

Abstract
Lung cancer is the leading cause of cancer-related mortality worldwide. The majority of lung cancers are sporadic, and familial cases are extremely rare. Previous studies have mainly focused on sporadic lung cancer and identified a large quantity of driver genes. However, familial lung cancers are rarer and studied less. The present study recruited a Chinese family in which multiple members had developed lung squamous carcinoma. To find the causative mutations, whole exome sequencing was conducted using a peripheral blood sample of one lung squamous carcinoma patient, and certain variants were validated in more samples. Whole exome sequencing analysis obtained ~2.0 Gb of data (an average of 60x depth for each targeted base), and further validation experiments identified two functional variants in two cancer-related genes (c.1218delA:p.E406fs in PDE4DIP and C1342A:p.L448I in CLTCL1). This study therefore provides useful sources for the further study of hereditary lung cancer.
Keywords: familial lung squamous cancer, whole exome sequencing, mutation
Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide. In 2012, World Health Organization statistical data reported that the numbers of global new cases and mortalities from lung cancer were 1,824,701 and 1,589,800, respectively (1). The majority of lung cancers are diagnosed at an advanced stage, so the 5-year survival rate of lung cancer patients is <15% (1). Based on the histological type, lung cancers are divided into two subtypes: Small cell lung cancer and non-small cell lung cancer (NSCLC). NSCLC accounts for >85% cases, and can be further classified into three main subtypes: Squamous cell carcinoma, adenocarcinoma and large cell carcinoma (2–4).
Previous studies have identified a large quantity of driver genes in lung carcinogenesis, genetic aberrations of which can promote tumor cell proliferation, survival, angiogenesis, invasion and metastasis, thus driving the development of lung cancer (5). For example, epidermal growth factor receptor (EGFR) mutations (6), EML4/ALK fusion genes (7), RAS mutations, BRAF mutations and PI3K/mTOR mutations are frequently observed in lung cancer tissues, and are predictive of response to targeted therapies (8).
The majority of lung cancers are sporadic, and familial cases are extremely rare. Unlike sporadic cancer, germline mutations may drive carcinogenesis in familial cases. For example, functional mutations in BRCA1/2 genes may cause familial breast or ovarian cancer. The family members who carry BRCA1/2 mutations have a 70–80% possibility of developing cancers (9,10). Another example in colorectal cancer demonstrates that DNA mismatch repair gene MSH2 and MLH1 mutations carriers have a high risk of developing colorectal cancer in their 40s (11,12).
To date, only three familial lung cancer cases have been studied. In one study, it was reported that a lung squamous cancer patient carried the EGFR R776H germline mutation, and that the patient's daughter, also a lung cancer patient, carried the same alteration (13). In another study, 3/6 individuals in a Japanese family developed lung adenocarcinoma. The germline EGFR V843I mutation was identified in all the lung adenocarcinoma-affected members of this family (14). In a study by Yamamoto et al, it was found that mutations in HER2 may be the causative variants of familial lung adenocarcinomas (15). The molecular mechanism of familial lung cancer remains largely unknown.
The present study recruited a family of Chinese descent in which multiple members were diagnosed with lung squamous carcinoma. To find the causative mutations, whole exome sequencing was conducted using a peripheral blood sample of one lung squamous carcinoma patient, and certain variants were validated in more samples. This study provides the basis for further studies, in which further hereditary lung cancer members will be sequenced.
Materials and methods
DNA extraction
The selected family were from Northwest China, Shaanxi province. All individuals were diagnosed with lung squamous cell carcinoma and treated at the Department of Thoracic Surgery, Second Affiliated Hospital, Medical School, Xi'an Jiaotong University (Xi'an, China) in August 2011. Peripheral blood samples were collected from 4 individuals, 3 of whom were lung cancer patients, and 1 of whom had no malignant disease history. DNA was extracted using a QIAamp DNA Mini kit (cat. no. 51106) according to the manufacturer's instruction (Qiagen, Hilden, Germany). Histological examination was performed by hematoxylin and eosin staining of the bronchoscopic biopsies, needle aspiration biopsies and surgical specimens.
The experiments were undertaken with the understanding and written consent of each subject, and the study conforms with The Code of Ethics of the World Medical Association (Declaration of Helsinki). This study was performed in accordance with the ethical standards of the Ethics Committee of the Second Affiliated Hospital, Medical School, Xi'an Jiaotong University (Xi'an, China).
Sanger sequencing
Specific primers were designed using Primer 5 (Premier Biosoft International, Palo Alto, CA, USA; Table I). The PCR materials included 0.25 mM dNTPs, 1 unit of HotStarTaq (Qiagen), 1X HotStarTaq buffer (Qiagen) and 0.4 µM primer. PCR conditions were 33 cycles of 95°C for 50 sec, 61°C for 40 sec and 72°C for 60 sec, following initial denaturation at 95°C for 5 min. PCR products were purified using SAP, and used as templates for the sequencing reaction using Big Dye v3.1 (Applied Biosystems Life Technologies, Foster City, CA, USA). Subsequent products were run on the ABI PRISM 3130×1 Genetic Analyzer (Applied Biosystems Life Technologies). Electropherograms were analyzed using Sequence Analysis Software version 5.2 (Applied Biosystems Life Technologies).
Table I.
Primer sequences.
| Primer | Sequence |
|---|---|
| EGFRrs2227983-F | GTGACCCACTCTGTCTCCG |
| EGFRrs2227983-R | GAAAACCCAAAACCTCCAA |
| NOTCH4-F | GGCTTCCTGGGTGAGACG |
| NOTCH4-R | TTGGTTGTGGGTAAGTGGATT |
| MAD1L18-F | ATGACCAGAGCAGGACCAA |
| MAD1L1-R | CACAGGGGAGGACAAACC |
| TNFRSF10A-F | CTGATGAAATGGGTCAACAAA |
| TNFRSF10A-R | TCCCGAGTAGCCGAGAATA |
| CDKN1Ars76-F | TGTGAGGTAGATGGGAGCG |
| CDKN1Ars76-R | GGGGAGGATTTGACGAGTG |
| CDKN1Ars18-F | GTGTCTAATCTCCGCCGTGAC |
| CDKN1Ars18-R | GCCTGCCTCCTCCCAACT |
| CDKN1Ars10-F | GCGTCCTCTTCTTCTTGGC |
| CDKN1Ars10-R | AAAGGAGAACACGGGATGAG |
| TP53rs1042522-F | ACCTGTGGGAAGCGAAA |
| TP53rs1042522-R | GGAAGGGACAGAAGATGACA |
| ATM-F | AATCTCCTCCTTTCTGCTGC |
| ATM-R | CCACTCTGCCAAGTATTATGCTAT |
| NBN-F | GGTTACCTCAGTGCCATTTAC |
| NBN-R | GCAGTGACCAAAGACCGA |
| XPC-F | TGGCAGCGGCTCTGATTT |
| XPC-R | GGCTCTGGTATGGTCTCAAGGTC |
| PDE4DIP-F | CCCATGACTGAAGAGTTGCT |
| PDE4DIP-R | CATCTCCCAAGTCCTACAAAG |
| CLTCL1-F | CAGAAAGATGCTTACCACCC |
| CLTCL1-R | GCTGCCTGATACTCACCGA |
| MLL3-F | TTTCATCATCAGCAGACATAAGC |
| MLL3-R | CACCCTACCTGTTTGGACC |
| ARID2-F | TTCTTTGCTTTCCATCCTTG |
| ARID2-R | CCTCCGTTTTACTCCTACCA |
| CLTCL1-F | TTATTGTTGAAGATAGAAGCAGTT |
| CLTCL1-R | AAAGTTAGGAGGCAGGTAGG |
| GNAS-F | CGGAGCCTTCAGTGGTG |
| GNAS-R | CTGTCGTCCCCAAATCCT |
| NACA-F | CAAAGGTCCCTCTACCATCTC |
| NACA-R | CAACAGAAGAAACGGGCAT |
| POT1-F | ATTTTAGGCAAGAAGTTCCACA |
| POT1-R | TTGAAAGCGGGAGAATACC |
| TFRC-F | TTCCTCAAGCCAAATAATGC |
| TFRC-R | CAAAGGCATAGATAATGTCAGC |
| NIN-F1 | GCTCTGGGATTGAAGGGAC |
| NIN-R1 | CAGGCTGGAGTACAGTGGC |
F, forward; R, reverse.
Whole exome sequencing
Whole exome sequencing was performed on the peripheral blood sample of the proband. Firstly, to construct a library, whole exome DNA from the patient's whole blood was treated using Agilent SureSelect Human All ExonV5 kits following the manufacturer's instructions (Agilent Technologies Inc., Santa Clara, CA, USA). Subsequent to the quality test, the qualified library was sequenced as 100-bp paired-end reads on an Illumina Hiseq 2000 platform (Illumina, San Diego, CA, USA) according to the manufacturer's instructions.
Data analysis
For whole exome sequencing, clean data was obtained after filtering the reads of low quality (reads with adapter sequence, reads with proportion of N >10% and reads with low quality base numbers of >5). Burrows-Wheeler transform methods (16) were adopted to map these reads in a human reference (UCSC hg19). Next, the Picard and Genome Analysis Toolkit (GATK) methods (17,18) were adopted for duplicate removal, local realignment and base quality recalibration. Finally, the GATK Unified Genotyper (version 3.0; Broad Institute, Cambridge, MA, USA) was used for single nucleotide variation (SNV)/InDel annotation.
Variants were annotated using the ANNOVAR software tool (http://www.openbioinformatics.org/annovar/). Annotations for function (exonic, intronic and untranslated region), reference genes, exonic function (synonymous, non-synonymous, stop-gain, frameshift and unknown), amino acid changes, 1000 Genomes Project data and dbSNP reference number were performed.
The whole exome sequencing generated large volumes of data, and several filtering criteria were applied to the data set. Firstly, variants with a low quality score (depth <20 or genotype quality <20) were filtered. Secondly, the variants with a reported frequency of >0.01 were filtered. Thirdly, synonymous changes were removed, taking only the protein-altering variants.
Results
Description of the pedigree
The lung cancer family were from Northwest China, Shaanxi province, and four members of the family were diagnosed with lung cancer (Fig. 1). The proband was a 65-year-old male who presented with a tumor in the inferior lobe of the right lung. Histological examination revealed individual cell keratinization, as well as intercellular bridge and squamous pearl formation, which resulted in a diagnosis of lung squamous cell carcinoma, stage IIIa (T3N2M0), according to the National Comprehensive Cancer Network TNM staging system (19). The patient was a heavy smoker, with 1.6 pack-year history of >10 years. Following lobectomy by thoracotomy with lymph node dissection, the patient underwent chemotherapy (120 mg/m2 cisplatin day 1, and 30 mg/m2 vinorelbine days 1 and 8 every 21 days for 4 cycles). A good response to chemotherapy was recorded. The proband had suffered from no respiratory or malignant diseases prior to the lung cancer diagnosis. Two elder brothers of the proband, who were also smokers, had succumbed to lung squamous carcinoma metastasis at the ages of 71 and 69 years old, respectively. One of the proband's nephews had been diagnosed with lung squamous cancer at the age of 51. All the lung cancer cases in the family were histologically confirmed. Other members in the family had no history of tumors or respiratory diseases. Peripheral blood samples were collected from four family members, including three of the cancer patients (I4, I6 and II3) and one healthy family member (II1). II1 was the only surviving healthy control who was elder than at least one of the lung cancer patients (Fig. 1).
Figure 1.

Pedigree chart of the family. The circles and boxes represent women and men, respectively.
Whole exome sequencing
Whole exome sequencing was conducted using the peripheral blood sample of the proband, obtaining ~2.0 Gb of data and achieving an average of 60x depth for each targeted base, with 98.3% of the exomic positions covered >10x. In all, 41,210 SNVs and 3,299 InDels were identified. As described in the Materials and methods section, 2,845 variations, including SNVs and InDels, were removed due to a low quality score.
Sanger sequencing technology was applied to validate the variants identified by high throughput sequencing. In all, 10 variants were selected randomly for testing. The sequence results by Sanger sequencing confirmed the variants identified by whole exome sequencing.
Candidate gene validation
To identify the causative variants, the no reported alterations, or the alterations with low frequency (<=0.01, or no reported frequency), and the alterations which were predicted to alter the protein product (non-synonymous SNVs, splice-site mutations and InDels) were selected. In all, 975 SNVs and 1,052 InDels in 767 genes satisfied the aforementioned criteria.
Next, the candidate alterations were further restricted to 11 variants, which were located in genes with definite functions and recorded in the COSMIC driver gene database (http://cancer.sanger.ac.uk/cosmic; Table II) (20).
Table II.
Variants for validation.
| Gene | Ref. | Alt. | Amino acid change | Mutation type | 1000G MAF | DP | GQ |
|---|---|---|---|---|---|---|---|
| ARID2 | A | G | NM_152641:exon14:c.A1759G:p.S587G | Non-synonymous SNV | 0.01 | 69 | 99 |
| GNAS | A | G | NM_080425:exon1:c.A484G:p.M162V | Non-synonymous SNV | 0.0014 | 116 | 99 |
| MLH1 | T | A | NM_001167619:exon11:c.T428A:p.V143D | Non-synonymous SNV | 0.01 | 83 | 99 |
| NACA | T | C | NM_001113203:exon3:c.A1247G:p.K416R | Non-synonymous SNV | 0.01 | 105 | 99 |
| NIN | G | A | NM_016350:exon28:c.C3856T:p.R1286C | Non-synonymous SNV | 0.0014 | 22 | 99 |
| POT1 | C | G | NM_001042594:exon13:c.G835C:p.D279H | Non-synonymous SNV | 0.0027 | 70 | 99 |
| TFRC | G | C | NM_001128148:exon6:c.C634G:p.L212V | Non-synonymous SNV | 0.01 | 273 | 99 |
| CLTCL1 | G | T | NM_001835:exon8:c.C1342A:p.L448I | Non-synonymous SNV | 0.01 | 61 | 99 |
| PDE4DIP | GT | G | NM_001002811:exon2:c.1218delA:p.E406fs | Frameshift deletion | – | 169 | 99 |
| CLTCL1 | A | AC | NM_001835:exon23:c.3601_3602insG:p.V1201fs | Frameshift insertion | – | 60 | 99 |
| MLL3 | G | GT | NM_170606:exon14:c.2447_2448insA: p.Y816_I817delinsX | Stop-gain SNV | – | 373 | 99 |
Ref, base in the human reference genome hg19; alt, variant base; 1000G MAF, minor allele frequency in the 1000 Genomes Project; DP, depth (read depth at a certain position); GQ, genotype quality; SNV, single nucleotide variation.
Subsequently, Sanger sequencing was conducted using four peripheral blood samples from this family, from the three patients and one non-cancer family member (II1), to validate the variations in the aforementioned genes. As shown in Fig. 2, for the CLTCL1 L448I mutation, the genotypes of the three affected individuals were heterozygous, while that of the healthy man was homozygous wild-type. For the PDE4DIPE406fs mutation, the three patients carried heterozygous frameshift mutations, while the healthy individual carried the homozygous wild-type. For variations in other genes, namely ARID2, CLTCL1, GNAS, MLH1, NACA, NIN, POT1, TFRC, PDE4DIP and MLL3, the variations did not segregate well.
Figure 2.
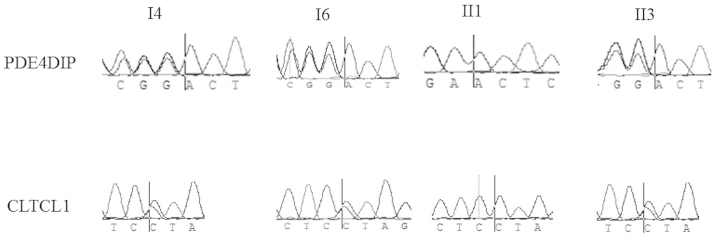
Sanger sequencing results. The vertical bars are at the mutation position. All the three patients (I4, I6, II3) carried heterozygous mutation in gene CLTCL1 and PDE4DIP, while the healthy individual (II1) carried homozygous wild type.
Discussion
The present study recruited a Chinese family in which four members had developed lung cancer, and is the first time a study of Chinese familial lung cancer has been reported. Lung cancer is one of the most common cancer types all over the world, and a number of previous studies have identified a large quantity of lung cancer driver genes, such as EGFR, Her2, AKT1, NRAS, PIK3CA, BRAF, ALK-fusion and RET-fusion (5–8). Somatic mutations of these genes have been recurrently identified in lung cancer samples (5–8). However, unlike in sporadic lung cancer, the molecular mechanisms behind inherited lung cancer remains largely unknown.
Previous studies reported the germline mutations of EGFR and HER2 as driver mutations in familial lung cancer patients (13–15). In the present study, whole exome sequencing identified 8 SNVs in the EGFR gene. However, 7 of these were synonymous, and 1 was a common variant with a frequency of >0.2. No variants that cause amino acid or splice changes were identified in the HER2 gene. The study also analyzed mutations in other inherited cancer-related genes, namely BRCA1/2, MSH2 and MLH1, but found no non-synonymous mutations in these genes. Thus, an unreported causative variation may drive lung cancer development in this family.
In this study, whole exome sequencing using a peripheral blood sample of one lung cancer member of the family totally identified >2,000 unreported or low frequency non-synonymous variants (975 SNVs and 1,052 InDels). Alterations were validated in 10 definitive cancer driver genes, and two plausible candidate genes, PDE4DIP and CLTCL1, were identified. For the PDE4DIP gene, all the patients carried a frameshift variation, while the non-cancer family member did not. The PDE4DIP protein acts as an anchor phosphodiesterase, localized in the Golgi/centrosome region of the cell. The protein is able to interact with a phosphodiesterase superfamily protein member (21,22). Functional mutations of PDE4DIP have been found to be associated with several disorders. In the study by DeWan et al, it was found that one mutation, I303L, in PDE4DIP may be the causative variant in hereditary asthma (23). CLTCL1 is a member of the clathrin family, which plays essential roles in intracellular traffic and centrosomal stabilization (24). Mutations or the abnormal expression of CLTCL1 have been reported to be associated with breast cancer, meningioma and pulmonary valve stenosis (25). Further functional studies are required to verify whether the mutations in the aforemenioned two genes cause functional change. Other variants in the ARID2, GNAS, MLH1, NACA, NIN, POT1, TFRC and MLL3 genes did not segregate perfectly with lung cancer. It is also possible that one or several alterations that were not validated are the causative mutations for this familial case.
Overall, the present study focused on a Chinese family in which four members had developed lung cancer. Whole exome sequencing was conducted using the peripheral blood sample of the proband, and 11 variants were validated in more samples. Two potentially functional variants were identified in two genes, PDE4DIP and CLTCL1. Although the present study has certain limitations, this study provides useful sources for the further study of hereditary lung cancer.
References
- 1.World Health Organization GLOBOCAN 2012, corp-author. http://globocan.iarc.fr/Default.aspx. Estimated cancer incidence, mortality and prevalence worldwide in 2012. 2014 Oct 10; Accessed. [Google Scholar]
- 2.Suh JH. Current readings: Pathology, prognosis, and lung cancer. Semin Thorac Cardiovasc Surg. 2013;25:14–21. doi: 10.1053/j.semtcvs.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Detterbeck FC, Postmus PE, Tanoue LT. The stage classification of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(Suppl):e191S–e210S. doi: 10.1378/chest.12-2354. [DOI] [PubMed] [Google Scholar]
- 4.VanSchil PE, Sihoe AD, Travis WD. Pathologic classification of adenocarcinoma of lung. J Surg Oncol. 2013;108:320–326. doi: 10.1002/jso.23397. [DOI] [PubMed] [Google Scholar]
- 5.Kris MG, Johnson BE, Kwiatkowski DJ, et al. Identification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: The NCI's lung cancer mutation consortium (LCMC) J Clin Oncol. 2011;29:CRA7506. [Google Scholar]
- 6.Palmer JD, Zaorsky NG, Witek M, Lu B. Molecular markers to predict clinical outcome and radiation induced toxicity in lung cancer. J Thorac Dis. 2014;6:387–398. doi: 10.3978/j.issn.2072-1439.2013.12.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ulivi P, Zoli W, Capelli L, Chiadini E, Calistri D, Amadori D. Target therapy in NSCLC patients: Relevant clinical agents and tumour molecular characterisation. Mol Clin Oncol. 2013;1:575–581. doi: 10.3892/mco.2013.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oxnard GR, Binder A, Jänne PA. New targetable oncogenes in non-small-cell lung cancer. J Clin Oncol. 2013;31:1097–1104. doi: 10.1200/JCO.2012.42.9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 10.Miki Y, Swensen J, ShattuckEidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 11.Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 12.Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J, Lindblom A, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 13.van Noesel J, van der Ven WH, van Os TA, Kunst PW, Weegenaar J, Reinten RJ, Kancha RK, Duyster J, van Noesel CJ. Activating germline R776H mutation in the epidermal growth factor receptor associated with lung cancer with squamous differentiation. J Clin Oncol. 2013;31:e161–e164. doi: 10.1200/JCO.2012.42.1586. [DOI] [PubMed] [Google Scholar]
- 14.Ohtsuka K, Ohnishi H, Kurai D, Matsushima S, Morishita Y, Shinonaga M, Goto H, Watanabe T. Familial lung adenocarcinoma caused by the EGFR V843I germ-line mutation. J Clin Oncol. 2011;29:e191–e192. doi: 10.1200/JCO.2010.31.4492. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto H, Higasa K, Sakaguchi M, Shien K, Soh J, Ichimura K, Furukawa M, Hashida S, Tsukuda K, Takigawa N, et al. Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. J Natl Cancer Inst. 2014;106:djt338. doi: 10.1093/jnci/djt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ettinger DS, Akerley W, Bepler G, et al. NCCN Non-Small Cell Lung Cancer Panel Members: Non-small cell lung cancer. J Natl Compr Canc Netw. 2010;8:740–801. doi: 10.6004/jnccn.2010.0056. [DOI] [PubMed] [Google Scholar]
- 20.Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:945–950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong KA, Wilson J, Russo A, et al. Intersectin (ITSN) family of scaffolds function as molecular hubs in protein interaction networks. PLoS One. 2012;7:e36023. doi: 10.1371/journal.pone.0036023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinayagam A, Stelzl U, Foulle R, et al. A directed protein interaction network for investigating intracellular signal transduction. Sci Signal. 2011;4:rs8. doi: 10.1126/scisignal.2001699. [DOI] [PubMed] [Google Scholar]
- 23.DeWan AT, Egan KB, Hellenbrand K, Sorrentino K, Pizzoferrato N, Walsh KM, Bracken MB. Whole-exome sequencing of a pedigree segregating asthma. BMC Med Genet. 2012;13:95. doi: 10.1186/1471-2350-13-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu SH, Towler MC, Chen E, Chen CY, Song W, Apodaca G, Brodsky FM. A novel clathrin homolog that co-distributes with cytoskeletal components functions in the trans-Golgi network. EMBO J. 2001;20:272–284. doi: 10.1093/emboj/20.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sens-Abuázar C, Napolitano E, Ferreira E, Osório CA, Krepischi AC, Ricca TI, Castro NP, da Cunha IW, Maciel Mdo S, Rosenberg C, Brentani MM. Down-regulation of ANAPC13 and CLTCL1: Early events in the progression of preinvasive ductal carcinoma of the breast. Transl Oncol. 2012;5:113–123. doi: 10.1593/tlo.11280. [DOI] [PMC free article] [PubMed] [Google Scholar]