

ABSTRACT
Activation of the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling pathway has been associated with numerous human malignancies, including primary effusion lymphomas (PELs). PEL, a cancerous proliferation of B cells, is caused by Kaposi's sarcoma-associated herpesvirus (KSHV). Previously we identified constitutive phosphorylation of STAT6 on tyrosine 641 (p-STAT6C) in PEL cell lines BC3 and BCBL1; however, the molecular mechanism leading to this activation remains unclear. Here we demonstrate that STAT6 activation tightly correlates with interleukin-13 (IL-13) secretion, JAK1/2 tyrosine phosphorylation, and reduced expression of SHP1 due to KSHV infection. Moreover, p-STAT6C and reduction of SHP1 were also observed in KS patient tissue. Notably, blockade of IL-13 by antibody neutralization dramatically inhibits PEL cell proliferation and survival. Taken together, these results suggest that IL-13/STAT6 signaling is modulated by KSHV to promote host cell proliferation and viral pathogenesis.
IMPORTANCE STAT6 is a member of signal transducer and activator of transcription (STAT) family, whose activation is linked to KSHV-associated cancers. The mechanism through which STAT6 is modulated by KSHV remains unclear. In this study, we demonstrated that constitutive activation of STAT6 in KSHV-associated PEL cells results from interleukin-13 (IL-13) secretion and reduced expression of SHP1. Importantly, we also found that depletion of IL-13 reduces PEL cell growth and survival. This discovery provides new insight that IL-13/STAT6 plays an essential role in KSHV pathogenesis.
INTRODUCTION
Cytokines play a critical role in many viral infections. Viruses not only manipulate host cytokine production to favor virus survival, replication, and infection but also help virus-infected cells to modulate the host immune response, which potentially results in the development of viral persistent infection, pathogenesis, or tumorigenesis (1). Kaposi's sarcoma-associated herpesvirus (KSHV), also named human herpesvirus 8 (HHV-8), is an oncogenic gammaherpesvirus that associates with several aggressive malignancies, including AIDS-related Kaposi's sarcoma (KS) (2), primary effusion lymphoma (PEL) (3), and multicentric Castleman's disease (MCD) (4). Increasing evidence has suggested that KSHV also deregulates an array of host cytokines, including interleukin-6 (IL-6), IL-8, and IL-1α, thereby inducing cell proliferation and malignant transformation (5–8).
Signal transducer and activator of transcription (STAT) proteins are a family of cytoplasmic transcription factors involved in cytokine signal transduction. STAT6 is a key member of the STAT family, whose role in the biology of cancer and immune cells has been firmly established (9, 10). STAT6 is activated by cytokine IL-4 or IL-13, via a common receptor chain, namely, IL-4Rα. Upon interleukin binding, IL-4Rα dimerizes with IL-4Rγ or IL-13Rα1 to form type I or type II IL-4R receptor, respectively. The dimerized receptor recruits and activates phosphorylation of Janus tyrosine kinases (JAK), including JAK1 and JAK2, which, in turn, phosphorylate tyrosine residues on IL-4R, providing a docking site for the recruitment of STAT6. STAT6 itself becomes phosphorylated at its conserved tyrosine residue Y641 (11) and subsequently translocates into the nucleus, where it regulates downstream gene expression through binding to distinct consensus TTCN3/4GAA regions within the gene promoter (12, 13). To date, at least 35 genes in physiological and pathophysiological processes are activated by STAT6 (12). Regulation of STAT6 signaling is governed by a variety of inhibitory signals, including SOCS1 (suppressor of cytokine signaling-1), and SHP1 (SH2-containing phosphatase-1). These proteins suppress IL-4/STAT6 and block STAT6 activation by dephosphorylating activated JAK, respectively (14).
Of significant importance is the identification of constitutive STAT6 activation in a number of human malignancies (9), including prostate carcinomas (15) and Hodgkin's lymphoma (16). Mechanistically, STAT6 is constitutively activated in primary mediastinal large B-cell lymphomas due to amplification of JAK2 (13), while in hepatocellular carcinoma, gastric carcinoma, colorectal cancer, and hematological malignancies, STAT6 activation results from promoter hypermethylation and silencing of SHP1 or SOCS1 (17–20). Interestingly, in virus-associated diseases, constitutive STAT6 activation occurs through different pathways (21–23). We and other colleagues recently found that in KSHV-associated cancers, IL-4-mediated STAT6 activation is tightly regulated by the virus in order to switch life cycles from latency to lytic replication (24, 25). These observations strongly suggest that STAT6 may play a role in KSHV-induced oncogenesis.
However, the molecular mechanism leading to constitutive STAT6 activation in PELs remains unclear. In an attempt to better understand the role of constitutively phosphorylated STAT6 in KSHV pathogenesis, we explored the expression pattern of STAT6-related molecules in KSHV-positive and -negative B lymphoma cells. In this report, we demonstrate that constitutive activation of STAT6 correlates with IL-13 secretion and JAK1/JAK2 phosphorylation due to downregulation of SHP1. Furthermore, blockade of IL-13 by antibody neutralization dramatically inhibits PEL cell proliferation and survival. These findings provide new insight into how KSHV usurps the STAT6 signaling pathway to promote host cell proliferation and viral pathogenesis.
MATERIALS AND METHODS
Cell culture and transfection.
KSHV- and Epstein-Barr virus (EBV)-negative (BJAB and DG75) and KSHV-positive (BC1, BC3, BCP1, BCBL1, and JSC1) B-lymphoma cells, EBV-transformed B-cell line LCL1, KSHV-infected BJAB with a low passage number (K-BJABLow), and iSLK and iSLK-Bac16 (K-iSLK, a gift from S. J. Gao at University of South California) cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (Gibco-BRL). All cell lines were incubated at 37°C in a humidified environmental incubator with 5% CO2. B-cell transfection was performed with Lonza-4D nucleofector system in an optimized program, CA137.
Antibodies.
SOCS1, SHP1, SHP2, and Akt (C-20) antibodies were purchased from Santa Cruz Biotech, Inc. Rat antibodies to IL-4, IL-13, and κ isotype IgG1 were purchased from BioLegend. Antibodies to β-actin, JAK1 (6G4), JAK2 (D2E12), STAT6, p-STAT6 (Tyr-641), and phospho-Akt (pAkt; Ser473, 587F11) were from Cell Signaling Technology (Beverly, MA). Antibodies to IL-4R (clone 25463; R&D), phosphor-tyrosine (clone 4G10; Millipore), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; G8140-01; U.S. Biological), and KAP1 (20C1; Abcam) were used according to the manufacturers' specifications.
IP and IB.
Cells were harvested, washed with ice-cold phosphate-buffered saline (PBS), and lysed in ice-cold RIPA buffer (10 mM Tris-HCl [pH 7.5], 1% Nonidet P-40, 150 mM NaCl, and 2 mM EDTA, with protease inhibitors). Cell lysates were individually subjected to immunoprecipitation (IP) and immunoblotting (IB) or direct immunoblotting with specific antibodies as described previously (26).
Virion purification and primary infection of KSHV or EBV.
HEK293-Bac36 (KSHV-green fluorescent protein [GFP]) cells or HEK293-GFP-EBV cells were individually induced with 20 ng/ml of tetradecanoyl phorbol acetate (TPA) and 1.5 mM sodium butyrate (Sigma-Aldrich, St. Louis, MO) for 2 days at 37°C with 5% CO2. After induction, the supernatant of culture medium was collected and filtered through a 0.45-μm filter, and viral particles were spun down at 25,000 rpm for 2 h at 4°C. The concentrated virus was collected and used for primary infection as described previously (27). The infection was evaluated by interrogating the expression of viral antigen (LANA for KSHV and EBNA1 for EBV) in addition to visualizing GFP expression using fluorescence microscopy.
Inhibition of tyrosine phosphorylation by chemical reagents.
The kinase inhibitor LY294002, PD98059, and AG490 from Upstate Biotechnology were reconstituted in dimethyl sulfoxide (DMSO). Cell lines were seeded at 0.4 × 105 to 0.6 × 105 cells/ml and treated for 24 h with different doses of LY294002, PD98059, or AG490. Control cells were treated with equal volumes of DMSO. After treatment, cells were washed in PBS and lysed as described for immunoblotting.
Flow cytometry of cell cycle.
Treated cells were harvested, washed in ice-cold PBS, and fixed in cold methanol-acetone, followed by staining with PBS containing propidium iodide (PI) and RNase A as described previously (27). Cell cycle profiles of stained cells were analyzed using FACScan (BD Biosciences, Foster, CA) and FlowJo software.
Nucleic acid extract.
Genomic DNA from B lymphoma cells was extracted by proteinase K digestion, phenol-chloroform extraction, and ethanol precipitation. Total RNA from cells was extracted by using TRIzol, and cDNA was made with a Superscript II reverse transcription kit (Invitrogen, Inc., Carlsbad, CA).
Quantitative PCR.
The primers used for PCR are shown in Table 1. The standard real-time PCR protocol was followed as described previously (27). A melting-curve analysis was performed to verify the specificities of the amplified products. Some PCR products were subjected to DNA sequencing for sequence analysis.
TABLE 1.
Primers used for PCRa
| Target | Primer sequence | Product (bp) |
|---|---|---|
| SHP1 | Sense: 5′-GTCGGAGTACGGGAACATCACC-3′ | 387 |
| Antisense: 5′-CCCAGGGATTTATTTACAAGAGGAG-3′ | ||
| SHP2 | Sense: 5′-ATGAGGAGACACGGGTAGGACT-3′ | 303 |
| Antisense: 5′-GCTATGTGTGAAAGTTGATCCC-3′ | ||
| SOCS1 | Sense: 5′-GACTGCTAGCCATGGTAGCACACAACCAGGTGGCAG-3′ | 640 |
| Antisense: 5′-AGTCCTCGAGTCAAATCTGGAAGGGGAAGGAGCTC-3′ | ||
| SOCS3 | Sense: 5′-GACTGCTAGCCATGGTCACCCACAGCAAGTTTCCC-3′ | 687 |
| Antisense: 5′-AGTCCTCGAGTTAAAGCGGGGCATCGTACTGGTC-3′ | ||
| GAPDH | Sense: 5′-TGCACCACCAACTGCTTAG-3′ | 190 |
| Antisense: 5′-GATGCAGGGATGATGTTC-3′ | ||
| IL-4 | Sense: 5′-ACCTCCCAACTGCTTCCC-3′ | 298 |
| Antisense: 5′-GCTGCTTGTGCCTGTGGA-3′ | ||
| IL-13 | Sense: 5′-GTGGACCCAGGGATGACA- 3′ | 297 |
| Antisense: 5′-CTCCTGGTGTCCACTGCT- 3′ | ||
| SHP1p | Sense: 5′- ATAGGTACCTTGGTTTGGCGGTGTTGATGTTT-3′ | 1,132 |
| Antisense: 5′-ATAAGCTTGGGAATGAGGAGGTGCAGCTAGTCT-3′ | ||
| IL-13p | Sense: 5′-CCGTTACATAAGGCCACCCCC-3′ | 1,596 |
| Antisense: 5′-TCCAGTGTCGCATAAAGGAAAGAGTT-3′ | ||
| IL-4-ChIP | Sense: 5′-GGCCTCTCCCTTCTATGCAAA-3′ | 211 |
| Antisense: 5′-GGGCCAATCAGCACCTCTCT-3′ | ||
| IL-13-ChIP | Sense: 5′-GCCCTCCACAGCACTCATTC-3′ | 237 |
| Antisense: 5′-GTGGCTGGAAGTAGTGTGCAC-3′ |
For primers for EBV latent and lytic genes, see reference 31.
ChIP.
Chromatin immunoprecipitation (ChIP) experiments were done as previously described (27). Briefly, aliquots were incubated with each specific antibody or IgG control overnight at 4°C. Immune complexes were separated into bound and unbound complexes with protein A-agarose, and cross-links were reversed by treatment at 65°C overnight. After treatment with RNase A and proteinase K, samples were extracted once with phenol-chloroform, and the DNA was precipitated and resuspended. The ChIP DNA and 10% input were amplified by qPCR using specific primers.
Immunohistochmistry.
Immunohistochemistry for p-STAT6 and SHP1 was performed on deparaffinized, formalin-fixed tissue sections using an indirect immunoperoxidase method with an automated immunostainer. Three micrometers of paraffin-embedded KS patient tissue was provided from the public health clinical center of Fudan University. Usage of redundant cancer samples for research purpose was recognized by the hospital according to medical ethics practices. Briefly, after deparaffination in xylene for 10 min three times and rehydration, antigen was retrieved with EDTA (pH 8.0) for 10 min at 95°C. Slides were treated with 0.3% hydrogen peroxide to block endogenous peroxidase activity. Subsequently, the appropriate primary antibody of LANA, SHP1, or p-STAT6 was added and incubated overnight at 4°C. The next day, after extensive washing with PBS, the goat anti-mouse or -rabbit secondary antibody IgG conjugated with horseradish peroxidase (kit PV8000; ZSGB-BIO Co., Beijing, China) was added and incubated for 30 min at 25°C. 3,3′-Diaminobenzidine (DAB) was added to stain the slide at room temperature and counterstained by hematoxylin.
Statistical analysis.
Statistical significance of differences between means of at least 3 experiments was determined using Student's t test.
RESULTS
KSHV induces constitutive phosphorylation of STAT6 (p-STAT6C).
To answer whether the constitutive activation of STAT6 on Y-641 (p-STAT6C) in both the BC3 and BCBL1 cell lines, which we observed previously (24), is associated with KSHV infection, we detected levels of p-STAT6C in more KSHV-positive (BC1, BC3, BCP1, BCBL1, and JSC1) and KSHV-negative (DG75, BJAB, iSLK, and LCL1) cell lines, including the same genotype cells with and without KSHV infection (K-BJABLow and K-iSLK). Interestingly, the results of immunoblotting against p-STAT6C show that although p-STAT6C is not highly activated in all KSHV-positive cell lines, the presence of KSHV in both BJAB and iSLK cell lines does induce p-STAT6C (1.8-fold and 2.4-fold increases, respectively) to some extent (Fig. 1A). In contrast, LCL1 cells with EBV infection alone did not present p-STAT6C.
FIG 1.

KSHV induces constitutive phosphorylation of STAT6 (p-STAT6C). (A) Relative levels of p-STAT6C/STAT6 in KSHV-positive (BC1, BC3, BCP1, BCBL1, JSC1, K-BJABLow, and K-iSLK) and KSHV/EBV-negative (DG75, BJAB, LCL1, and iSLK) cell lines. (B) p-STAT6C is blocked by JAK inhibitor in PEL cells. Representative data for immunoblotting of treated BC3 cells are shown at the bottom. (C) The levels of IL-4Rα expression and phosphorylated JAK (p-JAK1 and p-JAK2) are elevated in KSHV-infected cells. The relative protein level is quantitated in the histogram (bottom). RD, relative density.
To address whether activation of JAK kinases or other kinases is important for the constitutive activation of p-STAT6C, we individually treated BC3 and BCBL1 cells with the JAK inhibitor AG490, phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002, or MEK inhibitor PD98059, followed by immunoblotting assays against p-STAT6C and phosphorylated Akt. As shown in Fig. 1B, the results indicate that JAK but not PI3K or MEK kinases contribute to the constitutive activation of STAT6. Consistent with previous reports that JAK is able to phosphorylate PI3K, which, in turn, phosphorylates Akt, we observed reduction of Akt phosphorylation at serine 473 following AG490-induced JAK inhibition. Conversely, PI3K inhibitor only blocks phosphorylation of Akt instead of STAT6 (Fig. 1B). These findings indicate that constitutive activation of STAT6 is due to the cascade phosphorylation of JAK.
To further validate our findings, we looked at levels of JAK1/2 and the phosphorylated JAK1/2 in the KSHV-positive and -negative cell lines by immunoprecipitation and immunoblotting assays. While there was no significant difference in native JAK1 or JAK2 expression in KSHV-positive and -negative cells, a higher level of phosphorylated JAK1 or JAK2 was consistently observed in all the KSHV-positive cell lines (except in BC1, the absence of p-JAK could be due to lower expression of IL-4Rα as described below), including EBV-negative BJAB cells with KSHV infection, than in the KSHV-negative cell lines (Fig. 1C). Intriguingly, we also observed that an activated form of the IL-4Rα receptor (a key upstream regulator of JAK) was consistently elevated in all KSHV-positive cell lines (Fig. 1C, graph), which further supports our hypothesis that constitutive p-STAT6 in PEL cells stems from activation of JAK and is due to KSHV infection.
Low expression of SHP1 in PEL cells associates with p-STAT6C.
To further elucidate pathway changes contributing to constitutive phosphorylation of STAT6 in PEL cells, we performed an immunoblotting assay to detect the protein levels of three key negative regulators: SOCS1, SHP1, and SHP2. As shown in Fig. 2A, similar to the pattern of phosphorylated JAK1 and JAK2 observed in the KSHV-positive and -negative cell lines, lower levels of expression of SHP1 but not SHP2 or SOCS1 were consistently observed in KSHV-positive cells, particularly about 2.6-fold and 2.1-fold decreases in BJAB and iSLK cells after KSHV infection, respectively (Fig. 2A, right side). This led to our speculation that the absence of p-STAT6C (Fig. 1A) in the KSHV- and EBV-positive cell lines, including BC1 and JSC1, could be specimen specific, resulting from coinfection of EBV, which may directly block the constitutive phosphorylation of STAT6 induced by KSHV.
FIG 2.
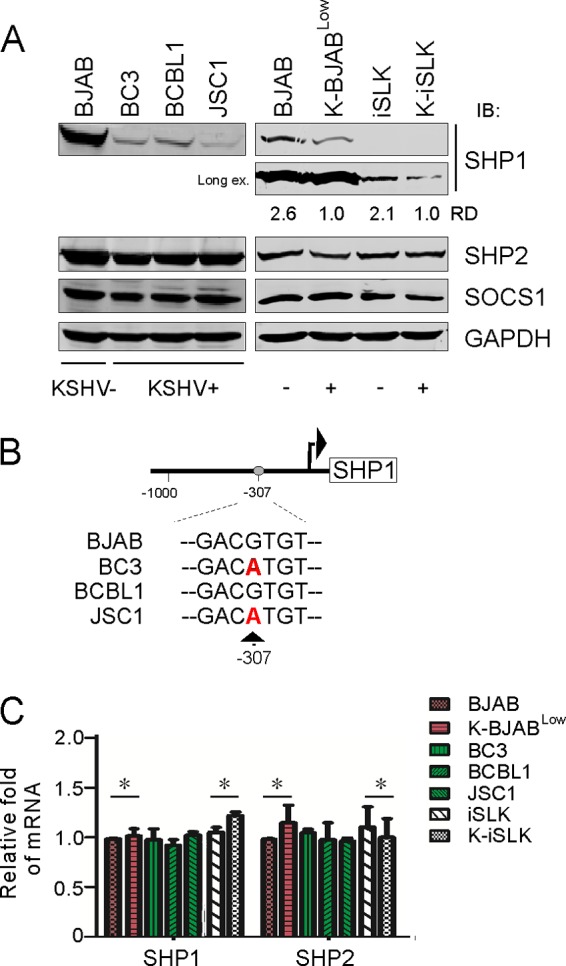
KSHV downregulates SHP1 expression. (A) Low expression of SHP1 in PEL cells. Cell lysates from equal amounts of KSHV-positive and -negative cells were subjected to immunoblotting analysis with antibodies as indicated. GAPDH was used as an internal control. (B) Analysis of SHP1 promoter sequence from B lymphoma and PEL cells. (C) Transcriptional level of SHP1 and SHP2 in KSHV-positive and -negative cells. Total RNA was extracted from cultured cells for quantitative PCR analysis for SHP1 and SHP2. GAPDH was used as an internal control. RD, relative density.
To exclude the possibility that the reduced expression of SHP1 was due to mutation or a deletion within its promoter instead of KSHV infection, we analyzed the transcriptional level and promoter sequences of SHP1 along with SHP2, SOCS1, and SOCS3 as controls in KSHV-positive and -negative cells. Surprisingly, the sequence analysis revealed that no recurrent mutations or deletions up to 1 kb upstream of the SHP1 promoter occurred in the KSHV-positive cells, although a site mutation at −307 position from the SHP1 promoter from BC3 and JSC1 cells was observed (Fig. 2B). In contrast, no mutation within the SHP2, SOCS1, or SOCS3 promoter was observed (data not shown). Unexpectedly, the results of analysis of transcriptional levels of SHP1 and SHP2 showed that there was no significant difference in SHP1 or SHP2 expression in KSHV-positive and -negative cells (Fig. 2C). This finding supports the notion that constitutively activated STAT6 in PEL cell lines is associated with KSHV-mediated reduced SHP1 expression at the protein, rather than transcriptional, level.
Activation of p-STAT6C is induced by KSHV in PEL cells.
Our previous study showed that inhibition of KAP1 results in reduction of KSHV episome copy number and viral protein expression in PEL cells (27). To prove that constitutively activated STAT6 is due to KSHV in PEL cells, we compared the p-STAT6C levels in BC3 cells with and without loss of KSHV induced by KAP1 knockdown. In agreement with our hypothesis, our results demonstrate that KAP1 knockdown mediates loss of KSHV associated with subsequent reduction of p-STAT6C (Fig. 3A, compare lanes 1 and 4 with lane 2 and 5, respectively). Moreover, p-STAT6C was only partially rescued when the endogenous level of knocked-down KAP1 was supplemented by transient expression of exogenous KAP1 (Fig. 3A, compare lane 2 and 5 with lanes 3 and 6, respectively). The reduced expression of SHP1 was reversed only by loss of KSHV and not introduction of exogenous KAP1 (Fig. 3A, compare lane 2 and 5 with lanes 3 and 6, respectively), further supporting the direct link between KSHV and p-STAT6C activation in PEL cells.
FIG 3.
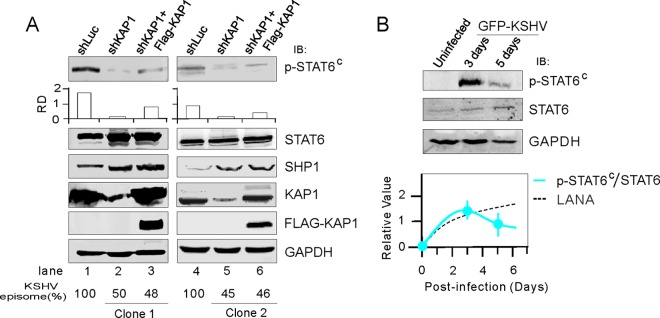
p-STAT6C is associated with SHP1 downregulation by KSHV infection. (A) Loss of the KSHV episome due to inhibition of KAP1 reduced the level of p-STAT6C. Cell lysates from BC3 cells (clone 1 and 2) with constitutive knockdown of KAP1 (shKAP1), a luciferase control (shKAP1), or supplementation with exogenous KAP1 with a FLAG tag were subjected to immunoblotting as indicated. The relative density (RD) of p-STAT6C was quantitated and is shown in the graph in the middle. (B) Immunoblotting analysis. Whole-cell lysates of human PBMCs subjected to GFP-KSHV infection for 3 or 5 days or left uninfected were immunoblotted with antibodies against p-STAT6C, STAT6, and GAPDH. The relative density of p-STAT6C/STAT6 and LANA is individually quantitated, and relative trend line is predicted and shown at the bottom.
Previous studies showed that KSHV primary infection usually involves a mixture of latent and lytic replication statuses within 7 days, shuts off lytic replication at about 3 days postinfection, and establishes dominant latency at about 5 days postinfection (26, 28–30). To further confirm that phosphorylated STAT6 is regulated by KSHV infection, we performed primary infection of human peripheral blood mononuclear cells (PBMCs), followed by immunoblotting analysis against STAT6 and phosphorylated STAT6. KSHV dramatically induced phosphorylation of STAT6 within 5 days following infection; however, the phosphorylated STAT6 was relatively lower on day 5 (Fig. 3B). In KS patient tissues, we also observed moderately higher levels of phosphorylated STAT6 and lower expression of SHP1 than in normal tissue (Fig. 4).
FIG 4.

Expression levels of p-STAT6C and SHP1 in KS patient tissues. (A) Representative images of immunohistochemistry assays of KS patient tissue and normal skin tissue against p-STAT6C, SHP1, and LANA. A larger magnification (×400) of the image is also shown. (B) The relative intensities of p-STAT6C and SHP1 in KS (n = 6) and normal skin tissue (n = 3) were individually quantitated by nuclear and cytoplasmic staining (double positive percentage: +, 10 to 20%; +/−, 1 to 10%; −, <1%) of 100 cells.
Given p-STAT6C in both BC3 and BCBL1 cells infected with KSHV alone but not in JSC1 or BC1 cells infected with both KSHV and EBV, we speculate that the discrepancy in p-STAT6C stems from EBV infection. To prove our hypothesis, BC3 and BCBL1 cells were individually infected with EBV carrying a GFP in vitro, followed by immunoblotting against p-STAT6C, STAT6, and SHP1. We observed a reduction in levels of p-STAT6C in BC3 cells after EBV infection, although SHP1 expression was not significantly affected (Fig. 5A and B; similar data for BCBL1 cells are not shown). Consistent with the previous studies (31), we also could observe that EBV rapidly induced both latent and lytic genes expression during early infection and maintained relatively higher expression of latent genes to establish dominant latency, which is similar to the case with LCL cells at 21 days postinfection (Fig. 5C). Therefore, our results suggest that coinfection with EBV reduces KSHV-induced p-STAT6C activation.
FIG 5.

EBV infection reduces p-STAT6C expression in PEL cells. (A) Lysates from BC3 cells infected with GFP-EBV for 2, 7, or 21 days or left uninfected (mock) were subjected to immunoblotting as indicated. (B) Relative ratio of p-STAT6C to STAT6 during EBV infection. The results are the average relative fold compared with that for uninfected cells from 3 independent experiments. A representative image of BC3 cells with GFP-EBV infection at each time point is shown at the top. (C) Latent (EBNA1, EBNA2, and LMP1) and lytic (BZLF1, BALF5, and BcLF1) gene expression during GFP-EBV infection. The relative mRNA levels of the genes, including GAPDH as a control, were examined by quantitative PCR. The fold change was calculated by the threshold cycle (ΔΔCT) method.
IL-13 is essential for p-STAT6C activation and proliferation of PEL cells.
Since phosphorylation of STAT6 signaling is primarily activated through IL-4 or IL-13, to determine if these two cytokines contribute to p-STAT6C, we performed an immunoblotting analysis for these interleukins in KSHV-positive -and negative cells. Strikingly, our results demonstrate that IL-13 but not IL-4 was highly expressed and correlated with p-STAT6C in the BC3 and BCBL1 cells, as well as BJAB and iSLK cells with KSHV infection (Fig. 6A and B). To further confirm these findings, we performed in vitro antibody neutralization against human IL-13 or IL-4 in culture media and monitored the phosphorylation level of STAT6. Neutralization of IL-13 in BC3 and BCBL1 strongly blocked p-STAT6C activation, whereas no significant changes were observed following IL-4 neutralization (Fig. 6C). Altogether, these findings imply that constitutive activation of STAT6 by KSHV in PEL cells is driven by IL-13 expression and secretion, along with low expression of SHP1 and phosphorylated JAK status.
FIG 6.

IL-13 expression is correlated with p-STAT6C in PEL cells with KSHV infection alone. (A) Equal amounts of KSHV-positive and-negative B lymphoma cells were subjected to immunoblotting analysis with antibodies as indicated. (B) The transcription level of IL-13 but not IL-4 is upregulated by KSHV. (C) IL-13 but not IL-4 depletion reduces the level of p-STAT6C. PEL cells were individually incubated with or without antibodies against IL-4, IL-13, or the same isotype IgG control for 12 h, followed by immunoblotting analysis as indicated. (D) The putative STAT6-binding sites within IL-13 and IL-4 promoters are indicated in the top portion. DNA sequencing reveals four hot mutation spots within the IL-13 promoter from PEL cells. nd, not determined. (E) p-STAT6C has a higher affinity for the IL-13 promoter than for the IL-4 promoter. Chromatin immunoprecipitation (ChIP) with p-STAT6C from BC3 cells was performed, and the relative density (RD) of p-STAT6C bound to IL-13 or IL-4 promoter was detected by quantitative PCR. The specific amplicon was verified by agarose gel electrophoresis (top).
To explore whether mutations of IL-13 promoter leads to high expression of IL-13 in both BC3 and BCBL1 cells, we amplified and sequenced DNA isolated from KSHV-positive (BC3, BCBL1, and JSC1) and KSHV-negative (BJAB) B lymphoma cells. While several mutations within IL-13 promoter were detected, there was no correlation between the mutation observed and IL-13 expression (Fig. 6D). To elucidate why expression of IL-13 and not IL-4 was associated with p-STAT6 activation, we hypothesized that p-STAT6C may selectively bind to the IL-13 promoter through specific binding sites. To evaluate our hypothesis, we analyzed the promoter sequences of both IL-13 and IL-4; we identified four potential STAT6-binding sites located at the IL-13 promoter but only one site at the IL-4 promoter (Fig. 6D). We validated this finding using chromatin immunoprecipitation (ChIP) assays, which demonstrated increased affinity of p-STAT6 antibody toward the IL-13 promoter (Fig. 6E).
To determine the biological consequences of IL-13-mediated STAT6 activation, we evaluated the effect of IL-13 or IL-4 neutralization along with nonspecific antibody control on the proliferation of KSHV-positive PEL cells (BC3 and BCBL1) as well as BJAB and BJAB cells with KSHV infection (K-BJABLow). The growth rate of PEL cells and K-BJABLow (but not BJAB) cells following IL-13 but not IL-4 neutralization was significantly lower (P < 0.05) than that in response to a nonspecific IgG isotype or untreated control, as shown in Fig. 7A. The inhibitory effect of IL-13 neutralization on PEL cell proliferation causes a moderate increase in cell apoptosis (30.2% versus 38.8%), although this effect was further enhanced (38.8% versus 61.5%) following cell starvation (Fig. 7B). Taken together, these results indicate that IL-13 is essential for activation of STAT6 and PEL cell proliferation.
FIG 7.

IL-13 is crucial for triggering PEL cell proliferation and survival. (A) Equal amounts (2 million) of BC3, BCBL1, BJAB, and K-BJABLow cells were individually treated with either 20 μg/ml of anti-IL-4, anti-IL-13 antibody or an IgG isotype control. Proliferation was measured at 24, 48, and 72 h by cell vitality counter. The proliferation rate of the treated cells is presented as a percentage of the corresponding untreated control and was calculated as the mean of triplicate samples. The statistical significance was evaluated, and P values of <0.05 are indicated by double asterisks. (B) IL-13 enhances PEL cell survival. BC3 cells were individually subjected to treatment with anti-IL-13 or control antibody (as for panel A) in combination or not with sera starved (0.1%) overnight, followed by analysis of the cell cycle profile. The average percentages of different phases (sub-G1, G1, S, G2/M) from three repeats are presented in a histogram.
DISCUSSION
JAK-STAT is one of the most important pathways induced by cytokines. Upon ligand binding to its cognate receptor, receptor-associated JAKs are phosphorylated and, in turn, successively activate STAT (32). It has been reported that constitutive activation of STAT6 is widely associated with tumor development, including virus-associated cancers (13, 15, 16, 33, 34). KSHV, a recently discovered oncogenic herpesvirus, has been shown to escape host immune surveillance through modulating the JAK-STAT pathway (35, 36). However, the specific molecular mechanism through which signal pathway modulation occurs remains largely uncharacterized. Our previous studies showed that KSHV blocks IL-4-mediated STAT6 activation (24). In the present investigation, we demonstrated that STAT6 was constitutively activated (p-STAT6C) in KSHV-associated PEL cells to some extent. Moreover, the moderate activation of p-STAT6C was tightly associated with IL-13 autocrine secretion and reduced expression of SHP1 caused by KSHV infection. Together, these pathway changes lead to PEL cell proliferation and survival (Fig. 8). Consistent with previous reports that the STAT6-associated IL-4 receptor is highly expressed in KS tissues (37, 38), we found that the IL-4 receptor was highly expressed in all PEL cells. These findings indicate that constitutive activation of STAT6 is a common feature in KSHV-associated cancers, although the level of p-STAT6C is relatively lower than that stimulated by IL-4.
FIG 8.
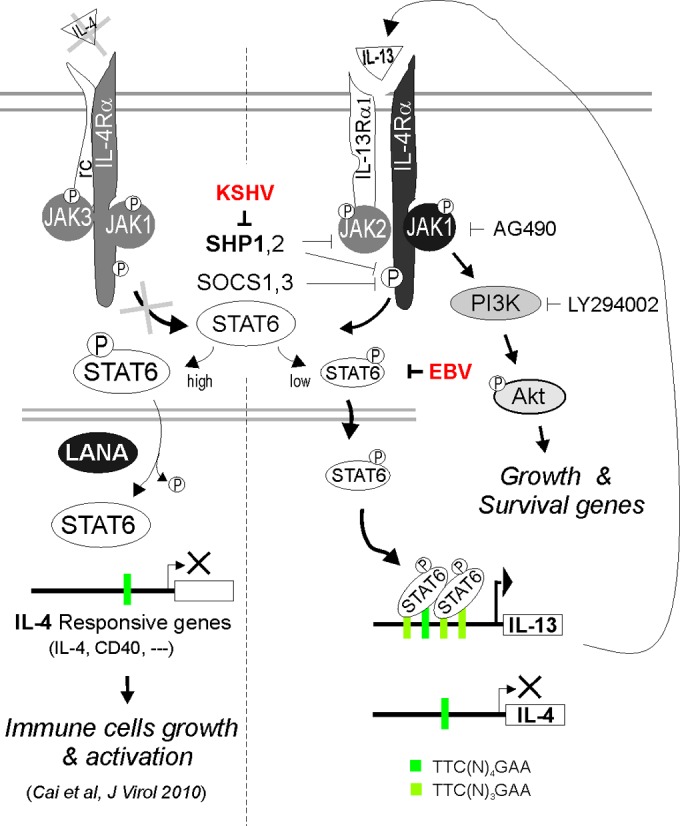
Schematic representation of constitutive activation of IL-13/STAT6 signaling in PEL cells. In KSHV-associated B lymphoma cells, KSHV not only significantly blocks IL-4-induced activation of STAT6 (high) for suppressing immune cell growth and activation (24) but also downregulates SHP1 and constitutively activates IL-13-mediated phosphorylation of STAT6 (low) to a certain extent via selective induction of IL-13 but not IL-4 expression, for enhancing host cell proliferation and survival. However, EBV coinfection dramatically blocks KSHV-induced activation of IL-13/STAT6 signaling.
Recent publications have shown that cytokine induction of a Th2 cell immune response is one of the mechanisms that inhibiting an antitumor immune response. Among Th2 cytokines, IL-4 has been documented as the most critical cytokine for Th2 induction. However, another related cytokine, IL-13, has also been identified as a critical mediator of antitumor immunity (39). The phosphorylation of STAT6 is primarily activated by cytokines IL-4 and IL-13. To decipher how STAT6 is constitutively activated in different tumors, several groups have shown that IL-13 is frequently expressed and antibody neutralization of IL-13 results in a dramatic decrease of phosphorylated STAT6 (15, 40). In this study, we also demonstrate that constitutive activation of STAT6 is induced by highly expressed IL-13 in PEL cells independent of IL-4; moreover, treatment of PEL cells with antibodies against IL-13 diminished phosphorylated STAT6. In addition, these results suggest that neutralization with IL-13 antibodies results in reduction of phosphorylated STAT6 and blocks the proliferation and survival of PEL cells. This work therefore supports the hypothesis that activated STAT6 could accelerate cell proliferation by downregulating cyclin-dependent kinase inhibitor p27 (9).
Despite IL-13's role in the regulation of STAT6 activation, functional dysregulation of its upstream activator or negative regulator may also contribute to constitutively active STAT6. In MedB-1 cells derived from primary mediastinal large B-cell lymphoma, phosphorylated STAT6 was a result of a mutation of negative regulator SOCS1, an upstream activator of JAK2 (34, 41). We found that the negative regulator SHP1 is significantly downregulated in KSHV-associated PEL cell lines compared to controls. In contrast, no difference of SOCS1 and SHP2 protein expression was observed in these cells. This implies that inhibition of SHP1 may contribute to the activation of STAT6 in PEL cells. Alternatively, increased STAT6 phosphorylation has been associated with promoter hypermethylation of SHP1 or SOCS1, as reported for many cancers (42). To further determine whether hypermethylation or promoter mutation of negative regulator resulted in activation of STAT6 in PEL cells, we amplified and sequenced SOCS1, SOCS3, SHP1, or SHP2 promoters, as well as treated PEL cells with demethylation reagent at different concentrations, and analyzed the transcriptional profile of SOCS1, SOCS3, SHP1, or SHP2. Our results demonstrate that demethylation did not affect gene expression, and no promoter mutation with consistent impact on gene expression was identified (data not shown).
It has been reported that virus could directly or indirectly activate STAT6 upon infection. For instances, herpesvirus saimiri (HVS)-encoded Tip (tyrosine kinase-interacting protein) could interact with STAT6 and induce phosphorylation in T cells (22); SHP1 expression was selectively downregulated in human T-cell leukemia virus 1 (HTLV-1)-transformed T-cell lines, and Tax transactivates IL-13 overexpression (43, 44). In addition, EBV Zta protein is able to induce the expression of IL-13 and promote the proliferation of EBV-infected B cells (45). To address whether KSHV infection triggered constitutive activation of STAT6 in PEL cells, we downregulated the KSHV episome by knocking down KAP1 and determined the level of phosphorylated STAT6. The results confirmed our hypothesis and showed that loss of KSHV due to KAP1 knockdown reduced STAT6 phosphorylation, while rescuing KAP1 expression via transfection only partially recovered the levels of phosphorylated STAT6. We verified these findings by infecting PBMCs with KSHV. Interestingly, due to the dynamic status of latency and lytic replication at different stages during primary infection (26, 28–30), STAT6 phosphorylation was upregulated at the early stages following infection and subsequently downregulated, suggesting that higher levels of phosphorylated STAT6 could be required for establishment but not maintenance of KSHV latency. Consistent with our and others' previous works (24, 25), the robust activation of STAT6 at early stages during KSHV primary infection could contribute to viral lytic replication and gradually be reduced to a lower extent (probably due to shutoff of potent IL-4 induction and continued mild IL-13 induction, as we have observed so far) along establishment of latency. Altogether, these results highlight a critical role for constitutive activation of STAT6 in PEL cells following KSHV infection.
Recent studies have shown that coinfection of two pathogens could compete for cytokine-mediated regulation of STAT6 (25). In this study, although we did not see any phosphorylated STAT6 in JSC1 cells with both KSHV and EBV infection, immunoblotting results showed a decrease in SHP1 in JSC1 which is consistent with KSHV-positive PEL cell lines BC3 and BCBL1. Moreover, infection of BC3 or BCBL1 with EBV in vitro reduced phosphorylated STAT6. These results imply that EBV coinfection could alter KSHV-induced STAT6 activation, and the PEL cells dually positive for EBV and KSHV may have different regulatory pathway to bypass the effect of IL-13/STAT6. However, details of this mechanism require a more thorough investigation.
In summary, we demonstrated that STAT6 is constitutively activated in PEL cells due to secretion of IL-13 and KSHV-mediated downregulation of SHP1. This indicates that constitutive activation of IL-13/STAT6 signaling is another mechanism utilized by KSHV to promote pathogenesis and tumorigenesis during latency infection, and STAT6 could be a candidate target for tumor therapy.
ACKNOWLEDGMENTS
We are grateful to Erle Robertson from the University of Pennsylvania for providing reagents.
This work was supported by the Research and Innovation Key Project of the Shanghai Municipal Education (13zz011) and S&T Commission (15YF1400900), the Natural Science Foundation of China (81471930 and 81402542), and the National Key Basic Research “973” Program (2012CB519001) of China.
C.Z. is a scholar of Yangfan Talents in Shanghai, China. F.W. is a scholar of Pujiang Talents in Shanghai. Q.C. is a scholar of New Century Excellent Talents at the University of China.
REFERENCES
- 1.Mogensen TH, Paludan SR. 2001. Molecular pathways in virus-induced cytokine production. Microbiol Mol Biol Rev 65:131–150. doi: 10.1128/MMBR.65.1.131-150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 3.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280. [PubMed] [Google Scholar]
- 4.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 5.Abend JR, Ramalingam D, Kieffer-Kwon P, Uldrick TS, Yarchoan R, Ziegelbauer JM. 2012. Kaposi's sarcoma-associated herpesvirus microRNAs target IRAK1 and MYD88, two components of the Toll-like receptor/interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. J Virol 86:11663–11674. doi: 10.1128/JVI.01147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abend JR, Uldrick T, Ziegelbauer JM. 2010. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi's sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J Virol 84:12139–12151. doi: 10.1128/JVI.00884-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCormick C, Ganem D. 2005. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 307:739–741. doi: 10.1126/science.1105779. [DOI] [PubMed] [Google Scholar]
- 8.Moore PS, Boshoff C, Weiss RA, Chang Y. 1996. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 274:1739–1744. doi: 10.1126/science.274.5293.1739. [DOI] [PubMed] [Google Scholar]
- 9.Bruns HA, Kaplan MH. 2006. The role of constitutively active Stat6 in leukemia and lymphoma. Crit Rev Oncol Hematol 57:245–253. doi: 10.1016/j.critrevonc.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Ansel KM, Djuretic I, Tanasa B, Rao A. 2006. Regulation of Th2 differentiation and Il4 locus accessibility. Annu Rev Immunol 24:607–656. doi: 10.1146/annurev.immunol.23.021704.115821. [DOI] [PubMed] [Google Scholar]
- 11.Wurster AL, Tanaka T, Grusby MJ. 2000. The biology of Stat4 and Stat6. Oncogene 19:2577–2584. doi: 10.1038/sj.onc.1203485. [DOI] [PubMed] [Google Scholar]
- 12.Hebenstreit D, Wirnsberger G, Horejs-Hoeck J, Duschl A. 2006. Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev 17:173–188. [DOI] [PubMed] [Google Scholar]
- 13.Guiter C, Dusanter-Fourt I, Copie-Bergman C, Boulland ML, Le Gouvello S, Gaulard P, Leroy K, Castellano F. 2004. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood 104:543–549. doi: 10.1182/blood-2003-10-3545. [DOI] [PubMed] [Google Scholar]
- 14.Rane SG, Reddy EP. 2002. JAKs, STATs and Src kinases in hematopoiesis. Oncogene 21:3334–3358. doi: 10.1038/sj.onc.1205398. [DOI] [PubMed] [Google Scholar]
- 15.Ni Z, Lou W, Lee SO, Dhir R, DeMiguel F, Grandis JR, Gao AC. 2002. Selective activation of members of the signal transducers and activators of transcription family in prostate carcinoma. J Urol 167:1859–1862. doi: 10.1016/S0022-5347(05)65249-4. [DOI] [PubMed] [Google Scholar]
- 16.Skinnider BF, Elia AJ, Gascoyne RD, Patterson B, Trumper L, Kapp U, Mak TW. 2002. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 99:618–626. doi: 10.1182/blood.V99.2.618. [DOI] [PubMed] [Google Scholar]
- 17.Yoshikawa H, Matsubara K, Qian GS, Jackson P, Groopman JD, Manning JE, Harris CC, Herman JG. 2001. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet 28:29–35. doi: 10.1038/ng0501-29. [DOI] [PubMed] [Google Scholar]
- 18.Oshimo Y, Kuraoka K, Nakayama H, Kitadai Y, Yoshida K, Chayama K, Yasui W. 2004. Epigenetic inactivation of SOCS-1 by CpG island hypermethylation in human gastric carcinoma. Int J Cancer 112:1003–1009. doi: 10.1002/ijc.20521. [DOI] [PubMed] [Google Scholar]
- 19.Fujitake S, Hibi K, Okochi O, Kodera Y, Ito K, Akiyama S, Nakao A. 2004. Aberrant methylation of SOCS-1 was observed in younger colorectal cancer patients. J Gastroenterol 39:120–124. doi: 10.1007/s00535-003-1262-0. [DOI] [PubMed] [Google Scholar]
- 20.Valentino L, Pierre J. 2006. JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol 71:713–721. doi: 10.1016/j.bcp.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 21.Mazumder ED, Jardin C, Vogel B, Heck E, Scholz B, Lengenfelder D, Sticht H, Ensser A. 2012. A molecular model for the differential activation of STAT3 and STAT6 by the herpesviral oncoprotein Tip. PLoS One 7:e34306. doi: 10.1371/journal.pone.0034306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim Y, Kwon EK, Jeon JH, So I, Kim IG, Choi MS, Kim IS, Choi JK, Jung JU, Cho NH. 2012. Activation of the STAT6 transcription factor in Jurkat T-cells by the herpesvirus saimiri Tip protein. J Gen Virol 93:330–340. doi: 10.1099/vir.0.036087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H, Sun H, You F, Sun W, Zhou X, Chen L, Yang J, Wang Y, Tang H, Guan Y, Xia W, Gu J, Ishikawa H, Gutman D, Barber G, Qin Z, Jiang Z. 2011. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 147:436–446. doi: 10.1016/j.cell.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 24.Cai Q, Verma SC, Choi JY, Ma M, Robertson ES. 2010. Kaposi's sarcoma-associated herpesvirus inhibits interleukin-4-mediated STAT6 phosphorylation to regulate apoptosis and maintain latency. J Virol 84:11134–11144. doi: 10.1128/JVI.01293-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reese TA, Wakeman BS, Choi HS, Hufford MM, Huang SC, Zhang X, Buck MD, Jezewski A, Kambal A, Liu CY, Goel G, Murray PJ, Xavier RJ, Kaplan MH, Renne R, Speck SH, Artyomov MN, Pearce EJ, Virgin HW. 2014. Helminth infection reactivates latent gamma-herpesvirus via cytokine competition at a viral promoter. Science 345:573–577. doi: 10.1126/science.1254517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai Q, Xiao B, Si H, Cervini A, Gao J, Lu J, Upadhyay SK, Verma SC, Robertson ES. 2012. Kaposi's sarcoma herpesvirus upregulates Aurora A expression to promote p53 phosphorylation and ubiquitylation. PLoS Pathog 8:e1002566. doi: 10.1371/journal.ppat.1002566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Zhu C, Guo Y, Wei F, Lu J, Qin J, Banerjee S, Wang J, Shang H, Verma SC, Yuan Z, Robertson ES, Cai Q. 2014. Inhibition of KAP1 enhances hypoxia-induced Kaposi's sarcoma-associated herpesvirus reactivation through RBP-Jkappa. J Virol 88:6873–6884. doi: 10.1128/JVI.00283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Purushothaman P, Thakker S, Verma SC. 2015. Transcriptome analysis of Kaposi's sarcoma-associated herpesvirus during de novo primary infection of human B and endothelial cells. J Virol 89:3093–3111. doi: 10.1128/JVI.02507-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu J, Verma SC, Cai Q, Saha A, Dzeng RK, Robertson ES. 2012. The RBP-Jkappa binding sites within the RTA promoter regulate KSHV latent infection and cell proliferation. PLoS Pathog 8:e1002479. doi: 10.1371/journal.ppat.1002479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu J, Verma SC, Cai Q, Robertson ES. 2011. The single RBP-Jkappa site within the LANA promoter is crucial for establishing Kaposi's sarcoma-associated herpesvirus latency during primary infection. J Virol 85:6148–6161. doi: 10.1128/JVI.02608-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halder S, Murakami M, Verma SC, Kumar P, Yi F, Robertson ES. 2009. Early events associated with infection of Epstein-Barr virus infection of primary B-cells. PLoS One 4:e7214. doi: 10.1371/journal.pone.0007214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imada K, Leonard WJ. 2000. The Jak-STAT pathway. Mol Immunol 37:1–11. doi: 10.1016/S0161-5890(00)00018-3. [DOI] [PubMed] [Google Scholar]
- 33.Benekli M, Baer MR, Baumann H, Wetzler M. 2003. Signal transducer and activator of transcription proteins in leukemias. Blood 101:2940–2954. doi: 10.1182/blood-2002-04-1204. [DOI] [PubMed] [Google Scholar]
- 34.Melzner I, Bucur AJ, Bruderlein S, Dorsch K, Hasel C, Barth TF, Leithauser F, Moller P. 2005. Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line. Blood 105:2535–2542. doi: 10.1182/blood-2004-09-3701. [DOI] [PubMed] [Google Scholar]
- 35.Hu Z, Usherwood EJ. 2014. Immune escape of gamma-herpesviruses from adaptive immunity. Rev Med Virol 24:365–378. doi: 10.1002/rmv.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alibek K, Baiken Y, Kakpenova A, Mussabekova A, Zhussupbekova S, Akan M, Sultankulov B. 2014. Implication of human herpesviruses in oncogenesis through immune evasion and supression. Infect Agents Cancer 9:3. doi: 10.1186/1750-9378-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Husain SR, Gill P, Kreitman RJ, Pastan I, Puri RK. 1997. Interleukin-4 receptor expression on AIDS-associated Kaposi's sarcoma cells and their targeting by a chimeric protein comprised of circularly permuted interleukin-4 and Pseudomonas exotoxin. Mol Med 3:327–338. [PMC free article] [PubMed] [Google Scholar]
- 38.Husain SR, Kreitman RJ, Pastan I, Puri RK. 1999. Interleukin-4 receptor-directed cytotoxin therapy of AIDS-associated Kaposi's sarcoma tumors in xenograft model. Nat Med 5:817–822. doi: 10.1038/10541. [DOI] [PubMed] [Google Scholar]
- 39.Terabe M, Park JM, Berzofsky JA. 2004. Role of IL-13 in regulation of anti-tumor immunity and tumor growth. Cancer Immunol Immunother 53:79–85. doi: 10.1007/s00262-003-0445-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skinnider BF, Elia AJ, Gascoyne RD, Trumper LH, von Bonin F, Kapp U, Patterson B, Snow BE, Mak TW. 2001. Interleukin 13 and interleukin 13 receptor are frequently expressed by Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 97:250–255. doi: 10.1182/blood.V97.1.250. [DOI] [PubMed] [Google Scholar]
- 41.Ritz O, Guiter C, Dorsch K, Dusanter-Fourt I, Wegener S, Jouault H, Gaulard P, Castellano F, Moller P, Leroy K. 2008. STAT6 activity is regulated by SOCS-1 and modulates BCL-XL expression in primary mediastinal B-cell lymphoma. Leukemia 22:2106–2110. doi: 10.1038/leu.2008.85. [DOI] [PubMed] [Google Scholar]
- 42.Xu SB, Liu XH, Li BH, Zhang Y, Yuan J, Yuan Q, Li PD, Yang XZ, Li F, Zhang WJ. 2009. DNA methylation regulates constitutive expression of Stat6 regulatory genes SOCS-1 and SHP-1 in colon cancer cells. J Cancer Res Clin Oncol 135:1791–1798. doi: 10.1007/s00432-009-0627-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng J, Zhang D, Zhou C, Marasco WA. 2004. Down-regulation of SHP1 and up-regulation of negative regulators of JAK/STAT signaling in HTLV-1 transformed cell lines and freshly transformed human peripheral blood CD4+ T-cells. Leuk Res 28:71–82. doi: 10.1016/S0145-2126(03)00158-9. [DOI] [PubMed] [Google Scholar]
- 44.Wäldele K, Schneider G, Ruckes T, Grassmann R. 2004. Interleukin-13 overexpression by tax transactivation: a potential autocrine stimulus in human T-cell leukemia virus-infected lymphocytes. J Virol 78:6081–6090. doi: 10.1128/JVI.78.12.6081-6090.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsai SC, Lin SJ, Chen PW, Luo WY, Yeh TH, Wang HW, Chen CJ, Tsai CH. 2009. EBV Zta protein induces the expression of interleukin-13, promoting the proliferation of EBV-infected B cells and lymphoblastoid cell lines. Blood 114:109–118. doi: 10.1182/blood-2008-12-193375. [DOI] [PMC free article] [PubMed] [Google Scholar]