

Abstract
The zoonotic Chagas’ disease is caused by infections with the hemoflagellate Trypanosoma cruzi (T. cruzi) which is endemic in Latin America. Despite recent advances in our understanding of the pathogenesis of the disease, the underlying molecular processes involved in host-parasite interactions are only poorly understood. In particular, the mechanisms for parasite persistence in host cells remain largely unknown. Cytokine-driven transcription factors from the family of STAT (signal transducer and activator of transcription) proteins appear to play a central role in the fight against T. cruzi infection. However, amastigotes proliferating in the cytoplasm of infected host cells develop effective strategies to circumvent the attack executed by STAT proteins. This review highlights the interactions between T. cruzi parasites and human host cells in terms of cytokine signaling and, in particular, discusses the impact of STATs on the balance between parasite invasion and clearance.
Keywords: Chagas’ disease, serine phosphorylation, STAT proteins, Trypanosoma cruzi, tyrosine phosphorylation
Life Cycle and Parasite Replication of Trypanosoma Cruzi
Trypanosoma cruzi (T. cruzi) is the causative agent of Chagas’ disease, a parasitic infection discovered by the Brazilian physician and scientist Carlos Chagas at the beginning of the 20th century, which affects 7 to 8 million people in Central and South America.1 This protozoan is frequently transmitted by blood-sucking triatomine insects and persists in various tissues for years or even decades before dilated cardiomyopathy and/or megavisceral disease finally develop as the characteristic features of chronic Chagas’ disease. Although Chagas’ disease is the main cause of heart failure among people living in poverty in Latin America, it is generally regarded as a neglected tropical disease with an immense socio-economic burden.2-4 At present, no vaccination against T. cruzi is available and chemotherapeutic intervention is limited and may cause severe side effects.
The sylvatic or domestic life cycle of T. cruzi was elucidated more than a century ago.5 In Chagas-endemic areas, the cycle starts when a reduviid bug bites to draw blood while defecating on the skin in the vicinity of the bite wound.6-9 Humans and other mammals are infected when metacyclic trypomastigotes are released with the faeces and rubbed or scratched into the wound or onto mucosal surfaces such as eyes or mouth. Upon entry into the body, the highly motile metacyclic trypomastigotes of T. cruzi can actively invade local tissues. Although any nucleated host cell can be infected, those preferably parasitized include cardiac myocytes, skeletal and smooth muscle cells, endothelial cells, neuroglial cells and cells of the peripheral nervous system as well as the reticulo-endothelial system including Kupffer cells.10-12 Inside the host cell, trypomastigotes transform to amastigotes within the first 24 h after cell penetration and replicate every 6–8 h by binary fission until the cytoplasm of the host cell is nearly completely filled.13 Then, amastigotes transform to trypomastigotes which, after rupture of the host cell, are released into the circulation and spread within the organism to subsequently infect other host cells. The trypomastigote does not divide in the blood but carries the infection to all parts of the body. The life cycle of T. cruzi is accomplished when the vector ingests trypomastigotes as it takes a blood meal from the infected mammalian host. Trypomastigotes transform in the mid-gut of the insect to the epimastigote form, which also divides by binary fission and then after 8 to 10 d develops into a metacyclic trypomastigote. Infectious metacyclic trypomastigotes are passed in the faeces to infect mammals during the next blood meal, when rubbed into the insect's puncture wound or onto exposed mucous membranes. Currently, non-vectorial transmissions are increasingly recognized as occurring through congenital transmission, blood transfusion, organ transplantation or orally through incidental ingestion of parasite-contaminated food or drink.14
The clinical course of Chagas’ disease can be divided into the acute, indeterminate and chronic stage. The acute stage of the disease is the consequence of the first encounter of the patient's immune system with the protozoan, whereas the chronic phase results from long-term challenge. In the acute and the chronic phase, there are intense interactions between the host's immune system and the invading parasite, whereas in the indeterminate phase patients are asymptomatic and trypomastigotes are seldom detected in the peripheral blood, although patients remain infectious. Symptoms of the initial acute phase of Chagas’ disease reflect the interaction of the innate, and later the adaptive, immune system of the host with the intruding parasites. Patients show a broad range of clinical signs ranging from local inflammation and swelling at the site of inoculation (chagoma), local myalgia, conjunctivitis (Romaña's sign), a generalized morbilliform eruption (schizotrypanides), and systemic infections including meningoencephalitis and acute myocarditis both of which may lead to death.15 Chronic Chagas’ disease may develop in up to one third of infected patients and is associated with a variety of clinical symptoms, such as heart failure, megacolon and megaesophagus.16 The most common cardiac manifestation of chronic American trypanosomiasis is progressive dilated cardiomyopathy characterized by cardiomegaly and conduction disturbances which occurs in 40–50% of cases.17
IFNγ-Mediated STAT1 Signaling During Infection with Trypanosoma Cruzi
Whereas STAT1 signaling in toxoplasmosis has been well studied, much less is known about the contribution made by this pathway to resist infection with T. cruzi. A better understanding of the impact of STAT1 in Chagas’ disease requires further research on the anti-parasitic effects of host cytokines and the immune evasion strategies of the parasites. The pro-inflammatory cytokine IFNγ plays an essential role in mediating protective immune responses in experimental Chagas’ disease.18-21 As reported by Ferreira and colleagues, IFNγ exerts a pleiotropic mode of action in both acute and chronic Chagas’ disease by acting as an immunological mediator in tissue remodeling.22 In a previous study, we showed that STAT1 played a major role in the first line of defense against invading trypomastigotes of T. cruzi.23 Furthermore, we demonstrated a protective effect of IFNγ against both entry of trypomastigotes into host cells and intracellular multiplication of amastigotes which was based on the activation of STAT1 by tyrosine phosphorylation23. Pre-incubation of host cells with IFNγ led to a lower parasite burden, while simultaneous incubation of cells with IFNγ and trypomastigotes had neither a beneficial effect on the number of infected cells nor on the number of intracellular parasites. This finding showed that IFNγ was active in controlling the parasitism caused by infection with T. cruzi amastigotes. We found that infection of host cells with T. cruzi trypomastigotes in the absence of IFNγ treatment led to STAT1 phosphorylation of both tyrosine residue 701 and serine residue 727 and, consequently, up-regulated STAT1-mediated gene expression. The observation that the STAT1 signaling pathway was activated in host cells after infection with T. cruzi leading to a significantly elevated STAT1 expression confirmed previous results by Vaena de Avalos and colleagues.24 Notably, Cobb, Hambright, and Smeltz reported that IFNγ-deficient and STAT1-deficient mice generated increased numbers of IL-17-producing cells and that STAT1 was required to inhibit Th17 response to T. cruzi infection.25 The authors adoptively transferred purified CD4+ T cells from donor STAT1−/− mice into Rag-2−/− recipient mice and observed a normal increase in Th17 cells, indicating that the inhibitory effects of STAT1 on Th17 development are T cell-intrinsic and not a result of STAT1-dependent functions of antigen-presenting cells.
Bergeron and Olivier demonstrated that parasite infection potentiated up-regulation of inducible NO synthase (iNOS) expression in macrophages and enhanced NO production upon stimulation of macrophages with IFNγ.26 Furthermore, the authors reported the transcription of a stable inos mRNA species in macrophages which resulted from a decreased rate of inos mRNA degradation. The increased inos mRNA stability in T. cruzi-infected cells probably resulted from activation of ERK1/ERK2 (extracellular-signal-regulated kinase 1/2) mitogen-activated protein kinases (MAPK) and stress-activated protein kinase pathways, while maximal inos mRNA expression was achieved through NF-κB activation. In summary, Bergeron and Olivier concluded that signaling pathway cross-talk was required for maximal NO generation in macrophages at both pre- and post-transcriptional levels. We confirmed that, in experimental infection with T. cruzi, some of the upregulated STAT1-dependent target genes, such as inos and ido, were most probably involved in the first line of the innate immune response23 Figure 1..
Figure 1.
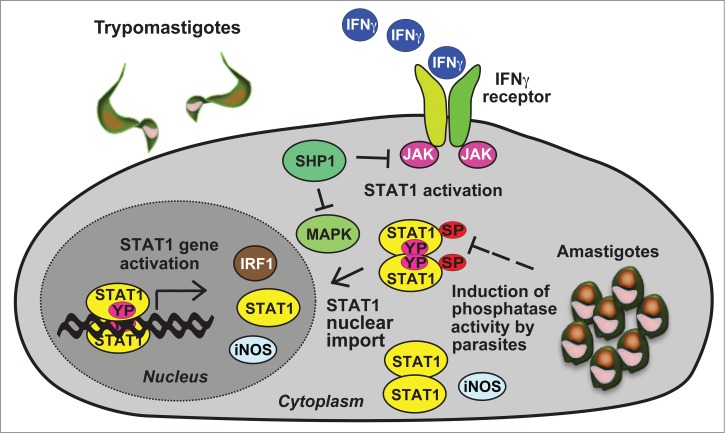
Intracellular trypanosomatid parasites evade the protective role of IFNγ signaling by targeting essential components of the JAK-STAT pathway. Gene induction by serine(SP)- and tyrosine(YP)-phosphorylated STAT1 including interferon-regulatory factor 1 (IRF1), inducible NO synthase (iNOS) and STAT1 is crucial for the anti-parasite actions of this pathway. The model depicts the inhibition of STAT1 transcriptional activity by parasite-mediated dephosphorylation of STAT1 serine 727 (as shown for T. cruzi.23) and activation of the protein tyrosine phosphatase SHP1 (in L. donovani-infected cells51).
Hölscher and colleagues demonstrated that reduced NO production or a proximal defect in the IFNγ signaling pathway both led to an enhanced susceptibility of mice infected with T. cruzi. Interferonγ receptor and iNOS knock-out mice were unable to survive the inoculation of sublethal doses of trypomastigotes, indicating a major role of the IFNγ/STAT1-driven NO production in defending host cells against invasion by this parasite.27 The authors observed enhanced parasite dissemination and extensive necrotic lesions in numerous organs in the 2 transgenic mouse lines with defective NO production, when challenged with low doses of T. cruzi, whereas most wild-type animals survived without histopathological lesions. Macrophages from mouse lines deficient for the IFNγ receptor or iNOS showed impaired trypanocidal activities and were unable to respond to T. cruzi infection by producing NO.27 In contrast to inos knockout-mice which displayed a rather normal pro-inflammatory cytokine response after infection with T. cruzi, animals lacking the IFNγ receptor showed elevated levels of IFNγ but reduced concentrations of tumor necrosis factor-α (TNFα) and interleukin-1α (IL-1α).
Despite the protective effects of IFNγ signaling in terms of parasitic burden, T. cruzi parasites, once in the cytoplasm of the host cell, continue to successfully replicate. We have recently shown that T. cruzi amastigotes selectively decrease the level of STAT1 serine 727 phosphorylation, suggesting an effective mechanism, whereby the parasite may circumvent the innate immune reaction following STAT1 activation (Fig. 1).23 In a previous study using the knock-in mouse line STAT1S727A/S727A it was well established that phosphorylation of serine 727 is required for fully-fledged transcriptional response in IFNγ-dependent innate immunity.28,29 The observation of impaired STAT1 activation may be an explanation for the persistence of this intracellular parasite and is in line with findings established for other parasites including Trypanosoma brucei and Leishmania donovani (discussed below) as well as viral and bacterial pathogens.30-32 However, further research is required to decipher the mechanistic pathway of serine 727 dephosphorylation used by the parasite.
Regulating Host Cell Apoptosis by STATs in Trypanosoma Cruzi Infection
It is still not clear whether STAT1 or STAT3 is engaged in regulating programmed cell death in host cells infected by T. cruzi. Expression of pro-apoptotic, as well as, anti-apoptotic genes in cardiomyocytes and its impact on apoptosis following infection with T. cruzi has been described by various groups who have come to partially contrary conclusions. Some studies have suggested that STATs regulate apoptosis of cardiomyocytes upon infection with T. cruzi trypomastigotes and amastigotes.33 De Souza and colleagues found host cell apoptosis after infection with different strains of T. cruzi.34 Likewise, Zhang et al. detected apoptotic cell death in a canine model of experimental acute chagasic myocarditis characterized by the presence of amastigotes and differentiating trypomastigotes of T. cruzi within the cytoplasm of cardiac myocytes.35 However, Aoki et al. reported on a protective effect of cruzipain against cardiomyocyte apoptosis after infection with T. cruzi.36 Another analysis also came to the conclusion that neonatal cardiomyocytes of the rat did not show apoptotic cells after T. cruzi infection.37 Based on their findings, de Souza and colleagues concluded that parasites of T. cruzi succumb to apoptosis during infection of murine cardiomyocytes.38 Apoptosis of host cells in combination with intracellular elimination of parasites may affect long-term parasite survival by counterbalancing the pathogenic mechanisms of the invading parasites.
Ponce and co-workers reported that secretion of endogenous IL-6 or addition of recombinant IL-6 protect cultured cardiomyocytes from apoptotic cell death during T. cruzi infection.39 In addition, the authors demonstrated phosphorylation of STAT3 by IL-6 in response to infection with T. cruzi. Expression of the STAT3-regulated anti-apoptotic factor Bcl2 was increased in cardiac tissue, indicating that, during the acute phase of the infection, active STAT3 acts as a mediator of cell survival. This observation suggests that STAT3 executes pro-survival effects in cardiomyocytes evoked by the parasite. Furthermore, the authors showed that inactive cruzipain, which is the main cysteine protease secreted by the parasite, specifically triggers the release of IL-6 via a toll-like receptor 2 (TLR2)-dependent pathway and is required to induce cardiomyocyte survival. In contrast, enzymatic activity of cruzipain critically interferes with IL-6-induced STAT3 phosphorylation by means of cleavage of the ectodomain of gp130, the shared receptor of several IL-6-type cytokines. Cleavage of glycoprotein gp130 is inhibited, when cruzipain binds to the parasite cysteine protease inhibitor chagasin. In summary, the authors have demonstrated that the pro-inflammatory IL-6 response in T. cruzi-infected cells may be modified by cysteine protease activity.
STAT3 is known to be an inductor of genes whose products have been identified as suppressors of cytokine signaling (SOCS) which modulate the immune response by decreasing the amount of inflammatory cytokines through inhibition of the JAK-STAT pathway.40 It is well known that STAT3 can be activated by IL-6 or IL-10,41,42 while SOCS3 expression is up-regulated by the anti-inflammatory cytokine IL-10 in T. cruzi-infected cardiomyocytes.43 During infection with T. cruzi SOCS2 is expressed and most probably plays an important role in the pathogenesis of Chagas’ heart disease by influencing the progress of heart damage.44
The transcription factor STAT4 is activated in response to the pro-inflammatory cytokine IL-12 and provides essential signals to drive Th cells along a Th1 lineage, whereas STAT6 is activated by receptor binding of IL-4 and IL-13, 2 cytokines with anti-inflammatory properties, which provides the alternative signal for development along a Th2 lineage. Tarleton, Grusby, and Zhang reported that STAT4-deficient mice were highly susceptible to infection, while STAT6-deficient mice showed enhanced resistance with fewer parasites and little or no evidence of inflammatory disease in the heart and skeletal muscle upon infection as compared to their wild-type littermates.45 The obvious absence of disease in chronically infected STAT6-deficient mice is remarkable in spite of the lack of complete clearance of parasites. These findings suggest that the severity of the inflammatory response critically depends on the STAT4- and STAT6-mediated cytokine-driven immune response and the ability of this response to limit tissue parasite load.45 The authors concluded, that parasite clearance may not be required to prevent the progression of disease in chronic infection with T. cruzi.
Lessons Learned from Other Kinetoplastids
For Trypanosoma brucei (T. brucei), the causative agent of African trypanosomiasis, also known as sleeping sickness, Coller and colleagues showed that its surface molecules significantly inhibit IFNγ-dependent STAT1 phosphorylation in macrophages. They demonstrated that pre-treatment of cells with GIP-sVSG (the soluble form of variant surface glycoprotein containing the glycosylinositolphosphate substituent that is released by parasites) from T. brucei reduced IFNγ-mediated STAT1 tyrosine phosphorylation and resulted in decreased nitric oxide production.46 The authors reported that the timing of exposure to IFNγ versus GIP-sVSG ultimately determined the macrophage response at the level of gene induction. While pre-treatment of macrophages with IFNγ followed by GIP-sVSG activated gene expression of TNFα and IL-6, treatment of macrophages with GIP-sVSG before IFNγ stimulation resulted in a markedly reduced transcription of the inducible NO synthase and secretion of NO. Using IFNγ-knock-out mice, Hertz and colleagues revealed that IFNγ plays a key role for host resistance to T. brucei.47 However, susceptibility of IFNγ knockout-mice to T. brucei was not due to a defect of inducible nitric oxide production, implicating factors other than iNOS and NO as being responsible for resistance to this vector-borne trypanosomatid.48 It will be interesting to know whether also glycosylphosphatidylinositol-anchored proteins from T. cruzi exert similar effects in American trypanosomiasis.
STAT1 signaling has been well studied in infections with obligate intracellular parasites of the genus Leishmania, which also belongs to the class of kinetoplastids. Ray et al. showed a significant inhibition of the IFNγ-receptor complex after infection of phorbol-differentiated U937 human promonocytic cells with Leishmania donovani (L. donovani), the pathogenic agent of visceral leishmaniasis. Reduced tyrosine phosphorylation of the IFNγ-receptor resulted in subsequently impaired activation of STAT1, regardless of whether promastigote or amastigote forms of L. donovani were used for infection.49 These findings confirmed a previous study by Nandan and Reiner indicating that L. donovani infection of both human mononuclear phagocytes and differentiated U-937 cells selectively impaired IFNγ-induced JAK1 and JAK2 activation as well as tyrosine phosphorylation of STAT1.50 The authors suggested that unresponsiveness to IFNγ might explain the impaired transcriptional response in Leishmania-infected cells.
Blanchette and colleagues demonstrated that infection of macrophages with L. donovani triggered protein tyrosine phosphatase (PTP) activity and resulted in dephosphorylation of tyrosine residues in molecules such as JAK2. In particular, the PTP Src-homology-2-domain-containing phosphatase 1 (SHP1) was activated upon infection with L. donovani, then interacted with JAK2 and critically impaired IFNγ signaling.51 IFNγ-stimulated STAT1 activity was reduced in SHP1-deficient macrophages following infection with L. donovani.52 Notably, Kar et al. demonstrated an upregulation of several protein phosphatases (MKP1, MKP3, and PP2A) during infection of macrophages with L. donovani and their impact on parasite survival.53 The authors showed that specific MAPK-directed phosphatases and protein phosphatase 2A (PP2A) inhibited ERK1/2 MAPK resulting in reduced expression of iNOS mRNA. Inhibition of phosphatases in L. donovani-infected BALB/c mice by cystatin, a therapeutic agent for experimental visceral leishmaniasis, shifted the cytokine balance in favor of the host by inducing TNFα and iNOS expression.53 The repression of MAPK and JAK/STAT cascades in macrophages infected with Leishmania parasites may be an effective strategy for the parasites to survive antimicrobial activities.54
Promastigotes of L. donovani specifically induced the transient expression of SOCS3 mRNA in human macrophages which suppressed macrophage activation.49,55 Moreover, it was reported by Matte and Descoteaux that amastigotes of L. donovani inhibit the expression of IRF-1 (interferon-regulatory factor 1). In addition, the authors suggested a novel mechanism for preventing IFNγ-induced STAT1 nuclear translocation due to infection with L. donovani amastigotes, namely by blocking STAT1 association with importin-α5.56 Rosas et al. reported that STAT1 knock-out mice were highly resistant to L. donovani infection and developed minimal liver pathology.57 In summary, there is evidence of multiple pathways and mechanisms used by L. donovani to interfere with IFNγ-activated macrophage functions.58,59
Likewise, STAT1-mediated IFNγ signaling is required also for the development of protective immunity against cutaneous leishmaniasis which is caused by Leishmania major (L. major).60 However, Späth and colleagues found evidence of a cytokine-independent physiological function of STAT1 in acidification of the host cell phago-lysosomes in L. major-infected cells. The increased susceptibility to Leishmania infection in STAT1-deficient mice may be linked to higher pH levels in phago-lysosomes of macrophages which most probably resulted from a chloride channel dysfunction.61 Thus, cytokine-independent STAT1 functions in innate antimicrobial resistance may have a greater impact on host-pathogen interactions than currently appreciated.
Johnson and Scott reported a requirement of STAT1 expression in dendritic cells, but not T cells, for immunity against L. major.62 In contrast, Barbi and colleagues demonstrated that STAT1 signaling in CD4+ T cells is essential for the development of an antimicrobial state against L. major, whereas it is not essential in CD8+ T cells.63 Additional results have suggested that STAT1 expression is critical for preventing systemic dissemination of parasite infection, most probably by controlling the migration of Th1 cells to sites of inflammation rather than their generation.63
Concluding Remarks
In summary, human pathogen species of the order Trypanosomatidae (T. cruzi, T. brucei and Leishmania spp.) have evolved complex and still undefined mechanisms to circumvent IFNγ/STAT1 signaling and to reside in mammalian hosts for years or even decades after infection. The bypassing of this protective signal pathway may account for the long-term persistence and intracellular multiplication of these protozoan parasites in the host. Further research efforts will shed light on the clinical importance of STAT1 dephosphorylation for parasite survival. Identification of novel targets in the JAK-STAT pathway used by intracellular parasites will promote new pharmacological strategies for the treatment of patients with acute and chronic Chagas’ disease and also of other parasitic diseases.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by a grant from the Deutsche Forschungsgemeinschaft to TM, a scholarship from the Roland und Elfriede Schauer-Stiftung to PS, and grants from the Deutsches Zentrum für Herz- und Kreislaufforschung (to RTS and TM) and the Deutsche Gesellschaft fü̈r Kardiologie (to PS, RTS and TM).
References
- 1. Rassi A. Jr., Rassi A, Marin-Neto JA. Chagas disease. Lancet 2010; 375:1388-402; PMID:20399979; http://dx.doi.org/ 10.1016/S0140-6736(10)60061-X [DOI] [PubMed] [Google Scholar]
- 2. Marin-Neto JA, Cunha-Neto E, Maciel BC, Simões MV. Pathogenesis of chronic Chagas heart disease. Circulation 2007; 115:1109-23; PMID:17339569; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.106.624296 [DOI] [PubMed] [Google Scholar]
- 3. Gutierrez FR, Guedes PM, Gazzinelli RT, Silva JS. The role of parasite persistence in pathogenesis of Chagas heart disease. Parasite Immunol 2009; 31:673-85; PMID:19825107; http://dx.doi.org/ 10.1111/j.1365-3024.2009.01108.x [DOI] [PubMed] [Google Scholar]
- 4. Jabari S, de Oliveira EC, Brehmer A, da Silveira AB. Chagasic megacolon: enteric neurons and related structures. Histochem Cell Biol 2014; 142:235-44; PMID:25059649; http://dx.doi.org/ 10.1007/s00418-014-1250-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chagas C. Nova tripanozomiaze humana: estudos sobre a morfolojia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., ajente etiolojico de nova entidade morbida do homem. Mem Inst Oswaldo Cruz 1909; 1:159-218; http://dx.doi.org/ 10.1590/S0074-027619-09000200008 [DOI] [Google Scholar]
- 6. Brener Z. Biology of Trypanosoma cruzi. Annu Rev Microbiol 1973; 27:347-82; PMID:4201691; http://dx.doi.org/ 10.1146/annurev.mi.27.100173.002023 [DOI] [PubMed] [Google Scholar]
- 7. De Souza W. Cell biology of Trypanosoma cruzi. Int Rev Cytol 1984; 86:197-283; PMID:6368447; http://dx.doi.org/ 10.1016/S0074-7696(08)60180-1 [DOI] [PubMed] [Google Scholar]
- 8. Vickerman K. Developmental cycles and biology of pathogenic trypanosomes. Br Med Bull 1985; 41:105-14; PMID:3928017 [DOI] [PubMed] [Google Scholar]
- 9. De Souza W. Basic cell biology of Trypanosoma cruzi. Curr Pharm Des 2002; 8:269-85; PMID:11860366; http://dx.doi.org/ 10.2174/1381612023396276 [DOI] [PubMed] [Google Scholar]
- 10. Tan H, Andrews NW. Don't bother to knock–the cell invasion strategy of Trypanosoma cruzi. Trends Parasitol 2002; 18:427-8; PMID:12377585; http://dx.doi.org/ 10.1016/S1471-4922(02)02368-1 [DOI] [PubMed] [Google Scholar]
- 11. Barrias ES, de Carvalho TM, De Souza W. Trypanosoma cruzi: Entry into mammalian host cells and parasitophorous vacuole formation. Front Immunol 2013; 4:186; PMID:23914186; http://dx.doi.org/ 10.3389/fimmu.2013.00186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Machado FS, Dutra WO, Esper L, Gollob K, Teixeira MM, Factor SM, Weiss LM, Nagajyothi F, Tanowitz HB, Garg NJ. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Semin Immunopathol 2012; 34:753-70; PMID:23076807; http://dx.doi.org/ 10.1007/s00281-012-0351-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burleigh BA, Andrews NW. The mechanisms of Trypanosoma cruzi invasion of mammalian cells. Annu Rev Microbiol 1995; 49:175-200; PMID:8561458; http://dx.doi.org/ 10.1146/annurev.mi.49.100195.001135 [DOI] [PubMed] [Google Scholar]
- 14. Sánchez LV, Ramírez JD. Congenital and oral transmission of American trypanosomiasis: an overview of physiopathogenic aspects. Parasitology 2013; 140:147-59; PMID:23010131; http://dx.doi.org/ 10.1017/S0031182012001394 [DOI] [PubMed] [Google Scholar]
- 15. Carod-Artal FJ. American trypanosomiasis. Handb Clin Neurol 2013; 114:103-23; PMID:23829903; http://dx.doi.org/ 10.1016/B978-0-444-53490-3.00007-8 [DOI] [PubMed] [Google Scholar]
- 16. Rossi MA, Bestetti RB. The challenge of chagasic cardiomyopathy. The pathologic roles of autonomic abnormalities, autoimmune mechanisms and microvascular changes, and therapeutic implications. Cardiology 1995; 86:1-7; PMID:7728781; http://dx.doi.org/ 10.1159/000176822 [DOI] [PubMed] [Google Scholar]
- 17. Higuchi Mde L, Benvenuti LA, Martins Reis M, Metzger M. Pathophysiology of the heart in Chagas’ disease: current status and new developments. Cardiovasc Res 2003; 60:96-107; PMID:14522411; http://dx.doi.org/ 10.1016/S0008-6363(03)00361-4 [DOI] [PubMed] [Google Scholar]
- 18. Wirth JJ, Kierszenbaum F, Sonnenfeld G, Zlotnik A. Enhancing effects of gamma interferon on phagocytic cell association with and killing of Trypanosoma cruzi. Infect Immun 1985; 49:61-6; PMID:3924832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Antúnez MI, Cardoni RL. IL-12 and IFN-γ production, and NK cell activity, in acute and chronic experimental Trypanosoma cruzi infections. Immunol Lett 2000; 71:103-9; PMID:10714437; http://dx.doi.org/ 10.1016/S0165-2478(99)00172-8 [DOI] [PubMed] [Google Scholar]
- 20. Michailowsky V, Silva NM, Rocha CD, Vieira LQ, Lannes-Vieira J, Gazzinelli RT. Pivotal role of interleukin-12 and interferonγ axis in controlling tissue parasitism and inflammation in the heart and central nervous system during Trypanosoma cruzi infection. Am J Pathol 2001; 159:1723-33; PMID:11696433; http://dx.doi.org/ 10.1016/S0002-9440(10)63019-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Müller U, Köhler G, Mossmann H, Schaub GA, Alber G, Santo JP Di, Brombacher F, Hölscher C. IL-12-independent IFNγ production by T cells in experimental Chagas’ disease is mediated by IL-18. J Immunol 2001; 167:3346-53; PMID:11544324; http://dx.doi.org/ 10.4049/jimmunol.167.6.3346 [DOI] [PubMed] [Google Scholar]
- 22. Ferreira LR, Frade AF, Baron MA, Navarro IC, Kalil J, Chevillard C, Cunha-Neto E. Interferon-γ and other inflammatory mediators in cardiomyocyte signaling during Chagas disease cardiomyopathy. World J Cardiol 2014; 6:782-90; PMID:25228957; http://dx.doi.org/ 10.4330/wjc.v6.i8.782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stahl P, Ruppert V, Schwarz RT, Meyer T. Trypanosoma cruzi evades the protective role of interferon-gamma-signaling in parasite-infected cells. PloS One 2014; 9:e110512; PMID:25340519; http://dx.doi.org/ 10.1371/journal.pone.0110512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vaena de Avalos S, Blader IJ, Fisher M, Boothroyd JC, Burleigh BA. Immediate/early response to Trypanosoma cruzi infection involves minimal modulation of host cell transcription. J Biol Chem 2002; 277:639-44; PMID:11668183; http://dx.doi.org/ 10.1074/jbc.M109037200 [DOI] [PubMed] [Google Scholar]
- 25. Cobb D, Hambright D, Smeltz RB. T-bet-independent effects of IL-12 family cytokines on regulation of Th17 responses to experimental T. cruzi infection. J Leukoc Biol 2010; 88:965-71; PMID:20807701; http://dx.doi.org/ 10.1189/jlb.0410238 [DOI] [PubMed] [Google Scholar]
- 26. Bergeron M, Olivier M. Trypanosoma cruzi-mediated IFN-γ-inducible nitric oxide output in macrophages is regulated by iNOS mRNA stability. J Immunol 2006; 177:6271-80; PMID:17056557; http://dx.doi.org/ 10.4049/jimmunol.177.9.6271 [DOI] [PubMed] [Google Scholar]
- 27. Hölscher C, Köhler G, Müller U, Mossmann H, Schaub GA, Brombacher F. Defective nitric oxide effector functions lead to extreme susceptibility of Trypanosoma cruzi-infected mice deficient in gamma interferon receptor or inducible nitric oxide synthase. Infect Immun 1998; 66:1208-15; PMID:9488415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wen Z, Zhong Z, Darnell JE, Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995; 82:241-50; PMID:7543024; http://dx.doi.org/ 10.1016/0092-8674(95)90311-9 [DOI] [PubMed] [Google Scholar]
- 29. Varinou L, Ramsauer K, Karaghiosoff M, Kolbe T, Pfeffer K, Müller M, Decker T. Phosphorylation of the Stat1 transactivation domain is required for full-fledged IFN-γ-dependent innate immunity. Immunity 2003; 19:793-802; PMID:14670297; http://dx.doi.org/ 10.1016/S1074-7613(03)00322-4 [DOI] [PubMed] [Google Scholar]
- 30. Ramachandran A, Horvath CM. Paramyxovirus disruption of interferon signal transduction: STATus report. J Interferon Cytokine Res 2009; 29:531-7; PMID:19694544; http://dx.doi.org/ 10.1089/jir.2009.0070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parisien JP, Lau JF, Rodriguez JJ, Ulane CM, Horvath CM. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of α/β interferon signal transduction. J Virol 2002; 76:4190-8; PMID:11932384; http://dx.doi.org/ 10.1128/JVI.76.9.4190-4198.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hussain S, Zwilling BS, Lafuse WP. Mycobacterium avium infection of mouse macrophages inhibits IFN-γ Janus kinase-STAT signaling and gene induction by down-regulation of the IFN-γ receptor. J Immunol 1999; 163:2041-8; PMID:10438942 [PubMed] [Google Scholar]
- 33. Stahl P, Ruppert V, Meyer T, Schmidt J, Campos MA, Gazzinelli RT, Maisch B, Schwarz RT, Debierre-Grockiego F. Trypomastigotes and amastigotes of Trypanosoma cruzi induce apoptosis and STAT3 activation in cardiomyocytes in vitro. Apoptosis 2013; 18:653-63; PMID:23435997; http://dx.doi.org/ 10.1007/s10495-013-0822-x [DOI] [PubMed] [Google Scholar]
- 34. De Souza EM, Araújo-Jorge TC, Bailly C, Lansiaux A, Batista MM, Oliveira GM, Soeiro MN. Host and parasite apoptosis following Trypanosoma cruzi infection in in vitro and in vivo models. Cell Tissue Res 2003; 314:223-35; PMID:12928860; http://dx.doi.org/ 10.1007/s00441-003-0782-5 [DOI] [PubMed] [Google Scholar]
- 35. Zhang J, Andrade ZA, Yu ZX, Andrade SG, Takeda K, Sadirgursky M, Ferrans VJ. Apoptosis in a canine model of acute chagasic myocarditis. J Mol Cell Cardiol 1999; 31:581-96; PMID:10198189; http://dx.doi.org/ 10.1006/jmcc.1998.0893 [DOI] [PubMed] [Google Scholar]
- 36. Aoki MP, Guiñazú NL, Pellegrini AV, Gotoh T, Masih DT, Gea S. Cruzipain, a major Trypanosoma cruzi antigen, promotes arginase-2 expression and survival of neonatal mouse cardiomyocytes. Am J Physiol Cell Physiol 2004; 286:C206-212; PMID:13679306; http://dx.doi.org/ 10.1152/ajpcell.00282.2003 [DOI] [PubMed] [Google Scholar]
- 37. Petersen CA, Krumholz KA, Carmen J, Sinai AP, Burleigh BA. Trypanosoma cruzi infection and nuclear factor kappa B activation prevent apoptosis in cardiac cells. Infect Immun 2006; 74:1580-7; PMID: 16495529; http://dx.doi.org/ 10.1128/IAI.74.3.1580-1587.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Souza EM, Nefertiti AS, Bailly C, Lansiaux A, Soeiro MN. Differential apoptosis-like cell death in amastigote and trypomastigote forms from Trypanosoma cruzi-infected heart cells in vitro. Cell Tissue Res 2010; 341:173-80; PMID:20495825; http://dx.doi.org/ 10.1007/s00441-010-0985-5 [DOI] [PubMed] [Google Scholar]
- 39. Ponce NE, Cano RC, Carrera-Silva EA, Lima AP, Gea S, Aoki MP. Toll-like receptor-2 and interleukin-6 mediate cardiomyocyte protection from apoptosis during Trypanosoma cruzi murine infection. Med Microbiol Immunol 2012; 201:145-55; PMID:21984337; http://dx.doi.org/ 10.1007/s00430-011-0216-z [DOI] [PubMed] [Google Scholar]
- 40. Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997; 387:917-21; PMID:9202125; http://dx.doi.org/ 10.1038/43206 [DOI] [PubMed] [Google Scholar]
- 41. Zhong Z, Wen Z, Darnell JE. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994; 264:95-8; PMID:8140422; http://dx.doi.org/ 10.1126/science.8140422 [DOI] [PubMed] [Google Scholar]
- 42. Riley JK, Takeda K, Akira S, Schreiber RD. Interleukin-10 receptor signaling through the JAK-STAT pathway. requirement for two distinct receptor-derived signals for anti-inflammatory action. J Biol Chem 1999; 274:16513-21; PMID:10347215; http://dx.doi.org/ 10.1074/jbc.274.23.16513 [DOI] [PubMed] [Google Scholar]
- 43. Hovsepian E, Penas F, Siffo S, Mirkin GA, Goren NB. IL-10 inhibits the NF-κB and ERK/MAPK-mediated production of pro-inflammatory mediators by up-regulation of SOCS-3 in Trypanosoma cruzi-infected cardiomyocytes. PloS One 2013; 8:e79445; PMID:24260222; http://dx.doi.org/ 10.1371/journal.pone.0079445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Esper L, Roman-Campos D, Lara A, Brant F, Castro LL, Barroso A, Araujo RR, Vieira LQ, Mukherjee S, Gomes ER, et al. Role of SOCS2 in modulating heart damage and function in a murine model of acute Chagas disease. Am J Pathol 2012; 181:130-40; PMID:22658486; http://dx.doi.org/ 10.1016/j.ajpath.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tarleton RL, Grusby MJ, Zhang L. Increased susceptibility of Stat4-deficient and enhanced resistance in Stat6-deficient mice to infection with Trypanosoma cruzi. J Immunol 2000; 165:1520-5; PMID: 10903759; http://dx.doi.org/ 10.4049/jimmunol.165.3.1520 [DOI] [PubMed] [Google Scholar]
- 46. Coller SP, Mansfield JM, Paulnock DM. Glycosylinositolphosphate soluble variant surface glycoprotein inhibits IFN-γ-induced nitric oxide production via reduction in STAT1 phosphorylation in African trypanosomiasis. J Immunol 2003; 171:1466-72; PMID:12874239; http://dx.doi.org/ 10.4049/jimmunol.171.3.1466 [DOI] [PubMed] [Google Scholar]
- 47. Hertz CJ, Filutowicz H, Mansfield JM. Resistance to the African trypanosomes is IFN-γ dependent. J Immunol 1998; 161:6775-83; PMID:9862708 [PubMed] [Google Scholar]
- 48. Hertz CJ, Mansfield JM. IFN-γ-dependent nitric oxide production is not linked to resistance in experimental African trypanosomiasis. Cell Immunol 1999; 192:24-32; PMID:10066343; http://dx.doi.org/ 10.1006/cimm.1998.1429 [DOI] [PubMed] [Google Scholar]
- 49. Ray M, Gam AA, Boykins RA, Kenney RT. Inhibition of interferon-γ signaling by Leishmania donovani. J Infect Dis 2000; 181:1121-8; PMID:10720539; http://dx.doi.org/ 10.1086/315330 [DOI] [PubMed] [Google Scholar]
- 50. Nandan D, Reiner NE. Attenuation of gamma interferon-induced tyrosine phosphorylation in mononuclear phagocytes infected with Leishmania donovani: selective inhibition of signaling through Janus kinases and Stat1. Infect Immun 1995; 63:4495-500; PMID:7591091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blanchette J, Racette N, Faure R, Siminovitch KA, Olivier M. Leishmania-induced increases in activation of macrophage SHP-1 tyrosine phosphatase are associated with impaired IFN-γ-triggered JAK2 activation. Eur J Immunol 1999; 29:3737-44; PMID:10556830; http://dx.doi.org/ 10.1002/(SICI)1521-4141(199911)29:11%3c3737::AID-IMMU3737%3e3.0.CO;2-S [DOI] [PubMed] [Google Scholar]
- 52. Forget G, Gregory DJ, Whitcombe LA, Olivier M. Role of host protein tyrosine phosphatase SHP-1 in Leishmania donovani-induced inhibition of nitric oxide production. Infect Immun 2006; 74:6272-9; PMID:17057094; http://dx.doi.org/ 10.1128/IAI.00853-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kar S, Ukil A, Sharma G, Das PK. MAPK-directed phosphatases preferentially regulate pro- and anti-inflammatory cytokines in experimental visceral leishmaniasis: involvement of distinct protein kinase C isoforms. J Leukoc Biol 2010; 88:9-20; PMID:20200403; http://dx.doi.org/ 10.1189/jlb.0909644 [DOI] [PubMed] [Google Scholar]
- 54. Olivier M, Gregory DJ, Forget G. Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin Microbiol Rev 2005; 18:293-305; PMID:15831826; http://dx.doi.org/ 10.1128/CMR.18.2.293-305.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bertholet S, Dickensheets HL, Sheikh F, Gam AA, Donnelly RP, Kenney RT. Leishmania donovani-induced expression of suppressor of cytokine signaling 3 in human macrophages: a novel mechanism for intracellular parasite suppression of activation. Infect Immun 2003; 71:2095-101; PMID:12654831; http://dx.doi.org/ 10.1128/IAI.71.4.2095-2101.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Matte C, Descoteaux A. Leishmania donovani amastigotes impair gamma interferon-induced STAT1α nuclear translocation by blocking the interaction between STAT1α and importin-α5. Infect Immun 2010; 78:3736-43; PMID:20566692; http://dx.doi.org/ 10.1128/IAI.00046-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rosas LE, Snider HM, Barbi J, Satoskar AA, Lugo-Villarino G, Keiser T, Papenfuss T, Durbin JE, Radzioch D, Glimcher LH, et al. Cutting edge: STAT1 and T-bet play distinct roles in determining outcome of visceral leishmaniasis caused by Leishmania donovani. J Immunol 2006; 177:22-5; PMID:16785492; http://dx.doi.org/ 10.4049/jimmunol.177.1.22 [DOI] [PubMed] [Google Scholar]
- 58. Shadab M, Ali N. Evasion of host defence by Leishmania donovani: subversion of signaling pathways. Mol Biol Int 2011; 2011:343961; PMID:22091401; http://dx.doi.org/ 10.4061/2011/343961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shio MT, Olivier M. Editorial: Leishmania survival mechanisms: the role of host phosphatases. J Leukoc Biol 2010; 88:1-3; PMID:20591873; http://dx.doi.org/ 10.1189/jlb.0210088 [DOI] [PubMed] [Google Scholar]
- 60. Rosas LE, Keiser T, Pyles R, Durbin J, Satoskar AR. Development of protective immunity against cutaneous leishmaniasis is dependent on STAT1-mediated IFN signaling pathway. Eur J Immunol 2003; 33:1799-805; PMID:12811839; http://dx.doi.org/ 10.1002/eji.200323163 [DOI] [PubMed] [Google Scholar]
- 61. Späth GF, Schlesinger P, Schreiber R, Beverley SM. A novel role for Stat1 in phagosome acidification and natural host resistance to intracellular infection by Leishmania major. PLoS Pathog 2009; 5:e1000381; PMID:19381261; http://dx.doi.org/ 10.1371/journal.ppat.1000381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Johnson LM, Scott P. STAT1 expression in dendritic cells, but not T cells, is required for immunity to Leishmania major. J Immunol 2007; 178:7259-66; PMID:17513775; http://dx.doi.org/ 10.4049/jimmunol.178.11.7259 [DOI] [PubMed] [Google Scholar]
- 63. Barbi J, Snider HM, Bhardwaj N, Lezama-Dávila CM, Durbin JE, Satoskar AR. Signal transducer and activator of transcription 1 in T cells plays an indispensable role in immunity to Leishmania major by mediating Th1 cell homing to the site of infection. FASEB J 2009; 23:3990-9; PMID:19641143; http://dx.doi.org/ 10.1096/fj.09-138057 [DOI] [PMC free article] [PubMed] [Google Scholar]