Clinical and Translational Science Award, NIH/NCRR 5UL1RR026314. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Disclosures
Tandon owns stock in GE and Medtronic. There are no other relevant disclosures for any of the authors of this study.
References
- Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003; 2:731-740. [DOI] [PubMed] [Google Scholar]
- Connuck DM, Sleeper LA, Colan SD, Cox GF, Towbin JA, Lowe AM, Wilkinson JD, Orav EJ, Cuniberti L, Salbert BA, Lipshultz SEPediatric Cardiomyopathy Registry Study G. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: a comparative study from the Pediatric Cardiomyopathy Registry. Am Heart J. 2008; 155:998-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002; 12:926-929. [DOI] [PubMed] [Google Scholar]
- Passamano L, Taglia A, Palladino A, Viggiano E, D'Ambrosio P, Scutifero M, Rosaria Cecio M, Torre V, DE Luca F, Picillo E, Paciello O, Piluso G, Nigro G, Politano L. Improvement of survival in Duchenne muscular dystrophy: retrospective analysis of 835 patients. Acta Myol. 2012; 31:121-125. [PMC free article] [PubMed] [Google Scholar]
- Schram G, Fournier A, Leduc H, Dahdah N, Therien J, Vanasse M, Khairy P. All‐cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J Am Coll Cardiol. 2013; 61:948-954. [DOI] [PubMed] [Google Scholar]
- Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology. 2003; 99:1-19. [DOI] [PubMed] [Google Scholar]
- Kirchmann C, Kececioglu D, Korinthenberg R, Dittrich S. Echocardiographic and electrocardiographic findings of cardiomyopathy in Duchenne and Becker‐Kiener muscular dystrophies. Pediatr Cardiol. 2005; 26:66-72. [DOI] [PubMed] [Google Scholar]
- Frankel KA, Rosser RJ. The pathology of the heart in progressive muscular dystrophy: epimyocardial fibrosis. Hum Pathol. 1976; 7:375-386. [DOI] [PubMed] [Google Scholar]
- Puchalski MD, Williams RV, Askovich B, Sower CT, Hor KH, Su JT, Pack N, Dibella E, Gottliebson WM. Late gadolinium enhancement: precursor to cardiomyopathy in Duchenne muscular dystrophy? Int J Cardiovasc Imaging. 2009; 25:57-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MC, Meira ZM, Gurgel Giannetti J, da Silva MM, Campos AF, Barbosa Mde M, Starling Filho GM, Ferreira Rde A, Zatz M, Rochitte CE. Myocardial delayed enhancement by magnetic resonance imaging in patients with muscular dystrophy. J Am Coll Cardiol. 2007; 49:1874-1879. [DOI] [PubMed] [Google Scholar]
- Walcher T, Steinbach P, Spiess J, Kunze M, Gradinger R, Walcher D, Bernhardt P. Detection of long‐term progression of myocardial fibrosis in Duchenne muscular dystrophy in an affected family: a cardiovascular magnetic resonance study. Eur J Radiol. 2011; 80:115-119. [DOI] [PubMed] [Google Scholar]
- Mewton N, Liu CY, Croisille P, Bluemke D, Lima JA. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol. 2011; 57:891-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hor KN, Taylor MD, Al‐Khalidi HR, Cripe LH, Raman SV, Jefferies JL, O'Donnell R, Benson DW, Mazur W. Prevalence and distribution of late gadolinium enhancement in a large population of patients with Duchenne muscular dystrophy: effect of age and left ventricular systolic function. J Cardiovasc Magn Reson. 2013; 15:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hor KN, Wansapura J, Markham LW, Mazur W, Cripe LH, Fleck R, Benson DW, Gottliebson WM. Circumferential strain analysis identifies strata of cardiomyopathy in Duchenne muscular dystrophy: a cardiac magnetic resonance tagging study. J Am Coll Cardiol. 2009; 53:1204-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur W, Hor KN, Germann JT, Fleck RJ, Al‐Khalidi HR, Wansapura JP, Chung ES, Taylor MD, Jefferies JL, Woodrow Benson D, Gottliebson WM. Patterns of left ventricular remodeling in patients with Duchenne muscular dystrophy: a cardiac MRI study of ventricular geometry, global function, and strain. Int J Cardiovasc Imaging. 2012; 28:99-107. [DOI] [PubMed] [Google Scholar]
- Hagenbuch SC, Gottliebson WM, Wansapura J, Mazur W, Fleck R, Benson DW, Hor KN. Detection of progressive cardiac dysfunction by serial evaluation of circumferential strain in patients with Duchenne muscular dystrophy. Am J Cardiol. 2010; 105:1451-1455. [DOI] [PubMed] [Google Scholar]
- Cerqueira MD, Weissman NJ, Dilsizian V, Jacobs AK, Kaul S, Laskey WK, Pennell DJ, Rumberger JA, Ryan T, Verani MSImaging AHAWGoMSaRfC. Standardized myocardial segmentation and nomenclature for tomographic imaging of the heart. A statement for healthcare professionals from the Cardiac Imaging Committee of the Council on Clinical Cardiology of the American Heart Association. Circulation. 2002; 105:539-542. [DOI] [PubMed] [Google Scholar]
- Bish LT, Yarchoan M, Sleeper MM, Gazzara JA, Morine KJ, Acosta P, Barton ER, Sweeney HL. Chronic losartan administration reduces mortality and preserves cardiac but not skeletal muscle function in dystrophic mice. PLoS One. 2011; 6:e20856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun JL, O'Brien R, Berry SE. Cardiac dysfunction and pathology in the dystrophin and utrophin‐deficient mouse during development of dilated cardiomyopathy. Neuromuscul Disord. 2012; 22:368-379. [DOI] [PubMed] [Google Scholar]
- Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004; 14:491-496. [DOI] [PubMed] [Google Scholar]
- Rafael‐Fortney JA, Chimanji NS, Schill KE, Martin CD, Murray JD, Ganguly R, Stangland JE, Tran T, Xu Y, Canan BD, Mays TA, Delfin DA, Janssen PM, Raman SV. Early treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in Duchenne muscular dystrophy mice. Circulation. 2011; 124:582-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilchick KC, Salerno M, Plitt D, Dori Y, Crawford TO, Drachman D, Thompson WR. Prevalence and distribution of regional scar in dysfunctional myocardial segments in Duchenne muscular dystrophy. J Cardiovasc Magn Reson. 2011; 13:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurney CF, Sali A, Guerron AD, Iantorno M, Yu Q, Gordish‐Dressman H, Rayavarapu S, van der Meulen J, Hoffman EP, Nagaraju K. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin‐deficient mdx mice. J Cardiovasc Pharmacol Ther. 2011; 16:87-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham LW, Kinnett K, Wong BL, Woodrow Benson D, Cripe LH. Corticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophy. Neuromuscul Disord. 2008; 18:365-370. [DOI] [PubMed] [Google Scholar]
- Barber BJ, Andrews JG, Lu Z, West NA, Meaney FJ, Price ET, Gray A, Sheehan DW, Pandya S, Yang M, Cunniff C. Oral corticosteroids and onset of cardiomyopathy in Duchenne muscular dystrophy. J Pediatr. 2013; 163:1080-1084.e1. [DOI] [PubMed] [Google Scholar]
- Mavrogeni S, Papavasiliou A, Douskou M, Kolovou G, Papadopoulou E, Cokkinos DV. Effect of deflazacort on cardiac and sternocleidomastoid muscles in Duchenne muscular dystrophy: a magnetic resonance imaging study. Eur J Paediatr Neurol. 2009; 13:34-40. [DOI] [PubMed] [Google Scholar]
- Silversides CK, Webb GD, Harris VA, Biggar DW. Effects of deflazacort on left ventricular function in patients with Duchenne muscular dystrophy. Am J Cardiol. 2003; 91:769-772. [DOI] [PubMed] [Google Scholar]
- Duboc D, Meune C, Pierre B, Wahbi K, Eymard B, Toutain A, Berard C, Vaksmann G, Weber S, Becane HM. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years' follow‐up. Am Heart J. 2007; 154:596-602. [DOI] [PubMed] [Google Scholar]
- Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, Neish SR, Smith EO, Towbin JA. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005; 112:2799-2804. [DOI] [PubMed] [Google Scholar]
- Kwon HW, Kwon BS, Kim GB, Chae JH, Park JD, Bae EJ, Noh CI. The effect of enalapril and carvedilol on left ventricular dysfunction in middle childhood and adolescent patients with muscular dystrophy. Korean Circ J. 2012; 42:184-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaciotti C, Heistein LC, Coursey M, Lemler MS, Eapen RS, Iannaccone ST, Scott WA. Left ventricular function and response to enalapril in patients with Duchenne muscular dystrophy during the second decade of life. Am J Cardiol. 2006; 98:825-827. [DOI] [PubMed] [Google Scholar]
- Politano L, Nigro G. Treatment of dystrophinopathic cardiomyopathy: review of the literature and personal results. Acta Myol. 2012; 31:24-30. [PMC free article] [PubMed] [Google Scholar]
- Hor KN, Mazur W, Taylor MD, Al‐Khalidi HR, Cripe LH, Jefferies JL, Raman SV, Chung ES, Kinnett KJ, Williams K, Gottliebson WM, Benson DW. Effects of steroids and angiotensin converting enzyme inhibition on circumferential strain in boys with Duchenne muscular dystrophy: a cross‐sectional and longitudinal study utilizing cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2011; 13:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwath ML, Jacobs IB, Crowe CA, Ashwath RC, Super DM, Bahler RC. Left ventricular dysfunction in Duchenne muscular dystrophy and genotype. Am J Cardiol. 2014; 114:284-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brilla CG. Aldosterone and myocardial fibrosis in heart failure. Herz. 2000; 25:299-306. [DOI] [PubMed] [Google Scholar]
- Bosch X, Rovira M, Sitges M, Domenech A, Ortiz‐Perez JT, de Caralt TM, Morales‐Ruiz M, Perea RJ, Monzo M, Esteve J. Enalapril and carvedilol for preventing chemotherapy‐induced left ventricular systolic dysfunction in patients with malignant hemopathies: the OVERCOME trial (preventiOn of left Ventricular dysfunction with Enalapril and caRvedilol in patients submitted to intensive ChemOtherapy for the treatment of Malignant hEmopathies). J Am Coll Cardiol. 2013; 61:2355-2362. [DOI] [PubMed] [Google Scholar]
- Tham EB, Haykowsky MJ, Chow K, Spavor M, Kaneko S, Khoo NS, Pagano JJ, Mackie AS, Thompson RB. Diffuse myocardial fibrosis by T1‐mapping in children with subclinical anthracycline cardiotoxicity: relationship to exercise capacity, cumulative dose and remodeling. J Cardiovasc Magn Reson. 2013; 15:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman SV, Hor KN, Mazur W, Halnon NJ, Kissel JT, He X, Tran T, Smart S, McCarthy B, Taylor MD, Jefferies JL, Rafael‐Fortney JA, Lowe J, Roble SL, Cripe LH. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double‐blind, placebo‐controlled trialLancet Neurol. 2014. 10.1016/S1474‐4422(14)70318‐7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assomull RG, Prasad SK, Lyne J, Smith G, Burman ED, Khan M, Sheppard MN, Poole‐Wilson PA, Pennell DJ. Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J Am Coll Cardiol. 2006; 48:1977-1985. [DOI] [PubMed] [Google Scholar]
- Gulati A, Jabbour A, Ismail TF, Guha K, Khwaja J, Raza S, Morarji K, Brown TD, Ismail NA, Dweck MR, Di Pietro E, Roughton M, Wage R, Daryani Y, O'Hanlon R, Sheppard MN, Alpendurada F, Lyon AR, Cook SA, Cowie MR, Assomull RG, Pennell DJ, Prasad SK. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA. 2013; 309:896-908. [DOI] [PubMed] [Google Scholar]
- Green JJ, Berger JS, Kramer CM, Salerno M. Prognostic value of late gadolinium enhancement in clinical outcomes for hypertrophic cardiomyopathy. JACC Cardiovasc Imaging. 2012; 5:370-377. [DOI] [PubMed] [Google Scholar]
- O'Hanlon R, Grasso A, Roughton M, Moon JC, Clark S, Wage R, Webb J, Kulkarni M, Dawson D, Sulaibeekh L, Chandrasekaran B, Bucciarelli‐Ducci C, Pasquale F, Cowie MR, McKenna WJ, Sheppard MN, Elliott PM, Pennell DJ, Prasad SK. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010; 56:867-874. [DOI] [PubMed] [Google Scholar]
- Rubinshtein R, Glockner JF, Ommen SR, Araoz PA, Ackerman MJ, Sorajja P, Bos JM, Tajik AJ, Valeti US, Nishimura RA, Gersh BJ. Characteristics and clinical significance of late gadolinium enhancement by contrast‐enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail. 2010; 3:51-58. [DOI] [PubMed] [Google Scholar]
- Kuruvilla S, Adenaw N, Katwal AB, Lipinski MJ, Kramer CM, Salerno M. Late gadolinium enhancement on cardiac magnetic resonance predicts adverse cardiovascular outcomes in nonischemic cardiomyopathy: a systematic review and meta‐analysis. Circ Cardiovasc Imaging. 2014; 7:250-258. [DOI] [PMC free article] [PubMed] [Google Scholar]