

ABSTRACT
Adenovirus E4-ORF3 and E1B-55K converge in subverting critical overlapping cellular pathways to facilitate virus replication. Here, we show that E1B-55K and E4-ORF3 induce sumoylation and the assembly of SUMO2/3 viral genome replication domains. Using a conjugation-deficient SUMO2 construct, we demonstrate that SUMO2/3 is recruited to E2A viral genome replication domains through noncovalent interactions. E1B-55K and E4-ORF3 have critical functions in inactivating MRN and ATM to facilitate viral genome replication. We show that ATM kinase inhibitors rescue ΔE1B-55K/ΔE4-ORF3 viral genome replication and that the assembly of E2A domains recruits SUMO2/3 independently of E1B-55K and E4-ORF3. However, the morphology and organization of SUMO2/3-associated E2A domains is strikingly different from that in wild-type Ad5-infected cells. These data reveal that E1B-55K and E4-ORF3 specify the nuclear compartmentalization and structure of SUMO2/3-associated E2A domains, which could have important functions in viral replication. We show that E4-ORF3 specifically targets and sequesters the cellular E3 SUMO ligase PIAS3 but not PIAS1, PIAS2, or PIAS4. The assembly of E4-ORF3 into a multivalent nuclear matrix is required to target PIAS3. In contrast to MRN, PIAS3 is targeted by E4-ORF3 proteins from disparate adenovirus subgroups. Our studies reveal that PIAS3 is a novel and evolutionarily conserved target of E4-ORF3 in human adenovirus infections. Furthermore, we reveal that viral proteins not only disrupt but also usurp SUMO2/3 to transform the nucleus and assemble novel genomic domains that could facilitate pathological viral replication.
IMPORTANCE SUMO is a key posttranslational modification that modulates the function, localization, and assembly of protein complexes. In the ever-escalating host-pathogen arms race, viruses have evolved strategies to subvert sumoylation. Adenovirus is a small DNA tumor virus that is a global human pathogen and key biomedical agent in basic research and therapy. We show that adenovirus infection induces global changes in SUMO localization and conjugation. Using virus and SUMO mutants, we demonstrate that E1B-55K and E4-ORF3 disrupt and usurp SUMO2/3 interactions to transform the nucleus and assemble highly structured and compartmentalized viral genome domains. We reveal that the cellular E3 SUMO ligase PIAS3 is a novel and conserved target of E4-ORF3 proteins from disparate adenovirus subgroups. The induction of sumoylation and SUMO2/3 viral replication domains by early viral proteins could play an important role in determining the outcome of viral infection.
INTRODUCTION
Viruses usurp and trigger cellular signaling cascades that have dynamic and system-wide consequences for host-pathogen interactions. Protein posttranslational modifications have the power to alter the functions, structural interactions, and localization of cellular and viral proteins to determine the outcome of infection. For example, studies with polyomavirus middle T antigen and v-Src-associated kinase activities led to the discovery of protein tyrosine phosphorylation (1, 2). Phosphorylation subsequently was found to be a critical signaling nexus that is deregulated in response to viral and cellular oncogenes (3). Interferon signaling cascades and the induction of ISGylation, ubiquitination, or sumoylation also can be triggered by viral infection as critical host antiviral signaling defenses that engage innate and systemic immunity (4–8).
Sumoylation, i.e., the conjugation of a small ubiquitin-like modifier, can affect a protein's activity, intracellular localization, stability, and interaction partners. Changes in SUMO levels and conjugation can be triggered by the cell cycle (9), differentiation (10), heat shock (11), DNA damage (12), and viral infection (6, 7). The regulation of sumoylation can occur at the level of transcription, translation, or degradation of different SUMO pathway components (13). In vertebrates, there are two SUMO subfamilies, SUMO1 and SUMO2/3. SUMO1 and SUMO2/3 share only about 50% sequence identity, while SUMO2 and SUMO3 share 97% sequence similarity and appear to be functionally indistinguishable. SUMO1 is predominantly conjugated with target proteins, while SUMO2 and SUMO3 are found in a larger pool of free, unconjugated SUMO, which is readily available for responding to external stimuli (14). SUMO-conjugated proteins are recognized and bound by proteins containing SUMO interacting motifs (SIMs) (13). The interactions between sumoylated proteins and SIM-containing proteins can act as a scaffold to promote the self-assembly of large multiprotein complexes. For example, PML is conjugated to SUMO and contains SIM motifs that drive PML assembly into nuclear bodies (15). Sumoylation is critical for the assembly of PML nuclear bodies and the recruitment of additional SUMO-SIM proteins, such as DAXX and Sp100 (16–18). SUMO-SIM-driven assembly creates subnuclear compartments that are physically distinct from the surrounding nucleoplasm despite the lack of a defining membrane. SUMO-SIM nuclear bodies can function as hubs for a particular activity, such as transcription, or induce the local concentration of components within the nucleoplasm (19). Thus, sumoylation is a means to induce the dynamic structural organization and compartmentalization of molecular interactions.
The potential for controlling transcriptional regulation and immunity makes it unsurprising that many viral proteins usurp the SUMO pathway (6, 7). For example, to evade the immune response, some viral proteins target the PIAS family of E3 SUMO ligases, which are associated with the suppression of innate immune signaling through the inhibition of STAT proteins, interferon-regulatory factors, and NF-κB (20, 21). There is also mounting evidence for viruses targeting the SUMO pathway to promote nuclear reorganization. One well-characterized example of viruses modulating nuclear organization is the disruption of nuclear PML bodies, which are targeted by many DNA tumor virus proteins via SUMO-mediated mechanisms (7). Thus, an interesting question is whether viral proteins utilize SUMO to assemble new nuclear bodies such as viral genome replication domains.
Many viruses utilize cytoplasmic membranes and cytoskeletal components to create a physical boundary to concentrate components involved in viral genome replication (22, 23). Adenovirus is a small double-stranded DNA tumor virus that replicates in the cell nucleus. After the initial rounds of DNA replication, single- and double-stranded virus genomes assemble with the viral E2A DNA-binding protein (DBP) to form specialized replication compartments that concentrate viral genomes, proteins, and RNA (24). E2A viral replication domains change in morphology and size at different stages in the viral life cycle (25). Therefore, an interesting question is whether sumoylation and/or SUMO-SIM interactions are associated with the assembly of adenovirus replication centers in the nucleus.
In adenovirus infection, the early viral proteins E1B-55K and E4-ORF3 have overlapping cellular targets and functions in viral replication. E1B-55K and E4-ORF3 converge in disrupting the MRE11/RAD50/NBS1 (MRN) complex and preventing p53-activated transcription via independent mechanisms (26, 27). E1B-55K targets p53 and MRE11 for ubiquitination and degradation in the proteasome (28, 29). E4-ORF3 assembles a multivalent nuclear scaffold that sequesters MRN and induces repressive heterochromatin silencing at the p53 target gene (26, 27). In addition to their overlapping roles in inactivating MRN and p53, E1B-55K and E4-ORF3 also have been implicated in the SUMO pathway (Fig. 1A). E1B-55K has been shown to function as an E3 SUMO ligase toward p53 (30, 31) and to interact with the E2 SUMO ligase UBC9 (32). E4-ORF3 forms a nuclear polymer that disrupts PML bodies (33) and mislocalizes TRIM24 and TRIM33 (26, 34, 35), all of which are sumoylated. Recently, E4-ORF3 was reported to modulate the sumoylation of MRN and other substrates (36, 37). The crystal structure of E4-ORF3 indicates that it is not a structural homolog of cellular proteins involved in sumoylation (38). Thus, a key question is whether E4-ORF3 targets cellular SUMO enzymes to modulate SUMO conjugation and interactions in viral infection (Fig. 1A).
FIG 1.

In Ad5-infected cells, SUMO2/3 is mislocalized by E4-ORF3 and recruited to E2A viral genome replication centers. (A) Schematic of premise for these studies and role of adenovirus proteins E1B-55K and E4-ORF3 in subverting SUMO to facilitate viral replication. (B) U2OS cells were transfected with Flag-SUMO2 or Flag-SUMO3 and then infected with wild-type (WT) Ad5 virus. Cells were fixed at 0, 12, 24, and 36 h p.i. and immunostained for Flag (green), E4-ORF3 (white), and E2A (red). Nuclei were counterstained with Hoechst-33342. (C) Uninfected (UI) and WT Ad5-infected U2OS cells were fixed at 36 h p.i. and immunostained for SUMO2/3 (green), E4-ORF3 (white), and E2A (red). Nuclei were counterstained with Hoechst-33342. (D) U2OS cells were transfected with GFP-SUMO2 or GFP-4xSUMO2AA and left uninfected or were infected with WT Ad5. Cells were fixed at 36 h p.i. and immunostained for E4-ORF3 (red) and E2A (white) Box indicates 4x zoom. (E) Additional images of WT Ad5-infected U2OS cells transfected with either GFP-SUMO2 or GFP-4×SUMO2AA. (F) U2OS cells were cotransfected with either GFP-SUMO2 or GFP-4×SUMO2AA and E4-ORF3 and then fixed 24 h later and immunostained for E4-ORF3 (red). Nuclei were counterstained with Hoechst-33342. (G) Quantitative analysis of colocalization of transfected E4-ORF3 with GFP-SUMO2 and GFP-4×SUMO2AA. Pearson's correlation coefficients were calculated using Imaris colocalization (see Materials and Methods) (41). Bars represent the means ± standard errors of the means. ***, P < 0.001, which indicates a significant difference between GFP-SUMO2 and GFP-4xSUMO2AA by t test.
Here, we show that E1B-55K and E4-ORF3 are required for the induction of sumoylation and the assembly of SUMO2/3-associated viral genome replication domains in Ad5-infected cells. We show that the assembly of E2A viral genome replication domains is sufficient to recruit SUMO2/3 independently of E1B-55K/E4-ORF3 and SUMO2/3 conjugation. However, E1B-55K/E4-ORF3 determine the structure and nuclear organization of SUMO2/3-associated E2A domains. We show that E4-ORF3 specifically targets and mislocalizes the cellular E3 SUMO ligase PIAS3 into a nuclear scaffold but not related protein family members PIAS1, PIAS2, or PIAS4. We show that in contrast to MRN, PIAS3 is a target of E4-ORF3 proteins from disparate human adenovirus subgroups, indicating that it is an important and conserved target in virus evolution.
MATERIALS AND METHODS
Cells, culturing conditions, and viral infections.
U2OS cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) without antibiotics. Infection was performed at an experimentally determined multiplicity of infection (MOI) of 30 PFU in DMEM containing 2% heat-inactivated FBS. Superinfection was performed at an MOI of 250 PFU.
Viruses.
Titers of viruses were determined on 293/E4/pIX cells as described previously (39). The wild-type (WT) virus is Ad5. The ΔE1B-55K virus (AdSyn-CO124) was created by mutating the start codon of E1B-55K (ATG to GTG) and I90 of E1B-55K to a stop codon (ATT to TAG). The ΔE4-ORF3 virus (AdSyn-CO118) was created by deleting the coding region of E4-ORF3. The ΔE1B-55K/ΔE4-ORF3 virus (AdSyn-CO140) is the same as ΔE1B-55K, except the E4-ORF3 coding region also is deleted (38).
Drugs.
KU-55933 (Calbiochem) was used at 10 μM and was added 2 h after adenovirus infection. Dimethyl sulfoxide (DMSO) was used as a vehicle control.
Plasmids and transfections.
SUMO2, SUMO3, UBC9, and PIAS family plasmids first were cloned into pDONR221 and then recombined into a cytomegalovirus expression vector with an N-terminal tag using the Gateway cloning system (Invitrogen). The GFP-4xSUMO2AA construct was obtained from Tony Hunter's laboratory. E4-ORF3 constructs were generated as described previously (38). Lipofectamine 2000 (Invitrogen) was used for transfection of U2OS cells according to the manufacturer's instructions.
Immunofluorescence.
Cells were fixed in 4% paraformaldehyde for 30 min at room temperature, permeabilized in 0.1% Triton X-100 and phosphate-buffered saline (PBS), and stained as described previously (39, 40). Primary antibodies were Flag (F742 from Sigma), E4-ORF3 (6A11), E2A (B6-8), SUMO2/3 (C terminus from Abgent), and NBS1 (NB 100 from Novus Biologicals). Alexa 488-, 555-, and 633-conjugated secondary antibodies (Molecular Probes) were used for the detection of primary antibodies. Hoechst-33342 was used to stain DNA. Images were acquired with a Zeiss LSM780 imaging system with a 63× objective. Images are single z-planes.
Quantification of immunofluorescence colocalization and statistics.
Colocalization was measured using Imaris software, which analyzes the intensity of each fluorescent label. The Pearson correlation coefficient was used as a measure of colocalization with values between −1 and +1, with positive values indicating a positive correlation (41). Statistical analyses consisted of Student's t tests with significance set at 0.05.
Protein lysates, dot blot analysis, and Western blot analysis.
Protein lysates were harvested in reducing SDS-PAGE sample buffer containing 50 mM Tris, pH 8, 2% SDS, 10% glycerol, 100 mM dithiothreitol, and 0.1% bromophenol blue. Lysates were boiled for 10 min and sonicated. For dot blotting, samples were spotted onto a nitrocellulose membrane. For Western blotting, samples were analyzed by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked using 5% milk. Primary antibodies were SUMO2/3 (8A2 from Abcam) and β-actin (AC-15 from Sigma). β-Actin expression was used as a loading control. Primary antibodies were detected with secondary antibodies labeled with either IRDye800 (Rockland) or Alexa Fluor 680 (Molecular Probes). Fluorescent antibodies were visualized using a LI-COR Odyssey scanner. The quantification of dot blots was performed using LI-COR Odyssey software (42).
RT-qPCR analysis.
Real-time quantitative PCR (RT-qPCR) quantification of SUMO2 and SUMO3 mRNA was performed using the Bio-Rad CFX96 and analyzed using Bio-Rad CFX Manager 3.0 software. RNA (1 μg) was reverse transcribed with an iScript cDNA synthesis kit (Bio-Rad). TaqMan primers and probe sets for SUMO2 and SUMO3 were obtained from Life Technologies. RT-qPCRs were set up using TaqMan Fast Mix (ABI) and run in triplicate. For 18S rRNA analysis, 10 ng input cDNA was used; for SUMO2 and SUMO3 analysis, 40 ng of input cDNA was used. Normalized gene expression (ΔΔCq) was determined using 18S rRNA as a reference gene, and fold change was determined by setting gene expression levels of uninfected samples to 1. Error bars represent standard errors of the means from triplicates.
Quantification of viral genome replication.
Total DNA was extracted using the QiaAMP DNA Micro kit (Qiagen) by following the manufacturer's protocol. TaqMan probes for quantifying adenovirus DNA were described previously (43). qPCRs were set up using TaqMan fast mix (ABI) and run in triplicate. Input DNA (5 ng) was used for Ad5 genomes and 18S rDNA analysis. qPCR quantification of viral genomes was performed using the Bio-Rad CFX96. Viral DNA was quantified relative to the 18S rDNA to obtain a change in threshold cycle (ΔCT) for each sample (44). Error bars represent standard deviations from triplicates.
RESULTS
In Ad5-infected cells, SUMO2/3 is mislocalized by E4-ORF3 and recruited to E2A viral genome replication centers.
Adenovirus infection and replication transforms the cell nucleus and remodels nuclear protein complexes, including PML bodies and MRN, both of which are targeted by sumoylation (26, 33, 36). We reasoned that adenoviral proteins could subvert SUMO conjugation and localization to reorganize cellular proteins and compartments within the nucleus to facilitate virus replication. SUMO2 and SUMO3 are the predominant SUMO conjugates that modify proteins in response to external stimuli and cell stress (14). SUMO2 and SUMO3 share 97% sequence homology and cannot be distinguished using endogenous cellular antibodies. Therefore, to analyze SUMO2 and SUMO3 in adenovirus infection, we transfected U2OS cells with either Flag-SUMO2 or Flag-SUMO3 and examined SUMO localization over the time course of wild-type (WT) Ad5 infection. In uninfected cells, Flag-SUMO2 and Flag-SUMO3 are distributed throughout the nucleus and PML bodies. At an early time, 12 h postinfection (h p.i.), E4-ORF3 colocalizes with Flag-SUMO2- and Flag-SUMO3-associated nuclear bodies. The latter is consistent with the well-established role of E4-ORF3 in targeting and disrupting sumoylated proteins at PML nuclear bodies (33). However, as the viral life cycle progresses, Flag-SUMO2 and Flag-SUMO3 are induced at viral genome replication domains demarcated by the viral E2A protein (24 h p.i. and 36 h p.i.) (Fig. 1B). Flag-tagged SUMO2 and SUMO3 have indistinguishable localization patterns. Therefore, for the remainder of these studies we used SUMO2, as it appears to be interchangeable with SUMO3. Furthermore, we confirmed that endogenous SUMO2/3 localizes to viral genome replication domains using an antibody that recognizes both SUMO2 and SUMO3 (Fig. 1C). We conclude that Ad5 infection mislocalizes sumoylated proteins into the E4-ORF3 nuclear matrix and induces SUMO2/3 at E2A viral genome replication domains, suggesting a larger role for SUMO2/3 in adenovirus infection than was previously appreciated.
SUMO2/3 is recruited to E2A viral genome replication domains independently of SUMO conjugation.
The modulation of SUMO2/3 localization in viral infection could be due to covalent SUMO2/3 conjugation or noncovalent SUMO2/3 interactions with cellular or viral SIM-containing proteins. To distinguish between these possibilities, we used a green fluorescent protein (GFP) construct that has four SUMO2 tandem repeats that can interact with SIM-containing proteins but cannot be conjugated to target proteins (GFP-4xSUMO2AA). In uninfected cells, GFP-4xSUMO2AA is diffuse and does not form nuclear bodies (Fig. 1D). The latter likely represents the failure of GFP-4xSUMO2AA to be conjugated to proteins at PML bodies. In WT virus-infected cells, GFP-4xSUMO2AA does not colocalize with E4-ORF3 but is induced at E2A viral genome replication domains (Fig. 1D and E). Furthermore, we show that E4-ORF3 does not mislocalize GFP-4xSUMO2AA in cotransfected cells (Fig. 1F and G). We conclude that SUMO2/3 localization at E2A viral genome replication domains does not require covalent SUMO conjugation and can be mediated through noncovalent SUMO interactions with cellular and/or viral components.
E1B-55K and E4-ORF3 determine the morphology and nuclear organization of SUMO2/3-associated E2A viral genome replication domains.
We hypothesized that SUMO localization at E2A viral genome replication domains is modulated by early viral oncoproteins. E1B-55K has been reported to have SUMO ligase activity (30, 31) and to interact with the cellular E2 SUMO ligase UBC9 (32). E4-ORF3 recently has been reported to modulate sumoylation of MRN and other substrates (36, 37), although the mechanism remains unknown. To determine if E1B-55K and E4-ORF3 modulate SUMO localization at E2A domains, we analyzed Flag-SUMO2 localization in cells infected with WT Ad5 or mutant viruses in which E1B-55K and/or E4-ORF3 sequences are deleted. In WT, ΔE1B-55K, and ΔE4-ORF3 virus-infected cells, Flag-SUMO2 is induced at E2A viral genome replication domains. However, ΔE1B-55K/ΔE4-ORF3 viruses fail to induce E2A- and SUMO2-associated viral genome replication domains (Fig. 2A). Similar results were observed with endogenous SUMO2/3 (data not shown).
FIG 2.

E1B-55K and E4-ORF3 determine the morphology and nuclear organization of SUMO2/3-associated E2A viral genome replication domains. (A) U2OS cells were transfected with Flag-SUMO2 and then infected as indicated. Cells were fixed at 36 h p.i. and immunostained for Flag (green), E4-ORF3 (white), and E2A (red). Nuclei were counterstained with Hoechst-33342. (B) U2OS cells were infected as indicated and then treated with DMSO or 10 μM KU-55933 at 2 h p.i. and harvested at 36 h p.i. Viral genomes were quantified by qPCR and normalized relative to 18S rDNA levels. Error bars indicate standard deviations from triplicate samples. (C) U2OS cells were transfected with Flag-SUMO2 and then infected as indicated. Cells were treated with DMSO or 10 μM KU-55933 at 2 h p.i. fixed at 36 h p.i., and immunostained for Flag (green) and E2A (red). Nuclei were counterstained with Hoechst-33342. (D) U2OS cells were transfected with Flag-SUMO2 and then infected with WT Ad5 or ΔE1B-55K/ΔE4-ORF3 virus. MOI 250 indicates a multiplicity of infection of 250 PFU. Cells were treated with 10 μM KU-55933 at 2 h p.i. as indicated, fixed at 36 h p.i., and immunostained for Flag (green) and E2A (red).
E1B-55K or E4-ORF3 is required to inactivate a critical early MRN-ATM checkpoint to viral genome replication (45). Therefore, the lack of SUMO2 at E2A domains could be an indirect consequence of an upstream block in viral genome replication in ΔE1B-55K/ΔE4-ORF3-infected cells. Alternatively, it could reflect a direct role of E1B-55K/E4-ORF3 in regulating SUMO2 localization and organization at E2A domains. To distinguish between these possibilities, we used the ATM kinase inhibitor KU-55933 (46) to rescue viral genome replication in ΔE1B-55K/ΔE4-ORF3-infected cells (45). We also analyzed ΔE1B-55K/ΔE4-ORF3 superinfected cells (MOI of 250), as previous studies have shown that high MOIs can rescue the replication defect of E4-deleted viruses (47–49).
KU-55933 has no effect on virus genome replication or the localization and morphology of SUMO2-associated viral genome replication domains in WT virus-infected cells. However, KU-55933 rescues viral genome replication by 15-fold in ΔE1B-55K/ΔE4-ORF3-infected U2OS cells (Fig. 2B). Similar to KU-55933, high MOIs also rescue viral genome replication and the assembly of SUMO2-associated E2A domains (Fig. 2C and D). We conclude that the assembly of E2A viral genome replication domains is sufficient to recruit SUMO2 independently of E1B-55K/E4-ORF3. However, the morphology of SUMO2/E2A domains in ΔE1B-55K/ΔE4-ORF3-infected cells is strikingly different from that in WT virus-infected cells (Fig. 2D). In contrast to WT virus-infected cells, the SUMO2-associated E2A domains are globular and amorphous in ΔE1B-55K/ΔE4-ORF3-infected cells treated with KU-55933 or infected at high MOI. The SUMO-associated E2A domains are fewer and larger in size in the absence of E1B-55K/E4-ORF3 and appear to be fused together into giant clusters as opposed to distinct compartments. These data indicate that E1B-55K and E4-ORF3 have important roles in determining the architecture and compartmentalization of SUMO2-associated E2A viral genome replication domains.
Ad5 infection induces SUMO2/3-modified proteins.
To determine if adenovirus infection induces SUMO2/3 levels and conjugation, we first used a dot blot analysis. There is an almost 5-fold increase in total SUMO2/3 levels in protein lysates from Ad5-infected cells relative to that of uninfected control U2OS cells (Fig. 3A). SUMO2 and SUMO3 share 97% sequence homology, but their transcription is differentially regulated (50). For example, the transcription of SUMO3, but not SUMO2, is downregulated in response to oxidative stress (51). To determine if adenovirus infection induces the transcription of SUMO2 or SUMO3, we compared mRNA levels of SUMO2 and SUMO3 in infected versus uninfected U2OS cells. SUMO2 and SUMO3 mRNA levels are similar in both infected and uninfected cells (Fig. 3B).
FIG 3.

Ad5 infection induces SUMO2/3-modified proteins. (A) U2OS cells were infected as indicated, protein lysates were collected at 36 h p.i., and dot blotting for total SUMO2/3 was performed. β-Actin is a loading control. Quantification was performed using LI-COR Odyssey software with SUMO2/3 normalized to β-actin. (B) U2OS cells were infected as indicated and analyzed by RT-qPCR. SUMO2 and SUMO3 transcripts were normalized relative to cellular 18S rRNA. Error bars indicate the standard errors of the means from triplicate samples. (C) U2OS cells were infected as indicated, and then protein lysates were collected at 36 h p.i. and immunoblotted for SUMO2/3. β-Actin is a loading control.
Unconjugated forms of SUMO2 and SUMO3 are 8 kDa. However, SUMO2 and SUMO3 form higher-molecular-weight bands on SDS-PAGE gels when they are covalently attached to protein substrates (42). In WT Ad5-infected U2OS cells, there is a substantial increase in the levels of high-molecular-weight SUMO2/3-conjugated proteins compared to those of uninfected cells (Fig. 3C). Taken together, these data demonstrate that WT adenovirus infection induces SUMO2/3 conjugation of cellular and/or viral proteins.
E1B-55K and E4-ORF3 induce SUMO2/3-conjugated proteins in Ad5-infected cells.
We hypothesized that E1B-55K and E4-ORF3 are required to induce sumoylation as well as SUMO nuclear organization in Ad5-infected cells. To determine if SUMO2/3 levels and conjugated proteins are induced in WT- versus ΔE1B-55K/ΔE4-ORF3-infected cells, we performed dot blotting and Western blotting for SUMO2/3. Total SUMO2/3 levels are induced 3- to 4-fold in WT-, ΔE1B-55K-, and ΔE4-ORF3-infected cells. However, in ΔE1B-55K/ΔE4-ORF3-infected cells, SUMO2/3 levels are only nominally increased relative to those of uninfected cells (1.6-fold relative to that for uninfected cells) (Fig. 4A). By Western blotting, we show that compared to uninfected and ΔE1B-55K/ΔE4-ORF3 virus-infected cells, there is an increase in the levels of high-molecular-weight SUMO2/3-conjugated proteins in WT-, ΔE1B-55K-, and ΔE4-ORF3-infected cells (Fig. 4B). We conclude that either E1B-55K or E4-ORF3 is required to induce SUMO2/3-conjugated proteins in Ad5-infected cells.
FIG 4.

E1B-55K and E4-ORF3 induce SUMO2/3-conjugated proteins in Ad5-infected cells. (A) U2OS cells were infected as indicated, and then protein lysates were collected at 36 h p.i. and dot blotting for total SUMO2/3 was performed. β-Actin is a loading control. Quantification was performed using LI-COR Odyssey software with SUMO2/3 normalized to β-actin. (B) U2OS cells were infected as indicated, and then protein lysates were collected at 36 h p.i. and immunoblotted for SUMO2/3. β-Actin is a loading control.
E4-ORF3 does not mislocalize or destabilize the E2 SUMO ligase UBC9.
Our data demonstrate that E1B-55K and E4-ORF3 modulate SUMO2/3 localization and conjugation during Ad5 infection. E1B-55K has been reported to have SUMO ligase activity itself (30, 31) and to interact with the cellular E2 SUMO ligase UBC9 (32). The recently determined crystal structure of an Ad5 E4-ORF3 dimer indicates that it is not a structural homolog of sumoylation enzymes (38). Therefore, to determine if E4-ORF3 targets cellular proteins that regulate sumoylation, we conducted a candidate screen. Conjugation of SUMO proteins requires processing by a SUMO protease to reveal a diglycine motif, followed by conjugation by the E1 ligase SAE1/SAE2 and the E2 SUMO ligase UBC9. From here, SUMO can be conjugated directly to a substrate or be targeted through one of about a dozen E3 SUMO ligases, such as the PIAS protein family (Fig. 5A). The single E2 SUMO ligase UBC9 is a particularly compelling candidate due to its central role in the SUMO pathway. Some viral proteins modulate sumoylation by regulating UBC9 protein levels, such as the avian adenovirus protein Gam1 (52). To determine if UBC9 protein levels are modulated by E1B-55K or E4-ORF3 during adenovirus infection, we infected U2OS cells with WT Ad5, ΔE1B-55K, ΔE4-ORF3, or ΔE1B-55K/ΔE4-ORF3 viruses (Fig. 5B). UBC9 protein levels are not changed upon infection. To determine if E4-ORF3 mislocalizes UBC9, we cotransfected U2OS cells with Flag-UBC9 and E4-ORF3. Flag-UBC9 is not mislocalized by E4-ORF3 (Fig. 5C). We conclude that E4-ORF3 does not target the central E2 SUMO ligase UBC9.
FIG 5.
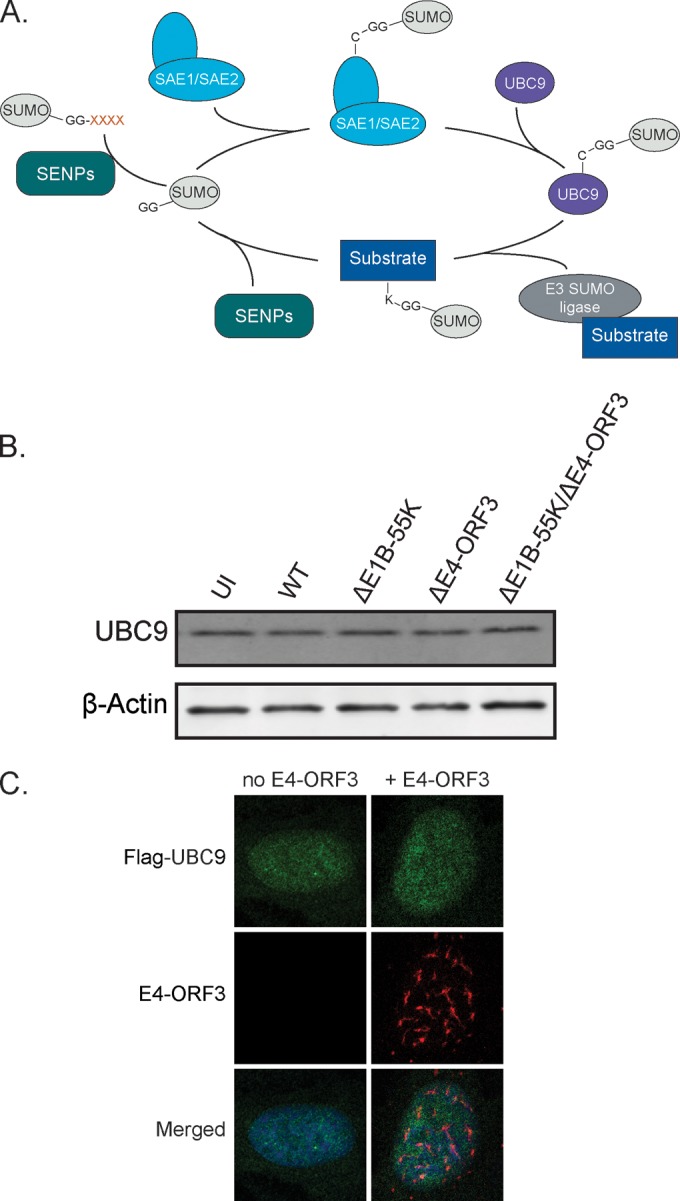
E4-ORF3 does not mislocalize or destabilize the E2 SUMO ligase UBC9. (A) Schematic representation of the sumoylation pathway. (B) U2OS cells were infected as indicated. Protein lysates were collected at 36 h p.i. and immunoblotted for UBC9. β-Actin is a loading control. (C) U2OS cells were cotransfected with Flag-UBC9 and E4-ORF3, fixed 24 h later, and immunostained for Flag (green) and E4-ORF3 (red). Nuclei were counterstained with Hoechst-33342.
E4-ORF3 specifically targets PIAS3 from the E3 SUMO ligase family of proteins.
E3 SUMO ligases determine the substrate specificity of sumoylation (53). The PIAS family of E3 SUMO ligases, including PIAS1, PIAS2 (PIASx), PIAS3, and PIAS4 (PIASy), has important roles in regulating transcription and immunity (54). To determine if the PIAS E3 SUMO ligases are targeted by E4-ORF3, we created epitope-tagged cDNA constructs of the four human PIAS family members and cotransfected them with E4-ORF3. E4-ORF3 fails to mislocalize Flag-PIAS1, Flag-PIAS2, and Flag-PIAS4 into nuclear track structures (Fig. 6A). In contrast, E4-ORF3 specifically targets and mislocalizes Flag-PIAS3 (Fig. 6B and C).
FIG 6.

E4-ORF3 specifically targets PIAS3 from the E3 SUMO ligase family of proteins. (A) U2OS cells were cotransfected with E4-ORF3 and either Flag-PIAS1, Flag-PIAS2, or Flag-PIAS4, fixed 24 h posttransfection, and immunostained for Flag (green) and E4-ORF3 (red). Nuclei were counterstained with Hoechst-33342. (B) U2OS cells were transfected with E4-ORF3 and Flag-PIAS3 as indicated, fixed 24 h posttransfection, and immunostained for Flag (green) and E4-ORF3 (red). Nuclei were counterstained with Hoechst-33342. (C) Zoomed (4×) images of the merged cell cotransfected with Flag-PIAS3 and E4-ORF3. (D) U2OS cells were transfected with Flag-PIAS3 and then infected as indicated. Cells were fixed 36 h p.i. and immunostained for Flag (green) and E4-ORF3 (red). Nuclei were counterstained with Hoechst-33342.
We also determined if PIAS3 is mislocalized by E4-ORF3 in the context of viral infection. Consistent with the conclusions of E4-ORF3 transfection experiments, PIAS3 is mislocalized by E4-ORF3 in WT- and ΔE1B-55K virus-infected cells. PIAS3 is not mislocalized in ΔE1B-55K/ΔE4-ORF3 virus-infected cells and has a localization pattern similar to that of uninfected cells. Interestingly, in ΔE4-ORF3-infected cells, although PIAS3 is not mislocalized by E4-ORF3, it appears to form larger, more fibrillar structures in the nucleus than uninfected and ΔE1B-55K/ΔE4-ORF3 virus-infected cells. These data suggest that virus infection and proteins could modulate PIAS3 distribution in the absence of E4-ORF3 (Fig. 6D). We conclude that PIAS3, but not other PIAS family members, is targeted and mislocalized by E4-ORF3.
E4-ORF3 higher-order assembly is required to mislocalize PIAS3 and is independent of binding to MRN.
E4-ORF3 assembles an insoluble nuclear polymer that makes conventional biochemical and structural analyses challenging. However, the structure of Ad5 E4-ORF3 was recently solved using N82 point mutations (26, 55, 56) that prevent the higher-order assembly of dimer subunits (38). E4-ORF3 forms a dimer with a central β-core that is sealed at the front and back by the C-terminal tail (residues 99 to 116) containing a short β4 strand (Fig. 7A and B). Multiple lines of evidence support a model in which N82 residue mutations lock the β-core into a closed conformation that prevents the further assembly of dimer subunits through C-terminal swapping (Fig. 7A) (38). E4-ORF3 higher-order assembly is required for interactions with PML, TRIM24, and MRN and inactivation of p53 target genes (26, 34, 38, 55, 56). To determine if the higher-order assembly of E4-ORF3 dimers is also required for targeting PIAS3, we cotransfected U2OS cells with Flag-PIAS3 and either WT E4-ORF3 or E4-ORF3 N82A. In contrast to WT E4-ORF3, E4-ORF3 N82A mutants do not assemble a higher-order polymer and exhibit a diffuse nuclear and cytoplasmic localization. Furthermore, we show that E4-ORF3 N82A dimers fail to mislocalize PIAS3 (Fig. 7C). We conclude that the higher-order assembly of E4-ORF3 dimer subunits is required for targeting and mislocalizing PIAS3.
FIG 7.

E4-ORF3 higher-order assembly is required to mislocalize PIAS3 and is independent of binding to MRN. (A) Model showing the assembly of E4-ORF3 dimer subunits through intermolecular exchanges of their C-terminal tails. N82* mutations prevent the further assembly of E4-ORF3 dimer subunits and lock the β-core in a closed configuration. (B) The crystal structure of an E4-ORF3 N82* dimer highlighting residues N82 and VHLIDL101-106. (C) U2OS cells were transfected with Flag-PIAS3 and wild-type (WT) E4-ORF3 or E4-ORF3 N82A. Cells were fixed 24 h posttransfection and immunostained for Flag (red), E4-ORF3 (green), and NBS1 (white). Nuclei were counterstained with Hoechst-33342. (D) Residues in the E4-ORF3 C-terminal tail required for mislocalizing MRN. (E) U2OS cells were cotransfected with Flag-PIAS3 and either WT E4-ORF3, E4-ORF3 V101A, or E4-ORF3 D105A/L106A. Cells were fixed 24 h posttransfection and immunostained for Flag (red), E4-ORF3 (green), and NBS1 (white). Nuclei were counterstained with Hoechst-33342.
The higher-order assembly of E4-ORF3 dimers creates avidity-driven interactions with PML and an emergent interface between residues V101 and D105 in the C-terminal tail that is required for mislocalizing MRN (Fig. 7D) (38). MRN mislocalization has been suggested to be a prerequisite for MRN sumoylation by E4-ORF3 (36). Therefore, we hypothesized that residues in the E4-ORF3 C-terminal tail required for mislocalizing MRN also are required to target PIAS3. To test this, we determined if WT E4-ORF3, E4-ORF3 V101A, and E4-ORF3 D105A/L106A mislocalize PIAS3 in cotransfected U2OS cells. Consistent with previous studies, E4-ORF3 V101A and E4-ORF3 D105A/L106A point mutants fail to mislocalize NBS1, which is part of the MRN complex (38, 55). However, E4-ORF3 V101A and D105A/L106A mutants behave analogously to WT E4-ORF3 with respect to their ability to mislocalize PIAS3 (Fig. 7E). Thus, the functions of Ad5 E4-ORF3 in mislocalizing MRN and PIAS3 can be biochemically separated. We conclude that Ad5 E4-ORF3 targets and mislocalizes PIAS3 independently of its interactions with MRN.
PIAS3 is a conserved target of E4-ORF3 proteins from disparate human adenovirus subgroups.
There are 68 human adenoviruses that are divided into 7 subgroups (A to G) based on sequence homology and biophysical and biochemical criteria. There is 37.6% pairwise amino acid identity and 55.2% pairwise similarity between Ad5 E4-ORF3 (subgroup C) and E4-ORF3 proteins from disparate adenoviral subgroups: Ad9 (subgroup D), Ad12 (subgroup A), and Ad34 (subgroup B) (Fig. 8A). The ability of E4-ORF3 to bind and mislocalize MRN appears to be peculiar to subgroup C virus proteins (57). However, interactions with PML, TRIM24, and TRIM33 are conserved functions of E4-ORF3 proteins across subgroups (34, 35, 58). To determine if PIAS3 is a conserved target of E4-ORF3 proteins from disparate adenovirus subgroups, we cotransfected Flag-PIAS3 with Myc-tagged Ad5, Ad9, Ad12, or Ad34 E4-ORF3 constructs. We show that E4-ORF3 proteins from Ad5, Ad9, Ad12, and Ad34 all target and mislocalize PIAS3 (Fig. 8B). We conclude that PIAS3 is an evolutionarily conserved cellular target of E4-ORF3 proteins from disparate human adenovirus subgroups.
FIG 8.

PIAS3 is a conserved target of E4-ORF3 proteins from disparate human adenovirus subgroups. (A) Alignment of E4-ORF3 protein sequences from subgroup A (Ad12), subgroup B (Ad34), subgroup C (Ad5), and subgroup D (Ad9). Blue, orange, and black letters denote conserved, semiconserved, and nonconserved residues, respectively. (B) U2OS cells were cotransfected with Flag-PIAS3 and Myc-tagged E4-ORF3 from Ad5, Ad9, Ad12, or Ad34. Cells were fixed 24 h posttransfection and immunostained for Flag (green) and Myc (red). Nuclei were counterstained with Hoechst-33342.
DISCUSSION
Our studies demonstrate that adenovirus infection induces SUMO2/3 conjugation and remodels SUMO2/3 subnuclear localization. We show that the induction of sumoylation and SUMO2/3-associated E2A viral genome replication centers in the nucleus requires the expression of either E1B-55K or E4-ORF3. Furthermore, we identify the E3 SUMO ligase PIAS3 as a novel target of E4-ORF3 proteins from disparate adenovirus subgroups, suggesting a conserved evolutionary role in adenovirus infection.
Our data reveal a striking change in the nuclear distribution of SUMO2/3 at different stages in adenovirus infection (Fig. 1B). While the disruption of sumoylated proteins at PML bodies by E4-ORF3 is well established (7), the association of SUMO in E2A viral genome replication domains has not been shown previously. Using conjugation-defective SUMO2 constructs, we reveal that SUMO can be recruited to E2A viral genome replication domains though noncovalent interactions with cellular and/or viral proteins (Fig. 1D). SUMO2/3 could be recruited through interactions with SIM containing viral and cellular proteins and/or through novel interactions with DNA, RNA, or other macromolecules concentrated at E2A domains.
In contrast to WT, ΔE1B-55K, and ΔE4-ORF3 viruses, ΔE1B-55K/ΔE4-ORF3 viruses fail to induce SUMO2/3-associated E2A viral genome replication domains (Fig. 2A). In addition to modulating sumoylation, E1B-55K and E4-ORF3 have a critical role in inactivating MRN and ATM to facilitate viral genome replication (45, 59–61). We show that the lack of SUMO2-associated E2A domains in ΔE1B-55K/ΔE4-ORF3-infected cells is due to an upstream MRN-ATM-mediated checkpoint that prevents viral genome replication. Using ATM kinase inhibitors and high MOIs to rescue ΔE1B-55K/ΔE4-ORF3 viral genome replication, we show that the assembly of E2A domains recruits SUMO2/3 independently of E1B-55K and E4-ORF3 (Fig. 2C and D). Thus, the assembly of viral genome replication domains is sufficient to induce the localization and recruitment of SUMO2/3 independently of E1B-55K/E4-ORF3. Taken together, these data reveal that SUMO2/3 is recruited through noncovalent interactions with additional adenoviral/cellular macromolecules concentrated at E2A viral replication domains.
E2A viral replication domains exhibit dramatic morphological differences and variations in size and number over the course of the virus life cycle (25). The structural basis for these differences in E2A domains and functional consequences is poorly understood. Intriguingly, the structure and nuclear organization of E2A domains in KU-55933-treated ΔE1B-55K/ΔE4-ORF3-infected cells is strikingly different from that in WT virus-infected cells. The E2A domains in KU-55933-treated ΔE1B-55K/ΔE4-ORF3 cells are larger in size and have a more amorphous, uniform morphology than WT virus-infected cells (Fig. 2D). Thus, although viral genome replication is rescued by KU-55933, the E2A domains lack internal structure and are not compartmentalized from each other and the surrounding nucleoplasm in the absence of E1B-55K/E4-ORF3. These data demonstrate that E1B-55K and E4-ORF3 have novel roles in determining the structure and nuclear compartmentalization of E2A viral replication domains. Interestingly, early confocal microscopy studies indicated that the morphologies of E2A domains associated with sites of single-strand and double-strand viral genome accumulation are distinct (62). In addition, these studies suggested that viral DNA replication and transcription/splicing are spatially and functionally separated by distinct E2A domains at specific sites in the nucleus (62). E1B-55K and E4-ORF3 have overlapping functions in facilitating transcription through late viral mRNA export and splicing (39, 40, 63). Thus, E1B-55K and E4-ORF3 may modulate sumoylation and SUMO-SIM interactions to specify the function and dynamic structural reorganization of viral replication and transcription/splicing centers. This could play an important role in compartmentalizing and catalyzing different activities in the course of the virus life cycle.
Adenovirus infection induces a global increase in SUMO2/3 conjugates (Fig. 3C). Sumoylation has strong links to transcriptional regulation and immunity. Therefore, the induction of sumoylation in Ad5-infected cells also could be a cellular antiviral response to productive virus replication. Many transcription factors and coregulators are proteins that are targeted and modulated by sumoylation, which usually results in transcriptional repression (64). E1B-55K and E4-ORF3 inhibit the interferon-mediated antiviral response (59). Thus, the induction of SUMO conjugation by E1B-55K and E4-ORF3 could inactivate a cellular antiviral transcriptional response to virus replication (Fig. 4B). For example, adenovirus-induced changes in sumoylation may regulate proteins that are involved in the transcriptional repression of p53 and antiviral genes (27).
Either E1B-55K or E4-ORF3 is sufficient to mediate changes in SUMO localization and conjugation in infected cells (Fig. 2A and 4B). E1B-55K functions as an E3 SUMO ligase that specifically conjugates SUMO to p53, inhibiting its transcriptional activity (30, 31), and likely targets other proteins for sumoylation. We hypothesized that E4-ORF3 usurps cellular SUMO enzymes to modulate the SUMO pathway. E1B-55K has been shown to interact with UBC9 (32). However, E4-ORF3 does not mislocalize UBC9 or modulate UBC9 protein levels in virus-infected cells (Fig. 5B and C). Instead, we show that E4-ORF3 specifically targets PIAS3 but not PIAS1, PIAS2, or PIAS4 E3 SUMO ligases (Fig. 6A and B).
The PIAS family of proteins share over 40% sequence identity and have an N-terminal SAP domain, a PINIT motif, a RING-type zinc-binding structure, a SIM, and a serine/threonine-rich (S/T) C-terminal tail (20). The least conserved region is in the C-terminal S/T region (65), which may account for the differences in E4-ORF3 specificity in targeting the PIAS family. However, the function of the C-terminal S/T region is poorly understood. The role of PIAS3 mislocalization by E4-ORF3 remains to be determined. One possibility is that E4-ORF3 mislocalizes PIAS3 to facilitate the disruption of sumoylated proteins in PML nuclear bodies and/or nuclear rearrangement, promoting the sumoylation of cellular targets that aid in the formation of E2A viral replication domains. An alternative but not mutually exclusive possibility is that PIAS3 plays a role in gene regulation during Ad5 infection, as it regulates the activity of many transcription factors, including those involved in interferon pathways (20). E4-ORF3 is key to suppressing the antiviral interferon response (59), but the mechanism is unknown. E4-ORF3 may utilize PIAS3 to overcome the cell's defenses and antiviral response. Alternatively, mislocalization of PIAS3 could be an indirect consequence of E4-ORF3 targeting a preexisting cellular protein complex that is associated with PIAS3. For example, if PIAS3 is involved in the formation or maintenance of PML bodies, the entire multiprotein complex could be disrupted by E4-ORF3.
Using oligomerization mutants, we show that the higher-order assembly of E4-ORF3 into a multivalent scaffold is required for targeting and mislocalizing PIAS3 (Fig. 7C). E4-ORF3 is necessary and sufficient for inducing the sumoylation of the MRN complex (36, 37). However, we show that E4-ORF3 V101A and D105A/L106A mutants that are defective for binding and sumoylating MRN still mislocalize PIAS3 (Fig. 7E). Thus, if E4-ORF3 induces MRN sumoylation through PIAS3, it first requires MRN mislocalization and sequestration in the E4-ORF3 nuclear scaffold. Alternatively, E4-ORF3-induced sumoylation of MRN could be mediated through a distinct E3 SUMO ligase or the inhibition of SUMO proteases. Lastly, in contrast to MRN, PIAS3 is a conserved target of E4-ORF3 proteins from disparate adenovirus subgroups (Fig. 8B). PML is also a conserved target of E4-ORF3. It will be interesting to determine if E4-ORF3's functions in targeting PML and PIAS3 can be biochemically or functionally separated. We conclude that PIAS3 is a conserved cellular target of E4-ORF3 that may play an important role in facilitating the productive infection of all human adenoviruses.
Our analysis of sumoylation during adenovirus infection demonstrates that early viral proteins disrupt and induce SUMO2/3-associated nuclear bodies and viral replication domains as well as SUMO conjugation. These data indicate an important role for SUMO2/3 in facilitating and/or responding to adenovirus replication that is targeted and modulated by E1B-55K and E4-ORF3 viral protein interactions. Our results reveal an increasingly important role for SUMO2/3 in orchestrating the assembly of intranuclear compartments and a new function for the SUMO pathway in pathological infection.
ACKNOWLEDGMENTS
We thank members of the O'Shea laboratory, Kristen Espantman, and J. Sebastian Gomez-Cavazos for their support, insights, and helpful comments. We thank Huaiyu Sun and Tony Hunter for discussions and for contributing the GFP-4xSUMO2AA construct.
This work was supported by R01 CA178932 and P30CA014195 from the National Cancer Institute. C.C.O. is supported by The Leona M. and Harry B. Helmsley Charitable Trust, grant 2012-PG-MED002, the Marshall Legacy Foundation, the William Scandling Trust, and the Price Family Foundation. J.M.H. was supported by the T32 GM008666 Genetics Training Program.
REFERENCES
- 1.Witte ON, Dasgupta A, Baltimore D. 1980. Abelson murine leukaemia virus protein is phosphorylated in vitro to form phosphotyrosine. Nature 283:826–831. doi: 10.1038/283826a0. [DOI] [PubMed] [Google Scholar]
- 2.Hunter T, Sefton BM. 1980. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci U S A 77:1311–1315. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reimand J, Wagih O, Bader GD. 2013. The mutational landscape of phosphorylation signaling in cancer. Sci Rep 3:2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skaug B, Chen ZJ. 2010. Emerging role of ISG15 in antiviral immunity. Cell 143:187–190. doi: 10.1016/j.cell.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy DE, Marie IJ, Durbin JE. 2011. Induction and function of type I and III interferon in response to viral infection. Curr Opin Virol 1:476–486. doi: 10.1016/j.coviro.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wimmer P, Schreiner S, Dobner T. 2012. Human pathogens and the host cell SUMOylation system. J Virol 86:642–654. doi: 10.1128/JVI.06227-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Everett RD, Boutell C, Hale BG. 2013. Interplay between viruses and host sumoylation pathways. Nat Rev Microbiol 11:400–411. doi: 10.1038/nrmicro3015. [DOI] [PubMed] [Google Scholar]
- 8.Rajsbaum R, Garcia-Sastre A. 2013. Viral evasion mechanisms of early antiviral responses involving regulation of ubiquitin pathways. Trends Microbiol 21:421–429. doi: 10.1016/j.tim.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azuma Y, Tan SH, Cavenagh MM, Ainsztein AM, Saitoh H, Dasso M. 2001. Expression and regulation of the mammalian SUMO-1 E1 enzyme. FASEB J 15:1825–1827. [DOI] [PubMed] [Google Scholar]
- 10.Talamillo A, Sanchez J, Barrio R. 2008. Functional analysis of the SUMOylation pathway in Drosophila. Biochem Soc Trans 36:868–873. doi: 10.1042/BST0360868. [DOI] [PubMed] [Google Scholar]
- 11.Golebiowski F, Matic I, Tatham MH, Cole C, Yin Y, Nakamura A, Cox J, Barton GJ, Mann M, Hay RT. 2009. System-wide changes to SUMO modifications in response to heat shock. Sci Signal 2:ra24. [DOI] [PubMed] [Google Scholar]
- 12.Morris JR. 2010. SUMO in the mammalian response to DNA damage. Biochem Soc Trans 38:92–97. doi: 10.1042/BST0380092. [DOI] [PubMed] [Google Scholar]
- 13.Gareau JR, Lima CD. 2010. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11:861–871. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saitoh H, Hinchey J. 2000. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem 275:6252–6258. doi: 10.1074/jbc.275.9.6252. [DOI] [PubMed] [Google Scholar]
- 15.Bernardi R, Pandolfi PP. 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 16.Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF III, Maul GG. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol 147:221–234. doi: 10.1083/jcb.147.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong S, Muller S, Ronchetti S, Freemont PS, Dejean A, Pandolfi PP. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 95:2748–2752. [PubMed] [Google Scholar]
- 18.Shen TH, Lin HK, Scaglioni PP, Yung TM, Pandolfi PP. 2006. The mechanisms of PML-nuclear body formation. Mol Cell 24:331–339. doi: 10.1016/j.molcel.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dundr M, Misteli T. 2010. Biogenesis of nuclear bodies. Cold Spring Harb Perspect Biol 2:a000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shuai K, Liu B. 2005. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol 5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- 21.Mattoscio D, Segre CV, Chiocca S. 2013. Viral manipulation of cellular protein conjugation pathways: the SUMO lesson. World J Virol 2:79–90. doi: 10.5501/wjv.v2.i2.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. 2005. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol Cell 97:147–172. doi: 10.1042/BC20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmid M, Speiseder T, Dobner T, Gonzalez RA. 2014. DNA virus replication compartments. J Virol 88:1404–1420. doi: 10.1128/JVI.02046-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoeben RC, Uil TG. 2013. Adenovirus DNA replication. Cold Spring Harb Perspect Biol 5:a013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyer GS, Leuchtenberger C, Ginsberg HS. 1957. Cytological and cytochemical studies of HeLa cells infected with adeno-viruses. J Exp Med 105:195–216. doi: 10.1084/jem.105.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stracker TH, Carson CT, Weitzman MD. 2002. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 27.Soria C, Estermann FE, Espantman KC, O'Shea CC. 2010. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 466:1076–1081. doi: 10.1038/nature09307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. 2001. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev 15:3104–3117. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harada JN, Shevchenko A, Shevchenko A, Pallas DC, Berk AJ. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J Virol 76:9194–9206. doi: 10.1128/JVI.76.18.9194-9206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller S, Dobner T. 2008. The adenovirus E1B-55K oncoprotein induces SUMO modification of p53. Cell Cycle 7:754–758. doi: 10.4161/cc.7.6.5495. [DOI] [PubMed] [Google Scholar]
- 31.Pennella MA, Liu Y, Woo JL, Kim CA, Berk AJ. 2010. Adenovirus E1B 55-kilodalton protein is a p53-SUMO1 E3 ligase that represses p53 and stimulates its nuclear export through interactions with promyelocytic leukemia nuclear bodies. J Virol 84:12210–12225. doi: 10.1128/JVI.01442-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wimmer P, Blanchette P, Schreiner S, Ching W, Groitl P, Berscheminski J, Branton PE, Will H, Dobner T. 2013. Cross-talk between phosphorylation and SUMOylation regulates transforming activities of an adenoviral oncoprotein. Oncogene 32:1626–1637. doi: 10.1038/onc.2012.187. [DOI] [PubMed] [Google Scholar]
- 33.Doucas V, Ishov AM, Romo A, Juguilon H, Weitzman MD, Evans RM, Maul GG. 1996. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev 10:196–207. doi: 10.1101/gad.10.2.196. [DOI] [PubMed] [Google Scholar]
- 34.Yondola MA, Hearing P. 2007. The adenovirus E4 ORF3 protein binds and reorganizes the TRIM family member transcriptional intermediary factor 1 alpha. J Virol 81:4264–4271. doi: 10.1128/JVI.02629-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forrester NA, Patel RN, Speiseder T, Groitl P, Sedgwick GG, Shimwell NJ, Seed RI, Catnaigh PO, McCabe CJ, Stewart GS, Dobner T, Grand RJ, Martin A, Turnell AS. 2012. Adenovirus E4orf3 targets transcriptional intermediary factor 1gamma for proteasome-dependent degradation during infection. J Virol 86:3167–3179. doi: 10.1128/JVI.06583-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sohn SY, Hearing P. 2012. Adenovirus regulates sumoylation of Mre11-Rad50-Nbs1 components through a paralog-specific mechanism. J Virol 86:9656–9665. doi: 10.1128/JVI.01273-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sohn SY, Bridges RG, Hearing P. 2014. Proteomic analysis of ubiquitin-like post-translational modifications induced by the adenovirus E4-ORF3 protein. J Virol 89:1744–1755. doi: 10.1128/JVI.02892-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ou HD, Kwiatkowski W, Deerinck TJ, Noske A, Blain KY, Land HS, Soria C, Powers CJ, May AP, Shu X, Tsien RY, Fitzpatrick JA, Long JA, Ellisman MH, Choe S, O'Shea CC. 2012. A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell 151:304–319. doi: 10.1016/j.cell.2012.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Shea CC, Johnson L, Bagus B, Choi S, Nicholas C, Shen A, Boyle L, Pandey K, Soria C, Kunich J, Shen Y, Habets G, Ginzinger D, McCormick F. 2004. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 6:611–623. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 40.O'Shea CC, Soria C, Bagus B, McCormick F. 2005. Heat shock phenocopies E1B-55K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy. Cancer Cell 8:61–74. doi: 10.1016/j.ccr.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 41.Manders EMM, Verbeek FJ, Aten JA. 1993. Measurement of co-localization of objects in dual-colour confocal images. J Microscopy 169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 42.Castoralova M, Ruml T, Knejzlik Z. 2012. Using dot blot with immunochemical detection to evaluate global changes in SUMO-2/3 conjugation. Biotechniques 2012:1–4. doi: 10.2144/000113925. [DOI] [PubMed] [Google Scholar]
- 43.Johnson L, Shen A, Boyle L, Kunich J, Pandey K, Lemmon M, Hermiston T, Giedlin M, McCormick F, Fattaey A. 2002. Selectively replicating adenoviruses targeting deregulated E2F activity are potent, systemic antitumor agents. Cancer Cell 1:325–337. doi: 10.1016/S1535-6108(02)00060-0. [DOI] [PubMed] [Google Scholar]
- 44.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 45.Shah GA, O'Shea CC. Viral and cellular genomes activate distinct DNA damage responses. Cell 162:987–1002. doi: 10.1016/j.cell.2015.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. 2004. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 47.Shepard RN, Ornelles DA. 2004. Diverse roles for E4orf3 at late times of infection revealed in an E1B 55-kilodalton protein mutant background. J Virol 78:9924–9935. doi: 10.1128/JVI.78.18.9924-9935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lakdawala SS, Schwartz RA, Ferenchak K, Carson CT, McSharry BP, Wilkinson GW, Weitzman MD. 2008. Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J Virol 82:8362–8372. doi: 10.1128/JVI.00900-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gautam D, Bridge E. 2013. The kinase activity of ataxia-telangiectasia mutated interferes with adenovirus E4 mutant DNA replication. J Virol 87:8687–8696. doi: 10.1128/JVI.00376-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watts FZ. 2013. Starting and stopping SUMOylation. What regulates the regulator? Chromosoma 122:451–463. [DOI] [PubMed] [Google Scholar]
- 51.Sang J, Yang K, Sun Y, Han Y, Cang H, Chen Y, Shi G, Wang K, Zhou J, Wang X, Yi J. 2011. SUMO2 and SUMO3 transcription is differentially regulated by oxidative stress in an Sp1-dependent manner. Biochem J 435:489–498. doi: 10.1042/BJ20101474. [DOI] [PubMed] [Google Scholar]
- 52.Boggio R, Colombo R, Hay RT, Draetta GF, Chiocca S. 2004. A mechanism for inhibiting the SUMO pathway. Mol Cell 16:549–561. doi: 10.1016/j.molcel.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 53.Jentsch S, Psakhye I. 2013. Control of nuclear activities by substrate-selective and protein-group SUMOylation. Annu Rev Genet 47:167–186. doi: 10.1146/annurev-genet-111212-133453. [DOI] [PubMed] [Google Scholar]
- 54.Schmidt D, Muller S. 2003. PIAS/SUMO: new partners in transcriptional regulation. Cell Mol Life Sci 60:2561–2574. doi: 10.1007/s00018-003-3129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evans JD, Hearing P. 2003. Distinct roles of the adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J Virol 77:5295–5304. doi: 10.1128/JVI.77.9.5295-5304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoppe A, Beech SJ, Dimmock J, Leppard KN. 2006. Interaction of the adenovirus type 5 E4 Orf3 protein with promyelocytic leukemia protein isoform II is required for ND10 disruption. J Virol 80:3042–3049. doi: 10.1128/JVI.80.6.3042-3049.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stracker TH, Lee DV, Carson CT, Araujo FD, Ornelles DA, Weitzman MD. 2005. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J Virol 79:6664–6673. doi: 10.1128/JVI.79.11.6664-6673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Forrester NA, Sedgwick GG, Thomas A, Blackford AN, Speiseder T, Dobner T, Byrd PJ, Stewart GS, Turnell AS, Grand RJ. 2011. Serotype-specific inactivation of the cellular DNA damage response during adenovirus infection. J Virol 85:2201–2211. doi: 10.1128/JVI.01748-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ullman AJ, Reich NC, Hearing P. 2007. Adenovirus E4 ORF3 protein inhibits the interferon-mediated antiviral response. J Virol 81:4744–4752. doi: 10.1128/JVI.02385-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ullman AJ, Hearing P. 2008. Cellular proteins PML and Daxx mediate an innate antiviral defense antagonized by the adenovirus E4 ORF3 protein. J Virol 82:7325–7335. doi: 10.1128/JVI.00723-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chahal JS, Qi J, Flint SJ. 2012. The human adenovirus type 5 E1B 55 kDa protein obstructs inhibition of viral replication by type I interferon in normal human cells. PLoS Pathog 8:e1002853. doi: 10.1371/journal.ppat.1002853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pombo A, Ferreira J, Bridge E, Carmo-Fonseca M. 1994. Adenovirus replication and transcription sites are spatially separated in the nucleus of infected cells. EMBO J 13:5075–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nordqvist K, Ohman K, Akusjarvi G. 1994. Human adenovirus encodes two proteins which have opposite effects on accumulation of alternatively spliced mRNAs. Mol Cell Biol 14:437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gill G. 2003. Post-translational modification by the small ubiquitin-related modifier SUMO has big effects on transcription factor activity. Curr Opin Genet Dev 13:108–113. doi: 10.1016/S0959-437X(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 65.Rytinki MM, Kaikkonen S, Pehkonen P, Jaaskelainen T, Palvimo JJ. 2009. PIAS proteins: pleiotropic interactors associated with SUMO. Cell Mol Life Sci 66:3029–3041. doi: 10.1007/s00018-009-0061-z. [DOI] [PMC free article] [PubMed] [Google Scholar]