

ABSTRACT
In measles virus (MV)-infected cells, the ribonucleoprotein (RNP) complex, comprised of the viral genome and the nucleocapsid (N) protein, phosphoprotein (P protein), and large protein, assembles at the perinuclear region and synthesizes viral RNAs. The cellular proteins involved in the formation of the RNP complex are largely unknown. In this report, we show that cofilin, an actin-modulating host protein, interacts with the MV N protein and aids in the formation of the RNP complex. Knockdown of cofilin using the short hairpin RNA reduces the formation of the RNP complex after MV infection and that of the RNP complex-like structure after plasmid-mediated expression of MV N and P proteins. A lower level of formation of the RNP complex results in the reduction of viral RNA synthesis. Cofilin phosphorylation on the serine residue at position 3, an enzymatically inactive form, is increased after MV infection and the phosphorylated form of cofilin is preferentially included in the complex. These results indicate that cofilin plays an important role in MV replication by increasing formation of the RNP complex and viral RNA synthesis.
IMPORTANCE Many RNA viruses induce within infected cells the structure called the ribonucleoprotein (RNP) complex in which viral RNA synthesis occurs. It is comprised of the viral genome and proteins that include the viral RNA polymerase. The cellular proteins involved in the formation of the RNP complex are largely unknown. In this report, we show that cofilin, an actin-modulating host protein, binds to the measles virus (MV) nucleocapsid protein and plays an important role in the formation of the MV RNP complex and MV RNA synthesis. The level of the phosphorylated form of cofilin, enzymatically inactive, is increased after MV infection, and the phosphorylated form is preferentially associated with the RNP complex. Our findings determined with cofilin will help us better understand the mechanism by which the RNP complex is formed in virus-infected cells and develop new antiviral drugs targeting the RNP complex.
INTRODUCTION
Measles is a highly contagious viral disease characterized by high fever, respiratory symptoms, conjunctivitis, and a macropapular rash. The patients exhibit immunosuppression, often resulting in secondary infections, the main cause of measles-associated mortality and morbidity. On rare occasions, measles virus (MV) persists in the central nervous system and causes subacute sclerosing panencephalitis with long incubation periods (1). Although mortality and morbidity have been greatly reduced thanks to worldwide vaccination efforts, the World Health Organization estimates that 145,700 people died of measles in 2013 (http://www.who.int/mediacentre/factsheets/fs286/en/).
MV, a member of the genus Morbillivirus in the family Paramyxoviridae, possesses 6 genes coding for the nucleocapsid (N) protein, phosphoprotein (P protein), matrix (M) protein, and hemagglutinin (HA), fusion, and large proteins. The two envelope glycoproteins, the hemagglutinin and fusion proteins, mediate receptor binding and membrane fusion, respectively (1). Pathogenic MVs use as cellular receptors the signaling lymphocyte activation molecule (SLAM) (2, 3) and nectin 4 (4, 5), which are expressed on immune and epithelial cells, respectively. The N, P, and large proteins, together with the viral genome, constitute the ribonucleoprotein (RNP) complex, which is found at the perinuclear region in MV-infected cells (6–8) and functions as the RNA-dependent RNA polymerase. The M protein is involved in virion assembly and the budding process (1).
Besides the P protein, the MV P gene encodes two additional proteins, C and V (9, 10). The V mRNA is produced through the insertion of a single nontemplated guanine at the editing site of the P gene during the transcription. The V protein possesses 231 amino acid residues in the N terminus which are identical to those possessed by the P protein and 68 unique residues in the C terminus. The C protein is synthesized from the P and V mRNA using the second start codon. The C protein counteracts the cellular interferon system by several different mechanisms (11–14), while the V protein interacts with many host proteins largely involved in innate immunity, including the protein encoded by melanoma differentiation-associated gene 5, signal transducer and activator of transcription 1/2, interferon regulatory factor 7, NOD-like receptor family, pyrin domain-containing 3, and nuclear factor-kappa B (15–22).
Cofilin is a member of the actin depolymerizing factor/cofilin protein family and has two isoforms: cofilin-1 (referred to as cofilin in this paper) and cofilin-2. The former is the major form in nonmuscle tissues, and the latter is predominant found in muscles (23). Cofilin binds and severs filamentous F-actin (24, 25), and its activity is controlled by the phosphorylation of the serine residue at position 3. LIM kinases and testicular protein kinases phosphorylate the serine residue and diminish the binding and severing activity of cofilin (26–29), while the members of the slingshot family of phosphatases dephosphorylate the serine residue and reactivate the severing activity (30, 31).
In this study, we searched for host factors interacting with the MV V protein by coprecipitating proteins from MV-infected cells with the glutathione S-transferase (GST)-tagged V protein, which were then subjected to mass spectrometry (MS) analysis. Cofilin was one of the cellular proteins thus identified. Our results indicate that cofilin plays an important role in the formation of the MV RNP complex and viral RNA synthesis.
MATERIALS AND METHODS
Cells and viruses.
Vero/human SLAM (hSLAM) cells (3), HeLa/hSLAM cells (32), and HEK293/hSLAM cells (33), which stably express human SLAM, were maintained in Dulbecco modified Eagle medium (DMEM; Wako Pure Chemical Industries) supplemented with 10% fetal bovine serum and penicillin-streptomycin (Gibco). IC323 is a recombinant MV based on the pathogenic IC-B strain (34). MV-ΔV, which lacks V protein expression, was generated by introducing four nucleotide substitutions into the RNA-editing motif in the P gene of IC323 (18). Virus stocks were prepared and titrated on Vero/hSLAM cells. All MV infections in the present study were carried out at a multiplicity of infection (MOI) of 0.1.
Plasmid constructions.
To construct expression plasmids, fragments encoding the GST-tagged MV N protein (GST-N), influenza virus hemagglutinin (HA)-tagged MV P protein (HA-P), HA-tagged MV V protein (HA-V), Flag-tagged cofilin (Flag-Cof), and Flag-tagged cofilin with the serine-to-alanine substitution at position 3 (Flag-CofS3A) were subcloned into expression plasmid pCA7, a derivative of pCAGGS (35). All tag sequences were added to the N terminus of the respective proteins. The fragment encoding the MV V protein was subcloned downstream of the GST tag in the pGEX-5X-1 bacterial expression plasmid (pGEX-MV V).
Gene knockdown by shRNAs.
Sequences targeting cofilin and luciferase mRNAs were designed using BLOCK-iT RNAi Designer (Invitrogen) and were inserted into pRS-U6/puro vector (Origene) along with the fragment encoding the cytomegalovirus enhancer, chicken beta-actin promoter, and Discosoma sp. red fluorescent protein (DsRed)-monomer gene (used to identify short hairpin RNA [shRNA]-expressing cells). The target shRNA sequences will be provided upon request. To analyze the effect of shRNAs, transfection was carried out 1 h after MV infection or simultaneously with processing of the expression plasmids, using Lipofectamine LTX and Plus reagent (Invitrogen) according to the manufacturer's recommendation.
GST pulldown assay.
The Escherichia coli XL1-Blue strain containing pGEX-MV V was grown in LB medium at 37°C. At an optical density at 600 nm (OD600) of 0.5, 100 μM IPTG (isopropyl-β-d-thiogalactopyranoside) was added and the culture was grown for a further 3 h at 30°C. The cells were pelleted and resuspended in phosphate-buffered saline (PBS) containing protease inhibitor cocktail (Sigma), sonicated, and then supplemented with Triton X-100 such that the final concentration in the solution was 1%. The lysate was clarified by centrifugation, and the supernatant was incubated with glutathione Sepharose beads (GE Healthcare) at 4°C for overnight with gentle shaking. After intensive washing with PBS containing 1% Triton X-100 and the protease inhibitor cocktail, the GST-tagged MV V protein (GST-MV V) was eluted with 50 mM reduced glutathione. The eluate was dialyzed against PBS, and insoluble debris was removed by centrifugation. The protein concentration was determined by 280-nm UV absorbance. Mock- or MV-infected cells were lysed at 24 h after infection in lysis buffer A (20 mM Tris-HCl [pH 7.2], 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA) containing the protease inhibitor cocktail. After insoluble debris was removed by centrifugation, the lysates were mixed with 100 μg of GST-MV V and glutathione Sepharose beads and incubated at 4°C for overnight with gentle shaking. After washing and elution were performed as described above, 25 μg of proteins was subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE).
Mass spectrometry assay.
Proteins pulled down with GST-MV V were separated by SDS-PAGE and visualized with silver staining. Polypeptide bands of interest were excised and diced into small pieces. The gel pieces were treated with 10 mM dithiothreitol (DTT) at 56°C for 45 min and incubated at room temperature for 30 min after the same volume of 55 mM iodoacetamide was added. This was followed by in-gel trypsin digestion at 37°C for 16 h. The digested polypeptides were extracted from the gel sequentially with 0.1% trifluoroacetic acid (TFA), with 33% acetonitrile (ACN)–0.1% TFA, and with 70% ACN–0.1% TFA, and extracted polypeptides were pooled. The pooled sample was concentrated by the use of a SpeedVac (Thermo Fisher Scientific), dissolved into 0.1% TFA–2% ACN, and analyzed by the use of a Finnigan LTQ mass spectrometer (Thermo Fisher Scientific). The obtained data were submitted to Mascot version 2.4.1 (Matrix Science) to search against the NCBInr 20070509 database. The tolerance level of tandem MS (MS/MS) ions was 0.8 Da. The proteins with a Mascot score greater than 47 were considered to be significant.
Coimmunoprecipitation and Western blot analysis.
Subconfluent monolayers of HEK293/hSLAM cells on six-well plates were infected with IC323 or left uninfected for 1 h at 37°C, washed with PBS, and supplied with complete DMEM. The cells were then transfected with 2 μg of pCA7-Flag-Cof together with 2 μg of pCA7-GST-N, 2 μg of pCA7-HA-P, or 2 μg of pCA7-HA-V, using PEI-Max (Polysciences). At 48 h posttransfection, the cells were washed with PBS and lysed in 1 ml of lysis buffer B (50 mM Tris-HCl [pH 8.0], 280 mM NaCl, 0.5% Nonidet P-40, 0.2 mM EDTA, 2 mM EGTA, 10% glycerol, 1 mM DTT) containing the protease inhibitor cocktail. After incubation for 1 h at 4°C, the lysates were centrifuged at 20,000 × g for 10 min at 4°C. A small amount (50 μl) of each supernatant was mixed with an equal volume of 2× SDS loading buffer (125 mM Tris-HCl [pH 6.8], 10% 2-mercaptoethanol, 4% SDS, 0.1% bromophenol blue, 20% glycerol) and kept as the lysate sample. The rest of the cleared supernatant was incubated with anti-Flag monoclonal antibody (MAb) (F1804; Sigma) or normal mouse IgG and with Dynabeads Pan Mouse IgG (Invitrogen) overnight at 4°C with gentle shaking. After intensive washing with lysis buffer B, the polypeptides in precipitated complexes were fractionated by SDS-PAGE and electroblotted onto polyvinylidene difluoride membranes (Immobilon-P; Millipore). The membranes were incubated with rabbit anti-HA polyclonal antibody (pAb) (561; Medical & Biological Laboratories) or goat anti-GST pAb (27457701V; GE Healthcare Life Sciences), followed by incubation with horseradish peroxidase-conjugated anti-rabbit IgG (Invitrogen) or anti-goat IgG (Jackson Immuno Research). Chemiluminescent signals (Chemi-Lumi One Super; Nacalai Tesque) were detected and visualized using a VersaDoc 5000 imager (Bio-Rad). To detect phosphorylated cofilin, subconfluent monolayers of HeLa/hSLAM cells on 24-well plates were infected with IC323 for 1 h at 37°C, washed with PBS, and cultured with complete DMEM for the indicated lengths of time. Cells were lysed in 1× SDS loading buffer, subjected to SDS-PAGE, and subsequently examined by blot analysis using antibody against phosphorylated cofilin (77G2; Cell Signaling Technology) or total cofilin (ab42824; Abcam). Tubulin (sc-5286; Santa Cruz Biotechnology) was used as a loading control.
Reverse transcription-quantitative PCR (RT-qPCR).
Total RNA was extracted from MV-infected and shRNA-transfected cells with TRIzol reagent (Life Technologies), treated with DNase I, and reverse transcribed into cDNAs using the M-MLV reverse transcriptase (Promega) and random hexamer primer. The MV N protein (36), cellular cofilin, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNAs were quantified using SYBR Premix Ex Taq II (TaKaRa) and a LightCycler (Roche). For quantification of the cofilin mRNA, we used primer pair 5′-ATGAAGGTGCGTAAGTCTTC-3′ and 5′-GTTGCATCATAGA GGGCATA-3′.
Immunofluorescence staining.
The following antibodies were used for immunofluorescence staining: rabbit anti-cofilin pAb (ab47401; Abcam), mouse anti-N MAb (clone E137; generously provided by T. Sato, National Institute of Infectious Diseases, Japan), goat anti-GST pAb, rabbit anti-HA pAb, or rabbit anti-Flag pAb (F7425; Sigma), Alexa Fluor 488-conjugated donkey anti-mouse IgG (H+L), Alexa Fluor 488-conjugated donkey anti-goat IgG (H+L), and Alexa Fluor 594-conjugated donkey anti-rabbit IgG (H+L) (Molecular Probes). HeLa/hSLAM cells seeded on coverslips were infected with IC323 or MV-ΔV for 1 h at 37°C, washed with PBS, and cultured with fresh medium. Transfection with shRNAs or expression plasmids was performed after MV infection, where indicated. At 24 h postinfection (p.i.) or posttransfection, cells were simultaneously fixed and permeabilized with PBS containing 2.5% formaldehyde and 0.5% Triton X-100. The cells were blocked with 10% normal donkey serum and incubated with appropriate combinations of primary and secondary antibodies. The nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) in some experiments. The stained cells were observed under a confocal microscope.
RESULTS
Pulldown assay with GST-MV V.
To search for cellular proteins interacting with the MV V protein, we performed the GST pulldown assay. The purified GST-MV V was mixed with lysates from mock- or MV-infected Vero/hSLAM cells. Proteins complexed with GST-MV V were separated by SDS-PAGE and visualized with silver staining. With lysates from mock-infected Vero/hSLAM cells, numerous protein bands appeared (Fig. 1), consistent with the previous report that the MV V protein interacts with more than 200 host proteins (15). Some bands were specifically detected or enhanced with lysates from MV-infected cells compared with those from mock-infected cells. We focused on an infected-cell-specific band (indicated by the arrow in Fig. 1) and analyzed it by mass spectrometry (Table 1). Some of the molecules identified in the band seemed to show contamination with degraded proteins, judging from their molecular masses. Considering its molecular mass and Mascot score, we decided to study cofilin further.
FIG 1.

Pulldown assay with GST-MV V. Lysates of MV-infected (+) or uninfected (−) Vero/hSLAM cells were subjected to the pulldown assay using GST-MV V and glutathione Sepharose beads. Proteins were eluted with reduced glutathione, separated by SDS-PAGE, and visualized with silver staining. The samples were electrophoresed on the same gel, and irrelevant intervening lanes were removed from the figure. Asterisks indicate GST-MV V, and the arrow indicates the peptide band analyzed in this study.
TABLE 1.
Proteins identified by mass spectrometrya
| Accession no. | Protein name | Molecular mass (Da) | Mascot scoreb | nc | Cov (%)d |
|---|---|---|---|---|---|
| gi|28317 | Unnamed product | 59,720 | 487 | 14 | 26 |
| gi|55956899 | Keratin 9 | 62,255 | 414 | 21 | 30 |
| gi|11935049 | Keratin 1 | 66,198 | 407 | 24 | 31 |
| gi|47132620 | Keratin 2 | 65,678 | 325 | 13 | 16 |
| gi|5031635 | Cofilin 1 (nonmuscle) | 18,719 | 252 | 10 | 46 |
| gi|232032 | Elongation factor 1-beta (EF-1-beta) | 24,616 | 93 | 3 | 12 |
| gi|5031595 | Actin-related protein 2/3 complex subunit 4 isoform a | 19,768 | 71 | 1 | 6 |
| gi|457464 | Dsc1a precursor | 93,668 | 64 | 1 | 1 |
| gi|4506597 | Ribosomal protein L12 | 17,979 | 50 | 3 | 18 |
| gi|7959325 | KIAA1529 | 196,058 | 48 | 5 | 1 |
Irrelevant proteins and nonhuman proteins are not included.
Mascot scores greater than 47 are significant (P < 0.05) and listed in the table.
n, numbers of matched peptides.
Cov (%), percent protein sequence coverage.
Cofilin interacts with the MV N protein.
We first examined the intracellular distribution of cofilin. Although cofilin was present throughout the cell (in both the nucleus and cytoplasm) in uninfected HeLa/hSLAM cells, it was colocalized with the MV N protein and formed dot-like or inclusion body-like structures mainly at the perinuclear region after MV infection (Fig. 2A). These structures in MV-infected multinucleated giant cells presumably represent the RNP complex (7, 8). When coexpressed by plasmid-mediated transfection, the MV N and P proteins also formed the dot-like structures at the perinuclear region, unlike those proteins individually expressed (Fig. 2B). When coexpressed with the N and P proteins, Flag-tagged cofilin was also colocalized with these proteins in the dot-like structures (Fig. 2B). In the coprecipitation assay, Flag-tagged cofilin was found to interact with any of the N, P, and V proteins in MV-infected HEK293/hSLAM cells (Fig. 2C, right panel). However, it interacted only with the N protein, but not with the P or V protein, when coexpressed with one of these MV proteins in uninfected cells (Fig. 2C, left panel). As it has been reported that the addition of the tag sequence at the N terminus of the N protein affects its function (37), we repeated the immunostaining and coprecipitation assay using the N protein tagged with 6×His at the C terminus and obtained similar results (data not shown). These findings indicate that cofilin directly associates with the N protein, thereby forming the complex with the P and V proteins in infected cells thanks to the ability of the N protein to interact with multiple partners (6, 38, 39). Indeed, cofilin formed the dot-like or inclusion body-like structures in cells infected with MV lacking the V protein, confirming that the interaction with the V protein is not required for the relocalization of cofilin (Fig. 2A, lower panel). Thus, although we originally identified cofilin through its coprecipitation with the V protein in MV-infected cells, it does not directly interact with the V protein. The interaction of cofilin with the V protein in MV-infected cells has also been reported in proteomics analysis (15).
FIG 2.

Cofilin interacts with the MV N protein. (A) HeLa/hSLAM cells were left uninfected or infected with MV or MV-ΔV at an MOI of 0.1. At 24 h p.i., the MV N protein (green) and cofilin (red) were detected by indirect immunofluorescence staining under a confocal microscope. The nuclei were counterstained with DAPI (blue). Infected cells formed syncytia. Dot-like or inclusion body-like structures were detected at the perinuclear region. (B) HeLa/hSLAM cells were transfected with the indicated combinations of expression plasmids. At 24 h after transfection, the N protein (green), the P protein (red), and cofilin (red) were immunostained with tag-specific antibodies and observed under a confocal microscope. Perinuclear dot-like structures were found. (C) A coimmunoprecipitation (Co-IP) assay was performed with lysates (Ly) from MV-infected or uninfected HEK293/hSLAM cells expressing Flag-cofilin plus GST-N, HA-P, or HA-V protein as indicated. At 48 h after transfection, cell lysates were prepared, and proteins were precipitated with anti-Flag antibody (Flag) or control mouse IgG (IgG) and examined by Western blotting (WB).
Cofilin knockdown affects the MV RNP complex formation and viral RNA synthesis.
To examine the role of cofilin in the RNP complex formation, HeLa/hSLAM cells were transfected with the expression plasmid encoding both DsRed (used to locate successfully transfected cells) and shRNA targeting the luciferase or human cofilin mRNA. The efficiency of cofilin knockdown at the protein level was 50% to 80% (Fig. 3A). Cofilin knockdown significantly reduced the level of RNP complex formation at the perinuclear region in MV-infected cells forming syncytia (Fig. 3B) as well as the formation of the dot-like structures in cells transfected with the N and P proteins (Fig. 3C). Next, we analyzed viral mRNA synthesis in shRNA-introduced cells. Expression of MV N mRNA was greatly suppressed compared with control cell results at 24 and 30 h postinfection (p.i.) (Fig. 3D, bottom panel), when that of cofilin mRNA was reduced to ∼50% (Fig. 3D, top panel). It is likely that suppression of the MV RNA synthesis was, at least partly, due to the poor formation of the RNP complex.
FIG 3.

Cofilin knockdown affects formation of the MV RNP complex and viral RNA synthesis. (A) Expression of cofilin in shRNA-transfected (sh) or untransfected (−) HeLa/hSLAM cells was analyzed by Western blotting. Tubulin was used as the loading control. Chemiluminescence signals were quantified, and the ratios of cofilin to tubulin in samples are shown. The value in untransfected cells is set to 1. (B) HeLa/hSLAM cells were infected with MV at an MOI of 0.1 and then transfected with the plasmid expressing both DsRed and shRNA targeting cofilin or luciferase mRNA at 1 h after infection. At 24 h after transfection, the N protein (green) was immunostained, and the nuclei were counterstained with DAPI. shRNA-transfected cells exhibited autofluorescent DsRed, and infected cells formed syncytia. (C) HeLa/hSLAM cells were transfected with expression plasmids encoding GST-N and HA-P proteins as well as the plasmid expressing DsRed and shRNA. At 24 h after transfection, the GST-N protein (green) was immunostained. The dots in individual DsRed-positive cells were counted. Data are expressed as the average and standard deviation of the dot number per cell. N, the number of DsRed-positive cells analyzed. ***, P < 0.001 (Student's t test). Luci, luciferase; Cof, cofilin. (D) HeLa/hSLAM cells were infected with MV at an MOI of 0.1 and then transfected with shRNA at 1 h after infection. Cofilin (top panel) and MV N (bottom panel) mRNAs were quantified by RT-qPCR at the indicated time points. GAPDH mRNA was used as the control. Samples were examined in triplicate, and the averages and standard deviations of the results are shown. *, P < 0.05; **, P < 0.01 (Student's t test). Data are representative of the results of three independent experiments.
Phosphorylation is required for cofilin to interact with the MV N protein.
The ability of cofilin to modulate reorganization of the actin cytoskeleton is regulated by the phosphorylation of its serine residue at position 3 (25, 40). MV infection did not greatly affect the expression of cofilin at the mRNA and protein levels (Fig. 3D, top panel; Fig. 4A, bottom panel). However, the level of phosphorylated cofilin increased after MV infection (Fig. 4A). When cotransfected with the N and P proteins, Flag-tagged cofilin with the serine-to-alanine substitution at position 3 (Flag-CofS3A) was not included in the dot-like structures, unlike the results seen with Flag-tagged wild-type cofilin (Flag-Cof) (Fig. 4B). Furthermore, experiments in MV-infected HEK293/hSLAM cells revealed that the amounts of the N and P proteins coprecipitated with Flag-CofS3A were reduced compared with those coprecipitated with Flag-Cof (Fig. 4C). These results indicate that the serine phosphorylation at position 3 is required for cofilin to interact with the N protein and to aid in the formation of the RNP complex.
FIG 4.
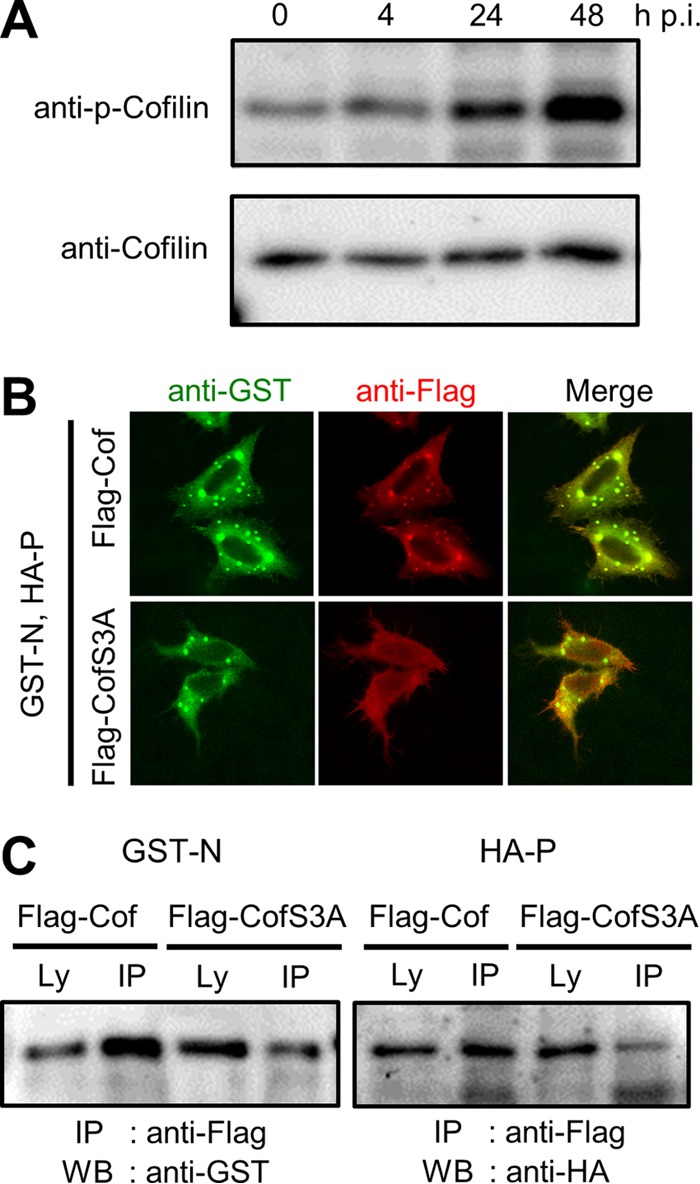
Phosphorylation is required for cofilin to interact with the MV N protein. (A) HeLa/hSLAM cells were infected with MV at an MOI of 0.1. Phosphorylated cofilin (p-Cofilin) and total cofilin were detected by immunoblotting at indicated time points. (B) HeLa/hSLAM cells were transfected with expression plasmids encoding GST-N, HA-P, and Flag-Cof or Flag-CofS3A. At 24 h after transfection, the GST-N protein (green) and Flag-Cof or Flag-CofS3A (red) were immunostained. (C) A Co-IP assay was performed with lysates from MV-infected HEK293/hSLAM cells expressing GST-N or HA-P plus Flag-Cof or Flag-CofS3A. At 48 h after transfection, cell lysates were prepared, and proteins were precipitated and examined by Western blotting.
DISCUSSION
Many mononegaviruses, including vesicular stomatitis virus (41), rabies virus (42), ebolavirus (43), and mumps virus (44), produce cytoplasmic inclusion bodies (equivalent to the MV RNP complex) where viral RNA synthesis occurs. In vesicular stomatitis virus-infected cells, primary RNA synthesis takes place throughout the host cell cytoplasm, and viral protein expression redirects viral RNA synthesis to cytoplasmic inclusions (41). Furthermore, in rabies virus-infected cells, Toll-like receptor 3 is involved in the formation of inclusion bodies (Negri bodies) and viral replication (45). Thus, both viral and host proteins are probably required for the formation of cytoplasmic inclusion bodies or RNP complexes, and different viruses may employ different host proteins for that purpose.
In this study, we demonstrated that the phosphorylated form of cofilin interacts with the MV N protein, thereby supporting the formation of the RNP complex at the perinuclear region. Cofilin modulates actin polymerization by binding and severing F-actin. It has been reported that F-actin stabilization or disruption by inhibitors decreases MV budding (cell-free virus titers) but does not affect viral protein synthesis, surface expression levels of MV glycoproteins, and cell-associated virus titers (46). In contrast, our results indicated that cofilin knockdown, which should affect actin reorganization, decreases formation of the RNP complex and MV RNA synthesis. Thus, actin may not be directly involved in the formation of the RNP complex. Alternatively, actin is indeed involved in the process, but stabilization or disruption by inhibitors may not sufficiently affect the function of actin in the MV RNP complex formation.
Interestingly, our study revealed that the level of the phosphorylated form of cofilin is increased after MV infection. Phosphorylated cofilin is enzymatically inactive, and the decrease in cofilin activity should result in enhanced F-actin formation. Intact actin filaments are required for transport of the MV M protein and nucleocapsids to the plasma membrane (46). Furthermore, F-actin was shown to bind to the MV M protein and modulate MV assembly and cell-cell fusion by controlling the interaction of the M protein with the cytoplasmic tail of the H protein (47). Thus, increased phosphorylation of cofilin after MV infection not only promotes RNP complex formation but also controls the later stages of the MV life cycle, including assembly, budding, and cell-cell fusion through actin remodeling. In herpes simplex virus 1 (HSV-1)-infected neuronal cells, the level of phosphorylated cofilin was found to decrease at an early stage of infection (∼2 h after infection) and then to continuously increase during the later stages of infection, causing biphasic F-actin dynamics (48). Knockdown of cofilin reduced virus production but did not affect protein expression in cells infected with HSV-1 or retroviruses (48, 49). In contrast, MV RNA synthesis was suppressed in cofilin knockdown cells, presumably because of its involvement in the MV RNP complex formation.
It has been reported that T cell activation and proliferation are impaired after contact with the MV glycoprotein complex and that the contact results in the inhibition of actin cytoskeletal remodeling and a collapse of membrane protrusions associated with reduced phosphorylation levels of cofilin and ezrin/radixin/moesin proteins (50). The authors propose that by interfering with cytoskeletal remodeling, the contact disturbs the ability of T cells to adhere, spread, and cluster receptors essential for sustained T-cell activation. Our results obtained with cofilin are not consistent with their observations. The discrepancy may be due to the difference in how the cells were treated: MV infection versus the contact with the MV glycoprotein complex (T cells were cocultured with MV in the presence of a fusion-inhibitory peptide to prevent infection) (50). The types of cells used may also be important, because contact-mediated cell arrest has been reported only in T cells. Furthermore, the time frame of the examinations of cofilin phosphorylation may account for the difference: 4 to 48 h after MV infection versus 5 to 60 min after CD3/CD28 stimulation of T cells pretreated with the MV glycoprotein complex (50). In fact, the levels of phosphorylated cofilin greatly change with time in HSV-1-infected neuronal cells, as described above (48).
It has been suggested that sites for virus replication and assembly in infected cells may share some features with aggresomes in which misfolded proteins are degraded or refolded (51, 52). The actin-stabilizing toxin jasplakinolide induced, in a microtubule-dependent manner, a single, large F-actin aggregate, which also contained cofilin (53). Although the RNP complexes found at the perinuclear region of MV-infected cells are apparently different from these aggresomes, the similar cell response may be responsible for inducing these structures. Further studies are required to clarify the mechanism by which the RNP complex is formed in MV-infected cells, and our finding with cofilin may be a first step toward that goal.
ACKNOWLEDGMENTS
We thank T. Sato for generously providing monoclonal antibody against the MV N protein. We appreciate the technical assistance from The Research Support Center, Kyushu University Graduate School of Medical Sciences. This work was partly performed in the Cooperative Research Project Program of the Medical Institute of Bioregulation, Kyushu University.
This study was supported by JSPS KAKENHI grant number 24115005 and by a grant from the Ministry of Health, Labor and Welfare of Japan (the Research Committee of Prion Disease and Slow Virus Infection).
REFERENCES
- 1.Griffin DE. 2013. Measles virus, p 1042–1069. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 3.Ono N, Tatsuo H, Hidaka Y, Aoki T, Minagawa H, Yanagi Y. 2001. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J Virol 75:4399–4401. doi: 10.1128/JVI.75.9.4399-4401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mühlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB Jr, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwasaki M, Takeda M, Shirogane Y, Nakatsu Y, Nakamura T, Yanagi Y. 2009. The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J Virol 83:10374–10383. doi: 10.1128/JVI.01056-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakatsu Y, Ma X, Seki F, Suzuki T, Iwasaki M, Yanagi Y, Komase K, Takeda M. 2013. Intracellular transport of the measles virus ribonucleoprotein complex is mediated by Rab11A-positive recycling endosomes and drives virus release from the apical membrane of polarized epithelial cells. J Virol 87:4683–4693. doi: 10.1128/JVI.02189-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakatsu Y, Takeda M, Ohno S, Shirogane Y, Iwasaki M, Yanagi Y. 2008. Measles virus circumvents the host interferon response by different actions of the C and V proteins. J Virol 82:8296–8306. doi: 10.1128/JVI.00108-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellini WJ, Englund G, Rozenblatt S, Arnheiter H, Richardson CD. 1985. Measles virus P gene codes for two proteins. J Virol 53:908–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cattaneo R, Kaelin K, Baczko K, Billeter MA. 1989. Measles virus editing provides an additional cysteine-rich protein. Cell 56:759–764. doi: 10.1016/0092-8674(89)90679-X. [DOI] [PubMed] [Google Scholar]
- 11.Fontana JM, Bankamp B, Bellini WJ, Rota PA. 2008. Regulation of interferon signaling by the C and V proteins from attenuated and wild-type strains of measles virus. Virology 374:71–81. doi: 10.1016/j.virol.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 12.Nakatsu Y, Takeda M, Ohno S, Koga R, Yanagi Y. 2006. Translational inhibition and increased interferon induction in cells infected with C protein-deficient measles virus. J Virol 80:11861–11867. doi: 10.1128/JVI.00751-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaffer JA, Bellini WJ, Rota PA. 2003. The C protein of measles virus inhibits the type I interferon response. Virology 315:389–397. doi: 10.1016/S0042-6822(03)00537-3. [DOI] [PubMed] [Google Scholar]
- 14.Yokota S, Saito H, Kubota T, Yokosawa N, Amano K, Fujii N. 2003. Measles virus suppresses interferon-alpha signaling pathway: suppression of Jak1 phosphorylation and association of viral accessory proteins, C and V, with interferon-alpha receptor complex. Virology 306:135–146. doi: 10.1016/S0042-6822(02)00026-0. [DOI] [PubMed] [Google Scholar]
- 15.Komarova AV, Combredet C, Meyniel-Schicklin L, Chapelle M, Caignard G, Camadro JM, Lotteau V, Vidalain PO, Tangy F. 2011. Proteomic analysis of virus-host interactions in an infectious context using recombinant viruses. Mol Cell Proteomics 10:M110.007443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Childs K, Randall R, Goodbourn S. 2012. Paramyxovirus V proteins interact with the RNA helicase LGP2 to inhibit RIG-I-dependent interferon induction. J Virol 86:3411–3421. doi: 10.1128/JVI.06405-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis ME, Wang MK, Rennick LJ, Full F, Gableske S, Mesman AW, Gringhuis SI, Geijtenbeek TB, Duprex WP, Gack MU. 2014. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe 16:19–30. doi: 10.1016/j.chom.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ikegame S, Takeda M, Ohno S, Nakatsu Y, Nakanishi Y, Yanagi Y. 2010. Both RIG-I and MDA5 RNA helicases contribute to the induction of alpha/beta interferon in measles virus-infected human cells. J Virol 84:372–379. doi: 10.1128/JVI.01690-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komune N, Ichinohe T, Ito M, Yanagi Y. 2011. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1beta secretion. J Virol 85:13019–13026. doi: 10.1128/JVI.05942-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, Horvath CM. 2003. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol 77:7635–7644. doi: 10.1128/JVI.77.13.7635-7644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfaller CK, Conzelmann KK. 2008. Measles virus V protein is a decoy substrate for IkappaB kinase alpha and prevents Toll-like receptor 7/9-mediated interferon induction. J Virol 82:12365–12373. doi: 10.1128/JVI.01321-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramachandran A, Parisien JP, Horvath CM. 2008. STAT2 is a primary target for measles virus V protein-mediated alpha/beta interferon signaling inhibition. J Virol 82:8330–8338. doi: 10.1128/JVI.00831-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernstein BW, Bamburg JR. 2010. ADF/cofilin: a functional node in cell biology. Trends Cell Biol 20:187–195. doi: 10.1016/j.tcb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishida E, Maekawa S, Sakai H. 1984. Cofilin, a protein in porcine brain that binds to actin filaments and inhibits their interactions with myosin and tropomyosin. Biochemistry 23:5307–5313. doi: 10.1021/bi00317a032. [DOI] [PubMed] [Google Scholar]
- 25.Bravo-Cordero JJ, Magalhaes MA, Eddy RJ, Hodgson L, Condeelis J. 2013. Functions of cofilin in cell locomotion and invasion. Nat Rev Mol Cell Biol 14:405–415. doi: 10.1038/nrm3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. 1998. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 27.Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. 1998. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 28.Toshima J, Toshima JY, Amano T, Yang N, Narumiya S, Mizuno K. 2001. Cofilin phosphorylation by protein kinase testicular protein kinase 1 and its role in integrin-mediated actin reorganization and focal adhesion formation. Mol Biol Cell 12:1131–1145. doi: 10.1091/mbc.12.4.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toshima J, Toshima JY, Takeuchi K, Mori R, Mizuno K. 2001. Cofilin phosphorylation and actin reorganization activities of testicular protein kinase 2 and its predominant expression in testicular Sertoli cells. J Biol Chem 276:31449–31458. doi: 10.1074/jbc.M102988200. [DOI] [PubMed] [Google Scholar]
- 30.Kousaka K, Kiyonari H, Oshima N, Nagafuchi A, Shima Y, Chisaka O, Uemura T. 2008. Slingshot-3 dephosphorylates ADF/cofilin but is dispensable for mouse development. Genesis 46:246–255. doi: 10.1002/dvg.20389. [DOI] [PubMed] [Google Scholar]
- 31.Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. 2002. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108:233–246. doi: 10.1016/S0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
- 32.Takeda M, Ohno S, Tahara M, Takeuchi H, Shirogane Y, Ohmura H, Nakamura T, Yanagi Y. 2008. Measles viruses possessing the polymerase protein genes of the Edmonston vaccine strain exhibit attenuated gene expression and growth in cultured cells and SLAM knock-in mice. J Virol 82:11979–11984. doi: 10.1128/JVI.00867-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iwasaki M, Yanagi Y. 2011. Expression of the Sendai (murine parainfluenza) virus C protein alleviates restriction of measles virus growth in mouse cells. Proc Natl Acad Sci U S A 108:15384–15389. doi: 10.1073/pnas.1107382108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takeda M, Takeuchi K, Miyajima N, Kobune F, Ami Y, Nagata N, Suzaki Y, Nagai Y, Tashiro M. 2000. Recovery of pathogenic measles virus from cloned cDNA. J Virol 74:6643–6647. doi: 10.1128/JVI.74.14.6643-6647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 36.Takeda M, Ohno S, Seki F, Nakatsu Y, Tahara M, Yanagi Y. 2005. Long untranslated regions of the measles virus M and F genes control virus replication and cytopathogenicity. J Virol 79:14346–14354. doi: 10.1128/JVI.79.22.14346-14354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devaux P, Cattaneo R. 2004. Measles virus phosphoprotein gene products: conformational flexibility of the P/V protein amino-terminal domain and C protein infectivity factor function. J Virol 78:11632–11640. doi: 10.1128/JVI.78.21.11632-11640.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huber M, Cattaneo R, Spielhofer P, Orvell C, Norrby E, Messerli M, Perriard JC, Billeter MA. 1991. Measles virus phosphoprotein retains the nucleocapsid protein in the cytoplasm. Virology 185:299–308. doi: 10.1016/0042-6822(91)90777-9. [DOI] [PubMed] [Google Scholar]
- 39.Tober C, Seufert M, Schneider H, Billeter MA, Johnston ICD, Niewiesk S, ter Meulen V, Schneider-Schaulies S. 1998. Expression of measles virus V protein is associated with pathogenicity and control of viral RNA synthesis. J Virol 72:8124–8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moriyama K, Iida K, Yahara I. 1996. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells 1:73–86. doi: 10.1046/j.1365-2443.1996.05005.x. [DOI] [PubMed] [Google Scholar]
- 41.Heinrich BS, Cureton DK, Rahmeh AA, Whelan SP. 2010. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog 6:e1000958. doi: 10.1371/journal.ppat.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lahaye X, Vidy A, Pomier C, Obiang L, Harper F, Gaudin Y, Blondel D. 2009. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J Virol 83:7948–7958. doi: 10.1128/JVI.00554-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoenen T, Shabman RS, Groseth A, Herwig A, Weber M, Schudt G, Dolnik O, Basler CF, Becker S, Feldmann H. 2012. Inclusion bodies are a site of ebolavirus replication. J Virol 86:11779–11788. doi: 10.1128/JVI.01525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katoh H, Kubota T, Kita S, Nakatsu Y, Aoki N, Mori Y, Maenaka K, Takeda M, Kidokoro M. 2015. Heat shock protein 70 regulates degradation of the mumps virus phosphoprotein via the ubiquitin-proteasome pathway. J Virol 89:3188–3199. doi: 10.1128/JVI.03343-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ménager P, Roux P, Megret F, Bourgeois JP, Le Sourd AM, Danckaert A, Lafage M, Prehaud C, Lafon M. 2009. Toll-like receptor 3 (TLR3) plays a major role in the formation of rabies virus Negri bodies. PLoS Pathog 5:e1000315. doi: 10.1371/journal.ppat.1000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dietzel E, Kolesnikova L, Maisner A. 2013. Actin filaments disruption and stabilization affect measles virus maturation by different mechanisms. Virol J 10:249. doi: 10.1186/1743-422X-10-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wakimoto H, Shimodo M, Satoh Y, Kitagawa Y, Takeuchi K, Gotoh B, Itoh M. 2013. F-actin modulates measles virus cell-cell fusion and assembly by altering the interaction between the matrix protein and the cytoplasmic tail of hemagglutinin. J Virol 87:1974–1984. doi: 10.1128/JVI.02371-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiang Y, Zheng K, Ju H, Wang S, Pei Y, Ding W, Chen Z, Wang Q, Qiu X, Zhong M, Zeng F, Ren Z, Qian C, Liu G, Kitazato K, Wang Y. 2012. Cofilin 1-mediated biphasic F-actin dynamics of neuronal cells affect herpes simplex virus 1 infection and replication. J Virol 86:8440–8451. doi: 10.1128/JVI.00609-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wen X, Ding L, Wang JJ, Qi M, Hammonds J, Chu H, Chen X, Hunter E, Spearman P. 2014. ROCK1 and LIM kinase modulate retrovirus particle release and cell-cell transmission events. J Virol 88:6906–6921. doi: 10.1128/JVI.00023-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Müller N, Avota E, Schneider-Schaulies J, Harms H, Krohne G, Schneider-Schaulies S. 2006. Measles virus contact with T cells impedes cytoskeletal remodeling associated with spreading, polarization, and CD3 clustering. Traffic 7:849–858. doi: 10.1111/j.1600-0854.2006.00426.x. [DOI] [PubMed] [Google Scholar]
- 51.Heath CM, Windsor M, Wileman T. 2001. Aggresomes resemble sites specialized for virus assembly. J Cell Biol 153:449–455. doi: 10.1083/jcb.153.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wileman T. 2006. Aggresomes and autophagy generate sites for virus replication. Science 312:875–878. doi: 10.1126/science.1126766. [DOI] [PubMed] [Google Scholar]
- 53.Lázaro-Diéguez F, Aguado C, Mato E, Sanchez-Ruiz Y, Esteban I, Alberch J, Knecht E, Egea G. 2008. Dynamics of an F-actin aggresome generated by the actin-stabilizing toxin jasplakinolide. J Cell Sci 121:1415–1425. doi: 10.1242/jcs.017665. [DOI] [PubMed] [Google Scholar]