

Abstract
Gastric cancer is one of the most malignant diseases and one of the leading causes of cancer-associated mortality worldwide. Although advances have been made in surgical techniques, perioperative management and the combined use of surgery with chemotherapy and/or radiotherapy, patients with advanced stage gastric cancer continue to face poor outcomes. Furthermore, it was reported that MET gene amplification and overexpression predicted the sensitivity to MET inhibitors in gastric cancer. However, the identification of drug-resistant tumors has encouraged the pre-emptive elucidation of the possible mechanisms of clinical resistance. The current study assessed a number of patient-derived gastric cancer models with MET amplification and overexpression, including CNGAS028. The tumor tissues were subjected to microarray analysis (using single nucleotide polymorphism 6.0 and human genome U133 arrays) followed by western blotting. The results demonstrated that CNGAS028 xenograft tumors did not respond to treatment with a selective MET inhibitor. Additional analysis indicated that FGFR2 overexpression contributed to the resistance to MET inhibitors. Furthermore, treatment with a combination of fibroblast growth factor receptor 2 and MET inhibitors inhibited the growth of CNGAS028 xenograft tumors in vivo. In conclusion, the current results aid in understanding the mechanism of inherent resistance to selective MET inhibitors as well as provide important information for patient selection and clinical treatment strategies.
Keywords: MET, fibroblast growth factor receptor 2, gastric cancer, resistance
Introduction
Gastric cancer is the second most prevalent cause of cancer-associated mortality worldwide (1). However, over the past 50 years, the total incidence rates of gastric cancer have gradually decreased, particularly in developed countries. Furthermore, the disease most commonly occurs within the male population in developing countries, predominantly East Asia, South America and Eastern Europe (2). Conventional therapeutic strategies for gastric cancer include surgery, chemotherapy and radiation therapy (3). However, as gastric cancer has few symptoms during the early stages, the majority of patients are typically diagnosed once the cancer has progressed to an advanced stage. Despite undergoing surgical resection, tumors recur in a large number of patients, in such cases the median survival time following cytotoxic chemotherapy is <1 year. Therefore, the diagnosis and effective treatment of advanced gastric cancer continues to be a challenge for oncologists (4). Although the use of molecular targeted therapy has been studied in other common types of solid tumors, including non-small cell lung cancer and breast cancer, it has yet to be fully explored in gastric cancer (5).
MET was initially identified as an oncogene encoding the receptor tyrosine kinase (RTK) for hepatocyte growth factor. The MET gene has been identified on chromosome 7q21-q31, where it encodes a single precursor that is digested and glycosylated post-transcriptionally, resulting in an extracellular α-chain (50-kDa) linked to a transmembrane β-chain (140-kDa) via disulfide bonds. Oncogenic activation of MET suppresses apoptosis and promotes cell survival, proliferation, migration and differentiation, as well as gene transcription and angiogenesis (6,7).
Gain-of-function mutations in MET are uncommon in gastric cancer (8), with MET activation predominantly attributed to gene amplification (9). A previous used fluorescence in situ hybridization analysis in order to detect MET amplification, which was reported to occur in ≤4% of patients with gastric cancer (10). Various MET inhibitors have been investigated in clinical trials, which showed promising initial results indicating that MET may be a potential therapeutic target for the treatment of gastric cancer (11,12). An increasing number of pharmaceutical companies are focusing on the identification of novel small molecular c-MET inhibitors, including PF2341066 (Pfizer Ltd., Surrey, UK) and ARQ197 (ArQule Inc., Woburn, MA, USA) (13,14). However, the identification of drug-resistant tumors has encouraged the pre-emptive elucidation of potential mechanisms of clinical resistance. The present study describes a patient-derived gastric cancer model resistant to a selective MET inhibitor and attempts to determine the underlying mechanism.
Materials and methods
Establishment of patient-derived gastric cancer xenograft models
Female athymic BALB/c nude mice (n=200), aged 6–7 weeks, were purchased from Shanghai Laboratory Animal Centre Co., Ltd. (Shanghai, China). Mice were maintained under super-specific pathogen-free conditions and housed in barrier facilities on a 12 h light/dark cycle, with food and water provided ad libitum. All animal experiments were performed in accordance with protocols approved by the Shandong Tumor Hospital Experimental Animal Care and Use Committee. Fresh human gastric tumor specimens obtained from 83 Chinese patients that had undergone surgery were received from Shandong Tumor Hospital (Jinan, China) by Shandong Tumor Hospital Experimental Animal Center (Shandong, China) within 1 h of removal from the patients. The samples were cut into 3×3×3-mm sections, soaked in 50% Matrigel™ (BD Biosciences, Franklin Lake, NJ, USA) and subcutaneously implanted into the flank of the nude mice. The tumors were passaged when the tumor volume reached ~300 mm3. Tumor volumes were calculated using the following standard formula: Tumor volume = (length × width2)/2. Written informed consent was obtained from all patients and the study was approved by the ethics committee of Shandong Tumor Hospital Experimental Animal Center.
Detection of gene copy number and expression by microarray in established gastric cancer xenograft models
The GeneChip® genome-wide human single nucleotide polymorphism (SNP) 6.0 and human genome U133 plus 2.0 arrays (Affymetrix, Inc., Santa Clara, CA, USA) were used to analyze the genomic gene copy number and gene expression levels in all established patient-derived gastric xenograft tumors, respectively. MAS5 software (Affymetrix, Inc.) was used to analyze the U133 results. Gene profiling comparison was performed by calculating the fold change of the copy number and gene expression between these tumors. The data were processed using the aroma.affymetrix R package (version 2.13.0; http:www.aroma-project.org/), according to the methods described by Bengtsson et al (15).
Efficacy studies in gastric cancer xenograft models with MET amplification and overexpression
Gastric tumors (2-cm diameter) were aseptically resected from established patient-derived gastric cancer xenografts with MET amplification and overexpression, then minced into 3×3×3 mm pieces. Host mice were then anesthetized with isoflurane and a section of tumor was implanted into the left flank of each mouse. Each gastric model that developed tumors reaching 150–200 mm3 in size were randomized into the following four treatment groups (10 mice per group): Group 1, once-daily dose with vehicle by intravenous (i.v.) tail injection; and groups 2, 3 and 4, once-daily dose with 10, 20 and 30 mg/kg PHA665752 by i.v. tail injection, respectively. PHA665752, a selective MET inhibitor, was purchased from Selleck Chemicals (Houston, TX, USA). In a subsequent experiment, the CNGAS028 model was also treated with vehicle, 15 mg/kg PHA665752, the pan-fibroblast growth factor receptor (FGFR2) selective inhibitor NVP-BGJ398 (15 mg/kg once-daily, oral administration; Selleck Chemicals) or 30 mg/kg PHA665752 in combination with 15 mg/kg NVP-BGJ398, respectively. All treatments were continued for 21 days and the mice were sacrificed by CO2 inhalation 2 h after the last treatment.
Western blot analysis
The tumor tissues were resected 2 h following the final treatment with PHA665752 or/and NVP-BGJ398 on day 21 of the efficacy studies. The tumor tissues were then homogenized and lysed in cell lysis buffer (Bio-Rad Laboratories, Hercules, CA, USA) containing phosphatase inhibitor cocktail and proteinase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA), and the protein concentrations were determined using the bicinchoninic acid protein assay kit (Pierce Biotechnology, Inc. Rockford, IL, USA). Subsequently, equal quantities of protein (30 µg) were separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis on 8% gels, blotted on polyvinylidene difluoride membranes (Invitrogen Life Technologies, Inc., Carlsbad, CA, USA), then probed with monoclonal phosphorylated (p)-MET (1:1,000; cat. no. 3126), polyclonal p-FGFR2 (1:1,000; cat no. af3285; R&D Systems, Inc., Minneapolis, MN, USA), monoclonal MET (1:1,000; cat. no. 4560) and monoclonal FGFR2 (1:1,000; cat. no. 11835) rabbit anti-human antibodies. Subsequently, the membranes were incubated with goat anti-rabbit horseradish peroxidase-conjugated secondary antibodies (1:1,000; cat. no. 7074) and detected by chemiluminescence. Gel Doc™ XR+ (Bio-Rad Laboratories, Inc., Hercules, CA, USA) was used to visualize the western blots. All antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA), unless otherwise stated.
Statistical analysis
All data are presented as the mean ± standard deviation for the indicated number of independently performed experiments. Statistical analyses were conducted using GraphPad InStat software (version 5.0; GraphPad Software, Inc., San Diego, CA, USA). Student's t tests were performed and P<0.05 was considered to indicate a statistically significant difference.
Results
MET gene amplification and expression in Chinese patient-derived gastric cancer models
Chinese patient-derived gastric cancer models (n=30) were established from 83 gastric cancer specimens. The established models were termed CNGAS001-030. The CNGAS001, CNGAS002 and CNGAS003 mouse models are indicated in Fig. 1A. Microarray data from the SNP 6.0 and U133 plus 2.0 gene chips were used to analyze the genomic gene copy number and gene expression levels of all established models, respectively. The microarray data demonstrated that MET was highly amplified and expressed in 16.7% (5/30) of the Chinese gastric cancer xenograft models (Fig. 1B and C). From the results, it was observed that MET amplification was positively correlated with MET overexpression in the xenograft models.
Figure 1.

MET gene amplification and expression in 30 patient-derived gastric cancer models. (A) Images of the CNGAS001, CNGAS002 and CNGAS003 gastric cancer models. Analysis of the established gastric cancer models using (B) single nucleotide polymorphism 6.0 and (C) U133 plus 2.0 gene chips.
High amplification and overexpression of MET predicts response to PHA665752 in patient-derived gastric cancer models
The present study analyzed the efficacy PHA665752 in the patient-derived gastric xenograft models with high MET amplification and overexpression (CNCAS005, CNCAS008, CNCAS015, CNCAS018 and CNCAS028). The results demonstrated that four gastric cancer xenograft models were significantly sensitive to PHA665752 treatment (P<0.05). However, the CNCAS028 model was resistant to PHA665752 (Fig. 2A–E; 30 mg/kg i.v. PHA665752 treatment group: P=0.008, P=0.006, P=0.004, P=0.007 and P=0.125, for the CNCAS005, 008, 015, 018 and CNCAS028 xenograft models, respectively).
Figure 2.

Tumor volumes of patient-derived gastric cancer models in response to PHA665752 or/and NVP-BGJ398 treatment. Nude mice bearing (A) CNCAS005, (B) CNCAS008, (C) CNCAS015, (D) CNCAS018 and (E) CNCAS028 tumors were treated with control vehicle or PHA665752 once-daily at the indicated doses by tail i.v. injection for 21 days. *P<0.05 and **P<0.01 vs. vehicle. i.v., intravenous; SD, standard deviation.
High FGFR2 amplification and expression in the CNCAS028 model
As indicated in Fig. 2A, the tumor growth of CNCAS028 was not inhibited by treatment with 30 mg/kg PHA665752 for 21 days. To explore the mechanism of the resistance to the selective MET inhibitor, the genome-wide gene profiles of CNCAS028 were compared with those of the other four gastric cancer models. It was identified that FGFR2 was highly amplified and expressed in the CNCAS028 model, whereas FGFR2 was expressed at a normal level and not amplified in the PHA665752-sensitive xenografts [normal FGFR2 expression, copy number <5 and expression level <4,000 (as determined by GeneChip®); Fig. 3A and B]. These results were confirmed by western blot analysis (Fig. 4).
Figure 3.
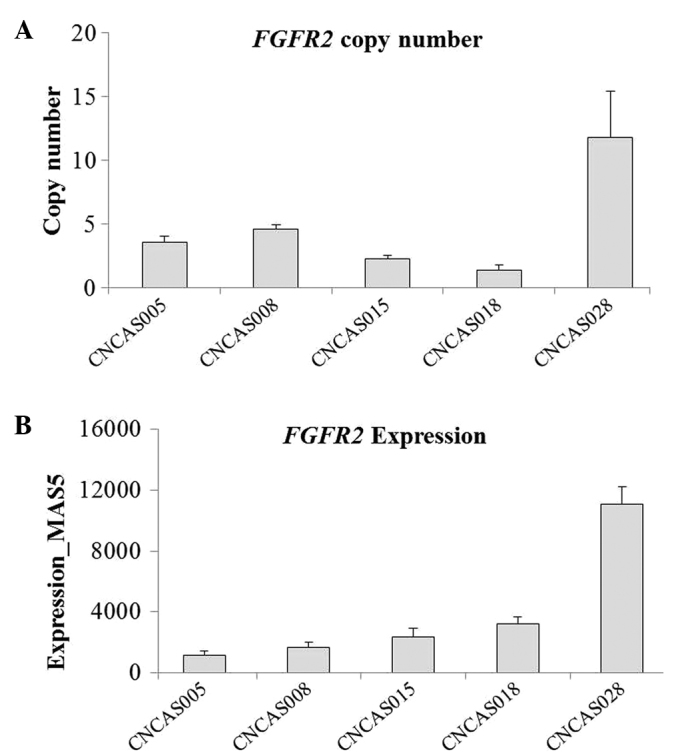
High FGFR2 amplification and expression in CNCAS028 xenografts. CNCAS005, CNCAS008, CNCAS015, CNCAS018 and CNCAS028 tumors were resected from xenografts and then subjected to (A) single nucleotide polymorphism 6.0 and (B) U133 plus 2.0 gene chips. Mean ± standard deviation; n=5.
Figure 4.

Expression level of p-MET, MET, p-FGFR2 and FGFR2 analyzed by western blotting in patient-derived gastric cancer models. (A) Expression levels of p-MET, MET, p-FGFR2 and FGFR2 in CNCAS005, CNCAS008, CNCAS015, CNCAS018 and CNCAS028 tumors. p-FGFR2, phosphorylated fibroblast growth factor receptor 2.
PHA665752 and NVP-BGJ398 combination treatment significantly inhibits tumor growth in the CNCAS028 model
To validate the association between FGFR2 amplification and overexpression as well as PHA665752 resistance, PHA665752 was combined with a selective pan-FGFR2 kinase inhibitor (NVP-BGJ398) to treat the CNCAS028 model (16). As indicated in Fig. 5, treatment with 30 mg/kg PHA665752 was not able to inhibit tumor growth in the CNCAS028 model. Furthermore, treatment with 15 mg/kg NVP-BGJ398 only marginally inhibited tumor growth. By contrast, combined treatment with these two compounds significantly inhibited tumor growth following 21 days of treatment (P<0.01).
Figure 5.

Tumor volumes of patient-derived CNCAS028 gastric cancer xenografts in response to PHA665752 or/and NVP-BGJ398 treatment, indicating the anti-tumor effect of each treatment. Nude mice bearing PHA665752-resistant CNCAS028 tumors were treated with 30 mg/kg PHA665752 once-daily by i.v. injection, 15 mg/kg NVP-BGJ398 orally or combination therapy for 3 weeks. **P<0.01 vs. vehicle. i.v., intravenous; SD, standard deviation.
Effect of PHA665752 and/or NVP-BGJ398 on signaling transduction in patient-derived gastric cancer models
To investigate the effect of PHA665752 and NVP-BGJ398 on downstream molecules of the phosphoinositide 3-kinase and RAS signaling pathways, western blot analysis was used to observe changes in the phosphorylation status and total protein expression levels of the tumor tissues. The results demonstrated that all five patient-derived gastric xenograft models highly expressed p-MET and the CNCAS028 xenograft also highly expressed p-FGFR2 (Fig. 4). Western blot analysis also identified that treatment with PHA665752 inhibited the phosphorylation of MET in all five gastric tumor models. In addition, the expression of p-FGFR2 was markedly inhibited by NVP-BGJ398 or combination treatment in the CNCAS028 model (Fig. 6).
Figure 6.

Expression level of p-MET, MET, p-FGFR2 and FGFR2 analyzed by western blotting in patient-derived gastric cancer models. Expression levels of p-MET, MET, p-FGFR2 and FGFR2 in CNCAS028 tumor tissues resected 2 h following the final treatment with PHA665752 and/or NVP-BGJ398 on day 21 of the efficacy study. p-FGFR2, phosphorylated fibroblast growth factor receptor 2.
Discussion
Gastric and gastroesophageal cancer affect 1 million individuals worldwide every year and are the second most common cause of cancer-associated mortality (17). Targeted therapies have been developed and incorporated into the standard treatment strategies for other types of solid cancer, such as lung or breast. However, such therapies (including MET inhibitors) are only now being examined in the context of gastric and gastroesophageal cancer (18). The current investigations identified MET gene amplification in 5/30 (16.7%) patient-derived gastric cancer xenografts. These results indicated the therapeutic potential of MET inhibitors in gastric cancer. Additional analysis identified that a selective MET inhibitor (PHA665742) was able to significantly inhibit tumor growth in 4/5 gastric cancer models with MET amplification. These results are consistent with a previous study, which demonstrated that MET amplification was associated with the response of the MKN45 gastric cancer cell line to PHA665752 treatment (19).
Aberrant RTK expression produces growth and survival signals that are essential for the pathogenesis and progression of various types of cancer. Furthermore, cancer patients treated with targeted inhibitors of key oncogenic kinase drivers, including imatinib, gefitinib and erlotinib, have exhibited promising clinical outcomes (20). However, based on the precedence set by agents such as imatinib in chronic myeloid leukemia and erlotinib in lung adenocarcinoma, inherent resistance may potentially limit the application of single agent therapies (21). The elucidation of novel oncogenic drivers may have extensive implications for targeted therapy. Corso et al (22) reported that activation of HER family members in MET-addicted cancer cells, subsequent to MET inactivation, resulted in increased cell viability in vitro and recovered tumorigenicity in vivo. In addition, Lee et al (23) reported that a novel SND1-BRAF fusion gene exhibited resistance to the MET inhibitor PF-04217903 in GTL16 cells via RAS/RAF/ERK signaling pathway activation. By contrast, the current study determined that a MET-amplified CNCAS028 model was resistant to MET inhibitor PHA66575 as a result of FGFR2 gene amplification and overexpression. Inhibition of FGFR2 signaling in this xenograft model recovered its sensitivity to PHA665752.
In conclusion, MET and FGFR2 coactivation may increase resistance to targeted therapy, possibly due to activation of multiple growth and survival signaling pathways. These findings indicate that combination therapy with MET and FGFR2 inhibitors may be a promising strategy to treat inherent resistance to MET inhibitors in cases of gastric cancer harboring MET and FGFR2 amplification. Future studies should be performed to investigate whether similar results could be obtained in an acquired resistant model.
Acknowledgements
The present authors would like to thank Dr Jianshe Wu and Dr Yanlin Wu for conducting the bioinformatic analysis of the GeneChip® genome-wide human single nucleotide polymorphism 6.0 and human genome U133 plus 2.0 array data.
References
- 1.Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality and prevalence across five continents: Defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol. 2006;24:2137–2150. doi: 10.1200/JCO.2005.05.2308. [DOI] [PubMed] [Google Scholar]
- 2.Camargo MC, Anderson WF, King JB, Correa P, Thomas CC, Rosenberg PS, et al. Divergent trends for gastric cancer incidence by anatomical subsite in US adults. Gut. 2011;60:1644–1649. doi: 10.1136/gut.2010.236737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamashita K, Sakuramoto S, Nemoto M, Shibata T, Mieno H, Katada N, et al. Trend in gastric cancer: 35 years of surgical experience in Japan. World J Gastroenterol. 2011;17:3390–3397. doi: 10.3748/wjg.v17.i29.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer HJ, Wilke H. Treatment strategies in gastric cancer. Dtsch Arztebl Int. 2011;108:698–705. doi: 10.3238/arztebl.2011.0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morishita A, Gong J, Masaki T. Targeting receptor tyrosine kinases in gastric cancer. World J Gastroenterol. 2014;20:4536–4545. doi: 10.3748/wjg.v20.i16.4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maestrini E, Tamagnone L, Longati P, Cremona O, Gulisano M, Bione S, et al. A family of transmembrane proteins with homology to the MET-hepatocyte growth factor receptor. Proc Natl Acad Sci USA. 1996;93:674–678. doi: 10.1073/pnas.93.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sattler M, Salgia R. c-Met and hepatocyte growth factor: Potential as novel targets in cancer therapy. Curr Oncol Rep. 2007;9:102–108. doi: 10.1007/s11912-007-0005-4. [DOI] [PubMed] [Google Scholar]
- 8.Lee JH, Han SU, Cho H, Jennings B, Gerrard B, Dean M, et al. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene. 2000;19:4947–4953. doi: 10.1038/sj.onc.1203874. [DOI] [PubMed] [Google Scholar]
- 9.Nakajima M, Sawada H, Yamada Y, Watanabe A, Tatsumi M, Yamashita J, et al. The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas. Cancer. 1999;85:1894–1902. doi: 10.1002/(SICI)1097-0142(19990501)85:9<1894::AID-CNCR3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 10.Hara T, Ooi A, Kobayashi M, Mai M, Yanagihara K, Nakanishi I. Amplification of c-myc, K-sam and c-met in gastric cancers: detection by fluorescence in situ hybridization. Lab Invest. 1998;78:1143–1153. [PubMed] [Google Scholar]
- 11.Sierra JR, Tsao MS. c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. 2011;3(Suppl):S21–S35. doi: 10.1177/1758834011422557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawakami H, Okamoto I, Arao T, Okamoto W, Matsumoto K, Taniguchi H, et al. MET amplification as a potential therapeutic target in gastric cancer. Oncotarget. 2013;4:9–17. doi: 10.18632/oncotarget.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6:3314–3322. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- 14.Abidoye O, Murukurthy N, Salgia R. Review of clinic trials: agents targeting c-Met. Rev Recent Clin Trials. 2007;2:143–147. doi: 10.2174/157488707780599357. [DOI] [PubMed] [Google Scholar]
- 15.Bengtsson H, Irizarry R, Carvalho B, Speed TP. Estimation and assessment of raw copy numbers at the single locus level. Bioinformatics. 2008;24:759–767. doi: 10.1093/bioinformatics/btn016. [DOI] [PubMed] [Google Scholar]
- 16.Pietrantonio F, Maggi C, Di Bartolomeo M, Facciorusso MG, Perrone F, Testi A, et al. Gain of ALK gene copy number may predict lack of benefit from anti-EGFR treatment in patients with advanced colorectal cancer and RAS-RAF-PI3KCA wild-type status. PLoS One. 2014;9:e92147. doi: 10.1371/journal.pone.0092147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartgrink HH, Jansen EP, van Grieken NC, van de Velde CJ. Gastric cancer. Lancet. 2009;374:477–490. doi: 10.1016/S0140-6736(09)60617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oshima T, Masuda M. Molecular targeted agents for gastric and gastroesophageal junction cancer. Surg Today. 2012;42:313–327. doi: 10.1007/s00595-011-0065-9. [DOI] [PubMed] [Google Scholar]
- 19.Funakoshi Y, Mukohara T, Tomioka H, Ekyalongo RC, Kataoka Y, Inui Y, et al. Excessive MET signaling causes acquired resistance and addiction to MET inhibitors in the MKN45 gastric cancer cell line. Invest New Drugs. 2013;31:1158–1168. doi: 10.1007/s10637-013-9959-2. [DOI] [PubMed] [Google Scholar]
- 20.Jänne PA, Gray N, Settleman J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat Rev Drug Discov. 2009;8:709–723. doi: 10.1038/nrd2871. [DOI] [PubMed] [Google Scholar]
- 21.SeboltLeopold JS, English JM. Mechanisms of drug inhibition of signalling molecules. Nature. 2006;441:457–462. doi: 10.1038/nature04874. [DOI] [PubMed] [Google Scholar]
- 22.Corso S, Ghiso E, Cepero V, Sierra JR, Migliore C, et al. Activation of HER family members in gastric carcinoma cells mediates resistance to MET inhibition. Mol Cancer. 2010;9:121. doi: 10.1186/1476-4598-9-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee NV, Lira ME, Pavlicek A, Ye J, Buckman D, Bagrodia S, et al. A novel SND1-BRAF fusion confers resistance to c-Met inhibitor PF-04217903 in GTL16 cells though [corrected] MAPK activation. PLoS One. 2012;7:e39653. doi: 10.1371/journal.pone.0039653. [DOI] [PMC free article] [PubMed] [Google Scholar]