

Abstract
Chemoresistance allows for disease to recur and ultimately causes the death of most breast cancer patients. This scenario is particularly relevant in patients harboring triple-negative breast cancer (TNBC) tumors for which there are no effective FDA-approved drugs. However, a recent study determined that TNBCs can be segregated into 6 genetically distinct subtypes that do in fact exhibit differential rates of pathological complete response (pCR) to standard-of-care chemotherapies. Of these, the mesenchymal and mesenchymal stem-like subtypes of TNBCs exhibit the lowest rates of pCR when treated with standard-of-care chemotherapies. WAVE3 is an actin-cytoskeleton remodeling protein, and recent studies have highlighted a potential role for WAVE3 in promoting tumor progression and metastasis in TNBC. However, whether WAVE3 activity is involved in the development of chemoresistance in TNBCs remains unclear. Here we show that loss of WAVE3 expression resensitizes human TNBC cells to doxorubicin and docetaxel, as measured by increased apoptosis and cell death. We also show that WAVE3 knockdown in the chemotherapy-treated TNBC cells results in inhibition of STAT1 phosphorylation, as well as a significant decrease in expression levels of its downstream effector HIF-1α. Since HIF-1α is a major activator of VEGF-A production, and therefore a stimulator of tumor angiogenesis, loss of HIF-1α in the WAVE3-knockdown cells resulted in the inhibition the chemotherapy-mediated VEGF-A secretion and the downstream activation of angiogenesis, a phenomenon that often accompanies chemoresistance. Our data identify a critical role of WAVE3 in sensitizing TNBC to chemotherapy by inhibiting the STAT1→HIF-1α→VEGF-A signaling axis, and support the possibility that WAVE3 inhibition may be a promising target for TNBC cancer therapy.
Keywords: apoptosis, angiogenesis, chemoresistance, HIF-1α, metastasis, STAT1, triple-negative breast cancer, WAVE3
Abbreviations
- BC
breast cancer
- cDNA
complementary-DNA
- Doc
Docetaxel
- Dox
Doxorubicin
- ELISA
enzyme-linked immunosorbent assay
- EMT
epithelial to mesenchymal transition
- ER
estrogen receptor
- HIF-1α
Hypoxia inducible factor 1 α
- HUVEC
human umbilical vascular endothelial cell
- JAK
janus kinase
- M TNBC
mesenchymal TNBC
- mRNA
mRNA
- MS-L TNBC
Mesenchymal Stem-like
- pCR
pathological complete response
- PCR
polymerase Chain Reaction
- PR
progesterone receptor
- RNA
Ribonucleic Acid
- RT-PCR
Reverse Transcriptase PCR
- STAT
signal transducer and activator of transcription
- TNBC
triple-negative breast cancer
- VEGF
vascular endothelial growth factor
- WAVE3
Wsckott Aldrich Verprolin Family member 3
Introduction
Metastasis is responsible for the deaths of ∼90% of patients with solid tumors, including those originating in the breast.1 In fact, metastatic breast cancer (BC) is the 2nd leading cause of death in woman in the United States, annually accounting for nearly 40,000 deaths and 228,000 new cases of invasive disease.1 BC metastasis is incurable and results in a median survival of only 1.5 to 3 y Moreover, disseminated BC cells can escape clinical detection by remaining dormant for years before reemerging as incurable secondary tumors that are resistant to the chemotherapies that appeared to be originally effective against the primary tumor.2,3 This problem is magnified in human BCs, which are heterogeneous and comprised of at least 5 genetically distinct subtypes,4-7 and perhaps as many as 10 distinct molecular subtypes.8,9 Among individual BC subtypes, those classified as TNBCs are especially lethal due to their highly metastatic behavior and propensity to rapidly recur.10-14 As a group, TNBCs lack expression of hormone receptors (ER-α and PR) and fail to exhibit amplification at the ErbB2/HER2 locus.10-14 These unique molecular features of TNBCs have precluded the development of FDA-approved targeted therapies against this BC subtype.15-21 Moreover, the acquisition of chemoresistance is the main cause of disease recurrence and eventual death of BC patients,22-25 particularly those harboring TNBC tumors.15-21 Indeed, TNBCs are highly proficient at acquiring chemoresistant phenotypes and recurring through mechanisms that remain incompletely understood. Likewise, a recent study determined that TNBCs can be segregated into 6 genetically distinct subtypes8 that exhibit differential rates of pathological complete response (pCR) to standard-of-care chemotherapies.9 Interestingly, the mesenchymal (M) and mesenchymal stem-like (MS-L) subtypes of TNBCs exhibit the lowest rates of pCR when treated with standard-of-care chemotherapies.9
WAVE3 belongs to the WASP/WAVE family of actin-binding proteins that play broad roles in regulating cell shape/morphology, actin polymerization, and cytoskeleton remodeling, all of which are coupled to cell motility and invasion.26-34 We demonstrated the necessity of WAVE3 expression to drive the invasion and metastasis of human TNBCs in part by stimulating: (i) expression of MMPs 1, 3, and 9; (ii) formation of invadopodia; and (iii) activation of p38 MAPK.27-32,35 Moreover, we also identified WAVE3 as a novel substrate for the nonreceptor protein kinase c-Abl. When c-Abl phosphorylates tyrosine residues in WAVE3, TNBC lamellipodia formation and invasion in TNBC are enhanced.31 Along these lines, we also observed that WAVE3 activated EMT and metastasis in TNBCs.33,34,36,37 Clinically, aberrant WAVE3 expression is a strong indicator of human BC progression, as well as predictor for TNBC size, stage, and lymph node metastasis.26,38 We have found that WAVE3 expression levels determine the sensitivity of TNBC cells to apoptosis and cell death driven by TNFα,35 as knockdown of WAVE3 by siRNA or shRNA approaches enhances susceptibility of MDA-MB-231 TNBC cells to the TNFα-driven apoptosis and cell death.35 How WAVE3 drives chemoresistance in TNBC, however, remains poorly understood. Because the acquisition of metastatic phenotypes coincides with development of chemoresistance,22-25 we sought to investigate the molecular mechanisms whereby WAVE3 determines the sensitivity of TNBC subtypes to chemotherapy. We found that WAVE3 expression regulates tumor angiogenesis, a response that supports chemoresistance and metastasis. It does so by encouraging a WAVE3→STAT1→HIF-1α→VEGF-A signaling axis, thereby imparting chemoresistance to TNBCs.
Results
Knockdown of WAVE3 expression sensitizes TNBC cells to chemotherapy-driven apoptosis and cell death
Our recently published data had shown that loss of WAVE3 expression in MDA-MB-231 cells that belong to the MS-L of TNBC subtype resulted in a significant increase in cell apoptosis and cell death by re-sensitizing these cells to the cytotoxic effect of TNF-α.35 We therefore sought to determine whether this sensitivity extended to other chemotherapeutic regimens that are commonly used to treat patients with TNBC, namely doxorubicin and docetaxel. MDA-MB-231 cells engineered to express nontargeting shRNA or that against WAVE3 were treated with either doxorubicin (50 nM)39,40 or docetaxel (5 nM)41,42 for 12h (Fig. 1A), and apoptosis or cell death was assessed by flow cytometry analysis of Annexin V and propidium iodide, respectively. Under these treatment conditions, the population of apoptotic cells in the WAVE3-knockdown cells was increased by 82% and 86 % (P < 0.05) by doxorubicin and docetaxel, respectively, as compared to the control non-targeting shRNA-transfected (Ctrl-sh) cells (Fig. 1B and 1C). Cell death was also increased by 76% and 40% (P < 0.05) in the WAVE3-knockdown population treated with doxorubicin and docetaxel, respectively, as compared to the control (Ctrl-sh) cells (Fig. 1B and 1C). These findings were duplicated in BT549, a mesenchymal BC cell line of TNBC subtype (Fig. 1D–F), which together with MDA-MB-231, represent the 2 subtypes of TNBC that were found to be the less responsive to chemotherapy.9 Of note, the chemotherapeutic agents had no effect on WAVE3 expression in either MDA-MB-231 or BT549 cells (Fig. 1A and 1D).
Figure 1.
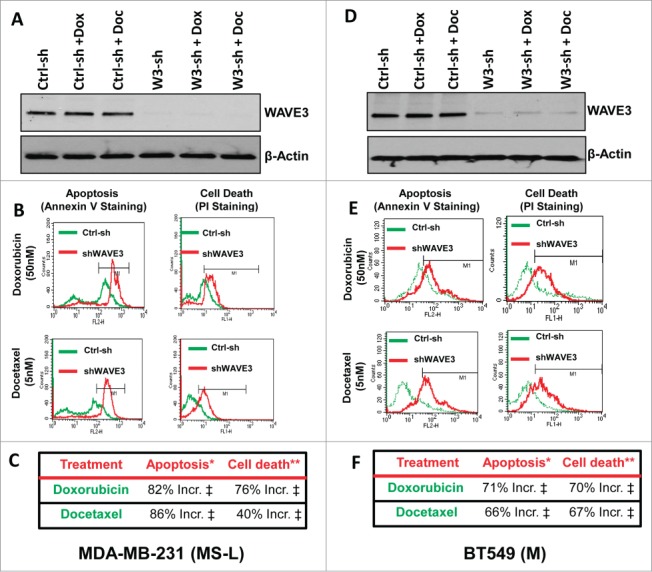
Knockdown of WAVE3 expression sensitizes TNBC cells to chemotherapy-driven apoptosis and cell death (flow cytometry assay). (A & D) Western blot analysis with the indicated antibodies of cell lysates from control shRNA (ctrl-sh)- or sh-WAVE3-expressing (W3-sh) MDA-MB-231 (A) or BT549 (D) cells after doxorubicin (Dox) or docetaxel (Doc) treatment. β-actin served as a loading control. (B & E) Representative histograms using flow cytometry of control shRNA (ctrl-sh, green)- or sh-WAVE3-expressing ( shWAVE3, red) MDA-MB-231 (B) of BT549 (E) cells after doxorubicin or docetaxel treatment and staining by Annexin V for apoptosis and by Propidium Iodide for cell death. (C & F) quantification of apoptosis and cell death in MDA-MB-231 (C) and BT549 (F). Data are the mean % increases in cell apoptosis (*) or death (**) elicited by WAVE3-deficiency normalized to the values found in their untreated control counterparts. Data are representative of 3 independent experiments (‡, P <0.05; Student's t-test).
Immunocytochemistry was used as an independent approach to demonstrate that WAVE3-knockdown sensitized the cells to the chemotherapeutic agents. Both Caspase 3 (Fig. 2A and 2B) and Annexin V (Fig. 2C and 2D) staining was increased in the WAVE3-knockdown cells treated with either doxorubicin or docetaxel. We also confirmed our findings using immunoblotting assay to assess for the amounts of cleaved Caspase 3, a further indication of increased apoptosis. We found the levels of cleaved Caspase 3 (Ccaspase3) to increase by at least fold5- in the WAVE3-knokdown MDA-MB-231 cells when treated with either doxorubicin or docetaxel, compared to the non-targeting shRNA control cells (Fig. 2E). Taken together, these data demonstrate that loss of WAVE3 is critical for the resensitization to the chemotherapy-induced apoptosis and cell death in TNBCs, and therefore, the overexpression of WAVE3 in TNBCs may play a major role in eliciting chemoresistance of these TNBC subtypes.
Figure 2.
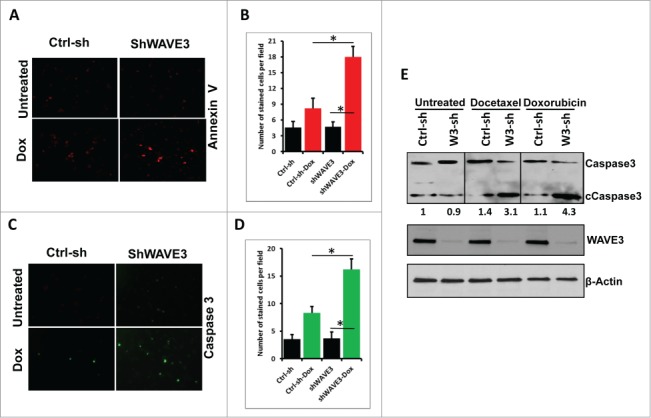
Knockdown of WAVE3 expression sensitizes TNBC cells to chemotherapy-driven apoptosis and cell death (immunofluorescence and immunoblotting assays). (A & C) Representative confocal images of Ctrl-sh and sh-W3 MDA-MB-231 cells stained Annexin V (A) and cleaved caspase3 (C) before and after treatment with doxorubicin (Dox). (B & D) Quantification of Annexin V staining levels (B) and Caspase 3 staining levels (D). Data are representative of 3 independent experiments (*, P <0.05; Student's t-test). (E) Western blot analysis with the indicated antibodies of cell lysates form the control shRNA (ctrl-sh)- or sh-WAVE3-expressing (W3-sh) MDA-MB-231 (A) of BT549 (B) cells after doxorubicin (Dox) or docetaxel (Doc) treatment. β-actin served as a loading control. The numbers below the cleaved Caspase 3 panel indicate the fold change of cleaved Caspase 3 levels, as compared to the untreated Ctrl-sh cells.
Loss of WAVE3 expression inhibits the chemotherapy-mediated induction of VEGF-A secretion in TNBC cells
Tumor growth at both primary and metastatic sites is driven by angiogenesis, which is heavily dependent upon VEGF-A production and secretion by the transformed cells (see43). Tumor angiogenesis has also been shown to be induced by chemotherapy in several cancers (see44), contributing to the failure of chemotherapy. We have previously shown that of WAVE3 is required for the progression and metastasis of BC by regulating VEGF-mediated activation of tumor angiogenesis.32 Hence, we sought to investigate whether WAVE3 is also involved in the chemotherapy-induced activation of angiogenesis by affecting secretion of VEGF-A by breast cancer cells. By ELISA, we found MDA-MB-231 cells treated with either doxorubicin or docetaxel secreted increased levels of VEGF-A (15–20% more, P < 0.05) compared to untreated cells (Fig. 3A), findings consistent with a previous report.45 Secreted VEGF-A levels were, however, significantly reduced (∼fold2-, P < 0.05) in WAVE3-knockdown cells (sh-W3) as compared to their parental counterparts (Ctrl-sh; Fig. 3A), which is concordant with our previously published study.32 Treatment of the WAVE3-knockdown cells with either doxorubicin (Dox) or docetaxel (Doc) failed to induce VEGF-A secretion, which remained as low as in the untreated WAVE3-knockdown cells (Fig. 3A). Thus, loss of WAVE3 impaired the chemotherapy-mediated induction of VEGF-A in cancer cells.
Figure 3.

Loss of WAVE3 expression inhibits the chemotherapy-mediated induction of VEGF-(A)secretion in TNBC cells. (A) Quantification of secreted VEGF-A in the conditioned media of control shRNA (ctrl-sh)- or sh-WAVE3-expressing (sh-W3) MDA-MB-231 cells after doxorubicin (Dox) or Docetaxel (Doc) treatment using ELISA. Data are representative of 3 independent experiments (*, P <0.05; Student's t-test). (B) Western blot analysis with the indicated antibodies of cell lysates form the control shRNA (ctrl-sh)- or sh-WAVE3-expressing (W3-sh) MDA-MB-231 cells after doxorubicin (Dox) or docetaxel (Doc) treatment. β-actin served as a loading control.
Loss of WAVE3 expression inhibits endothelial angiogenesis that is induced by chemotherapy
We next investigated the effect of loss of WAVE3 on inhibiting the chemotherapy-induced VEGF-A secretion by cancer cells using an in vitro angiogenesis assay. Early passage HUVECs were seeded onto growth-factor-reduced Matrigel with or without VEGF-A supplementation. Over time, the HUVECs supplemented with VEGF-A (VEGF) organized in tube-like networks (Fig. 4A). The HUVECs that were supplemented with the conditioned media derived from the non-targeting sh-RNA (Ctrl-sh) cells formed closed and regularly shaped tube-like structures in the absence of added VEGF-A, resembling those formed with addition of VEGF-A. Similarly, HUVECs supplemented with conditioned media from the Ctrl-sh treated with doxorubicin (Dox) or docetaxel (Doc), formed typical tube-like structures. The number of tube-like structures was 10 to 20% higher (P <0.05) in the cells treated with conditioned-media produced by cells treated with chemotherapeutic agents as compared to their untreated counterparts. In contrast, HUVECs supplemented with the conditioned media derived from the WAVE3-knockdown cells formed significantly fewer (∼fold4- decrease, P < 0.05) tubes and, in most cases, failed to fully close (Fig. 4A and 4B). More importantly, treatment with chemotherapeutic drugs failed to rescue the inhibitory effect that is caused by loss of WAVE3. These results are consistent with our findings that the WAVE3-knockdown cells secrete lower VEGF-A levels and that treatment with chemotherapeutic agents was unable to negate this effect (Fig. 4A and 4B). Additionally, pre-incubation of HUVECs supplemented with conditioned media from control cells with a soluble VEGFR inhibitor (sFLT) completely inhibited tube formation by the HUVECs (Fig. 4A and 4B), verifying that VEGF-A is the major angiogenic factor within by the conditioned media.
Figure 4.

Loss of WAVE3 expression inhibits endothelial angiogenesis that is induced by Chemotherapy. (A) Bright field microscopy of tube formation structures of HUVECs treated with the serum-free media (SFM) supplemented with VEGF-A (VEGF), SFM supplemented with VEGF-A and sFLT, a soluble VEGFR inhibitor (VEGF + sFLT), or conditioned media from MDA-MB-231 cells with the indicated treatments. (B) Quantification of tube formation under the indicated conditions. The number of closed tubes was counted in 12 different fields and plotted as the average of number of closed tubes per field ±SE . Each assay was repeated at least 3 times. (* and **, P <0.05; Student's t-test).
Loss of WAVE3 inhibits activation of the STAT1-HIF-1α signaling cascade by chemotherapy in TNBCs
Hypoxia-inducible factor-1α (HIF-1α) is a well-known inducer of VEGF-A, which in turn activates angiogenesis in both physiological and pathological settings. A recent study has shown that resistance to chemotherapy in cancer was in part mediated through the activation the HIF1-α downstream of STAT1, even under normoxic conditions.45 We therefore sought to investigate the possible involvement of WAVE3 in the chemotherapy-mediated activation of STAT1 and its downstream effector HIF-1α. Treatment of either MDA-MB-231 or BT549 cells with doxorubicin or docetaxel resulted in more than fold3- increase in phosphorylated levels of STAT1 and more than fold2- increase in expression levels of HIF-1α in both cell lines (Fig 5A and 5D, respectively). However, knockdown of WAVE3 expression in either MDA-MB-231 (Fig. 5C) or BT549 (Fig. 5F) inhibited the doxorubicin- and docetaxel-mediated activation of STAT1 and its downstream effector HIF1-α (Fig. 5B and 5E). In fact, in the doxorubicin and the docetaxel treated cells, the levels of phosphorylated STAT1 were reduced by at least fold2- in the WAVE3-knockdown (W3-sh) cells compared to the non-targeting shRNA (Ctrl-sh) counterparts. Concordant with the reduction in the levels phospho-STAT1, HIF-1α expression levels were also reduced by at least fold2- in the doxorubicin- and docetaxel-treated WAVE3-knockdown cells (Fig. 5B and 5E). Therefore, our data implicate WAVE3 in the chemotherapy-mediated activation of STAT1 and HIF-1α, which in turn stimulates VEGF-A secretion and angiogenesis. We further demonstrated that the chemotherapy induction of HIF-1α is mediated specifically downstream of STAT1. siRNA knockdown of STAT1 in MDA-MB-231 cells resulted in more than fold5- reduction (P < 0.05) in the doxorubicin-mediated activation levels of STAT1 (pSTAT1), when compared to pSTAT1 levels in the non-targeting siRNA (NT-si) cells treated with doxorubicin (Fig. 6).
Figure 5.

Loss of WAVE3 inhibits the STAT1-HIF-1α signaling cascade that is usually activated by chemotherapy in TNBCs. Western blot analysis with the indicated antibodies of cell lysates form the control shRNA (ctrl-sh)- or sh-WAVE3-expressing (W3-sh) MDA-MB-231 cells after doxorubicin (Dox) or docetaxel (Doc) treatment. β-actin served as a loading control. The numbers below the HIF-1α, p-STAT1 and WAVE3 panels indicate the fold change of HIF-1α, p-STAT1 and WAVE3 levels, respectively, as compared to the untreated Ctrl-sh cells.
Figure 6.

Loss of STAT1 expression inhibits the chemotherapy-mediated induction of HIF-1α. Western blot analysis with the indicated antibodies of cell lysates form the control siRNA (ctrl-si)- or siSTAT1-expressing MDA-MB-231 cells after doxorubicin (Dox) or docetaxel (Doc) treatment. β-actin served as a loading control.
Discussion
We previously demonstrated aberrant WAVE3 expression to be sufficient in driving the metastasis of TNBC subt-ypes.26-34,38,46-51 Specifically, we and others have shown that the WAVE3-mediated activation of the metastatic phenotype in TNBC is due in part to the stimulation of VEGF-A-secretion leading to enhanced tumor angiogenesis.32,46,52 We also reported the role of WAVE3 in mediating sensitization of TNBC cells to apoptosis and death.35 On the other hand, several studies have linked the JAK/STAT pathway to chemoresistance in different types of cancers, including breast cancer.45,53,54 In particular, a recent study has shown that a signaling axis involving STAT1/HIF-1α/VEGF-A was activated when breast cancer tumors became resistant to doxorubicin, leading to the stimulation of tumor angiogenesis and enhanced metastasis.45 Given the established role of WAVE3 in the regulation of both apoptosis and cell death,35 as well as tumor angiogenesis,32 we sought to investigate the link between WAVE3 and the STAT1/HIF-1α signaling axis in TNBC chemoresistance and tumor angiogenesis.
We applied a combination of genetic and pharmacological manipulations, as well as different biochemical and cell imaging assays, to investigate the role WAVE3 in the modulation of the STAT1/HIF-1α signaling axis. We showed that loss of WAVE3 in 2 TNBC cells results in: (i) resensitization to apoptosis and cell death that are driven by chemotherapeutics; (ii) resensitization to drug treatment is accompanied by inhibition of VEGF-induced angiogenesis, and, more importantly, (iii) inhibition of the STAT1→HIF-1α→VEGF-A signaling axis that is frequently activated during tumor chemoresistance and unfortunately results in tumor spread and the development of therapeutics-resistant metastasis (Fig. 7). Our data were observed in 2 different cell lines (MDA-MB-231 and BT549), providing support for the generality of our observations. BT549 and MDA-MB-231 cells belong to the mesenchymal and the mesenchymal-stem like subtypes of TNBCs, respectively.8 Interestingly, BC tumors generated from these 2 TNBC subtypes were shown to exhibit the lowest pCR when treated with conventional chemotherapy.9 Therefore, WAVE3 played a role in the resensitization of these 2 TNBC subtypes to chemotherapy. Whether, this effect could apply to other TNBC subtypes remains to be investigated.
Figure 7.

Model describing how WAVE3 modulates the molecular signaling pathway that regulates the chemoresistance-mediated activation of tumor angiogenesis and subsequent tumor recurrence and metastasis.
Our data show that therapeutic treatment has no effect of WAVE3 expression levels (Fig. 1) while both STAT1 and HIF-1α are activated by chemotherapy (Fig. 5). On the other hand, knockdown of WAVE3 has an inhibitory effect on STAT1 signaling (Fig. 6). Knockdown of STAT1 not only inhibited STAT1 expression and activity, but also that of HIF-1α, and did so without affecting WAVE3 expression levels (Fig. 6). Thus, the data clearly places WAVE3 upstream of STAT1 (Fig. 7). Inhibition of STAT1/HIF-1α pathway results in a significant activation of apoptosis and cell death, and inhibition of VEGF-A-induced angiogenesis after treatment with chemotherapy (Fig. 7). A study by Abasanz-Puig and colleagues55 showed that STAT1 has a negative effect on HIF-1α in the vascular smooth muscle cells, which seems opposite to our findings as well as in other published studies in cancer cells.45 One potential explanation for the differential effect of STAT1 on HIF-1α expression; i.e. inhibition in the vascular smooth muscle cells versus activation in cancer cells, is that the function of STAT1 may be different and even opposing depending on the cell type and the physiological conditions. The vascular smooth muscle cells represent the normal physiological environment, while the triple-negative breast cancer cells represent a pathological environment where many signaling pathways are dysregulated, including that of STAT1. Additionally, while doxorubicin treatment of the WAVE3-knockdown cells almost completely inhibited phosphorylation of STAT1 (Fig 5B), it did not have such a dramatic effect on HIF-1α expression levels. Therefore, additional mechanisms other than those requiring WAVE3-STAT1 axis are more likely involved in the regulation of HIF-1α expression. The intricate interplay between WAVE3 and the STAT1 signaling remains to be elucidated in future studies. Also remaining to be determined is how WAVE3 modulates the STAT1-mediated regulation of HIF-1α→VEGF-A→tumor angiogenesis during treatments with therapeutic agents.
In conclusion, we have identified a novel role of WAVE3 in STAT1 signaling and in the chemoresistance-induced inhibition of apoptosis and cell death via activation of tumor angiogenesis (Fig. 7). Extending our investigations to cell lines and human biopsy samples representative of all TNBC subtypes is the next logical step in these studies in the future when these reagents become available to us, and will certainly strengthen these lines of research. We therefore propose that the regulatory functions of WAVE3 may provide a novel treatment option for TNBCs, which could reduce metastasis-related mortality in TNBC patients.
Experimental Procedures
Antibodies
Rabbit anti-WAVE3 (1:1000), rabbit anti-Caspase 3 (1:1000), rabbit anti-HIF-α (1:1000), rabbit anti-phospho-STAT1 (Y701) (1:1000), and rabbit anti-total-STAT1 (1:1000) were from Cell Signaling. Goat horseradish peroxidase-conjugated anti-mouse IgG (1:5000) and goat horseradish peroxidase-conjugated anti-rabbit IgG (1:5000) from Calbiochem; and Alexa 488-conjugated anti-rabbit IgG and Alexa 568-conjugated anti-mouse IgG are from Invitrogen. Vecta-shield with 4′,6-diamidino-2-phenylindole was from Vector Laboratories. Gel electrophoresis reagents were from Bio-Rad. Doxorubicin and docetaxel purchased from Sigma as powders, were dissolved in DMSO and diluted at least 1000 fold as described.
siRNA and shRNA gene expression knockdown
We used the SignalSilence STAT1 siRNA (Cell Signaling Technology) for transient knockdown of STAT1 expression. Stable knockdown of WAVE3 was achieved through transfection of MDA-MB-231 and BT549 BC cells with WAVE3 MISSION shRNA clones (Sigma) followed by puromycin selection of positive clones as previously described.32,52 Non-targeting shRNA served as a control.
Cell culture
Human BT549 (Mesenchymal) and MDA-MB-231 (Mesenchymal stem-like) TNBC cells8 were purchased from American Type Culture Collection (ATCC) and were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units of penicillin/ml, 100 μg of streptomycin/ml.
Real-time quantitative–RT-PCR
Total RNA was extracted from cancer cell lines or tumor tissue using TRIzol reagent, following to the manufacturer's (Invitrogen) instructions. cDNA was generated and used as a template for quantitative RT-PCR performed as previously descr-ibed.26,33 qRT-PCR was perfor-med using the respective gene-specific primers (SABiosciences, Valencia, CA) and the RT2 SYBR Green/Fluorescein qPCR Master Mix (SABiosciences) following the manufacturer's instructions. qPCR was performed on the BioRad iCycler PCR system (BioRad, Berkeley, CA) where the reaction mixtures were incubated at 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The cycle threshold (Ct) values were calculated with SDS 1.4 software (Bio-Rad). The expression levels of each transcript were normalized using the 2−ΔΔCt method56,57 relative to GAPDH. The ΔCt was calculated by subtracting the Ct values of GAPDH from the Ct values of the transcript of interest. The ΔΔCt was then calculated by subtracting ΔCt of the matching normal human breast tissue from the ΔCt of cancer tissues, or the ΔCt of MCF10A cell line for the established cancer cell lines. Fold change in individual gene expression was calculated according to the equation 2-ΔΔCt.
Flow cytometry
MDA-MB-231 or BT549 cells, transfected with either the control non-targeting shRNA or the shWAVE3, were propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and puromycin (5 μg/mL). Subconfluent cell cultures were treated with either the diluent or doxorubicin (50 nM), or docetaxel (5 nM) for 12 h, after which cells were detached by trypsinization and washed with Hank's Balanced Salt Solution with Ca2+, Mg2+, and 0.1% BSA. Cells were processed for flow cytometry as described by the manufacturer (Roche In-Situ Cell Death Detection Kit, Fluorescein). Annexin V and propidium iodide was used to quantify apoptotic and dead cells, respectively. All data were acquired in a BD LSRII instrument and analyzed with FlowJo 7.6.3 (Treestar) software.
Immunoblot analysis
Whole cell lysates containing similar amounts of total protein (∼50 μg) were resolved on a 10% acrylamide gels in SDS, followed by transfer to nitrocellulose (Bio-Rad, Hercules, CA) or Immobilon-P (Millipore, Billerica, MA) membranes using the Bio-Rad gel and transfer apparatus. Membranes were incubated in 5% whole milk or bovine serum albumin for 1 hour at room temperature, washed with phosphate-buffered saline (PBS), followed by incubation with the primary antibody (as specified) overnight at 4°C. Membranes were then washed and incubated in the appropriate secondary antibody at room temperature for 1 hour, and immunocomplexes were visualized using the Western Lights chemiluminescence detection kit from Perkin-Elmer (Boston, MA). Signals were quantified using the ImageJ software according to the parameters described in ImageJ user guide (http://rsbweb.nih.gov/ij/docs/guide/146.html). Average values from 3 different blots are presented.
Immunofluorescence Microscopy- Cells were grown on glass coverslips, treated with either doxorubicin (50 nM) or docetaxel (5nM) for 6h, and fixed in 4% paraformaldehyde for 20 min in PBS at room temperature and washed with PBS. The cells were then permeabilized in 0.2% Triton X-100 in PBS for 15 min, washed again with PBS, and incubated in the blocking solution containing 5% bovine serum albumin (Sigma) in PBS for 2 h at room temperature. Primary as well as secondary antibodies were diluted to the recommended concentration in 5% bovine serum albumin in PBS. Cells were incubated with the primary antibody for 1 h, washed with PBS, and then incubated with the secondary antibody for 1 h. The coverslips were mounted on object slides using Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA). Fluorescence images were captured using a Nikon TE2000-E inverted microscopy. Signals (number of positively stained cells) were quantified using the ImageJ software according to the parameters described in ImageJ user guide (http://rsbweb.nih.gov/ij/docs/guide/146.html). Average values of at least 5 different fields were plotted.
Tube formation assay
Human umbilical vein endothelial cells (HUVECs)-based tube formation assays were performed as previously described.58 HUVECs were provided by Dr. Paul DiCorleto (Cleveland Clinic), supported with grant UL1TR000439 from NIH, and maintained in EGM Endothelial Cell Growth Medium. HUVECs were used between passages 2 and 4. Twelve-Multiwell culture plates were coated with 500 μl of growth factor-reduced Matrigel (BD Biosciences), and incubated at 37°C for 30 min. HUVECs were seeded at a density of 2.5 × 105 cells per well on polymerized Matrigel, treated as specified, and incubated at 37°C for 12 h. Bright-field images were obtained immediately after seeding and again after 18 h. Tube formation was analyzed and quantified using ImageJ (v1.34s).
VEGF ELISA assay
VEGF secreted in the conditioned media was quantified using the ELISA development kit and the polyclonal rabbit anti-human VEGF from PeproTech (Rocky Hill, NJ), following the manufacturer's instructions. In brief, 100 μl of capture antibody was transferred to a 96-well ELISA plate and incubated overnight at room temperature. Each well was washed 3 times and blocked by adding 300 μl of phosphate-buffered saline (PBS) containing 0.05% Tween 20, 5% sucrose and 0.05% NaN3 for a minimum of 1 hour. Following three washes, 100 μl of conditioned media was added per well, and the ELISA plate was incubated for 2 hours at room temperature. Subsequently, 100 μl of streptavidin HRP was added to each well, and the plate incubated for 30 minutes at room temperature. The plates were then washed and added 10 μl of ABTS substrate (Sigma-Aldrich) and the absorbance was measured after ∼20 min at 450 nm.
Statistical analyses
The data are presented as means ± standard deviations of at least 3 independent experiments. The results were tested for significance using an unpaired Student's t test. A p value less than 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Plow laboratory for critical comments and reading of the manuscript. We also thank Brian McCue for his technical assistance.
Funding
This work was supported in part by NIH grants P01 HL073311, R01 HL096062 and the Case Comprehensive Cancer Center grant (P30 CA43703).
Authors' Contributions
Conception and design was done by W Schiemann, EF Plow, K Sossey-Alaoui. Development of the methodology was done by G Davuluri. Analysis and interpretation of data was done by W Schiemann, EF Plow, K Sossey-Alaoui. Writing, review, and/or revision of the manuscript was done by G Davuluri, W Schiemann, EF Plow, K Sossey-Alaoui. The study was supervised by K Sossey-Alaoui.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012 Jan;62(1):10-29. [DOI] [PubMed] [Google Scholar]
- 2. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell 2006 Nov 17;127(4):679-95; PMID:17110329; http://dx.doi.org/ 10.1016/j.cell.2006.11.001 [DOI] [PubMed] [Google Scholar]
- 3. Li F, Tiede B, Massague J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res 2007 Jan;17(1):3-14; PMID:17179981; http://dx.doi.org/ 10.1038/sj.cr.7310118 [DOI] [PubMed] [Google Scholar]
- 4. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 2010 Sep;60(5):277-300; PMID:20610543 [DOI] [PubMed] [Google Scholar]
- 5. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature 2000 Aug 17;406(6797):747-52; PMID:10963602; http://dx.doi.org/ 10.1038/35021093 [DOI] [PubMed] [Google Scholar]
- 6. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001 Sep 11;98(19):10869-74. PMCID:PMC58566; PMID:11553815; http://dx.doi.org/ 10.1073/pnas.191367098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA 2003 Jul 8;100(14):8418-23. PMCID:PMC166244; PMID:12829800; http://dx.doi.org/ 10.1073/pnas.0932692100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011 Jul;121(7):2750-67. PMCID:PMC3127435; PMID:21633166; http://dx.doi.org/ 10.1172/JCI45014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, Valero V, Lehmann BD, Pietenpol JA, Hortobagyi GN, et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res 2013 Aug 15; 2013 Oct 1; 19(19):5533-5540; PMID:23948975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer 2009 Jun;9 Suppl 2:S73-S81. PMCID:PMC2919761; PMID:19596646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol 2010 Dec;7(12):683-92; PMID:20877296; http://dx.doi.org/ 10.1038/nrclinonc.2010.154 [DOI] [PubMed] [Google Scholar]
- 12. Finnegan TJ, Carey LA. Gene-expression analysis and the basal-like breast cancer subtype. Future Oncol 2007 Feb;3(1):55-63; PMID:17280502 [DOI] [PubMed] [Google Scholar]
- 13. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010 Nov 11;363(20):1938-48; PMID:21067385; http://dx.doi.org/ 10.1056/NEJMra1001389 [DOI] [PubMed] [Google Scholar]
- 14. Schneider BP, Winer EP, Foulkes WD, Garber J, Perou CM, Richardson A, Sledge GW, Carey LA. Triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res 2008 Dec 15;14(24):8010-8; PMID:19088017 [DOI] [PubMed] [Google Scholar]
- 15. Andre F, Dieci MV, Dubsky P, Sotiriou C, Curigliano G, Denkert C, Loi S. Molecular pathways: involvement of immune pathways in the therapeutic response and outcome in breast cancer. Clin Cancer Res 2013 Jan 1;19(1):28-33; PMID:23258741 [DOI] [PubMed] [Google Scholar]
- 16. Barton S, Swanton C. Recent developments in treatment stratification for metastatic breast cancer. Drugs 2011 Nov 12;71(16):2099-113; PMID:22035512; http://dx.doi.org/ 10.2165/11594480-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 17. Criscitiello C, Azim HA, Jr., Schouten PC, Linn SC, Sotiriou C. Understanding the biology of triple-negative breast cancer. Ann Oncol 2012 Aug;23 Suppl 6:vi13-vi18; PMID:23012296 [DOI] [PubMed] [Google Scholar]
- 18. Cristofanilli M. Advancements in the Treatment of Metastatic Breast Cancer (MBC): The Role of Ixabepilone. J Oncol 2012;2012:703858. PMCID:PMC3356906; PMID:22645612; http://dx.doi.org/ 10.1155/2012/703858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fojo T, Amiri-Kordestani L, Bates SE. Potential pitfalls of crossover and thoughts on iniparib in triple-negative breast cancer. J Natl Cancer Inst 2011 Dec 7;103(23):1738-40. PMCID:PMC3247787; PMID:22045362; http://dx.doi.org/ 10.1093/jnci/djr386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kerbel RS. Strategies for improving the clinical benefit of antiangiogenic drug based therapies for breast cancer. J Mammary Gland Biol Neoplasia 2012 Dec;17(3-4):229-39; PMID:23011602; http://dx.doi.org/ 10.1007/s10911-012-9266-0 [DOI] [PubMed] [Google Scholar]
- 21. Liedtke C, Kiesel L. Breast cancer molecular subtypes–modern therapeutic concepts for targeted therapy of a heterogeneous entity. Maturitas 2012 Dec;73(4):288-94; PMID:23020990; http://dx.doi.org/ 10.1016/j.maturitas.2012.08.006 [DOI] [PubMed] [Google Scholar]
- 22. Yardley DA. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int J Breast Cancer 2013;2013:137414. PMCID:PMC3707274; PMID:23864953; http://dx.doi.org/ 10.1155/2013/137414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adam MA. New prognostic factors in breast cancer. Adv Clin Exp Med 2013 Jan;22(1):5-15; PMID:23468257 [PubMed] [Google Scholar]
- 24. Droog M, Beelen K, Linn S, Zwart W. Tamoxifen resistance: From bench to bedside. Eur J Pharmacol 2013 Mar 29; 2013 Oct 5; 717(1-3):47-57; PMID: 23545365 [DOI] [PubMed] [Google Scholar]
- 25. Gnant M. Overcoming endocrine resistance in breast cancer: importance of mTOR inhibition. Expert Rev Anticancer Ther 2012 Dec;12(12):1579-89; PMID:23253223; http://dx.doi.org/ 10.1586/era.12.138 [DOI] [PubMed] [Google Scholar]
- 26. Kulkarni S, Augoff K, Rivera L, McCue B, Khoury T, Groman A, Zhang L, Tian L, Sossey-Alaoui K. Increased Expression Levels of WAVE3 Are Associated with the Progression and Metastasis of Triple Negative Breast Cancer. PLOS ONE 2012;7(8):e42895. PMCID:PMC3428347; PMID:22952619; http://dx.doi.org/ 10.1371/journal.pone.0042895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sossey-Alaoui K, Su G, Malaj E, Roe B, Cowell JK. WAVE3, an actin-polymerization gene, is truncated and inactivated as a result of a constitutional t(1;13)(q21;q12) chromosome translocation in a patient with ganglioneuroblastoma. Oncogene 2002 Aug 29;21(38):5967-74; PMID:12185600; http://dx.doi.org/ 10.1038/sj.onc.1205734 [DOI] [PubMed] [Google Scholar]
- 28. Sossey-Alaoui K, Head K, Nowak N, Cowell JK. Genomic organization and expression profile of the human and mouse WAVE gene family. Mamm Genome 2003 May;14(5):314-22; PMID:12856283; http://dx.doi.org/ 10.1007/s00335-002-2247-7 [DOI] [PubMed] [Google Scholar]
- 29. Sossey-Alaoui K, Li X, Ranalli TA, Cowell JK. WAVE3-mediated cell migration and lamellipodia formation are regulated downstream of phosphatidylinositol 3-kinase. J Biol Chem 2005 Jun 10;280(23):21748-55; PMID:15826941; http://dx.doi.org/ 10.1074/jbc.M500503200 [DOI] [PubMed] [Google Scholar]
- 30. Sossey-Alaoui K, Ranalli TA, Li X, Bakin AV, Cowell JK. WAVE3 promotes cell motility and invasion through the regulation of MMP-1, MMP-3, and MMP-9 expression. Exp Cell Res 2005 Aug 1;308(1):135-45; PMID:15907837; http://dx.doi.org/ 10.1016/j.yexcr.2005.04.011 [DOI] [PubMed] [Google Scholar]
- 31. Sossey-Alaoui K, Li X, Cowell JK. c-Abl-mediated phosphorylation of WAVE3 is required for lamellipodia formation and cell migration. J Biol Chem 2007 Sep 7;282(36):26257-65; PMID:17623672; http://dx.doi.org/ 10.1074/jbc.M701484200 [DOI] [PubMed] [Google Scholar]
- 32. Sossey-Alaoui K, Safina A, Li X, Vaughan MM, Hicks DG, Bakin AV, Cowell JK. Down-regulation of WAVE3, a metastasis promoter gene, inhibits invasion and metastasis of breast cancer cells. Am J Pathol 2007 Jun;170(6):2112-21. PMCID:PMC1899429; PMID:17525277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sossey-Alaoui K, Bialkowska K, Plow EF. The miR200 family of microRNAs regulates WAVE3-dependent cancer cell invasion. J Biol Chem 2009 Nov 27;284(48):33019-29. PMCID:PMC2785142; PMID:19801681; http://dx.doi.org/ 10.1074/jbc.M109.034553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sossey-Alaoui K, Downs-Kelly E, Das M, Izem L, Tubbs R, Plow EF. WAVE3, an actin remodeling protein, is regulated by the metastasis suppressor microRNA, miR-31, during the invasion-metastasis cascade. Int J Cancer 2011 Sep 15;129(6):1331-43. PMCID:PMC3081370; PMID:21105030; http://dx.doi.org/ 10.1002/ijc.25793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davuluri G, Augoff K, Schiemann WP, Plow EF, Sossey-Alaoui K. WAVE3-NFkappaB Interplay Is Essential for the Survival and Invasion of Cancer Cells. PLOS ONE 2014;9(10):e110627. PMCID:PMC4199728; PMID:25329315; http://dx.doi.org/ 10.1371/journal.pone.0110627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Augoff K, Das M, Bialkowska K, McCue B, Plow EF, Sossey-Alaoui K. miR-31 is a broad regulator of beta1-integrin expression and function in cancer cells. Mol Cancer Res 2011 Nov;9(11):1500-8. PMCID:PMC3219821; PMID:21875932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Augoff K, McCue B, Plow EF, Sossey-Alaoui K. miR-31 and its host gene lncRNA LOC554202 are regulated by promoter hypermethylation in triple-negative breast cancer. Mol Cancer 2012;11:5. PMCID:PMC3298503; PMID:22289355; http://dx.doi.org/ 10.1186/1476-4598-11-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sossey-Alaoui K. Surfing the big WAVE: Insights into the role of WAVE3 as a driving force in cancer progression and metastasis. Semin Cell Dev Biol 2012 Oct 29; 2013 Apr; 24(4):287-297; PMID:23116924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hannesdottir L, Tymoszuk P, Parajuli N, Wasmer MH, Philipp S, Daschil N, Datta S, Koller JB, Tripp CH, Stoitzner P, et al. Lapatinib and doxorubicin enhance the Stat1-dependent antitumor immune response. Eur J Immunol 2013 Jun 12; PMID:23843024 [DOI] [PubMed] [Google Scholar]
- 40. Rovida A, Castiglioni V, Decio A, Scarlato V, Scanziani E, Giavazzi R, Cesca M. Chemotherapy counteracts metastatic dissemination induced by antiangiogenic treatment in mice. MolCancer Ther 2013 Aug 5; 2013 Oct; 12(10):2237-2247; PMID:23918831 [DOI] [PubMed] [Google Scholar]
- 41. Hudachek SF, Gustafson DL. Incorporation of ABCB1-mediated transport into a physiologically-based pharmacokinetic model of docetaxel in mice. J Pharmacokinet Pharmacodyn 2013 Aug;40(4):437-49; PMID:23616082; http://dx.doi.org/ 10.1007/s10928-013-9317-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murakami M, Ernsting MJ, Undzys E, Holwell N, Foltz WD, Li SD. Docetaxel Conjugate Nanoparticles That Target α-Smooth Muscle Actin-Expressing Stromal Cells Suppress Breast Cancer Metastasis. Cancer Res 2013 Aug 1;73(15):4862-71; PMID:23907638; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0062 [DOI] [PubMed] [Google Scholar]
- 43. Gianni-Barrera R, Bartolomeo M, Vollmar B, Djonov V, Banfi A. Split for the cure: VEGF, PDGF-BB and intussusception in therapeutic angiogenesis. Biochem Soc Trans 2014 Dec 1;42(6):1637-42; PMID:25399582 [DOI] [PubMed] [Google Scholar]
- 44. Gacche RN, Meshram RJ. Angiogenic factors as potential drug target: efficacy and limitations of anti-angiogenic therapy. Biochim Biophys Acta 2014 Aug;1846(1):161-79; PMID:24836679 [DOI] [PubMed] [Google Scholar]
- 45. Cao Y, Eble JM, Moon E, Yuan H, Weitzel DH, Landon CD, Nien CY, Hanna G, Rich JN, Provenzale JM, et al. Tumor cells upregulate normoxic HIF-1alpha in response to doxorubicin. Cancer Res 2013 Oct 15;73(20):6230-42. PMCID:PMC3800255; PMID:23959856; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ghoshal P, Teng Y, Lesoon LA, Cowell JK. HIF1A induces expression of the WASF3 metastasis-associated gene under hypoxic conditions. Int J Cancer 2012 Sep 15;131(6):E905-E915. PMCID:PMC3629704; PMID:22581642; http://dx.doi.org/ 10.1002/ijc.27631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Teng Y, Ren MQ, Cheney R, Sharma S, Cowell JK. Inactivation of the WASF3 gene in prostate cancer cells leads to suppression of tumorigenicity and metastases. Br J Cancer 2010 Sep 28;103(7):1066-75. PMCID:PMC2965863; PMID:20717117; http://dx.doi.org/ 10.1038/sj.bjc.6605850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Teng Y, Liu M, Cowell JK. Functional interrelationship between the WASF3 and KISS1 metastasis-associated genes in breast cancer cells. Int J Cancer 2011 Dec 15;129(12):2825-35. PMCID:PMC3154992; PMID:21544801; http://dx.doi.org/ 10.1002/ijc.25964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Teng Y, Ngoka L, Mei Y, Lesoon L, Cowell JK. HSP90 and HSP70 proteins are essential for stabilization and activation of WASF3 metastasis-promoting protein. J Biol Chem 2012 Mar 23;287(13):10051-9. PMCID:PMC3323057; PMID:22315230; http://dx.doi.org/ 10.1074/jbc.M111.335000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Teng Y, Ghoshal P, Ngoka L, Mei Y, Cowell JK. Critical role of the WASF3 gene in JAK2/STAT3 regulation of cancer cell motility. Carcinogenesis 2013 Jun 17; 2013 Sep; 34(9):1994-1999; PMID:23677069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Teng Y, Mei Y, Hawthorn L, Cowell JK. WASF3 regulates miR-200 inactivation by ZEB1 through suppression of KISS1 leading to increased invasiveness in breast cancer cells. Oncogene 2013 Jan 14; PMID:23318438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Taylor MA, Davuluri G, Parvani JG, Schiemann BJ, Wendt MK, Plow EF, Schiemann WP, Sossey-Alaoui K. Upregulated WAVE3 expression is essential for TGF-β-mediated EMT and metastasis of triple-negative breast cancer cells. Breast Cancer Res Treat 2013 Nov;142(2):341-53. PMCID:PMC3888319; PMID:24197660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fryknas M, Dhar S, Oberg F, Rickardson L, Rydaker M, Goransson H, Gustafsson M, Pettersson U, Nygren P, Larsson R, et al. STAT1 signaling is associated with acquired crossresistance to doxorubicin and radiation in myeloma cell lines. Int J Cancer 2007 Jan 1;120(1):189-95; PMID:17072862; http://dx.doi.org/ 10.1002/ijc.22291 [DOI] [PubMed] [Google Scholar]
- 54. Roberts D, Schick J, Conway S, Biade S, Laub PB, Stevenson JP, Hamilton TC, O'Dwyer PJ, Johnson SW. Identification of genes associated with platinum drug sensitivity and resistance in human ovarian cancer cells. Br J Cancer 2005 Mar 28;92(6):1149-58. PMCID:PMC2361951; PMID:15726096; http://dx.doi.org/ 10.1038/sj.bjc.6602447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Albasanz-Puig A, Murray J, Namekata M, Wijelath ES. Opposing roles of STAT-1 and STAT-3 in regulating vascular endothelial growth factor expression in vascular smooth muscle cells. Biochem Biophys Res Commun 428, 179-184 2012; PMID:23068100; http://dx.doi.org/ 10.1016/j.bbrc.2012.10.037 [DOI] [PubMed] [Google Scholar]
- 56. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001 Dec;25(4):402-8; PMID:11846609; http://dx.doi.org/ 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 57. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008;3(6):1101-8; PMID:18546601; http://dx.doi.org/ 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- 58. Bialkowska K, Ma YQ, Bledzka K, Sossey-Alaoui K, Izem L, Zhang X, Malinin N, Qin J, Byzova T, Plow EF. The integrin co-activator Kindlin-3 is expressed and functional in a non-hematopoietic cell, the endothelial cell. J Biol Chem 2010 Jun 11;285(24):18640-9. PMCID:PMC2881789; PMID:20378539 [DOI] [PMC free article] [PubMed] [Google Scholar]