

ABSTRACT
Previously, no retroviral Gag protein has been highly purified in milligram quantities and in a biologically relevant and active form. We have purified Rous sarcoma virus (RSV) Gag protein and in parallel several truncation mutants of Gag and have studied their biophysical properties and membrane interactions in vitro. RSV Gag is unusual in that it is not naturally myristoylated. From its ability to assemble into virus-like particles in vitro, we infer that RSV Gag is biologically active. By size exclusion chromatography and small-angle X-ray scattering, Gag in solution appears extended and flexible, in contrast to previous reports on unmyristoylated HIV-1 Gag, which is compact. However, by neutron reflectometry measurements of RSV Gag bound to a supported bilayer, the protein appears to adopt a more compact, folded-over conformation. At physiological ionic strength, purified Gag binds strongly to liposomes containing acidic lipids. This interaction is stimulated by physiological levels of phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2] and by cholesterol. However, unlike HIV-1 Gag, RSV Gag shows no sensitivity to acyl chain saturation. In contrast with full-length RSV Gag, the purified MA domain of Gag binds to liposomes only weakly. Similarly, both an N-terminally truncated version of Gag that is missing the MA domain and a C-terminally truncated version that is missing the NC domain bind only weakly. These results imply that NC contributes to membrane interaction in vitro, either by directly contacting acidic lipids or by promoting Gag multimerization.
IMPORTANCE Retroviruses like HIV assemble at and bud from the plasma membrane of cells. Assembly requires the interaction between thousands of Gag molecules to form a lattice. Previous work indicated that lattice formation at the plasma membrane is influenced by the conformation of monomeric HIV. We have extended this work to the more tractable RSV Gag. Our results show that RSV Gag is highly flexible and can adopt a folded-over conformation on a lipid bilayer, implicating both the N and C termini in membrane binding. In addition, binding of Gag to membranes is diminished when either terminal domain is truncated. RSV Gag membrane association is significantly less sensitive than HIV Gag membrane association to lipid acyl chain saturation. These findings shed light on Gag assembly and membrane binding, critical steps in the viral life cycle and an untapped target for antiretroviral drugs.
INTRODUCTION
Many of the principles underlying retrovirus assembly are understood. For example, the retrovirus structural protein, Gag, is able to assemble into morphologically normal virus-like particles (VLPs) in cells (1) and as a purified protein in vitro (2). The structures and functions of the multiple Gag domains are known. Thus, the MA domain mediates binding to the plasma membrane (PM), where for most retroviruses the Gag lattice is formed and budding occurs, the CA domain and immediately adjoining sequences engage in critical contacts to form the hexagonal Gag lattice, the NC domain binds to and packages the viral genomic RNA, and short sequences called late domains recruit the cellular ESCRT machinery to finally pinch off the fully formed immature virus particle from the PM (1).
Despite the extensive knowledge about Gag, for at least two reasons a detailed mechanistic understanding of assembly is lacking. First, the three-dimensional structure of the multidomain Gag protein (also called a polyprotein) is unknown at an atomic level although electron cryo-tomography of VLPs has succeeded in visualizing the protein in the Gag lattice to a resolution of almost 8 Å (3). Second, purification of full-length biologically active Gag proteins, in the milligram amounts needed for many biochemical analyses, has not been reported. Most retroviral Gag proteins bear an N-terminal fatty acyl moiety, myristate, which is essential for PM binding in vivo. This modification apparently renders the protein poorly soluble after expression in Escherichia coli.
To date, most biochemical characterization of Gag has focused on HIV-1, with almost all studies using the unmyristoylated protein. This unmyristoylated version of Gag binds to membranes only weakly in vitro (4) and not at all in living cells (5–7). Based on size exclusion chromatography (SEC), small-angle neutron scattering (SANS), and other techniques, unmyristoylated HIV-1 Gag in solution is inferred to adopt a relatively compact, folded-over shape (8). This shape is quite different from the stretched-out or elongated shape seen in the immature virus particles of all retroviruses. The effect of the myristoylation on the shape of unassembled Gag remains unknown though unmyristoylated Gag can assemble in the cytoplasm under overexpression conditions, as well as in vitro in the presence of inositol phosphates and nucleic acid (5, 7, 9). The folded-over shape has led to a model in which both MA and NC domains can simultaneously bind to the PM or to nucleic acid (NA) since both domains are highly basic and thus can interact with negatively charged phospholipids or NA. According to the model, once both domains interact with their specific ligands, NC with NA and MA with phosphatidylinositol phosphates (PIPs) in the PM, Gag takes on the extended conformation needed to form the Gag lattice found in VLPs. This model is supported by neutron reflectometry applied to unmyristoylated HIV-1 Gag bound to a supported lipid bilayer at low ionic strength (10). However, since the fatty acyl modification is essential for PM interaction in cells, the biological relevance of the model remains to be established.
The Rous sarcoma virus (RSV) system has distinct advantages for studies of retrovirus assembly. First, an N-terminally truncated version of RSV Gag that is missing the membrane binding domain (MBD) of MA (and also is missing the C-terminal protease [PR] domain, which in other retroviruses is found in another reading frame) assembles robustly in vitro as a purified protein (here, ΔMBD) (11). This assembly reaction has been instrumental in elucidation of some of the principles of assembly (reviewed in reference 2) and has led to one of the highest-resolution structures of a retroviral Gag lattice yet determined, ∼8 Å (71). However, the absence of the membrane binding domain renders this protein irrelevant for studying the membrane interaction step in assembly. Second, and by contrast with HIV-1, RSV Gag does not carry any fatty acyl modification, and therefore this protein as obtained from E. coli expression should be biologically active.
We have purified RSV Gag (missing the C-terminal PR domain [ΔPR]) in milligram amounts after expression in E. coli as a SUMO fusion protein. N- and C-terminally truncated versions of the protein, as well as a protein with an internal deletion, were prepared in parallel as controls. After removal of the tag, purified RSV Gag is able to assemble into VLPs in vitro, verifying that it is biologically active. Using biophysical techniques, we determined that the overall shape of Gag in solution is extended and flexible, unlike that of HIV-1 Gag. RSV Gag binds to liposomes of various compositions with higher affinity than Gag proteins missing either the MA or NC (ΔMA or ΔNC, respectively) domain. Neutron reflectometry (NR) of Gag bound to a supported lipid bilayer is consistent with a model in which both MA and NC contact the membrane and contribute to membrane affinity. To our knowledge, the work reported here represents the first detailed biochemical characterizations of a purified, biologically active retroviral Gag protein.
MATERIALS AND METHODS
DNA vectors.
RSV Gag ΔPR (referred to as Gag throughout) and Gag truncations used in this study are shown in Fig. 1. All DNA constructs were cloned using standard subcloning techniques and propagated in DH5α cells. RSV Gag, MA, and MBD were cloned into pET-SUMO vector (12) by digesting the vector with BamHI and EagI and digesting the insert with BglII and EagI. SUMO-ΔNC was amplified and ligated into SUMO-Gag at SbfI and EagI sites. SUMO-ΔFlex (Gag with a deletion of amino acids 104 to 220) was amplified by two-step PCR and ligated into pET-SUMO at SacII sites. Glutathione S-transferase (GST)-NC was cloned by amplification and digestion of NC with BglII and EcoRI. The digested insert was ligated into pGEX at BamHI and EcoRI sites. All constructs were verified by sequencing.
FIG 1.
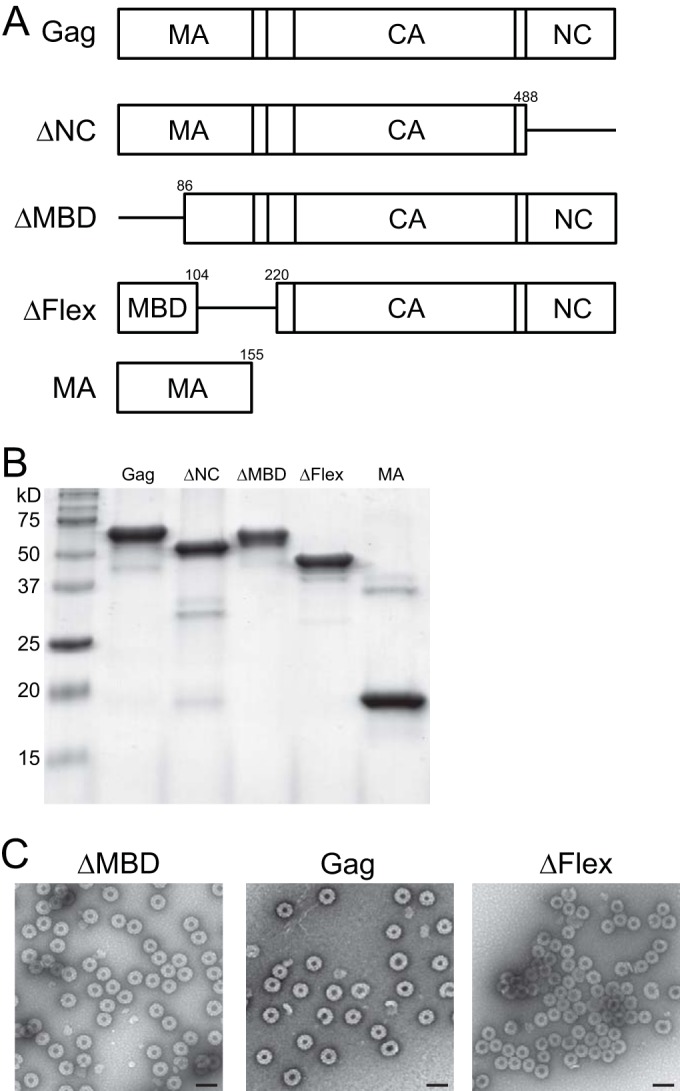
Proteins used in this study. (A) Schematic representation of proteins. Note that the proteins MBD and GST-NC are not shown. Numbers indicate amino acid positions. (B) Stained gel of the proteins used in liposome binding assays. For analysis by size exclusion chromatography (SEC), small angle X-ray scattering (SAXS), and neutron reflectometry (NR), the proteins were further purified by SEC. (C) Virus-like particles (VLPs) assembled in vitro and imaged by negative-stain electron microscopy. Scale bar, 100 nm.
Protein purification, VLP assembly, and electron microscopy (EM).
SUMO-tagged proteins were purified using standard bacterial expression and affinity column techniques. In brief, E. coli BL21 cultures were grown at 37°C to an optical density at 600 nm of 0.4 to 0.7, and isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to 0.5 mM to induce protein expression. Induced cells were harvested 4 to 6 h later. Pelleted cells were resuspended in lysis buffer [20 mM Tris-HCl, pH 8, 500 mM NaCl, 2 mM tris(2-carboxyethyl)phosphine (TCEP), 2 mM phenylmethylsulfonyl fluoride (PMSF) protease inhibitor, and 2 μM ZnCl2], lysed by sonication, and cleared by centrifugation in a TLA-110 Beckman rotor at 90,000 rpm for 45 min. Polyethyleneimine (PEI) was added to 0.3% to precipitate nucleic acid, which was spun down and removed. Ammonium sulfate was added (15 to 30% saturation, depending on the protein) to precipitate protein, followed by centrifugation. The pellet was resuspended in binding buffer (20 mM Tris-HCl, pH 8, 100 mM NaCl, 2 mM TCEP, and 2 μM ZnCl2) and further purified by cation exchange chromatography (HiTrap SP FF; GE Healthcare). Eluted protein was then purified by Ni+2 affinity chromatography (HisTrap HP; GE Healthcare). Protein bound to HisTrap columns was washed with high-salt buffer (20 mM Tris HCl, pH 8.0, 500 mM NaCl, 2 mM TCEP, and 2 μM ZnCl2) to remove residual nucleic acid. The SUMO tag was cleaved from the eluted protein overnight at 4°C during dialysis with SUMO-specific protease 1 (ULP protease) (12) (in dialysis buffer consisting of 20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10 mM dithiothreitol [DTT], 2 μM ZnCl2). The SUMO tag and ULP protease were removed by Ni+2 affinity chromatography. Purified protein at 2 to 5 mg/ml was flash frozen in aliquots and stored at −80°C. The protein at this stage had an A260/A280 ratio of ∼0.58, indicating the absence of measurable nucleic acid. Purified proteins subjected to hydrodynamic and small-angle X-ray scattering (SAXS) measurements were further purified by size exclusion chromatography (see Materials and Methods sections on hydrodynamic measurements, SAXS data collection and analysis, and low-angle neutron reflectometry [LANR] for additional protein handling). VLP assembly reactions were performed by diluting purified protein 1:5 in assembly buffer (20 mM morpholineethanesulfonic acid [MES], pH 6.5, 2 μM ZnCl2, 2 mM TCEP) to a final NaCl concentration of 100 mM, in the presence of 1:10 (nucleic acid to protein by mass) of 50-mer oligonucleotide (GT25) (2, 13), and incubated at 22°C for 1 h. Assembly reactions were screened for VLP formation by negative-stain (2% uranyl acetate) EM on a Morgagni 268 transmission electron microscope.
Lipids and liposome binding assay.
Large unilamellar vesicles (LUVs; diameter, 100 nm) in 20 mM Tris-HCl, pH 8.0, were prepared by rapid solvent exchange (RSE) as described by Buboltz and Feigenson (14), followed by extrusion (15, 16). All lipid stocks (purchased from Avanti Polar Lipids) were phosphate assayed (17) to determine concentration and subjected to thin-layer chromatography (TLC) to verify quality (lipids and abbreviations are given in Table 1). Membrane compositions are reported as mole percent throughout.
TABLE 1.
Lipids used to form large unilamellar vesicles
| Lipid name | Abbreviation | Composition |
|---|---|---|
| Palmitoyl-oleoyl-phosphatidylcholine | POPC | 16:0/18:1 |
| Dioleoyl-phosphatidylcholine | DOPC | 18:1/18:1 |
| Distearoyl-phosphatidylcholine | DSPC | 18:0/18:0 |
| Palmitoyl-oleoyl-phosphoethanolamine | POPE | 16:0/18:1 |
| Palmitoyl-oleoyl-phosphatidylserine | POPS | 16:0/18:1 |
| Dioleoyl-phosphatidylserine | DOPS | 18:1/18:1 |
| Dipalmitoyl-phosphatidylserine | DPPS | 16:0/16:0 |
| Phosphatidylinositol-4,5-bisphosphate | PI(4,5)P2 | 18:0/20:4 |
LUV flotations were performed as previously described (15). In the binding reaction mixture, 10 to 20 μg of purified protein was mixed with 50 μg of LUVs in a final reaction volume of 25 μl in 20 mM Tris-HCl (pH 8.0)–150 mM NaCl, and the mixture was incubated at room temperature for 10 min. Sucrose (75 μl of 67% [wt/wt]) was added to a final concentration of 50%, of which 80 μl was transferred to a 240-μl polycarbonate centrifuge tube (Beckman). Additional volumes of 120 μl of 40% sucrose and then 40 μl of 4% sucrose were layered on top of the 50% sucrose. All sucrose solutions were prepared in binding buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl). Tubes were subjected to centrifugation at 90,000 rpm in a TLA-100 (Beckman) rotor for 45 min at 4°C. Four 60-μl fractions were collected and subjected to SDS-polyacrylamide gel electrophoresis (PAGE). For pelleting assays, the total volume of the binding reaction mixture was brought to 240 μl with binding buffer, and the mixture was centrifuged at 90,000 rpm at 4°C for 45 min in a TLA-100 Beckman rotor. The supernatant was removed, and the pellet was resuspended and subjected to PAGE analysis. All liposome binding reactions were performed at 150 mM NaCl.
ESR and GUV analysis.
For electron spin resonance (ESR), multilamellar vesicles (MLVs) were prepared by freeze-thawing in buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl). Each sample contained ∼2,000 nmol lipid and 0.2 mol% of the spin label 16-DOXYL-steric acid (Sigma-Aldrich). ESR spectra were collected for all samples on a 9.4-GHz Bruker continuous wave (cw)-ESR electron momentum spectrometer (EMS) at room temperature (∼22°C). At least five scans were averaged for each sample. The maximum and minimum values of tensor A (Amax and Amin) were determined from each spectrum, and the order parameter was calculated according to Schorn and Marsh (18) using the hyperfine tensor (Axx, Ayy, Azz) = (5, 5, 32.8 G). Giant unilamellar vesicles (GUVs) containing the lipid dye Lissamine rhodamine-conjugated dioleoyl-phosphatidylethanolamine (DOPE) were prepared by gentle hydration (19) in 100 mM sugar buffer (sucrose inside, glucose outside) and imaged on a Nikon eclipse Ti-E microscope.
Neutron reflectometry. (i) Substrate preparation.
Silicon wafers of 3-in. diameter, 5,000-μm thick n-type Si:P[100] with one side polished to <5-Å roughness, were purchased from El-Cat, Waldwick, NJ. Surfaces were cleaned in a 5 vol% Hellmanex solution with thorough rinsing with ultrapure water (Millipore) and then 99.8% ethanol (EtOH) from Sigma and in an N2 gas stream. Metal deposition of Cr (∼20 Å) and Au (∼150 Å) was performed in a high-energy magnetron (Denton Vacuum Discovery 550) available at the NIST Center for Nanoscale Science and Technology, Nano-Fabrication facility. Wafers were immediately soaked overnight in a 7:3, mol/mol, ethanol solution of HC18 [Z-20-(Z-octadec-9-enyloxy)-3,6,9,12,15,18,22-heptaoxatetracont-31-ene-1-thiol] and β-mercaptoethanol (βME) at a total concentration of 0.2 mM forming a self-assembled monolayer (SAM). The wafer was assembled into a standard NIST Center for Neutron Research (NCNR) reflectometry flow cell, and a 7:3, mol/mol, dioleoyl-phosphatidylcholine/dioleoyl-phosphatidylserine (DOPC/DOPS) membrane was deposited through vesicle fusion/osmotic shock. Briefly, a lipid solution of the desired composition was dissolved in chloroform, the solvent was then evaporated, and the lipid film was resuspended in a 500 mM NaCl aqueous solution to a final concentration of 5 mg/ml. The lipid solution was incubated over the dry SAM layer for ∼2 h, after which the cell was flushed with 50 mM NaCl solution, resulting in an intact tethered bilayer lipid membrane (tBLM).
(ii) Data acquisition.
Neutron reflectivity was performed on an NG7 horizontal reflectometer and the MAGIK (multiple angle grazing incidence K vector) vertical reflectometer at the NIST Center for Neutron Scattering. A momentum transfer, qz, range between 0.008 and 0.258 Å−1 was accessed in most measurements. Typical measurements used two solvent isotopic contrasts per experimental condition, consisting of aqueous buffer prepared in either 100% D2O or H2O with a buffer composition of 50 mM NaCl–10 mM NaHPO4, pH 8.00. For each contrast, 6 h of data collection provided sufficient counting statistics to resolve signal over background counts, with counting times weighted toward the high q region of the scan. After measurement of the neat membrane, RSV Gag was introduced at a concentration of 0.5 μM in H2O buffer. After a 20-min incubation, NR spectra were collected. The same conditions were repeated for the D2O contrast.
(iii) Data analysis.
NR provides a one-dimensional (1D) profile along the axis normal to the membrane surface (z axis) spatially averaged over in-plane directions (i.e., the x-y plane). Data were analyzed by fitting an area-fraction distribution of substrate, membrane, and protein layers along the z axis. A composition space model that represents the lipid membrane in terms of overlapping distributions of the different molecular groups (lipid chain, head group, and tether) (20) was used to represent the tBLM. A Hermite spline was used to model the distribution of the protein along the z axis without any assumptions of the protein profile. The protein profile was allowed to partially or fully overlap the membrane where membrane volume was removed proportionally to satisfy a constraint of constant volume filling. All data sets, consisting of two solvent contrast and two experimental conditions (neat tBLM and binding of RSV Gag), were fit simultaneously. The substrate parameters (silicon oxide [SiOx], Cr, and Au) as well as the tether layer were conserved among all the models. Lipid thickness and the protein profile were allowed to vary between experimental conditions. Fitting was performed with the Refl1D software package (http://www.reflectometry.org/danse/docs/refl1d/index.html). A Monte Carlo error analysis procedure was used to determine the confidence intervals in the protein profile (21).
Hydrodynamic measurements.
To study the hydrodynamic properties of RSV Gag and related proteins, a Superose 12 (GE Healthcare) column was used in conjunction with a Rainin HPXL solvent delivery system connected to a Rainin Dynamax UV-1 detector and a Wyatt systems Dawn Helios static and quasi-elastic light scattering (SLS and QELS, respectively) detector. Proteins at concentrations of 0.8 to 2 mg/ml were filtered through 300-kDa centrifugal filtration devices (Pall Corporation) before injection onto the column, equilibrated in 20 mM Tris-HCl, pH 7.4, 0.3 M NaCl, and 1 mM TCEP. Solvent viscosity of 0.01035 cP and a temperature dependence of −1.95E−4 g/cm s K were assumed. The data collected simultaneously from light detectors 5 through 18, with the exception of detector 13, were used for SLS, while detector 13 was modified for QELS measurements. Protein standards (GE Healthcare), including beta amylase (radius of hydration [Rh] 54.2 Å), alcohol dehydrogenase (Rh 45.5 Å), bovine serum albumin (BSA; Rh 35.5 Å), carbonic anhydrase (Rh 23.5 Å), and cytochrome c (Rh 17.2 Å), were used for the determination of the radius of hydration from retention time. Retention time was used to calculate the parameter (KD)1/3 (where KD is the distribution constant), which has a linear relationship to the Rh of the eluting species (22). Eluting peaks from the column were collected and concentrated on Amicon Ultra 3K centrifugal devices before confirmatory Rh measurements on a Wyatt Titan dynamic light scattering (DLS) instrument.
SAXS data collection and analysis.
For SAXS measurements, the peak fractions of eluting species from a Superose 12 column, as described for hydrodynamic measurements, were used. X-ray scattering measurements were carried out at room temperature at the beamline 12ID-B of the Advanced Photon Source, at the Argonne National Laboratory. The setups were adjusted to achieve scattering q values of 0.006 < q < 1.0 Å−1, where q = (4π/λ)sinθ, and 2θ is the scattering angle. Thirty two-dimensional (2D) images were recorded for each buffer or sample solution using a flow cell, with the exposure time of 2 s to minimize radiation damage and to obtain a good signal-to-noise ratio. No radiation damage was observed, as confirmed by the absence of systematic signal changes in sequentially collected X-ray scattering images. The 2D images were reduced to one-dimensional scattering profiles using a Matlab script at the beamlines. The scattering profile of a sample solute was calculated by subtracting the buffer contribution from the sample buffer profile using the program PRIMUS (23) and standard procedures. Concentration series measurements (4- and 2-fold dilutions and stock solution) for the same sample were carried out to remove the scattering contribution due to interparticle interactions and to extrapolate the data to infinite dilution. The forward scattering intensity I(0) and the radius of gyration (Rg) were calculated from the data of infinite dilution at low q values in the range of qRg of <1.3, using the Guinier approximation: lnI(q) ≈ ln(I(0)) − Rg2q2/3. These parameters were also estimated from the scattering profile with a broader q range of 0.006 to 0.30 Å−1 using the indirect Fourier transform method implemented in the program GNOM (24), along with the pair distance distribution function (PDDF), p(r), and the maximum dimension of the protein, Dmax. The parameter Dmax (the upper end of distance r) was chosen so that the resulting PDDF has a short, near-zero-value tail to avoid underestimation of the molecular dimension and consequent distortion in low-resolution structural reconstruction. The molecular weights of solutes were calculated on a relative scale using SAXS MoW (25), in which estimation of molecular weights is independent of protein concentration and can be obtained with minimal user bias.
RESULTS
Conformation of Gag and Gag truncations in solution.
We have characterized the hydrodynamic properties of RSV Gag and of Gag mutants (Fig. 1A and B) by size exclusion chromatography (SEC) and multiangle light scattering (MALS) (Fig. 2A). All of the proteins were purified as SUMO fusions; the SUMO tag was then cleaved off, and the proteins were purified again to remove the SUMO moiety. ΔMBD has been described previously and has been used for years to study RSV assembly in vitro (11, 26–29). It is RSV Gag without the membrane binding domain (MBD) of MA and also without the PR domain at the C terminus (which is missing in all of the Gag proteins described here). ΔNC removes the mature NC sequence, thus ending at its C terminus with the sequence PLVM that terminates the serine protease (SP) domain. ΔFlex carries an internal deletion from residue 104 to 220, thus removing the putative flexible region, which appears to be unstructured by crystallography (R. Kingston, personal communication) or by secondary structure predictions. It was previously reported that deletion of a smaller stretch, amino acids 87 to 142 (Δ87–142), is compatible with budding and infectivity (30). ΔMBD, Gag, and ΔFlex assembled into VLPs, as determined by negative-stain EM analysis (Fig. 1C). MA is the 155-residue MA domain of RSV Gag. MBD comprises the N-terminal 86 residues of MA, which includes the basic residues known to be involved in membrane binding (31, 32).
FIG 2.
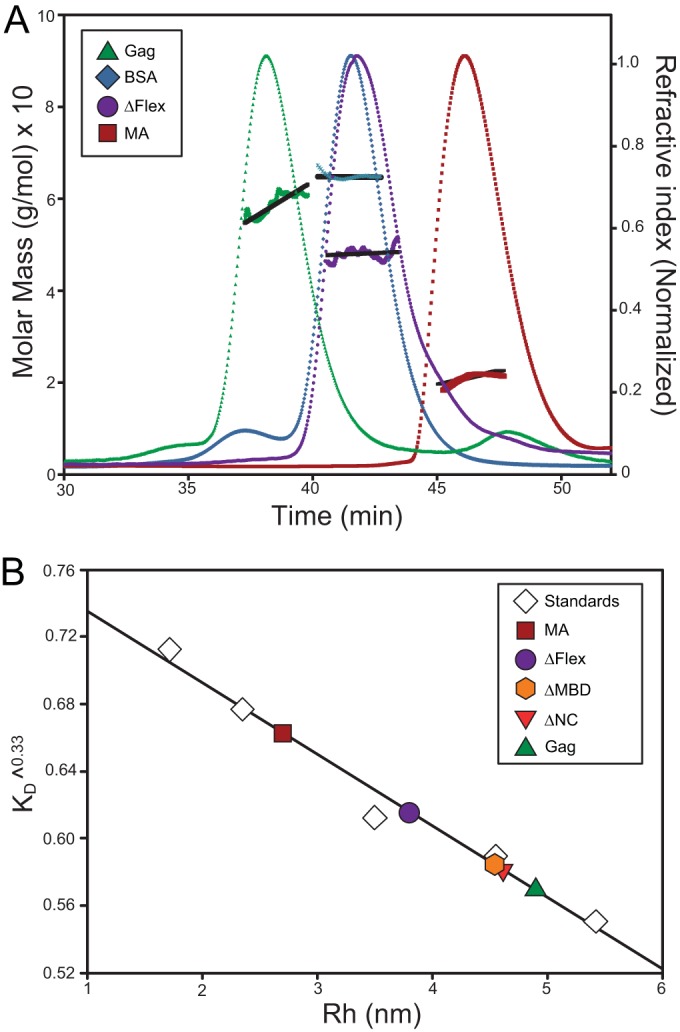
SEC and multiangle static light scattering (SLS) of RSV Gag and Gag-derived proteins. (A) RSV Gag, ΔFlex, and MA are shown with BSA. Gag solutions and BSA at 1.5 mg/ml were chromatographed on a Superose 12 column. Elution of protein was simultaneously monitored by determining the refractive index, giving rise to the elution profiles, and by SLS, yielding the data points above the elution profiles. The points indicate the molar mass of the protein in each fraction. These masses, in turn, were averaged over the breadth of each peak, producing the roughly horizontal lines above the elution profiles. (B) The Rh values of Gag and derived constructs determined from the elution time in panel A and in addition the Rh values for ΔMBD and ΔNC are shown. The column was calibrated with a series of proteins of known Rh values, as described in Materials and Methods. The graph shows the cube root of the column distribution coefficient KD plotted against the Rh value of the proteins.
By comparison with globular proteins, RSV Gag displayed anomalous mobility on a Superose 12 column (Fig. 2A, green profile). Even though it has a predicted molecular mass of ∼60 kDa, it eluted well before BSA (∼67 kDa; blue profile), at a volume comparable to that of dimeric BSA (small leading peak seen in the BSA elution profile). This elution position was constant over injection concentrations ranging between 25 μM and 5 μM (data not shown). The unexpectedly early elution of RSV Gag is due to its conformation and not oligomerization since by MALS the molar mass of the protein in the peak was determined as 6.0 × 104 Da (Fig. 2A). From the retention volume of proteins of known dimensions, we estimated the Stokes radius, Rh, of RSV Gag (22) to be 4.9 nm (Fig. 2B), considerably larger than that previously determined for monomeric HIV-1 Gag (3.6 nm) (Table 2). The Stokes radius is the radius of a smooth sphere which has the same frictional coefficient as the protein. It is related to the average volume the protein occupies in solution and is sensitive to the shape and physical properties, as well as size, of the protein.
TABLE 2.
Hydrodynamic parameters for RSV Gag and Gag-derived proteins
The results with the mutant and Gag-related proteins were all consistent with these data. The truncated Gag protein ΔFlex (∼49 kDa) (Fig. 2A, purple profile) eluted at a position comparable to that of BSA, while ΔNC and ΔMBD (∼51 and 50 kDa, respectively) (chromatograms not displayed) eluted between the monomeric and dimeric peaks of BSA. The small domains MA (∼16.4 kDa) (Fig. 2A, red profile) and MBD (∼10.6 kDa) (profile not displayed) eluted well after BSA. Similarly, the masses of eluting RSV ΔFlex, MA, and BSA were measured as 5.0 × 104 Da, 2.0 × 104 Da, and 6.5 × 104 Da, respectively, clearly demonstrating all of the proteins to be monomeric. From comparisons of the retention volumes of the Gag-derived proteins with proteins of known dimensions, we estimated the Rh values of ΔNC, ΔMBD, ΔFlex, and MA to be 4.6, 4.54, 3.8, and 2.7 nm, respectively (Fig. 2B). Comparable values were obtained by collecting the eluting peaks and measuring diffusion coefficients by dynamic light scattering (Table 2).
To gain further insight into the conformation of RSV Gag and its derivatives, we performed SAXS analysis (Fig. 3A). The scattering data showed no indication of aggregation and therefore were subjected to Guinier fitting (33) in order to obtain an estimate of the radius of gyration, Rg, of the scattering species (Fig. 3B and Table 3). The radius of gyration, which is the root mean square of distances between all parts of the molecule from its center of mass, gives an estimate of the molecular shape and dimensions. Elongated protein molecules typically have larger Rg values than those of compact proteins of identical molecular weights. To gain insight into the compactness and flexibility of the protein constructs, the SAXS data were analyzed to generate dimensionless Kratky plots (34, 35). The shape of the Kratky plot is an indicator of the degree of disorder, or “unfoldedness,” of a protein. It has therefore been used to monitor the folding of proteins (36) and also to characterize proteins that are intrinsically disordered (37). In the case of multidomain proteins, where domains are linked by flexible linkers, this method can be used to ascertain the degree of flexibility between the domains (38). The dimensionless Kratky plot for compact particles has a characteristic shape, with a peak at √3 (qRg). Deviations from this value suggest either asymmetry or particle flexibility.
FIG 3.

Analysis of proteins by X-ray scattering. (A) Experimental SAXS curves. (B) Guinier plots. (C) Dimensionless Kratky plots (34). (D) Pair distance distribution function p(r) derived from the intensity curve I(q) extrapolated to zero concentration of full-length GAG protein, ΔMBD, ΔFlex, MA, and MBD. au, arbitrary units.
TABLE 3.
Structural parameters derived from the SAXS data
| Protein | Predicted mass (kDa) | SAXS mass (kDa)a | aRg (Å)b | bRg (Å)c | Dmax (Å) |
|---|---|---|---|---|---|
| GAG | 60.8 | 57.4 | 54.1 ± 1.2 | 56.1 ± 0.6 | 192 ± 3 |
| ΔMBD | 50.2 | 52.2 | 46.9 ± 1.2 | 48.8 ± 0.4 | 164 ± 3 |
| ΔFlexd | 49.1 | 51.0 | 49.6 ± 1.6 | 49.5 ± 0.9 | 172 ± 4 |
| MA | 16.4 | 19.9 | 29.6 ± 0.4 | 31.9 ± 0.3 | 116 ± 3 |
| MBD | 10.6 | 9.4 | 16.5 ± 0.6 | 17.1 ± 0.5 | 60 ± 2 |
Calculated using the web portal SAXS MoW (www.if.sc.usp.br/∼SAXS).
TheaRg value was derived from Guinier fitting, using Igor Pro (Wavemetrics, Lake Oswego, OR, USA).
The bRg value was derived from GNOM fitting.
Deletion of Gag amino acids 104 to 220.
The dimensionless Kratky plot for MBD was bell shaped and showed a clear peak at ∼√3 (qRg) (Fig. 3C), indicative of its compact structure and consistent with the known nuclear magnetic resonance (NMR) structure (39). For MA, this peak was broadened and shifted to a larger value of ∼2.7 (qRg), suggesting asymmetry. The plots for ΔFlex and ΔMBD showed a loss of the characteristic peak and a plateauing of the curve at higher values of qRg. This shape for the modified Kratky plot is seen in multidomain proteins connected by flexible linkers (34). The plot for full-length Gag, similar to that of the intrinsically disordered protein PIR (40), suggests a very high degree of flexibility.
The pairwise distance distribution function [PDDF, or p(r)] is a weighted histogram of pair distances between scattering atoms in the protein. The shape of the PDDF plot gives an indication of the shape of the protein as well as a measure of its dimensions. The PDDF values for Gag and derived constructs were calculated using GNOM (Fig. 3D and Table 3). The PDDF for Gag showed a peak of ∼50 Å; this is the most common distance between two atoms in the molecule. There was a monotonic decrease in the frequency distribution of longer distances within the molecule. The asymmetric shapes of these distributions for Gag and for ΔFlex, ΔMBD, and MA suggest that these are elongated proteins. The PDDF distribution of globular proteins is more symmetric about the peak, as seen for MBD. The presence of a single broad maximum in the RSV Gag distribution, as well as its smooth shape, appears to preclude the presence of a single conformer and is consistent with a multidomain protein. Bimodal distributions in PDDF plots have been observed in some proteins with rigid, unique conformations (41, 42). Dmax, the distance r at which the p(r) drops to zero, is indicative of the longest dimension in the molecule. This value was estimated to be 193 ± 3 Å for Gag. By comparison, the length of Gag in immature virus-like particles as determined by electron cryo-tomography is about 180 Å. The estimates for the derived proteins are given in Table 3.
Shape of Gag on a membrane.
The membrane-bound structure of the full-length RSV Gag protein was evaluated using neutron reflectometry (NR). NR is unique in being able to characterize membrane proteins in their physiological environments (i.e., multicomponent, fluid lipid membranes in aqueous environments). In an NR experiment, a flat collimated beam is reflected off a planar membrane at very low grazing incidence angles, and the change in intensity of the reflected beam as a function of this angle is measured. Interpretation of the reflectivity spectra provides a 1D density profile of both lipid groups and protein distribution normal to the membrane plane. A tBLM with an intrinsically low roughness has been developed at NIST and is highly amenable to reflectivity and other surface-sensitive biophysical techniques. The tBLM consists of a thin Au film deposited onto either a glass or silicon substrate on which we form a lipid membrane separated from the underlying solid support by a 2-nm-thin aqueous reservoir generated by sparsely distributed tether molecules. Membranes can be formed of any composition and are both fluid and highly stable. An aqueous reservoir above the membrane surface allows the subsequent introduction of proteins and other biochemical factors to the system during measurements.
The binding affinity of RSV Gag to a tBLM consisting of DOPC/DOPS (70/30) was first characterized by surface plasmon resonance (SPR) (data not shown). The formation of the tBLM on Au chips provides a direct means to measure mass adsorption as a function of protein concentration. At 50 mM NaCl, Gag bound tightly and cooperatively to the membrane with a dissociation constant, Kd, of 207 nM and a Hill coefficient, n, of 2.3. At Gag concentrations up to 0.5 μM, the SPR signal remained stable for several hours, indicating that nonspecific binding and surface aggregation were not issues at these concentrations.
NR measurements were performed on a tBLM composed of DOPC/DOPS (70/30) at 50 mM NaCl and with a 0.5 μM solution concentration of RSV Gag. Reflectivity spectra of the neat tBLM and the tBLM after incubation with Gag were collected under both H2O and D2O isotopic buffer contrasts. Figure 4A shows spectra of the neat bilayer and the membrane after Gag incubation for the D2O buffer condition. Statistically significant changes in the spectra were observed upon protein addition, as indicated by the residual difference in the spectra (Fig. 4A, bottom panel). We interpreted the NR spectra using a composition space model, described in Materials and Methods, to derive a 1D distribution of both lipid and protein density. Figure 4B shows the distribution in terms of percent surface area coverage of the tBLM molecular groups (Au film, tether layer, lipid chain, and head groups) as well as the protein profile. The protein profile was embedded within the head group region of the membrane and extended ∼100 Å from the bilayer. By comparison, the dimensions of RSV Gag in virus-like particles (VLPs) assembled in vitro show the protein to be extended with a dimension of ∼180 Å (43). Thus, RSV Gag is more compact when bound to a membrane than when assembled into immature virus particles.
FIG 4.
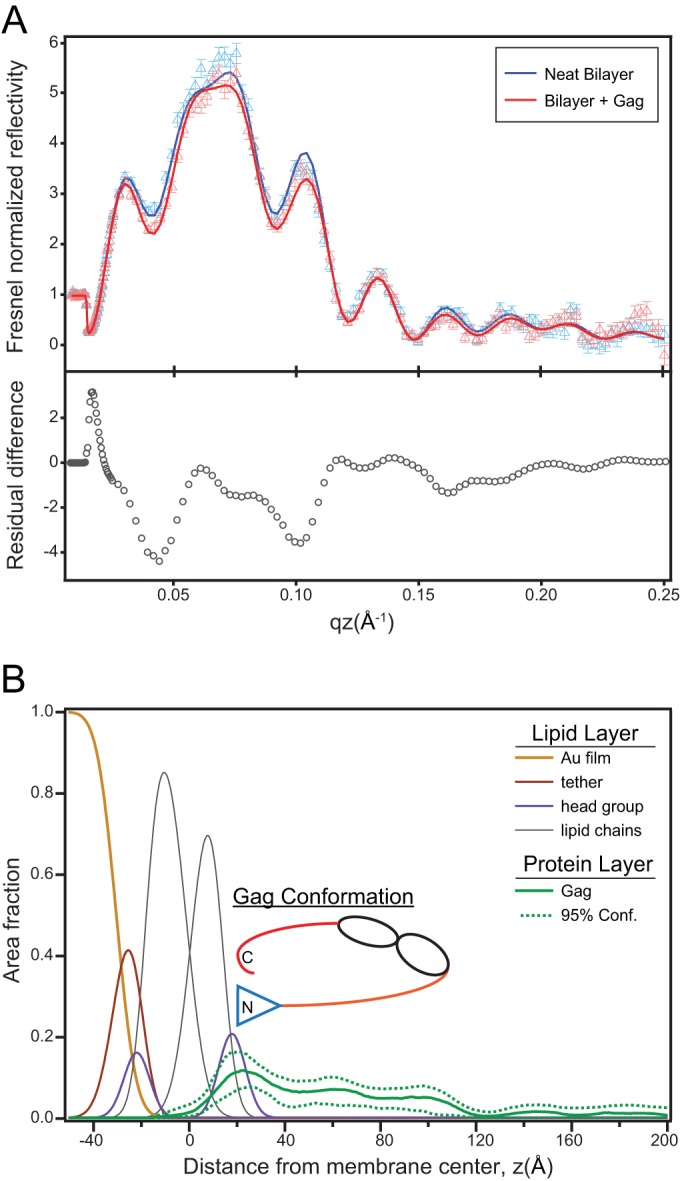
Structural characterization of Gag on a 70:30 mol% DOPC/DOPS membrane by neutron reflectivity. (A) Reflectivity spectra for the neat bilayer and the bilayer after incubation with 0.5 μM Gag in D2O buffer. Symbols represent the experimental data points, and solid lines are the fit to the data resulting in the profile shown in panel B. The bottom panel shows the residuals of the difference between the fit curves. (B) Area fraction profile showing the molecular distribution of the lipid layer and the Gag protein (solid green curve). The 95% confidence (Conf) bands determined by the fitting procedure are also shown. An interpretative cartoon is also included.
Efficient membrane binding requires fully intact Gag protein.
Previous membrane binding studies of retroviral Gag proteins have relied largely on radiochemically labeled but biochemically impure proteins translated in vitro, for example, in reticulocyte extracts (4, 16, 44–47). In contrast, in the present study we purified milligram quantities of assembly-competent Gag, thus allowing biochemical analyses in the absence of contaminating cellular components. We previously reported that liposome association of RSV Gag and HIV-1 Gag, both translated in vitro, is significantly greater than association of the N-terminal MA domain alone (16). We now have extended these findings to the purified system (examples of Gag flotation results are shown in Fig. 5A). Removal of either the N-terminal membrane binding domain (Fig. 1A, ΔMBD) or the C-terminal NC domain (Fig. 1A, ΔNC) nearly abrogated flotation of the proteins with liposomes at the ionic strength used here, 150 mM NaCl (Fig. 5B). Thus, both MA and NC contribute to binding, either directly or indirectly.
FIG 5.
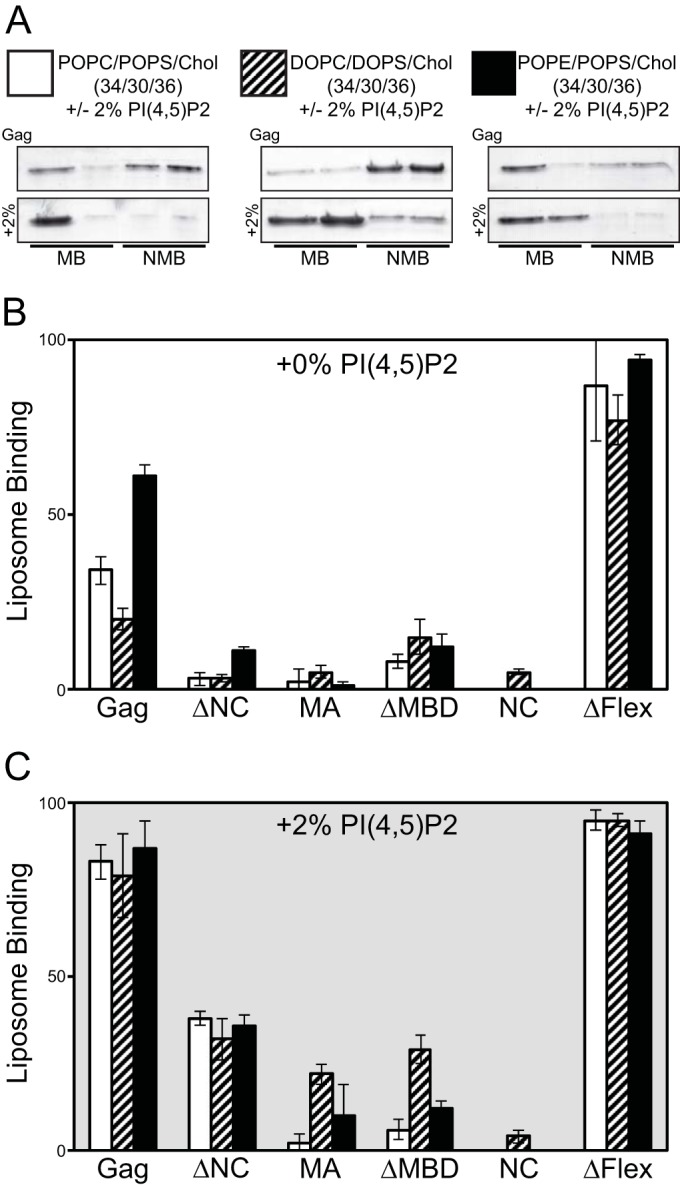
Flotation analysis of purified RSV Gag proteins. Large unilamellar vesicles (LUVs; 100 nm) were prepared with the following compositions, with or without (±) 2% PI(4,5)P2 (34/30/36 and 32/30/36/2, respectively): POPC/POPS/Chol, DOPC/DOPS/Chol, and POPE/POPS/Chol. PC/PS/Chol and PI(4,5)P2 ratios were the same for each acyl chain (DO and PO) and neutral lipid head group (PC and PE). (A) Representative stained gels of Gag flotation results for the six LUV compositions (MB, membrane bound; NMB, nonmembrane bound). Graphs show percentages of total protein associated with liposomes for lipid compositions without (B) and with (C) 2% PI(4,5)P2. For all graphs, bars represent the average of no less than three binding reactions at 150 mM NaCl. Error bars represent standard deviations from the means.
Full-length RSV Gag bound significantly better to liposomes containing the neutral lipid palmitoyl-oleoyl-phosphoethanolamine (POPE) than to liposomes containing the neutral lipid palmitoyl-oleoyl-phosphatidylcholine (POPC) or DOPC (Fig. 5B). Since in cellular membranes phosphoethanolamine (PE) is most abundant in the inner leaflet of the plasma membrane (PM) (48), we take this result as preliminary evidence that PE may play a role in the preference of RSV Gag for the PM. Surprisingly, the removal of the internal, putatively unstructured segment of Gag, in the protein ΔFlex, increased flotation to nearly 100% for all liposome compositions tested (Fig. 5B). This result suggests that the flexible domain between MA and CA somehow negatively regulates membrane interaction.
The minor PM inner leaflet signaling phospholipid phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2] has been shown to enhance binding of diverse retroviral Gag proteins to liposomes in vitro and also to enhance Gag-PM interaction and budding in vivo (4, 16, 46, 47, 49, 50). Addition of 2% PI(4,5)P2, a percentage approximating the amount of PI(4,5)P2 on the inner leaflet of the PM (51), to liposomes containing 30% phosphatidylserine (PS) resulted in a doubling of the amount of membrane-associated Gag (Fig. 5B, bottom panel). PI(4,5)P2 also increased the binding of ΔNC to liposomes from ∼5% to ∼40%, but notably it did not significantly enhance the binding of ΔMBD or NC. We interpret this result to suggest that the NC domain does not play a role in PI(4,5)P2 recognition by Gag. ΔFlex binding was nearly saturated in the absence of PI(4,5)P2, making it impossible to determine if PI(4,5)P2 enhances ΔFlex binding under these conditions. The addition of PI(4,5)P2 more than doubled the binding of MA to liposomes containing phospholipids with unsaturated dioleoyl (DO) acyl chains but did not significantly enhance the binding of MA to liposomes with the mixed acyl chain lipids POPC/palmitoyl-oleoyl-phosphatidylserine (POPS) or POPE/POPS (one saturated and one unsaturated acyl chain per lipid).
Acyl chain saturation does not influence RSV Gag membrane binding.
Previously we showed that HIV-1 Gag is highly sensitive to the saturation state of the acyl chains of lipids used to form liposomes (47). Liposomes composed of high-melting-temperature (H-Tm) lipids, i.e., carrying acyl chains with no double bonds, supported significantly less binding than liposomes composed of low-Tm (L-Tm) lipids, i.e., carrying unsaturated acyl chains. In addition, HIV-1 Gag sensitivity to H-Tm lipids appeared to depend only on the saturation state of the negatively charged PS lipid, not on that of the neutral lipid present in the same liposomes.
HIV Gag binds to membranes composed of the unsaturated lipid dioleoyl (DO; 18:1/18:1), which contains one double bond in each acyl chain, significantly better than it binds to membranes composed of the hybrid lipid palmitoyl-oleoyl (PO; 16:0/18:1), which contains one double bond in the SN-2 acyl chain (46). In contrast, RSV Gag showed no preference for membranes composed of DO or PO lipids (Fig. 6). The addition of 2% PI(4,5)P2 increased binding of RSV Gag to PO- and DO-based membranes from 3.3% to 54% and from 13% to 44%, respectively (Fig. 6A). Cholesterol (Chol) had a similar effect, increasing binding to 28% and 25% for PO and DO membranes, respectively. Increasing PS from 30% to 50% had a similar effect on RSV Gag membrane association for both PO and DO compositions (Fig. 6B). Membrane binding probed by the pelleting assay typically yielded less binding than what was observed by flotation (Fig. 5) for the same compositions. However, the trends observed were consistent for both pelleting and flotation. We previously reported that reticulocyte-generated HIV Gag membrane association favors membranes composed of DO lipids over membranes composed of PO lipids (46). Results presented here demonstrate that purified RSV Gag shows no such preference.
FIG 6.
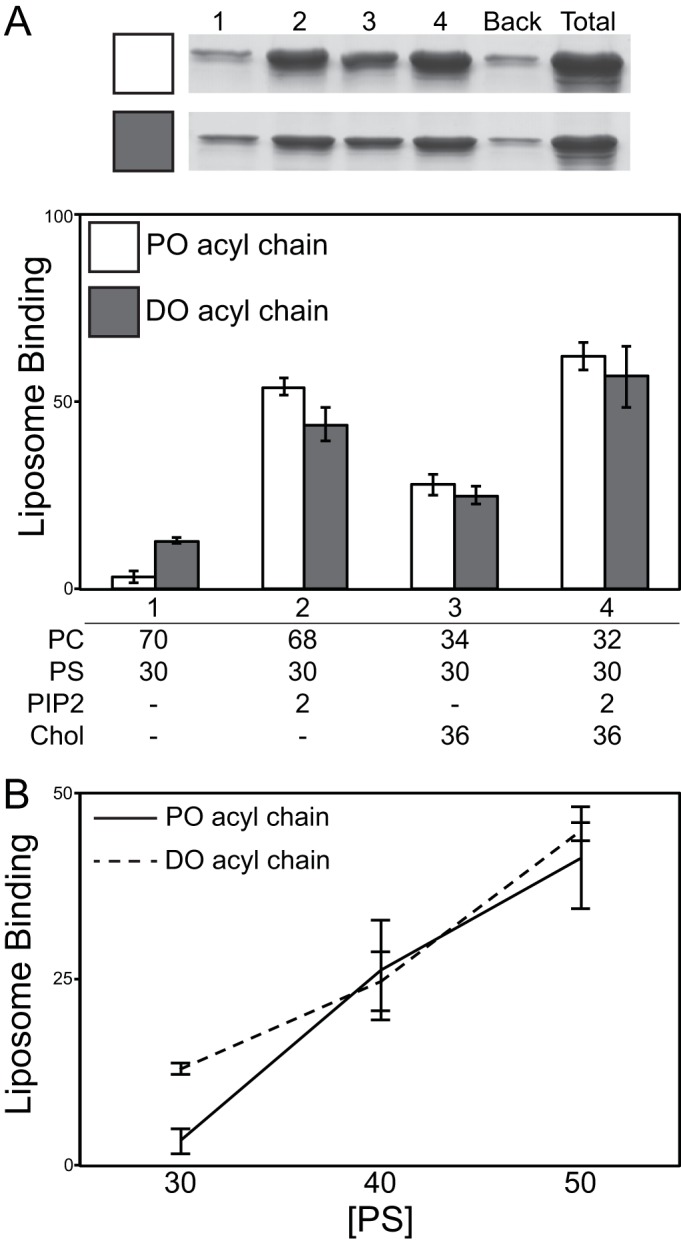
Gag and electrostatic sensor protein response to physiological concentrations of PI(4,5)P2 and cholesterol. (A) An example of stained gels for Gag pelleting results is shown at the top. Numbered lanes 1 to 4 correspond to bar graphs, with lipid compositions listed below the bar graphs. Back, background protein (pelleted protein in the absence of liposomes); Total, total protein. Effects of PI(4,5)P2 (PIP2) and cholesterol in POPC/POPS and DOPC/DOPS lipid mixtures on Gag membrane interaction are shown in the bar graph. (B) Percent of total Gag bound to LUVs with increasing PS concentration for PO acyl chain and DO acyl chain membranes.
To further probe the effects of acyl chain saturation on RSV Gag-liposome binding, we prepared liposomes composed of either H-Tm or L-Tm lipids. Based on known phase diagrams (52–54) and GUV analysis (data not shown), all of the lipid compositions yield membranes without phase separation (reviewed in reference 15). ESR measurements show that increasing the percentage of lipids with saturated acyl chains increased the order parameter, consistent with previously published results (46) (Fig. 7). The inclusion of PS in membranes (compare POPC/Chol, 60/40, to POPC/POPS/Chol, 30/30/40) had no significant effect on the order parameter. Liposomes with H-Tm phosphatidylcholine (PC) and L-Tm PS (DSPC/DOPS/Chol, 30/30/40) supported as much binding as liposomes with L-Tm PC and H-Tm PS (DOPC/DPPS/Chol, 30/30/40) (Fig. 7), in distinct contrast to the results of similar experiments with HIV-1 Gag translated in vitro. These two compositions supported approximately 20% more binding than hybrid acyl chain (POPC/POPS/Chol, 30/30/40) and unsaturated acyl chain (DOPC/DOPS/Chol, 30/30/40) membranes (Fig. 7). We also tested RSV Gag binding to liposomes with phospholipids carrying only fully saturated acyl chains, in which the membrane thus was highly ordered. In the presence of 40% cholesterol and 30% PS, ordered liposomes prepared with DSPC/DPPS/Chol supported levels of Gag binding similar to those of disordered liposomes prepared with DOPC/DOPS/Chol or POPC/POPS/Chol. It is unclear why RSV Gag does not show a strong preference for acyl chain composition, while HIV-1 Gag interacts more favorably with liposomes containing unsaturated PS. We speculate that the N-terminal myristoyl group inserts more efficiently into membranes that contain unsaturated lipids.
FIG 7.
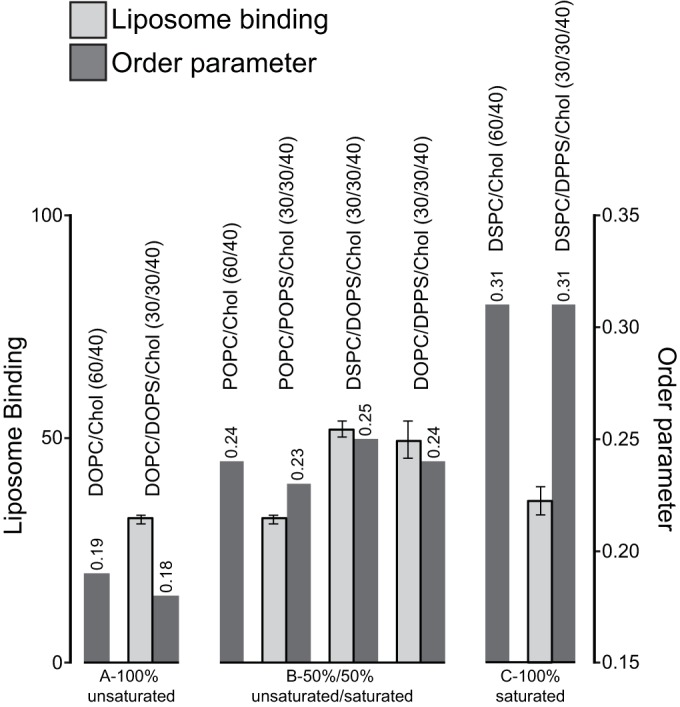
Effect of acyl chain saturation on Gag association with LUVs. Liposome binding (left axis) and membrane order parameter (right axis; values listed above bars) are shown. Membrane compositions are listed above the corresponding bars. Data are grouped into sets of saturated or unsaturated lipid acyl chains, as indicated below the graph.
DISCUSSION
The unavailability of substantial quantities of purified retroviral Gag proteins has limited progress in understanding the mechanisms underlying virus assembly and budding. Unmyristoylated HIV-1 Gag is readily purified after E. coli expression, and its biochemical properties have been studied extensively (8, 10, 55). Similarly, unmyristoylated murine leukemia virus (MLV) Gag also has been purified (56). However, the N-terminal fatty acylation of these and most other Gag proteins is essential for virus assembly and budding at the plasma membrane, making unmodified Gag less suitable for studying these parts of the life cycle. While E. coli can be engineered to add myristate to proteins that start with the canonical sequence Met-Gly, at least for HIV-1 Gag this fatty acylation renders the protein poorly soluble (57). Radiochemically pure HIV-1 Gag and other Gag proteins can be prepared by in vitro translation in reticulocyte or wheat germ systems (4), but the protein then is in a vast excess of cellular proteins, making many types of biochemical experiments difficult to interpret. RSV Gag is unusual among retroviruses in that its N terminus is naturally unmyristoylated. While RSV Gag is modified by an acetyl group on the initiating methionine residue (58), this modification is not essential for budding (59). We decided to purify and study the properties of this Gag protein (without the C-terminal protease domain, which is not needed for assembly) (60), with the aim of illuminating features of Gag that may also be applicable to other retroviruses. We found that this Gag protein is biologically active, being able to assemble in vitro into VLPs in the presence of nucleic acids. However, this reaction is less efficient and robust than that with RSV Gag lacking the membrane binding domain, ΔMBD, as determined by qualitative observation of multiple assembly reactions.
Our SEC and SAXS measurements lead to the firm conclusion that RSV Gag in solution is elongated and highly flexible. By comparison, unmyristoylated MLV Gag also is extended (56), but unmyristoylated HIV-1 Gag, while it is flexible, takes on compact conformations in solution (10, 55). Surprisingly, the longest stretch of RSV Gag predicted to be unstructured, about 117 residues including 18 Gly residues between the MBD and the C-terminal part of p10, can be deleted without abrogating the ability of the protein to assemble in vitro (Fig. 1C). Deletion of some of these residues is reported to be compatible with assembly and infectivity in cells (61). The only sequences with known functions in this part of RSV Gag are the two late domains that interact with the ESCRT machinery, PPPY (62–64) and LYPSL (62). Retroviral late domains are known to be located in unstructured regions (65, 66). Other biological functions of this long unstructured stretch of RSV Gag remain to be uncovered.
In contrast to its longest dimension in solution (∼190 Å), RSV Gag bound to anionic membranes extends only ∼100 Å from the surface of the bilayer. This is also different from RSV Gag assembled in immature virions, where its radial dimension is ∼180 Å (43). RSV Gag contains several disordered linkers, and the Kratky analysis of SAXS data (Fig. 3C) does indeed reveal the protein to be extremely flexible in solution. It is very likely that RSV Gag by virtue of its flexibility can sample many conformations, including compact or bent ones, and that interactions with the anionic membrane enrich compact forms. Under conditions where electrostatic interactions dominate, this could be mediated by simultaneous interactions between the anionic membrane and the basic MA and NC domains. Such a mode of interaction would be facilitated by the high degree of inherent flexibility in the molecule. These results suggest the importance of molecular context in determining Gag conformation. We previously demonstrated that membrane-bound HIV-1 Gag, in the presence of single-stranded nucleic acid, adopted only extended structures, congruent with dimensions found in the immature virion (10). Thus, the conformation of HIV-1 Gag can be modulated by the association of the MA domain and the NC domain with their preferred binding partners, lipids and nucleic acid, respectively. Similarly, RSV Gag organization may further depend on membrane composition, surface charge density, or nucleic acid interaction in order to form extended well-ordered arrays on the membrane surface.
A surprising result from our studies is that removing the NC domain of RSV Gag greatly attenuates the protein's ability to bind to acidic liposomes (Fig. 5). While Gag binds efficiently to liposomes at physiological ionic strengths (150 mM NaCl), Gag lacking either the MBD or NC shows significant binding only at a much lower ionic strength (50 mM NaCl) (16, 32; unpublished results), highlighting the strong electrostatic component of protein-membrane interaction at physiological ionic strength. Two models that are not mutually exclusive could account for these differences. In the first, both MA and NC domains contact the lipid bilayer. RSV MA (67) and other retrovirus MA proteins (67) all have a basic patch and are known to bind acidic bilayers. Published reports are difficult to compare because of the lack of uniformity in ionic strength, which is a critical parameter. For RSV MA, the reported binding constant to liposomes with 30% PS is in the millimolar range, measured at 75 mM NaCl (32), and for myristoylated HIV-1 MA, the value is similar (68). Since NC also is basic, it might be expected to interact with an acidic lipid bilayer, and for HIV-1 this interaction has been studied (10, 69). Simultaneous binding of NC and MBD would be expected to increase the apparent liposome affinity of Gag. According to the second model, the increased affinity of Gag for liposomes, compared with that of mutants lacking NC, is due to the known ability of NC to stimulate multimerization. Although it has been established that NC-mediated multimerization is based on NC binding to nucleic acids, thereby bringing Gag molecules into contact with each other (2), binding of Gag to membranes could also bring Gag molecules together and thus promote Gag-Gag interactions. Artificially induced multimerization of MA has been demonstrated experimentally to increase membrane affinity, both in vitro and in cells (16, 32, 68, 70).
We interpret the neutron reflectometry (NR) results to suggest that at the low ionic strength used for these experiments, the first model applies. Unfortunately, the surface-bound fraction of protein at physiological ionic strengths was below the sensitivity of NR, and hence the biological relevance of the NR results, as well as those previously published for unmyristoylated HIV-1 Gag (10), remains to be established. However, to account for the liposome binding experiments at physiological ionic strengths, we favor the second model, i.e., that Gag multimerizes on the two-dimensional sheet of a membrane. To support this model, it will be necessary to adduce direct evidence for multimerization on liposome membranes, independent of nucleic acid.
The mechanisms by which Gag proteins interact with membranes are poorly understood. HIV-1 Gag from an in vitro translation system can sense both the hydrophilic and the hydrophobic parts of the bilayer (47). For example, in the background of certain lipid compositions, the presence of cholesterol and also the saturation state of acyl chains can have major effects on binding. We have now for the first time carried out such studies with a purified Gag protein. We found that RSV Gag strongly prefers to bind to liposomes in which PE is the neutral lipid, as opposed to the more typically studied PC. The mechanistic explanation of this preference remains to be established, but it is tempting to interpret the preference to reflect the biological need of Gag to interact with the inner leaflet of the plasma membrane, which is highly enriched in PE compared with other biomembranes (48). But unlike HIV-1 Gag, RSV Gag appears to be indifferent to the nature of the acyl chains, binding equally well to liposomes with saturated and unsaturated chains. We hypothesize that this difference is due to the absence of the myristate fatty acyl modification on RSV Gag.
Taken together, our results demonstrate differences in the properties of retroviral Gag proteins, both in solution and in their membrane interaction behavior. The results also highlight the fact that retroviral Gag-membrane interaction is a complex process involving electrostatics, avidity, and hydrophobic forces. To fully account for Gag-membrane interactions, it will be necessary to address the effects of lipid head group, acyl chain, and cholesterol on binding. To that end, more quantitative assessments of the strength of membrane binding will be needed.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01GM107013 (to V.M.V.), NIH grant RO1GM101647 to Mathias Lösche, the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, the Intramural AIDS Targeted Antiviral Program (to A.R. and Y.-X.W.), and in part by the U.S. Department of Commerce (70NANB13H009).
We acknowledge Patrick Clark for critical discussions and suggestions. We thank the National Biomedical Center for Advanced ESR Technology at Cornell University and Siddarth Chandrasekaran and Milka Doktorova for help with ESR data collection and analysis.
REFERENCES
- 1.Sundquist WI, Krausslich HG. 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med 2:a006924. doi: 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bush DL, Vogt VM. 2014. In vitro assembly of retroviruses. Annu Rev Virol 1:561–580. doi: 10.1146/annurev-virology-031413-085427. [DOI] [PubMed] [Google Scholar]
- 3.Schur FK, Hagen WJ, Rumlova M, Ruml T, Muller B, Krausslich HG, Briggs JA. 2015. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature 517:505–508. doi: 10.1038/nature13838. [DOI] [PubMed] [Google Scholar]
- 4.Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A. 2008. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient Gag membrane binding. J Virol 82:2405–2417. doi: 10.1128/JVI.01614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Carroll IP, Soheilian F, Kamata A, Nagashima K, Rein A. 2013. Elements in HIV-1 Gag contributing to virus particle assembly. Virus Res 171:341–345. doi: 10.1016/j.virusres.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ono A, Freed EO. 1999. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J Virol 73:4136–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Carroll IP, Crist RM, Mirro J, Harvin D, Soheilian F, Kamata A, Nagashima K, Rein A. 2012. Functional redundancy in HIV-1 viral particle assembly. J Virol 86:12991–12996. doi: 10.1128/JVI.06287-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Datta SA, Curtis JE, Ratcliff W, Clark PK, Crist RM, Lebowitz J, Krueger S, Rein A. 2007. Conformation of the HIV-1 Gag protein in solution. J Mol Biol 365:812–824. doi: 10.1016/j.jmb.2006.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell S, Fisher R, Towler E, Fox S, Issaq H, Wolfe T, Phillips L, Rein A. 2001. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc Natl Acad Sci U S A 98:10875–10879. doi: 10.1073/pnas.191224698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Datta SA, Heinrich F, Raghunandan S, Krueger S, Curtis JE, Rein A, Nanda H. 2011. HIV-1 Gag extension: conformational changes require simultaneous interaction with membrane and Nucleic acid. J Mol Biol 406:205–214. doi: 10.1016/j.jmb.2010.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell S, Vogt VM. 1997. In vitro assembly of virus-like particles with Rous sarcoma virus Gag deletion mutants: identification of the p10 domain as a morphological determinant in the formation of spherical particles. J Virol 71:4425–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malakhov MP, Mattern MR, Malakhova OA, Drinker M, Weeks SD, Butt TR. 2004. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics 5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- 13.Campbell S, Vogt VM. 1995. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J Virol 69:6487–6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buboltz JT, Feigenson GW. 1999. A novel strategy for the preparation of liposomes: rapid solvent exchange. Biochim Biophys Acta 1417:232–245. doi: 10.1016/S0005-2736(99)00006-1. [DOI] [PubMed] [Google Scholar]
- 15.Dick RA, Vogt VM. 2014. Membrane interaction of retroviral Gag proteins. Front Microbiol 5:187. doi: 10.3389/fmicb.2014.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dick RA, Kamynina E, Vogt VM. 2013. Effect of multimerization on membrane association of Rous sarcoma virus and HIV-1 matrix domain proteins. J Virol 87:13598–13608. doi: 10.1128/JVI.01659-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kingsley PB, Feigenson GW. 1979. The synthesis of a perdeuterated phospholipid −1,2-dimyristoyl-sn-glycero-3-phosphocholine-D72. Chem Phys Lipids 24:135–147. doi: 10.1016/0009-3084(79)90083-5. [DOI] [Google Scholar]
- 18.Schorn K, Marsh D. 1997. Lipid mixing in dimyristoyl phosphatidylcholine-dimyristoyl glycerol dispersions: spin label ESR studies. Biochim Biophys Acta 1323:57–64. doi: 10.1016/S0005-2736(96)00175-7. [DOI] [PubMed] [Google Scholar]
- 19.Morales-Penningston NF, Wu J, Farkas ER, Goh SL, Konyakhina TM, Zheng JY, Webb WW, Feigenson GW. 2010. GUV preparation and imaging: minimizing artifacts. Biochim Biophys Acta 1798:1324–1332. doi: 10.1016/j.bbamem.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shekhar P, Nanda H, Losche M, Heinrich F. 2011. Continuous distribution model for the investigation of complex molecular architectures near interfaces with scattering techniques. J Appl Phys 110:102216–10221612. doi: 10.1063/1.3661986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinrich F, Ng T, Vanderah DJ, Shekhar P, Mihailescu M, Nanda H, Losche M. 2009. A new lipid anchor for sparsely tethered bilayer lipid membranes. Langmuir 25:4219–4229. doi: 10.1021/la8033275. [DOI] [PubMed] [Google Scholar]
- 22.Siegel LM, Monty KJ. 1966. Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim Biophys Acta 112:346–362. [DOI] [PubMed] [Google Scholar]
- 23.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36:1277–1282. doi: 10.1107/S0021889803012779. [DOI] [Google Scholar]
- 24.Svergun DI. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 25:495–503. doi: 10.1107/S0021889892001663. [DOI] [Google Scholar]
- 25.Fischer H, Neto MD, Napolitano HB, Polikarpov I, Craievich AF. 2010. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J Appl Crystallogr 43:101–109. doi: 10.1107/S0021889809043076. [DOI] [Google Scholar]
- 26.Ma YM, Vogt VM. 2002. Rous sarcoma virus Gag protein-oligonucleotide interaction suggests a critical role for protein dimer formation in assembly. J Virol 76:5452–5462. doi: 10.1128/JVI.76.11.5452-5462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma YM, Vogt VM. 2004. Nucleic acid binding-induced Gag dimerization in the assembly of Rous sarcoma virus particles in vitro. J Virol 78:52–60. doi: 10.1128/JVI.78.1.52-60.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bush DL, Monroe EB, Bedwell GJ, Prevelige PE, Phillips JM, Vogt VM. 2014. Higher-order structure of the Rous sarcoma virus SP assembly domain. J Virol 88:5617–5629. doi: 10.1128/JVI.02659-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phillips JM, Murray PS, Murray D, Vogt VM. 2008. A molecular switch required for retrovirus assembly participates in the hexagonal immature lattice. EMBO J 27:1411–1420. doi: 10.1038/emboj.2008.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelle TD, Wills JW. 1996. A large region within the Rous sarcoma virus matrix protein is dispensable for budding and infectivity. J Virol 70:2269–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Callahan E, Wills J. 2000. Repositioning basic residues in the M domain of the Rous sarcoma virus gag protein. J Virol 74:11222–11229. doi: 10.1128/JVI.74.23.11222-11229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalton A, Murray P, Murray D, Vogt V. 2005. Biochemical characterization of Rous sarcoma virus MA protein interaction with membranes. J Virol 79:6227–6238. doi: 10.1128/JVI.79.10.6227-6238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Putnam CD, Hammel M, Hura GL, Tainer JA. 2007. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys 40:191–285. [DOI] [PubMed] [Google Scholar]
- 34.Durand D, Vives C, Cannella D, Perez J, Pebay-Peyroula E, Vachette P, Fieschi F. 2010. NADPH oxidase activator p67(phox) behaves in solution as a multidomain protein with semi-flexible linkers. J Struct Biol 169:45–53. doi: 10.1016/j.jsb.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 35.Glatter O, Kratky O. 1982. Small angle X-ray scattering. Academic Press, London, United Kingdom. [Google Scholar]
- 36.Akiyama S, Takahashi S, Kimura T, Ishimori K, Morishima I, Nishikawa Y, Fujisawa T. 2002. Conformational landscape of cytochrome c folding studied by microsecond-resolved small-angle X-ray scattering. Proc Natl Acad Sci U S A 99:1329–1334. doi: 10.1073/pnas.012458999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Receveur-Brechot V, Durand D. 2012. How random are intrinsically disordered proteins? A small-angle scattering perspective. Curr Protein Pept Sci 13:55–75. doi: 10.2174/138920312799277901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sterckx YG, Volkov AN, Vranken WF, Kragelj J, Jensen MR, Buts L, Garcia-Pino A, Jove T, Van Melderen L, Blackledge M, van Nuland NA, Loris R. 2014. Small-angle X-ray scattering- and nuclear magnetic resonance-derived conformational ensemble of the highly flexible antitoxin PaaA2. Structure 22:854–865. doi: 10.1016/j.str.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 39.McDonnell JM, Fushman D, Cahill SM, Zhou W, Wolven A, Wilson CB, Nelle TD, Resh MD, Wills J, Cowburn D. 1998. Solution structure and dynamics of the bioactive retroviral M domain from Rous sarcoma virus. J Mol Biol 279:921–928. doi: 10.1006/jmbi.1998.1788. [DOI] [PubMed] [Google Scholar]
- 40.Moncoq K, Broutin I, Craescu CT, Vachette P, Ducruix A, Durand D. 2004. SAXS study of the PIR domain from the Grb14 molecular adaptor: a natively unfolded protein with a transient structure primer? Biophys J 87:4056–4064. doi: 10.1529/biophysj.104.048645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Receveur V, Czjzek M, Schulein M, Panine P, Henrissat B. 2002. Dimension, shape, and conformational flexibility of a two domain fungal cellulase in solution probed by small angle X-ray scattering. J Biol Chem 277:40887–40892. doi: 10.1074/jbc.M205404200. [DOI] [PubMed] [Google Scholar]
- 42.Graille M, Cladiere L, Durand D, Lecointe F, Gadelle D, Quevillon-Cheruel S, Vachette P, Forterre P, van Tilbeurgh H. 2008. Crystal structure of an intact type II DNA topoisomerase: insights into DNA transfer mechanisms. Structure 16:360–370. doi: 10.1016/j.str.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 43.Ako-Adjei D, Johnson MC, Vogt VM. 2005. The retroviral capsid domain dictates virion size, morphology, and coassembly of Gag into virus-like particles. J Virol 79:13463–13472. doi: 10.1128/JVI.79.21.13463-13472.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chukkapalli V, Oh SJ, Ono A. 2010. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci U S A 107:1600–1605. doi: 10.1073/pnas.0908661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inlora J, Chukkapalli V, Derse D, Ono A. 2011. Gag localization and virus-like particle release mediated by the matrix domain of human T-lymphotropic virus type-1 Gag are less dependent on phosphatidylinositol-(4,5)-bisphosphate than those mediated by the matrix domain of human immunodeficiency virus type-1 Gag. J Virol 85:3802–3810. doi: 10.1128/JVI.02383-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan J, Dick RA, Vogt VM. 2011. Rous sarcoma virus gag has no specific requirement for phosphatidylinositol-(4,5)-bisphosphate for plasma membrane association in vivo or for liposome interaction in vitro. J Virol 85:10851–10860. doi: 10.1128/JVI.00760-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dick RA, Goh SL, Feigenson GW, Vogt VM. 2012. HIV-1 Gag protein can sense the cholesterol and acyl chain environment in model membranes. Proc Natl Acad Sci U S A 109:18761–18766. doi: 10.1073/pnas.1209408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Meer G, Voelker DR, Feigenson GW. 2008. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol-(4,5)-bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A 101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nadaraia-Hoke S, Bann DV, Lochmann TL, Gudleski-O'Regan N, Parent LJ. 2013. Alterations in the MA and NC domains modulate phosphoinositide-dependent plasma membrane localization of the Rous sarcoma virus Gag protein. J Virol 87:3609–3615. doi: 10.1128/JVI.03059-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McLaughlin S, Wang J, Gambhir A, Murray D. 2002. PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct 31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 52.Heberle FA, Wu J, Goh SL, Petruzielo RS, Feigenson GW. 2010. Comparison of three ternary lipid bilayer mixtures: FRET and ESR reveal nanodomains. Biophys J 99:3309–3318. doi: 10.1016/j.bpj.2010.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Konyakhina TM, Wu J, Mastroianni JD, Heberle FA, Feigenson GW. 2013. Phase diagram of a 4-component lipid mixture: DSPC/DOPC/POPC/chol. Biochim Biophys Acta 1828:2204–2214. doi: 10.1016/j.bbamem.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao J, Wu J, Heberle FA, Mills TT, Klawitter P, Huang G, Costanza G, Feigenson GW. 2007. Phase studies of model biomembranes: complex behavior of DSPC/DOPC/cholesterol. Biochim Biophys Acta 1768:2764–2776. doi: 10.1016/j.bbamem.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Datta SA, Rein A. 2009. Preparation of recombinant HIV-1 gag protein and assembly of virus-like particles in vitro. Methods Mol Biol 485:197–208. doi: 10.1007/978-1-59745-170-3_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Datta SA, Zuo X, Clark PK, Campbell SJ, Wang YX, Rein A. 2011. Solution properties of murine leukemia virus gag protein: differences from HIV-1 gag. J Virol 85:12733–12741. doi: 10.1128/JVI.05889-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carlson LA, Hurley JH. 2012. In vitro reconstitution of the ordered assembly of the endosomal sorting complex required for transport at membrane-bound HIV-1 Gag clusters. Proc Natl Acad Sci U S A 109:16928–16933. doi: 10.1073/pnas.1211759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Palmiter RD, Gagnon J, Vogt VM, Ripley S, Eisenman RN. 1978. The NH2-terminal sequence of the avian oncovirus gag precursor polyprotein (Pr76gag). Virology 91:423–433. doi: 10.1016/0042-6822(78)90388-4. [DOI] [PubMed] [Google Scholar]
- 59.Parent LJ, Cairns TM, Albert JA, Wilson CB, Wills JW, Craven RC. 2000. RNA dimerization defect in a Rous sarcoma virus matrix mutant. J Virol 74:164–172. doi: 10.1128/JVI.74.1.164-172.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson MC, Scobie HM, Vogt VM. 2001. PR domain of Rous sarcoma virus Gag causes an assembly/budding defect in insect cells. J Virol 75:4407–4412. doi: 10.1128/JVI.75.9.4407-4412.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Joshi SM, Vogt VM. 2000. Role of the Rous sarcoma virus p10 domain in shape determination of gag virus-like particles assembled in vitro and within Escherichia coli. J Virol 74:10260–10268. doi: 10.1128/JVI.74.21.10260-10268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dilley KA, Gregory D, Johnson MC, Vogt VM. 2010. An LYPSL late domain in the gag protein contributes to the efficient release and replication of Rous sarcoma virus. J Virol 84:6276–6287. doi: 10.1128/JVI.00238-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wills JW, Cameron CE, Wilson CB, Xiang Y, Bennett RP, Leis J. 1994. An assembly domain of the Rous sarcoma virus Gag protein required late in budding. J Virol 68:6605–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parent LJ, Bennett RP, Craven RC, Nelle TD, Krishna NK, Bowzard JB, Wilson CB, Puffer BA, Montelaro RC, Wills JW. 1995. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J Virol 69:5455–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fossen T, Wray V, Bruns K, Rachmat J, Henklein P, Tessmer U, Maczurek A, Klinger P, Schubert U. 2005. Solution structure of the human immunodeficiency virus type 1 p6 protein. J Biol Chem 280:42515–42527. doi: 10.1074/jbc.M507375200. [DOI] [PubMed] [Google Scholar]
- 66.Kyere SK, Joseph PRB, Summers MF. 2008. The p12 domain is unstructured in a murine leukemia virus p12-CAN Gag construct. PLoS One 3:e1902. doi: 10.1371/journal.pone.0001902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murray P, Li Z, Wang J, Tang C, Honig B, Murray D. 2005. Retroviral matrix domains share electrostatic homology: models for membrane binding function throughout the viral life cycle. Structure 13:1521–1531. doi: 10.1016/j.str.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 68.Dalton AK, Ako-Adjei D, Murray PS, Murray D, Vogt VM. 2007. Electrostatic interactions drive membrane association of the human immunodeficiency virus type 1 Gag MA domain. J Virol 81:6434–6445. doi: 10.1128/JVI.02757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kempf N, Postupalenko V, Bora S, Didier P, Arntz Y, de Rocquigny H, Mely Y. 2015. The HIV-1 nucleocapsid protein recruits negatively charged lipids to ensure its optimal binding to lipid membranes. J Virol 89:1756–1767. doi: 10.1128/JVI.02931-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Keller H, Krausslich HG, Schwille P. 2013. Multimerizable HIV Gag derivative binds to the liquid-disordered phase in model membranes. Cell Microbiol 15:237–247. doi: 10.1111/cmi.12064. [DOI] [PubMed] [Google Scholar]
- 71.Shur FKM, Dick RA, Hagen WJH, Vogt VM, Briggs JAG. 2015. The structure of immature virus-like Rous sarcoma virus Gag particles reveals a structural role for the p10 domain in assembly. J Virol 89:10294–10302. doi: 10.1128/JVI.01502-15. [DOI] [PMC free article] [PubMed] [Google Scholar]