

Abstract
Background
Mammalian SWItch/Sucrose NonFermentable (SWI/SNF) adenosine triphosphate (ATP)‐dependent chromatin‐remodeling complexes play important roles in embryonic vascular development by modulating transcription of specific target genes. We sought to determine whether SWI/SNF complexes likewise impact postnatal physiological and pathological angiogenesis.
Methods and Results
Brahma‐related gene 1 (BRG1) and Brahma gene (BRM) are ATPases within mammalian SWI/SNF complexes and are essential for the complexes to function. Using mice with vascular‐specific mutations in Brg1 or with a global mutation in Brm, we employed 3 models to test the role of these ATPases in postnatal angiogenesis. We analyzed neonatal retinal angiogenesis, exercise‐induced angiogenesis in adult quadriceps muscles, and tumor angiogenesis in control and mutant animals. We found no evidence of defective angiogenesis in Brg1 or Brm mutants using these 3 models. Brg1/Brm double mutants likewise show no evidence of vascular defects in the neonatal retina or tumor angiogenesis models. However, 100% of Brg1/Brm‐double mutants in which Brg1 deletion is induced at postnatal day 3 (P3) die by P19 with hemorrhaging in the small intestine and heart.
Conclusions
Despite their important roles in embryonic vascular development, SWI/SNF chromatin‐remodeling complexes display a surprising lack of participation in the 3 models of postnatal angiogenesis we analyzed. However, these complexes are essential for maintaining vascular integrity in specific tissue beds before weaning. These findings highlight the temporal and spatial specificity of SWI/SNF activities in the vasculature and may indicate that other chromatin‐remodeling complexes play redundant or more essential roles during physiological and pathological postnatal vascular development.
Keywords: BRG1, BRM, exercise‐induced angiogenesis, retinal angiogenesis, tumor angiogenesis
Introduction
Mammalian SWItch/Sucrose NonFermentable (SWI/SNF) adenosine triphosphate (ATP)‐dependent chromatin remodeling complexes impact a number of developmental processes by modulating transcription of target genes.1–2 These multiprotein complexes utilize energy derived from ATP hydrolysis to transiently displace nucleosomes in gene‐regulatory regions and thereby impact the ability of large transcriptional machinery to access DNA.3–4 SWI/SNF chromatin‐remodeling complexes contain 1 of 2 catalytic ATPases, which are critical for their assembly and function: Brahma (Brm) and Brahma‐related gene 1 (Brg1). Genetic mutation of these ATPases in mice has revealed their disparate functions. Brm−/− mice are viable,5 but Brg1−/− embryos die at the peri‐implantation stage of development.6 Conditional inactivation of Brg1 in the early embryonic vasculature has revealed roles for this enzyme in various aspects of blood vessel development. Deletion of Brg1 with the endothelial and hematopoietic cell‐specific Tie2‐Cre transgenic (Tg) line has yielded information about roles for BRG1 in promoting yolk sac vascular Wnt signaling,7 embryonic and extraembryonic venous specification,8 and cardiac trabeculation.9 However, death of Brg1fl/fl;Tie2‐Cre embryos at embryonic day 10.5 (E10.5) from defective primitive erythropoiesis10 has prevented evaluation of the contribution of BRG1 to vascular development beyond midgestation.
Most vasculature is quiescent after birth, although some notable exceptions exist. For example, physiological angiogenesis occurs in the neonatal rodent retina,11 human female reproductive tract during ovulation and menstruation,12–13 uterus and placenta during pregnancy,14 and skeletal muscle after exercise.15 Physiological angiogenesis also occurs during bone growth,16 wound healing,17 and tissue repair.18 Pathological angiogenesis can occur in the diabetic retina,19 joints of patients with rheumatoid arthritis,20 skin of patients with psoriasis,21 and patients with hematological malignancies22 and solid tumors.23 Many of the same gene programs that contribute to embryonic vascular development are recapitulated during postnatal physio‐ and pathological angiogenesis. For example, vascular endothelial growth factor and Notch signaling impact embryonic vascular development as well as postnatal retinal and tumor angiogenesis.24–25 However, chromatin‐based mechanisms regulating these and other signaling pathways have been largely underexplored during postnatal vascular development. Specifically, it is unclear whether SWI/SNF chromatin‐remodeling complexes are required for postnatal angiogenesis under physiological or pathological conditions.
In order to determine whether SWI/SNF complexes impact postnatal angiogenesis, we assessed conventional, conditional, and inducible genetic mutants of Brg1 and Brm using 3 different models of postnatal angiogenesis. To our surprise, we found no evidence of a role for BRG1 or BRM during neonatal retinal vascular growth, exercise‐induced angiogenesis in quadriceps muscles of adult mice, or tumor angiogenesis in adult mice. However, infant mice deficient in both BRG1 and BRM die before weaning, with bleeding in the small intestine and heart. These findings illuminate the temporal specificity with which SWI/SNF complexes regulate vascular development and implicate other chromatin‐remodeling complexes in transcriptionally regulating the genes that are required to drive postnatal angiogenesis.
Methods
Mice
Brg1‐floxed mice (Brg1fl/fl),26 Brm null mice (Brm−/−),5 cVECad‐Cre Tg mice,27 Cdh5(PAC)‐CreERT2 Tg mice,28 and ROSA26RLacZ Tg mice29 were maintained on a mixed genetic background at the Oklahoma Medical Research Foundation (Oklahoma City, OK) animal facility. All animal use protocols were approved by the institutional animal care and use committee. Brg1‐floxed, Brm−/−, and ROSA26RLacZ mice were genotyped as previously described.7,10,29 cVECad‐Cre Tg mice were polymerase chain reaction (PCR) genotyped by amplifying a 300‐base‐pair (bp) fragment of the transgene using a gene‐specific forward primer (5′‐GCAGGCAGCTCACAAAGGAACAAT‐3′) and a Cre recombinase‐specific reverse primer (5′‐ATCACTCGTTGCATCGACCGGTAA‐3′). An additional gene‐specific reverse primer (5′‐TGTCCTTGCTGAGTGACAGTGGAA‐3′) was included in the reaction to amplify a 550‐bp internal control fragment. PCR was performed at an annealing temperature of 60°C. Cdh5(PAC)‐CreERT2 Tg mice were genotyped by amplifying a 473‐bp fragment of the transgene using a gene‐specific forward primer (5′‐TCCTGATGGTGCCTATCCTC‐3′) and a Cre‐specific reverse primer (5′‐CGAACCTGGTCGAAATCAGT‐3′). PCR was performed at an annealing temperature of 55°C. Cre‐mediated Brg1 excision was genotyped by amplifying a 300‐bp fragment using primers outside the 2 sets of loxP sites: forward (5′‐GATCAGCTCATGCCCTAAGG‐3′) and reverse (5′‐GCCTTGTCTCAAACTGATAAG‐3′). Positive control primers were used in the same reaction to amplify a 200‐bp fragment of the T‐cell receptor delta chain (Tcrd) gene: forward (5′‐CAAATGTTGCTTGTCTGGTG‐3′) and reverse (5′‐GTCAGTCGAGTGCACAGTTT‐3′). PCR was performed at an annealing temperature of 51°C.
For induction of Cdh5(PAC)‐CreERT2 in vivo to study exercise‐induced angiogenesis and Lewis lung carcinoma (LLC) tumor angiogenesis, adult mice at ≈6 weeks of age were injected intraperitoneally with 100 μL (10 mg/mL) of tamoxifen (Catalog No.: T5648; Sigma‐Aldrich, St. Louis, MO) every other day for a total of 5 injections. To induce Cdh5(PAC)‐CreERT2 for studying retinal angiogenesis, mouse pups were fed by placing a pipette tip on their tongues until they voluntarily ingested 2 μL (25 mg/mL) of tamoxifen. Animals were fed at postnatal day 3 (P3), P4, and P5. Tamoxifen was dissolved in a mixture of 95% peanut oil and 5% ethanol and sonicated as previously described,30 and all control and mutant animals were treated with tamoxifen for each study.
Immunohistochemistry
Tissues were fixed with 4% paraformaldehyde (PFA) overnight at 4°C and then transferred to 70% ethanol overnight at 4°C. Tissues were then paraffin embedded and sectioned on a microtome (8 μm). Sections were dewaxed and rehydrated, and then antigens were retrieved by boiling slides in sodium citrate buffer (10 mmol/L of sodium citrate and 0.05% Tween 20 [pH 6.0]) for 20 minutes. Once slides had cooled to room temperature, endogenous peroxidases were bleached with 3% H2O2 in methanol for 10 minutes at room temperature. Sections were washed with PBS and blocked using the Streptavidin/Biotin Blocking Kit (Catalog No.: SP‐2002; Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. Sections were washed with PBS and incubated with anti‐SNF2β/BRG1 (1:100, Catalog No.: 07‐478; Millipore, Billerica, MA) diluted in BRG1 staining buffer (1% normal goat serum, 0.1% Triton X‐100, and 1% BSA in 1× PBS) for 30 minutes at room temperature. Sections were washed with PBS and incubated with biotinylated goat anti‐rabbit immunoglobulin G (IgG; 1:500, Catalog No.: BA‐1000; Vector Laboratories) in BRG1 staining buffer for 10 minutes at room temperature. After sections were washed with PBS, VECTASTAIN Elite ABC reagent (Catalog No.: PK‐6100; Vector Laboratories) was added to sections and incubated for 5 minutes at room temperature. Sections were then washed with PBS, and 3,3′‐diaminobenzidine (DAB) Peroxidase (HRP) Substrate Kit solution without NiCl (Catalog No.: SK‐4100; Vector Laboratories) was applied for 5 minutes at room temperature. Sections were washed with water, counterstained with hematoxylin for 30 seconds, dehydrated with ethanol and xylenes, and mounted with Permount Mounting Medium (Catalog No.: SP15‐500; Fisher Scientific, Waltham, MA).
Hematoxylin and Eosin Staining
Tissues were dissected, immersion‐fixed in 4% PFA overnight, dehydrated, embedded in paraffin, sectioned (10 μm), and stained with hematoxylin (Catalog No.: 3536‐32; Ricca Chemical Co, Arlington, TX) and eosin (1%; Catalog No.: L088‐03; J.T. Baker, Center Valley, PA).
LacZ Staining
Hearts, quadriceps, and retinas were dissected and placed in Fix Solution (2% PFA and 0.2% glutaraldehyde in 1× PBS) for 6 hours. Tissues were washed in PBS for 30 minutes at room temperature to remove the Fix Solution and then placed in Staining Solution (5 mmol/L of potassium ferricyanide, 5 mmol/L of potassium ferrocyanide, 2 mmol/L of MgCl, 0.02% nonyl phenoxypolyethoxylethanol (NP‐40), 0.01% Na‐deoxycholate, 20 mmol/L of Tris [pH 7.4], and 1 mg/mL of X‐gal in PBS) for 2 days at room temperature protected from light. Tissues were then washed with PBS for 30 minutes at room temperature and post‐fixed in 4% PFA overnight at 4°C. Retinas were flat‐mounted and photographed at this point. After a brief wash in PBS, heart and quadriceps tissues were placed in 70% ethanol overnight at 4°C and then paraffin embedded. Paraffin blocks were sectioned (20 μm) and then counterstained with nuclear fast red.
Retina Studies
Whole eyes from P7 pups were enucleated and placed in 4% PFA for 20 minutes and then washed with PBS. Retina cups were dissected out and hyaloid vessels were carefully removed, as previously described.31 Retina cups were fixed again with 4% PFA for 1 hour at room temperature, then washed and placed in methanol for storage at −20°C. For isolectin staining, retinas were removed from methanol, rehydrated by washing in PBS, and blocked in Block/Permeabilization buffer (1% BSA and 0.5% Triton X‐100 in PBS) overnight at 4°C. Retinas were then washed 3 times in Pblec buffer (1 mmol/L of CaCl2, 1 mmol/L of MgCl2, 0.1 mmol/L of MnCl2, and 1% Triton X‐100 in PBS) and stained with isolectin B4 (IB4)/Alexa Fluor 488 (Catalog No.: I21411; Invitrogen, Grand Island, NY) overnight at 4°C. After staining, retinas were washed 3 times in wash buffer (PBS/0.3% Triton X‐100) at room temperature, then 2 times in PBS only, then flat mounted by cutting 4 radial slits in the retina, creating a flower petal arrangement. Retinas were mounted with DABCO (2.5% 1,4‐diazabicyclo[2.2.2]octane), and fluorescent images were taken.
For retina studies, all annotation was blinded, and fields were chosen at random throughout the entire retina. For measuring distance between the vascular front and the end of each retina, measurements were made using NIS‐Elements AR3.0 software (Nikon, Melville, NY), and 4 measurements were taken for each retina. For counting vascular branches, every branch was counted by hand in 10 fields for each retina, and values were averaged together. For quantifying vascular sprouting, every sprout was counted by hand in 10 fields for each retina. For measuring vascular branch width, NIS‐Elements AR3.0 software (Nikon) was used to measure 10 random branch widths in 10 fields for each retina. All values were then averaged together.
Exercise Studies
Eight‐week‐old male mice were used for all exercise studies. Exercised animals were housed in customized cages containing an exercise wheel (4.5 inch diameter, Catalog No.: 610‐0003‐00; Mini Mitter, Bend, OR) and a probe and digital activity counter that recorded wheel rotations (Catalog No.: 199‐0048‐00, ResKit Magnetic Switch Custom; Catalog No.: 130‐0023‐00, Digital Activity Counter; Mini Mitter). Before the experiment, animals were allowed to habituate to the cages for 3 days, with the wheel locked in a stationary position. After habituation, the wheel was released, and animals were allowed to run voluntarily for 3 weeks. Wheel rotations were recorded daily during the exercise period (0.36 m/wheel revolution). For studies with Brg1fl/fl;Cdh5(PAC)‐CreERT2 animals, control and mutant male mice received 5 tamoxifen injections (every other day) beginning at 6 weeks of age followed by 3 days of habituation and 3 weeks of exercise. Sedentary animals were induced comparably to exercised animals, but were housed in cages without wheels for the duration of the study.
Immediately after animals were euthanized, quadriceps muscles were removed from exercised and sedentary mice. Muscles were washed with PBS and immediately placed in 30% sucrose/PBS overnight at 4°C. Quadriceps muscles were then cryoembedded, and transverse sections (8 μm) were cut from proximal, medial, and distal regions of the total muscle. Sections were fixed in 4% PFA for 10 minutes at room temperature and blocked using the Streptavidin/Biotin Blocking Kit (Vector Laboratories), according to the manufacturer's instructions. Sections were washed with wash buffer (PBS/0.1% Triton X‐100) and blocked with 3% normal goat serum, 0.3% Triton X‐100, and 3% BSA in 1× PBS for 1 hour at room temperature. Sections were then incubated with anti‐platelet endothelial cell adhesion molecule 1 (PECAM‐1; 1:100; Catalog No.: 557355; BD Biosciences, San Jose, CA) diluted in PECAM staining buffer (1% normal goat serum, 0.1% Triton X‐100, and 1% BSA in 1× PBS) overnight at 4°C. After washing with wash buffer, sections were stained with Biotinylated Goat Anti‐Rat IgG (1:500; Catalog No.: BA‐9400; Vector Laboratories) secondary antibody for 30 minutes at room temperature protected from light. Sections were washed with wash buffer and then stained with DyLight 594‐Streptavidin (1:500; Catalog No.: SA‐5594‐1; Vector Laboratories) for 5 minutes at room temperature protected from light. After another wash, sections were again blocked with the Streptavidin/Biotin Blocking Kit, according to the manufacturer's instructions, and then blocked with 3% normal donkey serum, 0.3% Triton X‐100, and 3% BSA in 1× PBS for 1 hour at room temperature protected from light. Sections were stained with anti‐laminin antibody (1:500; Catalog No.: ab14055; Abcam, Cambridge, MA) diluted in laminin staining buffer (1% normal donkey serum, 0.1% Triton X‐100, and 1% BSA in 1× PBS) for 30 minutes at room temperature protected from light. After washing, sections were stained with Donkey Anti‐Chicken DyLight 488 antibody (1:500; Catalog No.: 703‐485‐155; Jackson ImmunoResearch, West Grove, PA) for 30 minutes at room temperature protected from light. Slides were then washed with wash buffer, washed again in PBS to remove the Triton X‐100, mounted with DABCO (in 9:1 glycerol/PBS [pH 8.6]), and fluorescent images were taken immediately. For exercise studies, all annotation was blinded. Ten random fields were chosen for analysis in the center of each muscle section, where the vasculature is most receptive to exercise‐induced angiogenesis.32 Muscle fibers and blood vessels in each section were counted by hand. Sections from the proximal, medial, and distal regions of the quadriceps muscle from each animal were stained independently, and values were averaged together.
Lewis Lung Carcinoma Tumor Studies
LLC (Catalog No.: CRL‐1642; ATCC, Manassas, VA) cells were maintained in DMEM containing 1500 mg/L of sodium bicarbonate (Catalog No.: 30‐2002; ATCC) supplemented with 10% FBS and 1% antibiotic‐antimycotic (Catalog No.: 15240‐062; Life Technologies, Grand Island, NY). For injections, cells were diluted in HBSS at a concentration of 1×106 cells/mL. Next, 100 μL of LLC cells were injected into the shaved flank of 8‐week‐old female mice. Tumor size was monitored by electronic calipers every other day for 3 weeks or until tumor size reached 2 cm. Tumors were then dissected out of animals and weighed. Tumors were fixed with 4% PFA overnight at 4°C, then cryoembedded and sectioned (8 μm). Sections were washed with PBS and placed in blocking buffer (3% normal donkey serum, 0.3% Triton X‐100, and 3% BSA in 1× PBS) for 2 hours at room temperature. Sections were then incubated with anti‐PECAM‐1 (1:100; BD Biosciences) diluted in PECAM staining buffer (1% normal goat serum, 0.1% Triton X‐100, and 1% BSA in 1× PBS) overnight at 4°C. Sections were washed with wash buffer (PBS/0.1% Triton X‐100), then stained with Donkey anti‐Rat Cy3 (1:500; Catalog No.: 712‐165‐153; Jackson ImmunoResearch) diluted in PECAM staining buffer for 1 hour at room temperature protected from light. Sections were washed with wash buffer, washed in PBS to remove the Triton X‐100, mounted with DABCO, and fluorescent images were taken.
For fluorescein isothiocyanate (FITC)/dextran injections, 100 μL of lysine fixable FITC/dextran (2000 kDa; Catalog No.: D‐7137; Life Technologies) at a concentration of 2 mg/mL in filtered PBS was injected into the tail vein of Brm−/−;Brg1fl/fl (control) and Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 (double‐mutant) LLC mice after 3 weeks or once tumors reached 2 cm in size. After 10 minutes, the animal was sacrificed and the tumor was immediately removed and placed in 4% PFA overnight at 4°C. Fixed tumors were then cryoembedded, sectioned (8 μm), and costained with anti‐PECAM‐1, as described above, for fluorescent imaging.
Fluorescent Imaging
Fluorescent images were obtained with a Nikon Eclipse 80i microscope, an X‐cite 120Q light source, and a Nikon DS‐Qi1Mc camera. NIS‐Elements AR3.0 (Nikon) software was used for all fluorescent image acquisition and annotation.
Statistical Analysis
All graphs were generated and statistical analyses were performed using GraphPad Prism software (6; GraphPad Software Inc., La Jolla, CA). Individual study designs and sample sizes are described in the figure legends. Data are presented as the mean±SEM. All statistics were generated using an unpaired, 2‐tailed Student t test. A probability of P<0.05 was considered to be statistically significant.
Results
Postnatal Vascular Brg1 Excision
Given that deletion of Brg1 with an endothelial Tie2‐Cre Tg line results in embryonic lethality at midgestation,9–10 we used 2 alternative Cre recombinase lines to bypass this lethality and assess the role of BRG1 in postnatal vascular development. First, we used a constitutive VE‐Cadherin‐Cre line (cVECad‐Cre), which begins to be expressed in embryonic endothelial cells (ECs) at E8.5 and reaches full penetrance in embryonic arteries, veins, capillaries, and lymphatic ECs by E14.5.27 This line is also active in postnatal vasculature in a variety of tissues, including the retina.27 In order to assess the efficiency of this Cre line for excising Brg1 in ECs, we immunostained sections of control (Brg1fl/fl) and mutant (Brg1fl/fl;cVECad‐Cre) E15.5 jugular veins for BRG1. We found a modest number of ECs with diminished or depleted BRG1 staining using this technique (Figure 1A, top), indicating that the cVECad‐Cre line excised BRG1 with mixed efficiency in embryonic ECs. We also evaluated Brg1 excision with this Cre line in a postnatal setting by staining for BRG1 in LLC tumors. We found robust EC excision of Brg1 in this model (Figure 1A, bottom).
Figure 1.

Validation of Brg1 excision using vascular‐specific cVECad‐Cre and Cdh5(PAC)‐CreERT2 transgenes. A, Immunohistochemistry of E15.5 jugular veins (JV; top images) from littermate control (Brg1fl/fl) and mutant (Brg1fl/fl;cVECad‐Cre) embryos immunostained for BRG1 (brown) and counterstained with hematoxylin (blue). Bottom images show BRG1 expression in blood vessels of control (Brg1fl/fl) and mutant (Brg1fl/fl;cVECad‐Cre) adult LLC tumors. Asterisk (*) highlights endothelial cells with reduced BRG1 expression. Scale bar=50 μm. B, Tissue samples of heart, quadriceps, and retina from ROSA26RLacZ control animals or ROSA26RLacZ;Cdh5(PAC)‐CreERT2 mutants were whole‐mount X‐gal stained (blue) to detect Cre activity and sectioned. Heart and quadriceps tissues were counterstained with nuclear fast red. Scale bar=50 μm. C, BRG1 expression (brown) in blood vessels of control (Brg1fl/fl) and mutant (Brg1flfl;Cdh5(PAC)‐CreERT2) adult LLC tumors. Sections were counterstained with hematoxylin (blue). Arrows denote endothelial cells with BRG1 expression; asterisks highlight endothelial cells with reduced BRG1 expression. Scale bar=50 μm. D, PCR genotyping of DNA isolated from tail clips of a litter of P7 pups (lanes 1 to 11) generated from a cross between a Brg1fl/fl female and a Brg1fl/fl;Cdh5(PAC)‐CreERT2 male. Top image shows PCR products using primers to detect Cdh5(PAC)‐CreERT2, and bottom image shows PCR products using primers to detect a Brg1fl/fl excision event (Brg1Δ, top band) and an internal control gene to validate sample quality (Tcrd gene; bottom band). Tissue from a Brg1fl/+;Cdh5(PAC)‐CreERT2 animal and sample lysis buffer are included as positive and negative controls, respectively. E, PCR on DNA isolated from various tissues of a 14‐week‐old Brg1fl/fl;Cdh5(PAC)‐CreERT2 female. Top image shows PCR products using primers to detect Cdh5(PAC)‐CreERT2, and bottom image shows PCR products using primers detecting a Brg1fl/fl excision event (Brg1Δ, top band) and an internal control gene to validate sample quality (Tcrd gene; bottom band). Tissue was collected from ear, heart, quadriceps, and whole eye as indicated. Tissue from a Brg1fl/+;Cdh5(PAC)‐CreERT2 animal and sample lysis buffer are included as positive and negative controls, respectively. Brg1 indicates Brahma‐related gene 1; LLC, Lewis lung carcinoma; PCR, polymerase chain reaction; Tcrd, T‐cell receptor delta chain gene.
As an alternative to the cVECad‐Cre line, we also employed a tamoxifen‐inducible Cdh5(PAC)‐CreERT2 line to excise Brg1 in postnatal vasculature. This line has been used extensively for deleting genes in postnatal ECs.28,33–37 In our hands, reporter analysis with this line confirmed efficient Cre activity in blood vessels within the heart, quadriceps, and adult retina (Figure 1B). To confirm that Brg1 could be excised with this line, we stained for BRG1 in LLC tumors from control and Brg1fl/fl;Cdh5(PAC)‐CreERT2 mice that had been induced with tamoxifen before tumor cell implantation at 8 weeks of age. As with the cVECad‐Cre line, we found robust excision of Brg1 in ECs from tumors in Brg1fl/fl;Cdh5(PAC)‐CreERT2 mice (Figure 1C). To confirm that Brg1 could be excised with this Cre line in young pups, we fed tamoxifen to a litter of control (Brg1fl/fl) and mutant (Brg1fl/fl;Cdh5(PAC)‐CreERT2) animals at P3, P4, and P5 and performed PCR for a Brg1 deletion product on tail clips harvested at P7. All of the animals carrying the Cre showed the Brg1 deletion product, and none of the Cre‐negative pups showed the deletion product (Figure 1D). In addition, we assessed Brg1 excision in various tissues harvested from a 14‐week‐old Brg1fl/fl;Cdh5(PAC)‐CreERT2 animal that had been induced with tamoxifen at 4 weeks of age. PCR analysis again showed Brg1 excision in all the tissues we analyzed (Figure 1E). Therefore, the Cdh5(PAC)‐CreERT2 line is capable of excising Brg1 in pups and mature mice after tamoxifen treatment.
Vascular BRG1 and BRM Are Not Required for Neonatal Retinal Angiogenesis
With 2 different methods of achieving postnatal vascular Brg1 excision in hand, we proceeded to study the effects of SWI/SNF ATPases on retinal vascular development. From birth until P7, blood vessels gradually extend in a two‐dimensional sheet from the optic nerve toward the periphery in mice.31 Because these vessels can be easily visualized by staining with isolectin B4 (IB4) and flat mounting, the retina is an excellent model for studying neonatal vascular development. We compared various parameters of retinal vascular development in 4 cohorts of control and mutant animals at P7. First, we compared retinal vascular development in Brg1fl/fl (control) and Brg1fl/fl;cVECad‐Cre (mutant) littermates. Second, we compared retinal vasculature in Brg1fl/fl (control) and Brg1fl/fl;Cdh5(PAC)‐CreERT2 (mutant) littermates after tamoxifen induction at P3, P4, and P5. Third, we compared wild‐type (WT) and Brm−/− littermates, and, finally, we compared Brm−/−;Brg1fl/fl (control) and Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 (mutant) littermates after tamoxifen induction at P3 to P5. Using these 4 cohorts of control and mutant genotypes, we made 4 separate measurements on a single retina from each animal analyzed (8 to 11 animals per genotype). First, we measured the distance from the edge of the vascular front to the edge of the retina, as a general readout of the rate of vascular growth (Figure 2A). Second, we counted vascular branches in 10 separate fields from each retina analyzed (Figure 2B). Third, we counted vascular sprouts in 10 separate fields from each retina (Figure 2C). And, finally, we measured the width of 10 random vessels in 10 separate fields from each retina (Figure 2D). We found no significant differences in any of the retinal vascular measurements we made, with the exception of a slight decrease in vascular branching in Brg1fl/fl;Cdh5(PAC)‐CreERT2 retinas, compared to littermate controls, and a slight increase in Brm−/− vascular branches, compared to littermate controls (Figure 2B). These small differences in vascular branching were neutralized in Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 double mutants (Figure 2B). Overall, the morphological effects of Brg1 and Brm deletion in retinal vasculature were unremarkable, indicating that SWI/SNF‐mediated chromatin remodeling does not significantly impact transcription of genes required for neonatal retinal vascular development.
Figure 2.

SWI/SNF mutants do not display major vascular defects in the neonatal retina. Retinas from postnatal day 7 (P7) pups were stained with isolectin B4 and flat mounted to visualize the vasculature. A, Distance (μm) was measured from the edge of the vascular front to the end of the retina (white bar). Four measurements were made for each retina. B, Retina vascular branches (white dots) were counted in 10 fields for each retina. C, Vascular sprouts (white circle) were counted in 10 fields for each retina. D, Branch widths were measured from 10 vessels in 10 different fields for each retina. For A through D, pictures show representative measurement criteria. Data represent averages±SEM from 8 to 11 animals for each genotype. *P<0.05; Student t test. Scale bars=500 μm (A); 50 μm (B through D). Brg1 indicates Brahma‐related gene 1; Brm, Brahma; SWI/SNF, SWItch/Sucrose NonFermentable.
Vascular Brg1/Brm Double Mutants Die With Multiorgan Hemorrhage
While generating SWI/SNF mutants for analysis of postnatal angiogenesis in adult quadriceps and tumors, we discovered that no Brg1/Brm double mutants survived to weaning. We analyzed 101 pups from 13 litters born to Brm−/−;Brg1fl/+;cVECad‐Cre fathers and Brm−/−;Brg1fl/fl mothers. Although we expected ≈25 Brm−/−;Brg1fl/fl;cVECad‐Cre+ double mutants to be produced from these litters, we did not detect any double mutants at weaning (Table 1). Further analysis indicated that these animals die before birth. Similarly, we noticed that no Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 pups induced with tamoxifen at P3 to P5 survived past weaning, although these double mutants were indistinguishable from their control littermates at P7 when we analyzed their retinal vasculature. Upon closer examination, we found that Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 animals died between P13 and P19. The most striking phenotype associated with these double mutants, and the likely cause of their death, was massive hemorrhage that occurred in the small intestine. We saw blood filling the lamina propria of the ileum of all dead and dying double mutants that we evaluated (Figure 3A). We also detected hemorrhage in the hearts of dead and dying double‐mutant pups (Figure 3B), although the bleeding in this tissue was not as extensive as in the small intestine. Interestingly, we did not detect hemorrhage in the kidneys, livers, or lungs of dead and dying Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 pups, highlighting the tissue specificity of this vascular integrity defect.
Table 1.
Live Offspring at Weaning (From Brm−/−;Brg1fl/+;cVECad‐Cre+×Brm−/−;Brg1fl/fl Matings)
| Genotype | No. of Observed* | No. of Expected |
|---|---|---|
| Brm −/− ;Brg1 fl/+ | 42 | 25 |
| Brm −/− ;Brg1 fl/fl | 30 | 25 |
| Brm −/− ;Brg1 fl/+ ;cVECad‐Cre + | 29 | 25 |
| Brm −/− ;Brg1 fl/fl ;cVECad‐Cre + | 0 | 25 |
Vascular Brg1/Brm double mutants do not survive until weaning. Thirteen litters of mice generated from crosses between Brm−/−;Brg1fl/+;cVECad‐Cre+ males and Brm−/−;Brg1fl/fl females were genotyped 18 to 20 days after birth. No Brg1/Brm double mutants were genotyped at this age [χ2(3dof): P<0.001]. Brg1 indicates Brahma‐related gene 1; Brm, Brahma.
P<0.001.
Figure 3.
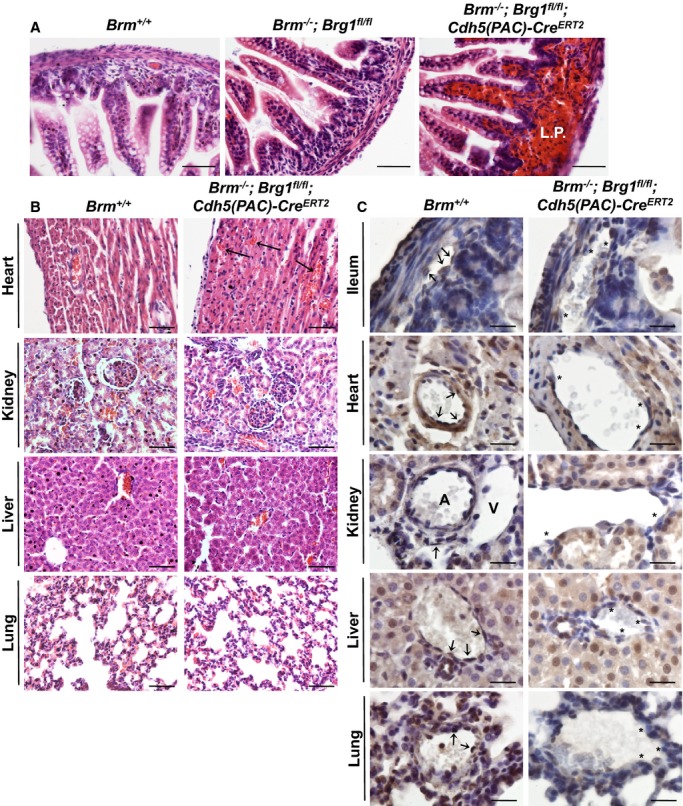
Characterization of Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 infant mice. A, P14 Brm+/+ (control), Brm−/−;Brg1fl/fl (control), and Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 (double‐mutant) ilea were hematoxylin and eosin stained. Hemorrhage is seen in the lamina propria (L.P.) of the double‐mutant ileum. B, Control and double‐mutant tissues from postnatal day 14 (P14) pups were stained with hematoxylin and eosin. Top images show hemorrhage in the intramuscular tissue of the double‐mutant heart (arrows). No hemorrhage was detected in double‐mutant kidney, liver, or lung tissues. C, Control and double‐mutant tissues from P14 pups were immunostained for BRG1 (brown) and counterstained with hematoxylin (blue). Arrows point to endothelial cells that express BRG1. Asterisks highlight endothelial cells where BRG1 expression is diminished. Scale bars=50 μm (A and B); 100 μm (C). A indicates artery; Brg1, Brahma‐related gene 1; Brm, Brahma; V, vein.
To determine whether the tissue‐specific hemorrhage we saw in Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 pups reflected differing levels of BRG1 expression or excision in infant mice, we immunostained various tissues from P14 control and Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 pups that had been induced for Cre excision at P3 to P5 (Figure 3C). We detected endothelial BRG1 expression (arrows) in all the WT tissues we examined, although expression appeared more uniform within ECs of the ileum than in the other tissues. The kidney displayed particularly limited BRG1 endothelial expression patterns, with negligible staining within arterial ECs. We detected ECs with diminished BRG1 expression in all the Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 tissues we examined, although the mosaic pattern of endothelial BRG1 expression in WT tissues makes it difficult to determine whether excision efficiency is equivalent in all tissues.
Willis et al. previously reported that Brg1/Brm double mutants in which Brg1 is deleted with an inducible Mx1‐Cre die within 1 month of Cre induction owing to EC death, leading to vascular leakage and cardiac hemorrhage.38 The Mx1‐Cre induction in that study was performed on 5‐ to 7‐week‐old mice, and death occurred within 1 month of Cre induction. When we induced Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 animals at 5 to 7 weeks of age, we saw low penetrance death in our double‐mutant animals (19%), despite the 100% lethality that occurred when we induced these mutants at P3 to P5. Therefore, although our data support the Willis et al. finding that Brg1/Brm double deficiency causes vascular integrity defects, we saw variable timing and tissue specificity associated with these defects on the Cdh5(PAC)‐CreERT2 line. In addition, we saw no evidence of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) or activated caspase‐3 staining in P14 Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 ileum or heart ECs, indicating that the vascular integrity defects in our mutants are not associated with EC apoptosis.
Vascular BRG1 and BRM Do Not Individually Contribute to Exercise‐Induced Angiogenesis in Adult Quadriceps
Exercise is a powerful physiological trigger for angiogenesis in adult animals.39 The quadriceps femoris muscle is a convenient model for studying exercise‐induced angiogenesis in mice because it can undergo up to a 2‐fold increase in capillary density after voluntary wheel running.32 In order to determine whether SWI/SNF chromatin remodeling influences this physiological angiogenesis process, we examined 3 groups of age‐matched, male control and mutant animals: (1) Brg1fl/fl (control) and Brg1fl/fl;cVECad‐Cre (mutant); (2) Brg1fl/fl (control) and Brg1fl/fl;Cdh5(PAC)‐CreERT2 (mutant); and (3) WT and Brm−/−. In each case, 8‐week‐old male mice that were slated for exercise were habituated in a cage with a locked exercise wheel for 3 days. After 3 days, the wheel was unlocked and animals were allowed to run voluntarily for 3 weeks. Wheels were attached to an external activity monitor, which recorded the total distance run each night. No significant differences in distances run were observed between the control and mutant genotypes within each cohort (Figure 4A through 4C, left panels). Age‐matched sedentary animals of each genotype were housed in cages without running wheels for the course of the exercise study to provide a baseline for assessing angiogenesis. Four to 6 sedentary and exercised animals were assessed for each control and mutant genotype analyzed. For the studies involving animals carrying the Cdh5(PAC)‐CreERT2 transgene, control and mutant sedentary and exercised mice were induced with tamoxifen every other day for 5 injections starting at 6 weeks. To assess angiogenesis, quadriceps muscles were harvested from euthanized mice after 3 weeks of sedentary or running activity. Transverse muscle sections were stained with antibodies against PECAM‐1 for visualizing blood vessels and against laminin for outlining muscle fibers (Figure 4A through 4C, middle panels). Sections from proximal, medial, and distal regions of the quadriceps were stained from each animal, and vessels and fibers were counted in 10 fields from each stained section (Figure 4A through 4C, right panels). We saw significantly more vessels per fiber for exercised versus sedentary animals, indicating that exercise‐induced angiogenesis occurred in all genotypes. However, we saw no significant differences in the magnitude of the angiogenic response between control and mutant animals for exercised animals in the 3 different cohorts. Therefore, vascular BRG1 and BRM appear not to be required—at least independently—for exercise‐induced angiogenesis.
Figure 4.

BRG1 and BRM are not essential for exercise‐induced angiogenesis. A through C, left panels: Total distance traveled in kilometers by adult mice after 3 weeks of voluntary exercise. Data represent averages±SEM of 4 to 6 animals per genotype. A through C, middle panels: Representative images of stained quadriceps from sedentary and exercised control and SWI/SNF mutant mice. Quadriceps were stained with PECAM‐1 (red) to visualize blood vessels and laminin (green) to outline muscle fibers (insert). Scale bars=50 μm. A through C, right panels: Quantification of immunostaining experiments represented at left. Tissue from each animal was stained 3 separate times, and vessels and muscle fibers from 10 fields were counted for each staining experiment. Data represent averages±SEM of blood vessels per muscle fiber from 4 to 6 animals, normalized to sedentary controls. *P<0.05; Student t test. Brg1 indicates Brahma‐related gene 1; Brm, Brahma; N.S., not significant; PECAM‐1, platelet endothelial cell adhesion molecule 1; SWI/SNF, SWItch/Sucrose NonFermentable.
Vascular BRG1 and BRM Are Not Required for LLC Tumor Angiogenesis and Growth
To assess the role of SWI/SNF ATPases in a pathological angiogenesis context, we examined tumor‐induced angiogenesis in Brg1 and Brm mutant animals using the LLC model. We compared 4 groups of control and mutant mice: (1) Brg1fl/fl (control) and Brg1fl/fl;cVECad‐Cre (mutant); (2) Brg1fl/fl (control) and Brg1fl/fl;Cdh5(PAC)‐CreERT2 (mutant); (3) WT and Brm−/−; and (4) Brm−/−;Brg1fl/fl (control) and Brm−/−;Brg1fl/fl;Cdh5(PAC)‐CreERT2 (double mutant). For the studies with animals carrying the Cdh5(PAC)‐CreERT2 transgene, control and mutant mice were induced with tamoxifen every other day for 5 injections starting at 6 weeks of age. In all mice, LLC cells were subcutaneously injected into the flank of 8‐ to 10‐week‐old female mice and were allowed to grow until tumors reached a diameter of ≈2 cm. At this time, tumors were harvested, weighed, and embedded for sectioning. Tumor sections were stained with antibodies against PECAM‐1 to visualize vasculature and 4′,6‐diamidino‐2‐phenylindole (DAPI) to visualize cell nuclei. Eleven to 21 animals from each genotype were analyzed. We saw no quantitative differences in tumor growth rates between any of the sets of mutant and control animals (Figure 5, left panels). Likewise, we saw no qualitative differences in vascular density or morphology in any of the tumor sections (Figure 5, right panels). In addition, Brm/Brg1 double‐mutant mice that were intravenously injected with FITC/dextran before being sacrificed, showed no evidence of vascular leakage (Figure 5, bottom right panel). Therefore, vascular BRG1 and BRM do not play significant individual or redundant roles in LLC tumor growth and angiogenesis.
Figure 5.
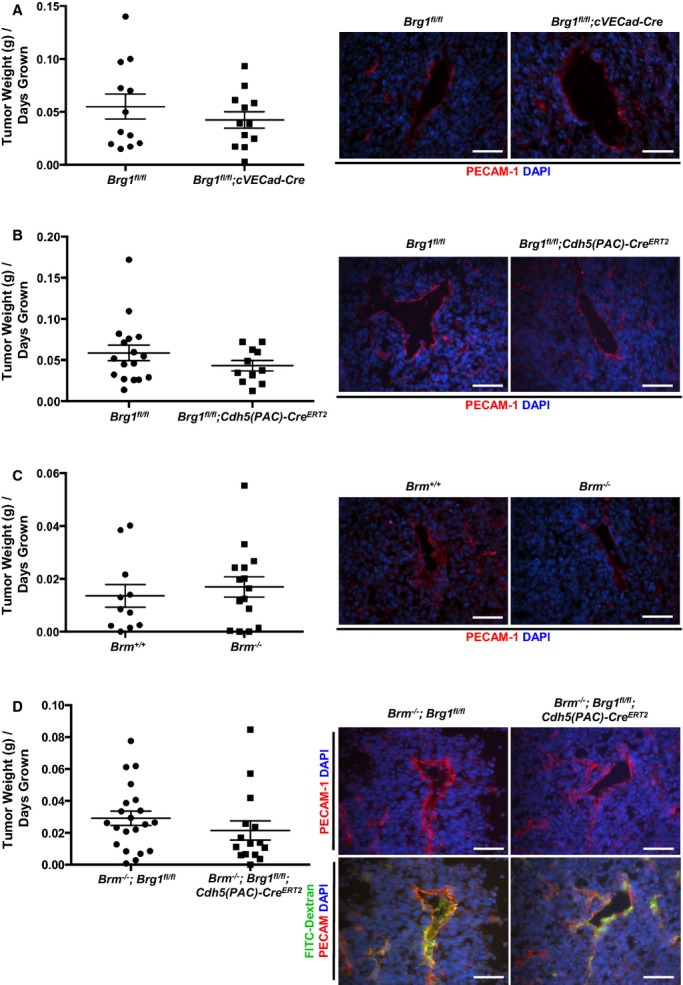
Lewis lung carcinoma (LLC) tumor growth and vascularization are comparable in control and SWI/SNF mutant animals. A through D, LLC cells were subcutaneously injected into the flank of 8‐ to 10‐week‐old female mice, and tumor growth was monitored every other day until tumors reached a diameter of ≈2 cm, at which time animals were sacrificed and tumors were weighed. Average tumor size (grams) per days grown for 11 to 21 animals from each genotype are shown on the left. Representative images of tumor sections stained for PECAM‐1 (red) for visualization of tumor vasculature are shown on the right. Nuclei are stained with DAPI (blue). Control and Brm/Brg1 double‐mutant animals were tail‐vein injected with FITC/dextran (2000 kDa, green; bottom panel of D) before animals were sacrificed to assess tumor vessel integrity. Scale bars=50 μm. Brg1 indicates Brahma‐related gene 1; Brm, Brahma; DAPI, 4′,6‐diamidino‐2‐phenylindole; FITC, fluorescein isothiocyanate; SWI/SNF, SWItch/Sucrose NonFermentable.
Discussion
BRG1 impacts angiogenesis in the midgestation embryo by promoting Wnt signaling and venous specification.7–8 We predicted that BRG1 would likewise perform these and other functions during postnatal vascular growth. This prediction was based, in part, on the finding that BRG1 can recapitulate its developmental cardiac gene regulatory roles in a postnatal pathological context.40 During embryonic development, BRG1 helps to promote transcription of the embryonic form of myosin heavy chain (β‐MHC) and repress transcription of the adult form (α‐MHC). Because BRG1 expression is down‐regulated after birth, myocardial cells switch their MHC production from the embryonic to the adult form. However, when the adult heart is stressed, BRG1 expression is re‐elevated and once again represses α‐MHC and activates β‐MHC, as it did in the embryo, thereby contributing to hypertrophic cardiomyopathy. In the current study, we used mice with Brg1 deleted from vascular ECs (VECs) to look for similarly repetitive roles for BRG1 in embryonic and postnatal vascular development. However, we did not find evidence that BRG1 is essential for promoting vascular development in the neonatal retina, exercise‐stimulated quadriceps, and solid tumors (summarized in Table 2).
Table 2.
Summary of Genotypes and Postnatal Angiogenesis Phenotypes
| Brm −/− | Brg1 fl/fl ;cVECad‐Cre | Brg1 fl/fl ;Cdh5(PAC)‐Cre ERT2 | Brm −/− ;Brg1 fl/fl ;Cdh5(PAC)‐Cre ERT2 * * | |
|---|---|---|---|---|
| Retinas* | Small increase in vascular branching | Small decrease in vascular branching | No phenotype | No phenotype |
| Exercised quadriceps* | No phenotype | No phenotype | No phenotype | Not assessed |
| LLC tumors* | No phenotype | No phenotype | No phenotype | No phenotype |
Brg1 indicates Brahma‐related gene 1; Brm, Brahma; LLC, Lewis lung carcinoma.
Infants induced at postnatal day 3 (P3) to P5 die between P13 and P19 with gut and heart hemorrhage.
Adults induced at 6 weeks have low penetrance lethality (19%).
Induced at P3 to P5 for Cdh5(PAC)‐CreERT2 animals; analyzed at P7 for all genotypes.
Induced at 6 weeks for Cdh5(PAC)‐CreERT2 animals; analyzed at 11 weeks for all genotypes.
The reason for this discrepancy between embryonic and postnatal roles for BRG1 in developing vasculature is unclear. It is possible that the 2 endothelial Cre lines we used did not excise Brg1 sufficiently to elicit phenotypes in the tissues we studied. The mosaic pattern of BRG1 expression in postnatal endothelium and its widespread expression in other cell types makes it difficult to quantify endothelial Brg1 excision efficiently. However, the lethality that we observe when Brg1 is excised with either cVECad‐Cre or Cdh5(PAC)‐CreERT2 on a Brm−/− background indicates that both of these established Cre lines are capable of excising Brg1 sufficiently to cause consistent phenotypes in certain contexts. Alternatively, we may not see roles for BRG1 in our postnatal angiogenesis models if BRG1 does not promote the same genes in postnatal retinal or tumor vasculature that it does in embryonic vasculature. We previously showed that BRG1 directly promotes expression of several Frizzled (Fzd) receptors in the yolk sac vasculature, including Fzd4, Fzd5, and Fzd8.7 Deletion of Fzd4 in ECs yields postnatal retinal vascular abnormalities, such as absence of intraretinal capillaries, dilated vessels forming arteriovenous anastomoses, and intraocular hemorrhages.41 However, we did not see any such phenotype in the retinas of our postnatal vascular Brg1 mutants. Therefore, it is possible that BRG1 does not regulate transcription of Fzd4 in postnatal retinal ECs or that the level of reduction of Fzd4 in our postnatal Brg1 mutants is not dramatic enough to yield retinal vascular phenotypes. Likewise, we previously showed that BRG1 promotes expression of the nuclear receptor, Coup‐TFII, which drives venous specification.8 Postnatal global deletion of Coup‐TFII reduces tumor angiogenesis and tumor growth in multiple mouse tumor models, including the LLC tumor model.42 Therefore, our lack of tumor angiogenesis phenotypes in Brg1 vascular mutants using the LLC model may indicate that BRG1 does not promote Coup‐TFII in tumor vasculature. On the other hand, it is possible that Coup‐TFII expression is not essential in tumor ECs to promote tumor angiogenesis, but is more important in tumor stromal cells for the production of the angiogenic growth factor, angiopoietin‐1.42
Simultaneous deletion of Brg1 and Brm did not yield retinal or tumor angiogenesis defects either, indicating that the enzymes do not play necessary and redundant roles in these angiogenic processes. However, we saw striking hemorrhage in the small intestine and hearts of Brm/Brg1 double‐mutant infant mice, which correlated with lethality. We found BRG1 to be expressed in ECs from a variety of tissues in P14 mice, although its expression was typically mosaic within ECs of a single vessel. However, BRG1 appeared to be expressed more consistently in ECs of the ileum, where we saw the most profound bleeding. We do not have a specific antibody to assess BRM staining in mouse tissues, but our phenotypic data indicate that BRM is likewise expressed in ileum and heart vascular endothelium, where it plays a critical role, together with BRG1, in maintaining vascular integrity in infant mice. The temporal and spatial specificity of this role for BRG1 and BRM in maintaining vascular integrity is consistent with similarly specific roles attributed to SWI/SNF complexes in embryonic vasculature. For example, we found that yolk sac vascular Wnt signaling is more impacted by Brg1 deletion at midgestation than embryonic vascular Wnt signaling.7 Such specificity may be impacted by redundant roles of other ATP‐dependent chromatin remodelers or by tissue‐specific expression of cofactors required for SWI/SNF recruitment and activity at target genes.43
We acknowledge the possibility that the 3 models of postnatal vascular growth we analyzed—physiological angiogenesis in the neonatal retina, exercise‐induced angiogenesis in the adult quadriceps, and tumor angiogenesis in an LLC tumor model—may not require SWI/SNF ATPases, but that BRG1 and BRM may play critical roles in other postnatal angiogenesis contexts. Anecdotally, we know that BRG1 is not essential for angiogenesis associated with pregnancy, given that Brg1fl/fl;cVECad‐Cre females are able to give birth to normal‐sized litters. However, if SWI/SNF complexes do impact angiogenesis in alternative contexts, such as wound healing or retinopathy, it would be consistent with their spatially specific actions in embryonic vasculature at midgestation.
Altogether, the current work provides surprising evidence that SWI/SNF chromatin‐remodeling complexes do not play critical roles in multiple models of postnatal physio‐ and pathological angiogenesis. However, the complexes do play a critical role in maintaining vascular integrity in ileum and heart of infant mice. This extends our understanding of the temporal and spatial specificity these remodeling complexes utilize in choosing their target genes. Future investigations will be focused on investigating other chromatin‐remodeling complexes or combinations of complexes that may regulate fundamental genes and signaling pathways involved in postnatal vascular development.
Acknowledgments
The authors thank Matthew Ward and Stefano Tarantini for their assistance with the exercise‐induced angiogenesis studies.
Sources of Funding
This work was supported by grants from the National Heart Lung and Blood Institute (R01HL111178) and the National Institute of General Medical Sciences (P20GM103441).
Disclosures
None.
References
- Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010; 463:474-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CS, Wagner D. Unwinding chromatin for development and growth: a few genes at a time. Trends Genet. 2007; 23:403-412. [DOI] [PubMed] [Google Scholar]
- Liu N, Balliano A, Hayes JJ. Mechanism(s) of SWI/SNF‐induced nucleosome mobilization. Chembiochem. 2011; 12:196-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009; 78:273-304. [DOI] [PubMed] [Google Scholar]
- Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha). EMBO J. 1998; 17:6979-6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G, Magnuson T. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000; 6:1287-1295. [DOI] [PubMed] [Google Scholar]
- Griffin CT, Curtis CD, Davis RB, Muthukumar V, Magnuson T. The chromatin‐remodeling enzyme BRG1 modulates vascular Wnt signaling at two levels. Proc Natl Acad Sci USA. 2011; 108:2282-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RB, Curtis CD, Griffin CT. BRG1 promotes COUP‐TFII expression and venous specification during embryonic vascular development. Development. 2013; 140:1272-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankunas K, Hang CT, Tsun ZY, Chen H, Lee NV, Wu JI, Shang C, Bayle JH, Shou W, Iruela‐Arispe ML, Chang CP. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev Cell. 2008; 14:298-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin CT, Brennan J, Magnuson T. The chromatin‐remodeling enzyme BRG1 plays an essential role in primitive erythropoiesis and vascular development. Development. 2008; 135:493-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl A, Connor KM, Sapieha P, Chen J, Dennison RJ, Krah NM, Seaward MR, Willett KL, Aderman CM, Guerin KI, Hua J, Lofqvist C, Hellstrom A, Smith LE. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci. 2010; 51:2813-2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin HG. Vascular morphogenesis in the ovary. Baillieres Best Pract Res Clin Obstet Gynaecol. 2000; 14:867-882. [DOI] [PubMed] [Google Scholar]
- Demir R, Yaba A, Huppertz B. Vasculogenesis and angiogenesis in the endometrium during menstrual cycle and implantation. Acta Histochem. 2010; 112:203-214. [DOI] [PubMed] [Google Scholar]
- Zygmunt M, Herr F, Munstedt K, Lang U, Liang OD. Angiogenesis and vasculogenesis in pregnancy. Eur J Obstet Gynecol Reprod Biol. 2003; 110suppl 1:S10-S18. [DOI] [PubMed] [Google Scholar]
- Waters RE, Rotevatn S, Li P, Annex BH, Yan Z. Voluntary running induces fiber type‐specific angiogenesis in mouse skeletal muscle. Am J Physiol Cell Physiol. 2004; 287:C1342-C1348. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Ferrara N. Angiogenesis and bone growth. Trends Cardiovasc Med. 2000; 10:223-228. [DOI] [PubMed] [Google Scholar]
- Tonnesen MG, Feng X, Clark RA. Angiogenesis in wound healing. J Investig Dermatol. 2000; 5:40-46. [DOI] [PubMed] [Google Scholar]
- Drixler TA, Vogten MJ, Ritchie ED, van Vroonhoven TJ, Gebbink MF, Voest EE, Borel Rinkes IH. Liver regeneration is an angiogenesis‐ associated phenomenon. Ann Surg. 2002; 236:703-711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford TN, Alfaro DV, III, Kerrison JB, Jablon EP. Diabetic retinopathy and angiogenesis. Curr Diabetes Rev. 2009; 5:8-13. [DOI] [PubMed] [Google Scholar]
- Paleolog EM. Angiogenesis in rheumatoid arthritis. Arthritis Res. 2002; 4suppl 3:S81-S90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidenreich R, Rocken M, Ghoreschi K. Angiogenesis drives psoriasis pathogenesis. Int J Exp Pathol. 2009; 90:232-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehler TM, Ho AD, Goldschmidt H, Barlogie B. Angiogenesis in hematologic malignancies. Crit Rev Oncol Hematol. 2003; 45:227-244. [DOI] [PubMed] [Google Scholar]
- Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011; 17:1359-1370. [DOI] [PubMed] [Google Scholar]
- Chung AS, Ferrara N. Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol. 2011; 27:563-584. [DOI] [PubMed] [Google Scholar]
- Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007; 21:2511-2524. [DOI] [PubMed] [Google Scholar]
- Gebuhr TC, Kovalev GI, Bultman S, Godfrey V, Su L, Magnuson T. The role of Brg1, a catalytic subunit of mammalian chromatin‐remodeling complexes, in T cell development. J Exp Med. 2003; 198:1937-1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P, Iruela‐Arispe ML. VE‐Cadherin‐Cre‐recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006; 235:759-767. [DOI] [PubMed] [Google Scholar]
- Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, Adams RH. Ephrin‐B2 controls VEGF‐induced angiogenesis and lymphangiogenesis. Nature. 2010; 465:483-486. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999; 21:70-71. [DOI] [PubMed] [Google Scholar]
- Feil S, Valtcheva N, Feil R. Inducible Cre mice. Methods Mol Biol. 2009; 530:343-363. [DOI] [PubMed] [Google Scholar]
- Pitulescu ME, Schmidt I, Benedito R, Adams RH. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc. 2010; 5:1518-1534. [DOI] [PubMed] [Google Scholar]
- Chinsomboon J, Ruas J, Gupta RK, Thom R, Shoag J, Rowe GC, Sawada N, Raghuram S, Arany Z. The transcriptional coactivator PGC‐1alpha mediates exercise‐induced angiogenesis in skeletal muscle. Proc Natl Acad Sci USA. 2009; 106:21401-21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, Penfold ME, Shido K, Rabbany SY, Rafii S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014; 505:97-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allinson KR, Lee HS, Fruttiger M, McCarty JH, Arthur HM. Endothelial expression of TGFbeta type II receptor is required to maintain vascular integrity during postnatal development of the central nervous system. PLoS One. 2012; 7:e39336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, Crystal RG, Simons M, Sato TN, Worgall S, Shido K, Rabbany SY, Rafii S. Endothelial‐derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell. 2011; 147:539-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung B, Obinata H, Galvani S, Mendelson K, Ding BS, Skoura A, Kinzel B, Brinkmann V, Rafii S, Evans T, Hla T. Flow‐regulated endothelial S1P receptor‐1 signaling sustains vascular development. Dev Cell. 2012; 23:600-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014; 507:323-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis MS, Homeister JW, Rosson GB, Annayev Y, Holley D, Holly SP, Madden VJ, Godfrey V, Parise LV, Bultman SJ. Functional redundancy of SWI/SNF catalytic subunits in maintaining vascular endothelial cells in the adult heart. Circ Res. 2012; 111:e111-e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior BM, Yang HT, Terjung RL. What makes vessels grow with exercise training? J Appl Physiol (1985). 2004; 97:1119-1128. [DOI] [PubMed] [Google Scholar]
- Hang CT, Yang J, Han P, Cheng HL, Shang C, Ashley E, Zhou B, Chang CP. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature. 2010; 466:62-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Wang Y, Cahill H, Yu M, Badea TC, Smallwood PM, Peachey NS, Nathans J. Norrin, frizzled‐4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009; 139:285-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Chen X, Xie X, Tsai MJ, Tsai SY. COUP‐TFII regulates tumor growth and metastasis by modulating tumor angiogenesis. Proc Natl Acad Sci USA. 2010; 107:3687-3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis CD, Davis RB, Ingram KG, Griffin CT. Chromatin‐remodeling complex specificity and embryonic vascular development. Cell Mol Life Sci. 2012; 69:3921-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]