

ABSTRACT
The human papillomavirus (HPV) capsid is composed of the major capsid protein L1 and the minor capsid protein L2. During entry, the HPV capsid undergoes numerous conformational changes that result in endosomal uptake and subsequent trafficking of the L2 protein in complex with the viral DNA to the trans-Golgi network. To facilitate this transport, the L2 protein harbors a number of putative motifs that, if capable of direct interaction, would interact with cytosolic host cell factors. These data imply that a portion of L2 becomes cytosolic during infection. Using a low concentration of digitonin to selectively permeabilize the plasma membrane of infected cells, we mapped the topography of the L2 protein during infection. We observed that epitopes within amino acid residues 64 to 81 and 163 to 170 and a C-terminal tag of HPV16 L2 are exposed on the cytosolic side of intracellular membranes, whereas an epitope within residues 20 to 38, which are upstream of a putative transmembrane region, is luminal. Corroborating these findings, we also found that L2 protein is sensitive to trypsin digestion during infection. These data demonstrate that the majority of the L2 protein becomes accessible on the cytosolic side of intracellular membranes in order to interact with cytosolic factors to facilitate vesicular trafficking.
IMPORTANCE In order to complete infectious entry, nonenveloped viruses have to pass cellular membranes. This is often achieved through the viral capsid protein associating with or integrating into intracellular membrane. Here, we determine the topography of HPV L2 protein in the endocytic vesicular compartment, suggesting that L2 becomes a transmembrane protein with a short luminal portion and with the majority facing the cytosolic side for interaction with host cell transport factors.
INTRODUCTION
Human papillomaviruses (HPVs) are nonenveloped DNA tumor viruses that infect the skin and mucosal epithelial cells. Infection by HPVs can induce hyper-proliferative lesions of the tissues, whereupon high-risk HPVs can progress to malignant tumors. Of the high-risk HPVs, 70% of cervical cancer can be attributed to HPVs 16 and 18 (1). These HPVs are associated with a variety of anogenital and oral carcinomas. Despite the success of the prophylactic vaccine against these high-risk HPVs, HPV-associated cancers will continue to be a global health burden in the future due to low vaccination rates and lack of therapeutic effect (2). Therefore, uncovering the conserved steps of how HPVs establish infection can be used to find antiviral targets that may be less virus restrictive than the current vaccines.
The HPV capsid is composed of 360 molecules of the major capsid protein, L1, and up to 72 molecules of the minor capsid protein, L2 (3–6). Much of our advancement in understanding HPV entry can be attributed to the efficient generation of pseudovirions. With respect to cellular entry, pseudovirions are considered to be indistinguishable from native virions but encapsidate a reporter plasmid (pseudogenome) in place of the HPV genome (7–9). By using these pseudovirions as a model to study HPV entry, significant progress has been made to understand how HPVs utilize distinct cellular pathways and rely on numerous host cell factors to gain access to the nucleus during a pseudoviral infection.
HPV virions initially bind to extracellular matrix or the cell surface by engaging the L1 protein with heparan sulfate proteoglycan (HSPG) (10–14). This engagement triggers specific conformational changes that affect both L1 and L2, whereupon host cell cyclophilin B, a chaperone protein localized on the cell surface and in the luminal compartments, facilitates the exposure of the N terminus of L2 (15–17). Furin convertase then cleaves off the first 12 amino acids of the L2 protein (18, 19). Next, the virion associates with tetraspanin-enriched microdomains (TEMs), which may serve as an entry platform in a process described as micropinocytosis (20–23). Furthermore, TEMs are enriched in proteins implicated as secondary uptake receptors (21–27). Once the virion is in the endosome, acidification triggers disassembly of the viral capsid, and host cell cyclophilins facilitate the dissociation of the majority of L1 protein molecules from the L2 protein, which remains in complex with the viral DNA (16, 28). The L1 protein is then degraded, and the L2/DNA complex is sorted out of the late endosomes, with L2/DNA subsequently transported to the trans-Golgi network (TGN) by the retromer complex (28–30). Additionally, this trafficking event requires γ-secretase activity, the specific substrate of which, however, has yet to be determined (31–33). Last, the incoming L2/DNA complex requires nuclear envelope breakdown during mitosis to enter the nucleus, where it will associate with promyelocytic leukemia protein (PML) nuclear bodies (34–36).
The HPV L2 protein harbors two putative transmembrane (TM)-like domains located on the N terminus (amino acid [aa] residues 45 to 67) and the C terminus (aa 445 to 467). Recent data from Bronnimann et al. have demonstrated that the N-terminal putative TM domain can function as a TM domain in a cellular system by tethering a fusion protein to the plasma membrane (37). Kämper et al. identified a membrane-destabilizing peptide at the C termini of HPV16 and HPV33 which is essential for efficient infection and inserts into membranes when it is fused to marker proteins like green fluorescent protein (GFP) (38).
The contributions of numerous groups suggest that cytosolic factors are important for papillomavirus infection by interacting with the L2 protein. Bossis et al. demonstrated that syntaxin 18, a retrograde trafficking fusion protein, binds to bovine papillomavirus type 1 (BPV1) L2 just upstream (aa 45 to 47) of the N-terminal TM-like domain (39). Bergant et al. have shown that sorting nexin 17 (SNX17), a retrograde sorting protein that binds the cytoplasmic tails of cargo, interacts with HPV16 L2 (16L2) residues 254 to 257 (40, 41). In addition, Florin et al. and Schneider et al. have reported that the dynein light chains DYNLT1 and DYNLT3 interact with the 40 C-terminal residues of HPV16 and HPV33 L2 (42, 43). Recently, Lipovsky et al. and Popa et al. demonstrated that L2 protein directly interacts with components of the retromer complex via motifs at the very C terminus of the L2 protein during infectious entry (residues 446 to 448 and 452 to 455) (30, 44). Each of these host cell factors, if capable of direct interaction, would recognize motifs on the cytosolic side of the membrane, thus implying that the L2 protein is accessible within the cytoplasm. While the reported evidence suggests that portions of L2 protein penetrate the endocytic membrane and become accessible to cytosolic factors, no attempt was made to determine the topography of the L2 protein during intracellular trafficking. Here, we utilized a low concentration of digitonin to selectively permeabilize plasma membranes to determine the accessibility of HPV16 L2 epitopes to epitope-mapped monoclonal antibodies and confirmed the findings by biochemical approaches.
MATERIALS AND METHODS
Generation of HPV16 and HPV18 pseudovirus.
Pseudovirus carrying an encapsidated pfwB plasmid was generated in the 293TT cell line as previously described using expression plasmids pShell16L1L2HA-3′ for HPV16 and puf18L1 and puf18L2 for HPV18 (3, 8). The pfwB plasmid was a kind gift from John Schiller, and codon-optimized L1 and L2 expression plasmids have been described previously (45). Mutant HPV18 pseudovirions harboring 18L2-R295/8A has been described previously (28). Virions were characterized by L1- and L2-specific Western blotting using monoclonal antibodies 312F and 33L2-1, respectively (46). The viral DNA was isolated using a NucleoSpin Blood QuickPure system (740569.250; Macherey-Nagel) supplemented with 4 μM EDTA and dithiothreitol (DTT). The genome copy number was quantified using real-time PCR. On average, pseudovirus preparations in our hands have a particle-to-infectivity ratio of 1:100 to 1:300. For pseudogenome detection by immunofluorescence (IF), the pseudogenome was labeled by supplementing the growth medium with 100 μM 5-ethynyl-2′-deoxyuridine (EdU) at 6 h posttransfection, as previously described, during generation of pseudovirus (47).
Cell lines.
293TT and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. HaCaT cells were grown in low-glucose DMEM containing 5% FBS and antibiotics.
Antibodies.
HPV16 L1-specific mouse monoclonal antibodies (MAbs) 312F and 33L1-7 were used for the specific detection of HPV16 L1 protein (48). HPV16 L2 protein was detected using MAb 33L2-1 (46) as well as 16L2-K4-18-30 and 16L2-K1-64-81 (49). The 33L2-1 epitope has been mapped to amino acid residues 163 to 170, whereas K4 and K1 have not yet been mapped, but the antibodies were generated against peptides with HPV16 L2 residues 18 to 30 and 64 to 81, respectively. MAbs 18L2-28F, 18L2-104E, 18L2-412F, and K4 were used to detect the HPV18 L2 protein. MAb 28F has not been epitope mapped but was generated against a glutathione S-transferase (GST) fusion protein comprising HPV18 L2 residues 81 to 254. Peroxidase-conjugated AffiniPure goat-anti mouse and anti-rabbit antibodies were used for the Western blot analyses (Jackson ImmunoResearch). The endoplasmic reticulum (ER) was stained using mouse MAb anti-calnexin (MAB3126; Chemicon). The lysosome was detected using rabbit polyclonal antibody (pAb) anti-Lamp1 (L1418; Sigma). The trans-Golgi network was detected using sheep pAb anti-TGN46 (AHP500GT; Serotec), pAb rabbit anti-TGN46 (PA5-23068; Thermo Scientific Pierce), and MAb mouse anti-p230 (611280; BD Biosciences). BiP protein was detected using pAb rabbit anti-BiP (ab21685; Abcam). Rat polyclonal anti-hemagglutinin (HA) (3F10; Roche) was used in the in vivo neutralization experiment. A Click-iT EdU imaging kit and Alexa Fluor (AF)-labeled secondary antibodies were used in immunofluorescence microscopy (C10338; Invitrogen).
IF microscopy.
HaCaT cells were grown on coverslips for 24 h to approximately 50% confluence and infected with EdU-labeled HPV16 or HPV18 at approximately 1 × 106 viral genome equivalents (vge) per coverslip. At 24 h postinfection (hpi), the cells were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature, washed with phosphate-buffered saline (PBS), pH 7.5, permeabilized with 0.5% Triton X-100 in PBS for 10 min, washed, and blocked with 5% normal goat serum for 30 min; the Click-iT reaction mixture containing Alexa Fluor 555 was used for the detection of the pseudogenome (47). After cells were washed, they were incubated with primary antibodies in 2.5% goat serum for 1 h at 37°C in a humidified chamber. After an extensive washing, cells were incubated with Alexa Fluor-tagged secondary antibodies for 1 h. We extensively washed cells again in PBS and mounted them with ProLong Gold and SlowFade Antifade containing 4′,6′-diamidino-2-phenylindole (DAPI) (P36931; Invitrogen). Single-slice images were acquired with a Leica TCS SP5 spectral confocal microscope.
Immunofluorescence microscopy after selective permeabilization.
HaCaT cells were grown on coverslips for 24 h to approximately 50% confluence and infected with EdU-labeled HPV16 or HPV18 at approximately 1 × 106 vge per coverslip. At 18 hpi (see Fig. 3E, F, G, and H) or 24 hpi (see Fig. 2, 4, and 6), the cells were fixed with 4% PFA for 15 min at room temperature, and washed with PBS (pH 7.5); they were then selectively permeabilized with 5 μg/ml digitonin in PBS for 10 min, washed, and blocked with 5% normal goat serum for 30 min. After being washed, cells were incubated with primary antibodies in 2.5% goat serum for 1 h at 37°C in a humidified chamber. Note that the staining shown in Fig. 3E to H and 4G and H was performed after the cells were treated with the Click-iT reaction mixture for 30 min at room temperature to denature the capsids, as previously described (16). After cells were washed extensively, they were incubated with Alexa Fluor-tagged secondary antibodies for 1 h, followed by another extensive wash in PBS. Then, we performed sequential staining for anti-calnexin for 1 h at 37°C. This was followed by extensive washing and incubation with the sequential secondary antibody for 1 h at 37°C. After washing off excess secondary antibody, we fixed the cells once more in 4% PFA in PBS. Next, we completely permeabilized the cells in 0.5% Triton X-100 for 10 min, followed by treatment with the Click-iT reaction mixture for 30 min using Alexa Fluor 555 to detect the total pseudogenome present. The coverslips were washed once more and mounted with 4′,6-diamidino-2-phenylindole. Single-slice images were acquired with a Leica TCS SP5 spectral confocal microscope. Images were quantified by determining the ratio of the signal strength in arbitrary units of L2 relative to that of EdU in intracellularly localized puncta (n = 50 to 100 puncta) (see Fig. 7A). Results are representative of data from two independent biological replicates.
FIG 3.

Low concentration of digitonin selectively permeabilizes the plasma membrane. (A and B) HaCaT cells were grown for 24 h and then fixed and permeabilized with either 1 mg/ml (A) or 5 μg/ml (B) digitonin. Lamp1, cytosol-facing late endosome marker Lamp1; calnexin (Calnx), luminal endoplasmic reticulum marker; DAPI, nuclear marker. (C and D) HaCaT cells were grown for 24 h and then fixed and permeabilized with either 0.5% Triton X-100 or 5 μg/ml digitonin. p230, cytosol-facing TGN marker; TGN46, luminal TGN marker; DAPI, nuclear marker. (E to H) HaCaT cells were infected with HPV16 pseudovirus for 18 h in the presence of 1 μM bafilomycin A1. Cells were fixed, permeabilized using 5 μg/ml digitonin, and stained using L2-specific antibodies (K4, K1, and 33L2-1) or an L1-specific antibody (33L1-7) following the Click-iT reaction. Colors are coded as indicated by the labels on the figure.
FIG 2.

Detection of L2 protein during infection. HaCaT cells were infected with HPV16 pseudovirus for 24 h and analyzed by immunofluorescence microscopy. The L2 protein (detected by K4, K1, or 33L2-1), EdU-labeled pseudogenome, TGN marker TGN46, and nucleus (DAPI) were stained following a Click-iT reaction. Intracellular L2 was detected using MAb K4 (aa 20 to 38) (A), MAb K1 (aa 64 to 81) (D), and MAb 33L2-1 (aa 163 to 170) (G). (B, E, and H) Colocalization was confirmed by analyzing the line profile of individual EdU puncta. (C, F, and I) Mock-infected HaCaT cells served as controls to assess the amount of background signal from each of the MAbs used. Arrowheads indicate the tri-color (white) colocalization of the L2-specific antibodies with TGN46 and EdU signals.
FIG 4.

Regions of 16L2 downstream of the N-terminal putative transmembrane domain become accessible in selectively permeabilized cells. HaCaT cells were infected for 24 h with HPV16 pseudovirus. Cells were fixed and permeabilized using 0.5% Triton X-100 (A, C, E, and G) or 5 μg/ml digitonin (B, D, F, and H) and stained using L2-specific MAbs (K4, K1, and 33L2-1) or an L1-specific MAb (33L1-7). The ER was stained using MAb against the luminal ER marker calnexin (Calnx). Cells were fixed again and permeabilized using 0.5% Triton X-100, followed by treatment with the Click-iT reaction mixture to detect EdU. Note that only the L2-specific MAb K4, but not K1 and 33L2-1 MAbs, fails to detect the L2 protein adjacent to the nucleus, probably residing in the TGN (arrows), under selective permeabilization. (G and H) As a control, we also did not detect 33L1-7 staining following the Click-iT reaction adjacent to the nucleus under selective permeabilization. Colors are coded as indicated by the labels on the figure.
FIG 6.

Regions of 18L2 downstream of the N-terminal putative transmembrane domain become accessible in selectively permeabilized cells. HaCaT cells were infected for 24 h with HPV18 pseudovirus. Cells were fixed and permeabilized using 0.5% Triton X-100 (A and C) or 5 μg/ml digitonin (B and D) and stained using L2-specific MAbs (K4 and 28F). The ER was stained using MAb against the luminal ER marker calnexin (Calnx). Cells were fixed again and permeabilized using 0.5% Triton X-100, followed by treatment with the Click-iT reaction mixture to detect EdU. Colors are coded as indicated by the labels on the figure. Note that only the L2-specific MAb K4 and not 28F fails to detect the L2 protein adjacent to the nucleus, probably residing in the TGN (arrows), under selective permeabilization.
FIG 7.

The majority of the L2 protein becomes exposed within the cytoplasm during infectious entry. (A) Cytoplasm-localized puncta were selected from the experiments shown in both Fig. 4 (PSV16, HPV16 pseudovirus) and 6 (PSV18, HPV18 pseudovirus) and quantified from infected HaCaT cells treated with either 0.5% Triton X-100 (TX) or 5 μg/ml digitonin (DIG). The ratio of L2 to EdU was determined by measuring the signal strength of each channel. (B) Histograms of signal strengths for all channels from select representative puncta from the experiments shown in both Fig. 4 and 6. Note the lack of L2-specific MAb K4 signal under selective permeabilization. (C) HeLa cells were infected with 18L2-R295/8A-harboring pseudovirions for 18 h. Cells were harvested, and lysates were prepared by mechanically disrupting the plasma membranes. Whole-cell extracts were treated with or without 1 μM bafilomycin A1 and/or 0.2% trypsin and/or Triton X-100 for 1 h. Western blotting was performed, and the L2 protein was detected using a cocktail of 18L2-specific MAbs. Detection of BiP protein served as an internal control. The protein levels were quantified by measuring the pixel intensity of each band using densitometry relative to their respective control lanes. Note that the L2 protein is sensitive to trypsin, regardless of the addition of 0.5% Triton X-100. ns, not significant.
Microinjection.
293TT cells were grown on coverslips for 24 h and infected with wild-type (wt) 16L2- or 16L2-HA-3′-harboring pseudovirions for 72 h. Cells were microinjected with purified monoclonal rat anti-HA antibody in vivo at 2 hpi using an InjectMan NI 2 micromanipulator connected to the FemtoJet, both from Eppendorf. Infectivity was scored by counting the number of GFP-expressing cells by immunofluorescence. Quantification is based on a single experiment (n > 100 cells).
Trypsin digestion.
HeLa cells were infected with 18L2-R295/8A-harboring pseudovirions for 18 h at 37°C with or without 1 μM bafilomycin A1 (BafA1). Cells were then washed with high-pH phosphate buffer (pH 10.25) and harvested using 0.25% trypsin, with trypsin deactivated with 10% FBS in DMEM and then aspirated. Prior to lysis, cells were again incubated with 0.025% trypsin in PBS for 1 h at 37°C, with trypsin deactivated as before; this step was performed to digest away any remaining pseudovirus on the cell surface. Cell lysis was performed by resuspending cells in hypotonic buffer (20 mM Tris-Cl, 10 mM EDTA, pH 8.5), incubating them on ice for 15 min, and then mechanically lysing cells via passage through a 25-5/8-gauge needle syringe 20 times. Next, whole-cell extracts were treated with or without 0.038% trypsin and/or 0.5% Triton X-100 for 1 h at 37°C. Trypsin was inactivated using soybean trypsin inhibitor (T6522; Sigma), and the samples were boiled for 10 min under reducing conditions (5% beta-mercaptoethanol). Presence of L2 protein was detected via Western blotting, using a cocktail of the 18L2-specific MAbs 28F, 104E, and 412F (1:200 dilution each). The protein levels were quantified by measuring the pixel intensity of each band using ImageJ.
RESULTS
Antibodies recognize epitopes up- and downstream of the putative N-terminal TM regions of HPV16 and HPV18 L2 proteins.
We sought to determine the topography of the L2 protein in the TGN compartment following infectious entry using an immunofluorescence microscopy approach combined with selective permeabilization of the plasma membrane. We utilized an array of epitope-mapped mouse monoclonal antibodies flanking the N-terminal putative TM domains of the HPV16 and HPV18 L2 proteins (Fig. 1A). Here, we used MAbs 16L2-K4-18-30 (K4), 16L2-K1-64-81 (K1), and 33L2-1 as well as MAb 18L2-28F (28F). The K4 epitope is directly upstream of the N-terminal TM domain; the K1 and 33L2-1 epitopes are downstream of the TM domain. K1 and K4 recognize epitopes that are identical in HPV16 and HPV18. 33L2-1 was generated using HPV33 L2; however, this MAb is cross-reactive with HPV16. MAb 28F has not been epitope-mapped but was generated against a GST fusion protein comprising HPV18 L2 residues 81 to 254. To test if these antibodies recognize their respective L2 proteins, we performed Western blot analysis on purified HPV16 and HPV18 pseudovirions. We readily detected HPV16 L2 protein at approximately 75 kDa with K1 and K4 (Fig. 1B). The use of 33L2-1 in Western blotting and immunofluorescence has been described elsewhere (16, 50). We were also able to detect the HPV18 L2 protein using K1, K4, and 28F (Fig. 1B).
FIG 1.
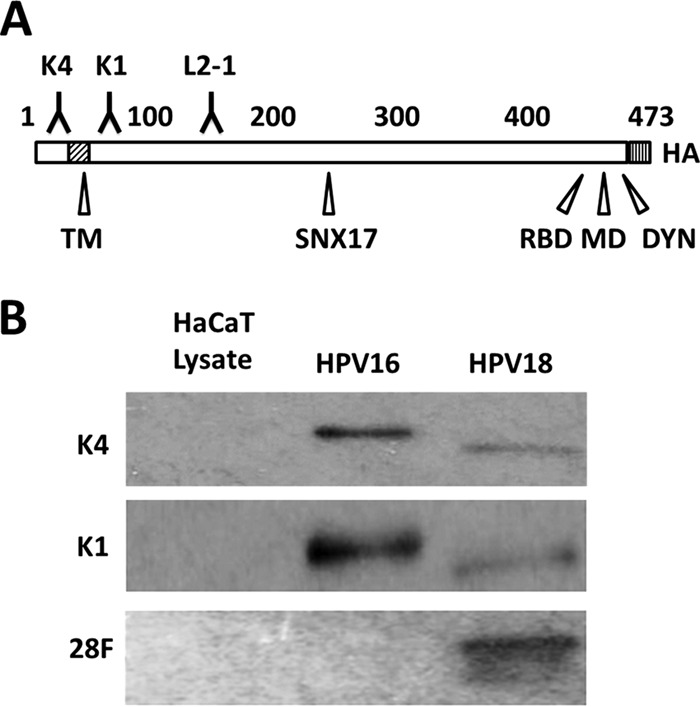
Characterization of L2-specific monoclonal antibodies. (A) Schematic of the L2 protein with labeled key features. Numbers refer to amino acid positions of HPV16 L2: K4, MAb K4 epitope (aa 20 to 38); TM, N-terminal putative transmembrane region (aa 45 to 67); K1, MAb K1 epitope (aa 64 to 81); L2-1, MAb 33L2-1 epitope (aa 163 to 170); SNX17, sorting nexin 17 binding domain (aa 245 to 257); RBD, retromer binding domain (aa 446 to 455); MD, membrane-destabilizing domain (aa 454 to 473); DYN, dynein binding domain (aa 456 to 461); HA, C-terminal HA epitope. (B) Western blot using L2-specific antibodies (listed on the left side) to detect L2 protein from purified HPV16 and HPV18 pseudoviruses. To exclude cross-reactivity, HaCaT whole-cell extracts were run as controls.
In order to test whether these selected antibodies can recognize TGN-localized L2 protein during an infection, HaCaT cells were infected with EdU-labeled HPV16 pseudovirus for 24 h. The cells were fixed and permeabilized using 0.5% Triton X-100, and L2 protein was stained using MAbs K1, K4, and 33L2-1. The TGN was also stained, and reactivity of the L2 protein within the TGN was observed. We found that all three antibodies tested recognized intracellular L2 protein localized within and around the TGN (Fig. 2A, D, and G). Representative puncta were selected to illustrate colocalization based on the signal strength intensities (Fig. 2B, E, and H). While K4 shares a similar epitope with MAb RG-1, unlike RG-1, K4 can be used to recognize intracellular L2 protein after the capsid has undergone disassembly. We note that we observed L2-only signal that does not colocalize with the EdU signal (Fig. 2A, D, and G). This is likely due to the presence of DNA-free particles present in our preparation. These L2-only puncta are mostly observed distally away from the nucleus of infected cells and not present in the mock-infected HaCaT cells (Fig. 2C, F, and I).
Digitonin permeabilizes the plasma membrane but not the endoplasmic reticulum and the TGN.
In order to determine the topography of intracellular L2 protein, we needed to selectively permeabilize the plasma membrane without disrupting intracellular compartments. Digitonin forms pores in cholesterol-rich membranes such as the plasma membrane, whereas cholesterol-poor membranes such as the endoplasmic reticulum are not affected. Low concentrations of digitonin have been used before to achieve this goal (51–53). Here, we fixed uninfected HaCaT cells, permeabilized with a high concentration (100 μg/ml) and low concentration (5 μg/ml) of digitonin. We then stained cells using polyclonal antibodies recognizing a Lamp1 epitope facing the cytosol and an anti-calnexin MAb. Calnexin is a luminal marker for the endoplasmic reticulum (ER) and has been used in the past to assess differential permeabilization. The mechanism of action for digitonin requires binding to cholesterol-rich membranes, whereupon digitonin oligomerizes and forms a pore complex (51). The plasma membrane and ER were permeabilized under the high concentration of digitonin (Fig. 3A). Under a low concentration of digitonin, only the plasma membrane, not the ER, was permeabilized, as seen by the positive staining for Lamp1 and lack of calnexin staining (Fig. 3B). To confirm that the TGN is also not permeabilized by digitonin, we repeated the experiment using antibodies raised against TGN-resident proteins (TGN46 and p230) that recognize epitopes predicted to face the luminal and cytosolic surfaces, respectively. Indeed, the antibody directed against a luminal epitope did not stain the TGN at low digitonin concentrations, whereas the antibody directed to a cytosolic domain strongly stained the TGN (Fig. 3D). Both antibodies were reactive with their respective TGN-resident proteins in cells permeabilized with Triton X-100 (Fig. 3C).
Next, we tested whether our antibodies can detect L2 protein within the early endosomes of selectively permeabilized cells. We infected HaCaT cells with EdU-labeled HPV16 pseudovirus for 18 h in the presence of 1 μM bafilomycin A1 (BafA1), which interrupts trafficking of early endosomes (54–56). Cells were fixed and permeabilized with a low concentration of digitonin. We performed the Click-iT reaction prior to staining to denature the capsid proteins and to allow the antibodies access to their respective epitopes. We observed that K4, K1, and 33L2-1 colocalized with the viral DNA under selective permeabilization (Fig. 3E to G). We do not find it surprising that the virus-containing endocytic vesicles are permeabilized under our conditions as the composition of these vesicles may be similar to that of the cholesterol-rich plasma membrane. We confirmed that the positive staining is mostly due to permeabilization of the early endosomes by digitonin rather than by L2 penetrating the endosomal membrane, based on detection of L1 protein in endosomes in BafA1-treated cells (Fig. 3H). We conclude that MAbs K4, K1, and 33L2-1 are able to detect L2 protein in selectively permeabilized cells when it is trapped in early endosomes by the use of bafilomycin A1.
The N-terminal 40 amino acids of TGN-resident L2 protein remain inaccessible under selective permeabilization.
Having established selective permeabilization and identified antibodies that recognize specific regions up- and downstream of the putative N-terminal TM-like domain of L2 protein for use in immunofluorescence studies, we were able to test the topography of the L2 protein during infection. HaCaT cells were infected with EdU-labeled HPV16 pseudovirus for 24 h. The HaCaT cells were fixed and selectively permeabilized with 5 μg/ml of digitonin or permeabilized using 0.5% Triton X-100 and stained with the MAbs K4, K1, and 33L2-1. Next, the cells were fixed again and permeabilized with 0.5% Triton X-100, and the viral DNA was detected using a Click-iT reaction. Under total permeabilization, K4 was able to detect L2 protein colocalized with the viral DNA in an intracellular compartment (Fig. 4A). However, K4 failed to detect the L2 protein under selective permeabilization (Fig. 4B), whereas both K1 and 33L2-1 were able to detect intracellular L2 protein in close proximity to the nucleus colocalized with the viral DNA, comparable to results in Triton X-100-permeabilized cells (Fig. 4C to F). We did observe some K4 staining colocalized with EdU signal localized distally from the nucleus, which probably corresponds to particles still residing in the early endosomes or on the cell surface. Since the epitopes for both K1 and 33L2-1 are downstream of the N-terminal TM-like domain, whereas the epitope of K4 is located upstream of the N-terminal TM-like domain, these data suggest that regions C-terminal to the TM-like domain are accessible on the cytosolic face of the membrane compartment, while the very N terminus is inaccessible under selective permeabilization. We also stained selectively permeabilized cells using L1-specific antibodies. It was recently shown that a small fraction of L1 colocalizes with L2 and viral genome in the TGN (28). Similar to results with the K4 staining, L1 was detected in the perinuclear region only under total, but not selective, permeabilization (Fig. 4G and H), confirming the validity of our approach.
Available HA tag antibodies do not stain well in IF and could thus not be used to determine the accessibility of the very C terminus of the HA-tagged versions of L2. However, we used microinjection of HA tag antibodies to determine whether HA-specific antibodies can block HPV16 infection. HEK 293TT cells were infected with HPV pseudovirions harboring an HA tag at the L2 C terminus (Fig. 5). Cells were microinjected with purified HA tag-specific antibody at 2 hpi, and GFP-expressing cells were counted at 72 hpi. HPV16 pseudovirions carrying an untagged L2 protein served as controls. Infection was impaired by 70% in HA-tagged pseudovirus compared to 24% in wt HPV16. Taken together, these data imply that the majority of L2 protein downstream of the putative TM domain, probably starting at residue 65 but definitely encompassing residues 65 to 81, 163 to 170, and the very C terminus, is facing the cytosolic side of intracellular membranes and that only a short N-terminal region, comprising residues 13 to 45, is on the luminal side of membranes during infectious entry.
FIG 5.

Microinjection of anti-HA antibody neutralizes infection in vivo. 293TT cells were infected with 16L2-expressing pseudovirus (PsV16 wt) or C-terminally tagged 16L2-HA-3′-expressing pseudovirus (HA) for 72 h. Cells were microinjected with purified MAb anti-HA antibody in vivo at 2 hpi using a Leica mechanical manipulator. Infectivity was quantified by counting the number of GFP-expressing cells (n > 100 cells each).
The N-terminal epitope of HPV18 also is inaccessible during infection under selective permeabilization.
In addition to determining the topography of HPV16 L2, we also tested the topography of HPV18 L2 protein under selective permeabilization using MAbs K4 and 28F. As demonstrated in the experiment shown in Fig. 1B, both MAbs can recognize the HPV18 L2 protein. 28F is not epitope mapped but was generated using a GST fusion protein encompassing HPV18 L2 residues 81 to 254. We used this antibody to recognize the epitope downstream of the HPV18 N-terminal TM-like domain. HaCaT cells were infected with EdU-labeled HPV18 pseudovirus for 24 h. Next, the cells were fixed and either permeabilized with 0.5% Triton X-100 or selectively permeabilized using 5 μg/ml digitonin. The cells were stained using K4 or 28F, fixed once more, and permeabilized using 0.5% Triton X-100, followed by treatment with the Click-iT reaction mixture. Under total permeabilization by Triton X-100, the K4 antibody was detected colocalized with the viral DNA within the cell (Fig. 6A). However, K4 antibody in digitonin-treated cells displayed no reactivity, indicating that the epitope was not accessible under selective permeabilization (Fig. 6B). In the cells stained using 28F, we observed intracellular staining of Triton X-100-permeabilized cells comparable to that of selectively permeabilized cells using digitonin (Fig. 6C and D). Quantification for these experiments is shown in Fig. 7A. We observed a significant reduction in K4 reactivity under selective permeabilization for both 16L2 and 18L2. We also observed a reduction in reactivity of 28F for 18L2 under selective permeabilization; however, this reduction was not as pronounced as that observed with K4. These data suggest that amino acid residues downstream of the N-terminal TM of HPV18 L2 protein are also accessible within the cytosol while the N terminus remains inaccessible.
The HPV16 L2 protein is sensitive to trypsin digestion following infection.
Our above analysis does not exclude the possibility that only a subfraction of L2 molecules penetrates intracellular membranes. Therefore, we wanted to quantify the percentages of L2 protein molecules that are accessible under our experimental conditions. For this quantification, we selected intracellular puncta that displayed colocalization of EdU and 33L2-1 signals and compared the signal strength ratios under Triton X-100 or low-concentration digitonin conditions. We found that the ratios of signal strength of L2/EdU under both conditions were comparable (Fig. 7A). Graphical representation of the signal intensities of MAbs K4, 33L2-1, and 28F compared to the intensities of EdU signals taken from representative EdU-positive puncta in cells permeabilized with Triton X-100 and digitonin is included in Fig. 7B. These data suggest that the majority of L2 molecules in the cytoplasmic space are accessible on the cytoplasmic side of the membrane. To further support these results, we next tested the sensitivity of the L2 protein to trypsin digestion at 18 hpi. To this end, HeLa cells were infected with HPV18 pseudovirus harboring 18L2-R295/8A for 18 h in the presence or absence of bafilomycin A1. We chose to use this mutant rather than wild-type HPV18 because it preferentially localizes within the TGN (28). Cell surface-resident particles were removed prior to harvest of cells by a short wash with an alkaline buffer (17). We performed a mechanical disruption of the plasma membrane by passing the whole cells through a small-gauge syringe. This results in plasma membrane disruption without disruption of the endosomes and other intracellular membrane compartments (57). Extracts were subsequently treated with trypsin to digest L2 protein facing the cytosolic side of the membranes. We were able to detect L2 protein in untreated controls. Furthermore, we observed that L2 protein was resistant to trypsin digestion when cells were infected in the presence of bafilomycin A1. Bafilomycin A1 blocks uncoating and retains viral particles in early endosomes (54, 55). In contrast, we observed a strong reduction in the amount of full-length L2 protein after trypsin treatment when cells were infected in the absence of bafilomycin A1 in both the presence and absence of Triton X-100. These data support our previous findings that the majority of the L2 protein molecules are accessible within the cytosol.
DISCUSSION
Here, we describe the topography of the L2 protein of HPV16 and HPV18 during an infection in vivo. We utilized a low concentration of digitonin to selectively permeabilize the plasma membrane while retaining the membrane integrity of other intracellular compartments, such as the ER and TGN. Low concentrations of digitonin have been previously used to map the topography of the HPV16 E5 oncoprotein (52). Using the same concentration of digitonin as previously described, we were able to achieve selective permeabilization of the plasma membrane without penetrating the ER or TGN (Fig. 3A to D). Under these conditions, we mapped the accessibility of antibody epitopes within the L2 protein during infection. We observed that epitopes on the opposite side of a putative amino-terminal TM region showed differential antibody reactivity in intracellularly localized pseudoviral particles (Fig. 4B, D, and F). In contrast, total permeabilization of the plasma membrane under Triton X-100 treatment showed comparable reactivity of antibodies on both sides of the N-terminal TM region (Fig. 4A, C, and E). We observed that the HPV16 L2 protein has amino acid residues, including but not limited to residues 64 to 81 and 163 to 170, exposed on the cytosolic side, whereas the N terminus residues, specifically residues 20 to 38, are luminal during infectious entry. These data support the idea that the L2 protein becomes a transmembrane protein during infectious entry and that the N-terminal TM region indeed separates accessible from nonaccessible epitopes. We note that the L2 protein may be snaking in and out of the intracellular membranes, exposing the identified epitopes on the cytosolic surface of the TGN. However, we find this an unlikely scenario since the L2 protein contains only two domains that have the propensity to become transmembrane, according to prediction software (58).
While data from numerous labs have proposed that portions of the L2 protein may become cytosolic during infection, this was mostly based on indirect evidence. Yeast two-hybrid screens using L2 protein as bait have identified factors involved in intracellular trafficking, like SNX17, which is involved in vesicular trafficking, and DYNLT1 and DYNLT3, components of the dynein motor protein complex (40–43). SNX17 binds to an NPXY motif (residues 254 to 257), which is well conserved in HPV L2 proteins. Dynein requires the approximately 40 C-terminal residues of L2 for interaction. Their involvement in HPV16 infection has been confirmed in the past by mutational as well as knockdown approaches. More recently, pulldown assays showed interaction of the C terminus (residues 446 to 448 and 452 to 455) of the L2 protein with the retromer complex (44), which we here support using microinjection of anti-HA antibodies into 293TT cells infected with C-terminally HA-tagged 16L2-expressing pseudovirions (Fig. 5). Taken together, our mapping of luminal and cytosolic L2 epitopes is consistent with the published literature. The only conflict to our findings may be syntaxin 18, which has been reported to interact with BPV1 L2 residues 45 to 47 (39). However, syntaxin 18 is a transmembrane protein, and the luminal portions might mediate this interaction. Furthermore, we do not see consistent colocalization of HPV16 and syntaxin 18 during infection, suggesting that HPV16 might not depend on this host factor (data not shown).
We were interested in estimating how many of the L2 molecules become cytosolic during infection. By selecting L2/EdU-colocalized intracellular pseudoviral particles and measuring the relative signal intensities, we were able to estimate that the majority of the L2 protein molecules are facing the cytosolic side during infection (Fig. 7A and B). Determining trypsin sensitivity of L2 protein at 18 hpi supported this finding (Fig. 7C). Both analyses suggest that a portion of L2 might not have penetrated the endocytic membranes. However, both analyses have their caveats. The microscopic analysis is confounded by various amounts of L2 molecules per pseudoviral particle and differences in reactivity between the different antibodies used. Use of Triton X-100 as opposed to digitonin might also alter accessibility of epitopes to antibodies other than by disrupting membrane bilayers. Furthermore, the biochemical analysis is complicated by the fact that HPV entry is asynchronous and that, at 18 hpi, some particles are still localizing to early endosomes. Other particles might be directed to lysosomes for degradation prior to uncoating.
The mechanism of how the L2 protein invades the membrane during entry is currently unknown. We can determine that this translocation event would most likely occur within the endosomal compartment after acidification and uncoating have occurred as trafficking to the TGN requires direct interaction with the retromer complex. One possible mechanism that has been proposed before would be that acidification and/or proteolytic processing triggers conformational changes in L2 that result in L2 molecules invading the endosomal membrane. The N-terminal TM domain region or the putative C-terminal membrane-destabilizing region would likely facilitate this event. Then, the L2 molecules may oligomerize within the membrane and form a pore, as previously proposed, whereupon L2 translocates itself to the cytosolic side of the endosomal membrane (37). This mechanism would be similar to that by which diphtheria toxin forms a pore and translocates itself across the endosomal membrane (59, 60). If the C-terminal membrane-destabilizing region is facilitating this invasion, then the putative N-terminal transmembrane region may act as a membrane retention signal, thus anchoring the protein within the endosomal membrane.
Not surprisingly, other nonenveloped DNA viruses also must utilize membrane translocation (61). After endocytic uptake, simian virus 40 (SV40) is trafficked to the ER, whereupon the virion uncoats and undergoes conformational changes leading to the exposure of VP2. It has been shown that VP2 and VP3 can integrate into membranes while only VP2 can perforate the membrane (60, 62). Through the cooperation of SV40 and ER-resident proteins, a partially disassembled SV40 capsid is translocated into the cytoplasm, whereupon the virus facilitates nuclear entry (63, 64). While it is unknown how HPV ultimately escapes into the cytoplasm, both the putative N-terminal transmembrane domain and the C-terminal membrane-destabilizing domain are required for infection. If either of these domains is mutated, nuclear entry is abrogated, thus illustrating the importance of these domains during entry (37, 38). Further research is needed to understand the mechanism of how these domains are involved in the translocation of the L2 protein across the membrane into the cytoplasm.
ACKNOWLEDGMENTS
The project described was supported by R01AI081809 from the National Institute of Allergy and Infectious Diseases to M.S. and in part by grants from the National Institute of General Medical Sciences (P20GM103433). S.D. was supported by a Carroll Feist Predoctoral Fellowship.
REFERENCES
- 1.Crow JM. 2012. HPV: the global burden. Nature 488:S2–S3. doi: 10.1038/488S2a. [DOI] [PubMed] [Google Scholar]
- 2.Forman D, de Martel C, Lacey CJ, Soerjomataram I, Lortet-Tieulent J, Bruni L, Vignat J, Ferlay J, Bray F, Plummer M, Franceschi S. 2012. Global burden of human papillomavirus and related diseases. Vaccine 30(Suppl 5):F12–F23. doi: 10.1016/j.vaccine.2012.07.055. [DOI] [PubMed] [Google Scholar]
- 3.Buck CB, Thompson CD, Pang YY, Lowy DR, Schiller JT. 2005. Maturation of papillomavirus capsids. J Virol 79:2839–2846. doi: 10.1128/JVI.79.5.2839-2846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finch JT, Klug A. 1965. The structure of viruses of the papilloma-polyoma type 3. Structure of rabbit papilloma virus, with an appendix on the topography of contrast in negative-staining for electron-microscopy. J Mol Biol 13:1–12. [DOI] [PubMed] [Google Scholar]
- 5.Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. 2008. Arrangement of L2 within the papillomavirus capsid. J Virol 82:5190–5197. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Modis Y, Trus BL, Harrison SC. 2002. Atomic model of the papillomavirus capsid. EMBO J 21:4754–4762. doi: 10.1093/emboj/cdf494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol Med 119:445–462. [DOI] [PubMed] [Google Scholar]
- 8.Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2004. Efficient intracellular assembly of papillomaviral vectors. J Virol 78:751–757. doi: 10.1128/JVI.78.2.751-757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unckell F, Streeck RE, Sapp M. 1997. Generation and neutralization of pseudovirions of human papillomavirus type 33. J Virol 71:2934–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knappe M, Bodevin S, Selinka HC, Spillmann D, Streeck RE, Chen XS, Lindahl U, Sapp M. 2007. Surface-exposed amino acid residues of HPV16 L1 protein mediating interaction with cell surface heparan sulfate. J Biol Chem 282:27913–27922. doi: 10.1074/jbc.M705127200. [DOI] [PubMed] [Google Scholar]
- 11.Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J Virol 75:1565–1570. doi: 10.1128/JVI.75.3.1565-1570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joyce JG, Tung JS, Przysiecki CT, Cook JC, Lehman ED, Sands JA, Jansen KU, Keller PM. 1999. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J Biol Chem 274:5810–5822. doi: 10.1074/jbc.274.9.5810. [DOI] [PubMed] [Google Scholar]
- 13.Selinka HC, Florin L, Patel HD, Freitag K, Schmidtke M, Makarov VA, Sapp M. 2007. Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces noninfectious uptake of human papillomavirus. J Virol 81:10970–10980. doi: 10.1128/JVI.00998-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Culp TD, Budgeon LR, Christensen ND. 2006. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology 347:147–159. doi: 10.1016/j.virol.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 15.Bienkowska-Haba M, Patel HD, Sapp M. 2009. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog 5:e1000524. doi: 10.1371/journal.ppat.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M. 2012. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J Virol 86:9875–9887. doi: 10.1128/JVI.00980-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards KF, Bienkowska-Haba M, Dasgupta J, Chen XS, Sapp M. 2013. Multiple heparan sulfate binding site engagements are required for the infectious entry of human papillomavirus type 16. J Virol 87:11426–11437. doi: 10.1128/JVI.01721-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards RM, Lowy DR, Schiller JT, Day PM. 2006. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci U S A 103:1522–1527. doi: 10.1073/pnas.0508815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Day PM, Schiller JT. 2009. The role of furin in papillomavirus infection. Future Microbiol 4:1255–1262. doi: 10.2217/fmb.09.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, Schiller JT, Helenius A. 2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog 8:e1002657. doi: 10.1371/journal.ppat.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, Florin L. 2008. Clathrin- and caveolin-independent entry of human papillomavirus type 16: involvement of tetraspanin-enriched microdomains (TEMs). PLoS One 3:e3313. doi: 10.1371/journal.pone.0003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scheffer KD, Gawlitza A, Spoden GA, Zhang XA, Lambert C, Berditchevski F, Florin L. 2013. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J Virol 87:3435–3446. doi: 10.1128/JVI.02906-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheffer KD, Berditchevski F, Florin L. 2014. The tetraspanin CD151 in papillomavirus infection. Viruses 6:893–908. doi: 10.3390/v6020893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abban CY, Meneses PI. 2010. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology 403:1–16. doi: 10.1016/j.virol.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoon CS, Kim KD, Park SN, Cheong SW. 2001. α6 Integrin is the main receptor of human papillomavirus type 16 VLP. Biochem Biophys Res Commun 283:668–673. doi: 10.1006/bbrc.2001.4838. [DOI] [PubMed] [Google Scholar]
- 26.Evander M, Frazer IH, Payne E, Qi YM, Hengst K, McMillan NA. 1997. Identification of the α6 integrin as a candidate receptor for papillomaviruses. J Virol 71:2449–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Surviladze Z, Dziduszko A, Ozbun MA. 2012. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog 8:e1002519. doi: 10.1371/journal.ppat.1002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DiGiuseppe S, Bienkowska-Haba M, Hilbig L, Sapp M. 2014. The nuclear retention signal of HPV16 L2 protein is essential for incoming viral genome to transverse the trans-Golgi network. Virology 458–459:93–105. doi: 10.1016/j.virol.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Day PM, Thompson CD, Schowalter RM, Lowy DR, Schiller JT. 2013. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J Virol 87:3862–3870. doi: 10.1128/JVI.03222-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipovsky A, Popa A, Pimienta G, Wyler M, Bhan A, Kuruvilla L, Guie MA, Poffenberger AC, Nelson CD, Atwood WJ, DiMaio D. 2013. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc Natl Acad Sci U S A 110:7452–7457. doi: 10.1073/pnas.1302164110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Kazakov T, Popa A, DiMaio D. 2014. Vesicular trafficking of incoming human papillomavirus 16 to the Golgi apparatus and endoplasmic reticulum requires gamma-secretase activity. mBio 5(5):e01777-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karanam B, Peng S, Li T, Buck C, Day PM, Roden RB. 2010. Papillomavirus infection requires gamma secretase. J Virol 84:10661–10670. doi: 10.1128/JVI.01081-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang HS, Buck CB, Lambert PF. 2010. Inhibition of gamma secretase blocks HPV infection. Virology 407:391–396. doi: 10.1016/j.virol.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Day PM, Baker CC, Lowy DR, Schiller JT. 2004. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A 101:14252–14257. doi: 10.1073/pnas.0404229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aydin I, Weber S, Snijder B, Samperio Ventayol P, Kuhbacher A, Becker M, Day PM, Schiller JT, Kann M, Pelkmans L, Helenius A, Schelhaas M. 2014. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog 10:e1004162. doi: 10.1371/journal.ppat.1004162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. 2009. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog 5:e1000318. doi: 10.1371/journal.ppat.1000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bronnimann MP, Chapman JA, Park CK, Campos SK. 2013. A transmembrane domain and GxxxG motifs within L2 are essential for papillomavirus infection. J Virol 87:464–473. doi: 10.1128/JVI.01539-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamper N, Day PM, Nowak T, Selinka HC, Florin L, Bolscher J, Hilbig L, Schiller JT, Sapp M. 2006. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J Virol 80:759–768. doi: 10.1128/JVI.80.2.759-768.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bossis I, Roden RBS, Gambhira R, Yang R, Tagaya M, Howley PM, Meneses PI. 2005. Interaction of tSNARE syntaxin 18 with the papillomavirus minor capsid protein mediates infection. J Virol 79:6723–6731. doi: 10.1128/JVI.79.11.6723-6731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bergant Marusic M, Ozbun MA, Campos SK, Myers MP, Banks L. 2012. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 13:455–467. doi: 10.1111/j.1600-0854.2011.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bergant M, Banks L. 2013. SNX17 facilitates infection with diverse papillomavirus types. J Virol 87:1270–1273. doi: 10.1128/JVI.01991-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Florin L, Becker KA, Lambert C, Nowak T, Sapp C, Strand D, Streeck RE, Sapp M. 2006. Identification of a dynein interacting domain in the papillomavirus minor capsid protein l2. J Virol 80:6691–6696. doi: 10.1128/JVI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneider MA, Spoden GA, Florin L, Lambert C. 2011. Identification of the dynein light chains required for human papillomavirus infection. Cell Microbiol 13:32–46. doi: 10.1111/j.1462-5822.2010.01515.x. [DOI] [PubMed] [Google Scholar]
- 44.Popa A, Zhang W, Harrison MS, Goodner K, Kazakov T, Goodwin EC, Lipovsky A, Burd CG, DiMaio D. 2015. Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog 11:e1004699. doi: 10.1371/journal.ppat.1004699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leder C, Kleinschmidt JA, Wiethe C, Muller M. 2001. Enhancement of capsid gene expression: preparing the human papillomavirus type 16 major structural gene L1 for DNA vaccination purposes. J Virol 75:9201–9209. doi: 10.1128/JVI.75.19.9201-9209.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Volpers C, Sapp M, Snijders PJF, Walboomers JMM, Streeck RE. 1995. Conformational and linear epitopes on virus-like particles of human papillomavirus type-33 identified by monoclonal-antibodies to the minor capsid protein L2. J Gen Virol 76:2661–2667. doi: 10.1099/0022-1317-76-11-2661. [DOI] [PubMed] [Google Scholar]
- 47.Ishii Y, Tanaka K, Kondo K, Takeuchi T, Mori S, Kanda T. 2010. Inhibition of nuclear entry of HPV16 pseudovirus-packaged DNA by an anti-HPV16 L2 neutralizing antibody. Virology 406:181–188. doi: 10.1016/j.virol.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 48.Sapp M, Kraus U, Volpers C, Snijders PJ, Walboomers JM, Streeck RE. 1994. Analysis of type-restricted and cross-reactive epitopes on virus-like particles of human papillomavirus type 33 and in infected tissues using monoclonal antibodies to the major capsid protein. J Gen Virol 75:3375–3383. doi: 10.1099/0022-1317-75-12-3375. [DOI] [PubMed] [Google Scholar]
- 49.Rubio I, Seitz H, Canali E, Sehr P, Bolchi A, Tommasino M, Ottonello S, Muller M. 2011. The N-terminal region of the human papillomavirus L2 protein contains overlapping binding sites for neutralizing, cross-neutralizing and non-neutralizing antibodies. Virology 409:348–359. doi: 10.1016/j.virol.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 50.Becker KA, Florin L, Sapp C, Maul GG, Sapp M. 2004. Nuclear localization but not PML protein is required for incorporation of the papillomavirus minor capsid protein L2 into virus-like particles. J Virol 78:1121–1128. doi: 10.1128/JVI.78.3.1121-1128.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulz I. 1990. Permeabilizing cells: some methods and applications for the study of intracellular processes. Methods Enzymol 192:280–300. doi: 10.1016/0076-6879(90)92077-Q. [DOI] [PubMed] [Google Scholar]
- 52.Krawczyk E, Suprynowicz FA, Sudarshan SR, Schlegel R. 2010. Membrane orientation of the human papillomavirus type 16 E5 oncoprotein. J Virol 84:1696–1703. doi: 10.1128/JVI.01968-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Labay V, Weichert RM, Makishima T, Griffith AJ. 2010. Topology of transmembrane channel-like gene 1 protein. Biochemistry 49:8592–8598. doi: 10.1021/bi1004377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bayer N, Schober D, Prchla E, Murphy RF, Blaas D, Fuchs R. 1998. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: implications for viral uncoating and infection. J Virol 72:9645–9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Selinka HC, Giroglou T, Sapp M. 2002. Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions. Virology 299:279–287. doi: 10.1006/viro.2001.1493. [DOI] [PubMed] [Google Scholar]
- 56.Engel S, Heger T, Mancini R, Herzog F, Kartenbeck J, Hayer A, Helenius A. 2011. Role of endosomes in simian virus 40 entry and infection. J Virol 85:4198–4211. doi: 10.1128/JVI.02179-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clarke MS, McNeil PL. 1992. Syringe loading introduces macromolecules into living mammalian cell cytosol. J Cell Sci 102:533–541. [DOI] [PubMed] [Google Scholar]
- 58.Kyte J, Doolittle RF. 1982. A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 59.Sharpe JC, London E. 1999. Diphtheria toxin forms pores of different sizes depending on its concentration in membranes: probable relationship to oligomerization. J Membr Biol 171:209–221. doi: 10.1007/s002329900572. [DOI] [PubMed] [Google Scholar]
- 60.Zalman LS, Wisnieski BJ. 1984. Mechanism of insertion of diphtheria toxin: peptide entry and pore size determinations. Proc Natl Acad Sci U S A 81:3341–3345. doi: 10.1073/pnas.81.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bilkova E, Forstova J, Abrahamyan L. 2014. Coat as a dagger: the use of capsid proteins to perforate membranes during non-enveloped DNA viruses trafficking. Viruses 6:2899–2937. doi: 10.3390/v6072899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daniels R, Rusan NM, Wadsworth P, Hebert DN. 2006. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: implications for DNA translocation out of the ER. Mol Cell 24:955–966. doi: 10.1016/j.molcel.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 63.Inoue T, Tsai B. 2011. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog 7:e1002037. doi: 10.1371/journal.ppat.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rainey-Barger EK, Magnuson B, Tsai B. 2007. A chaperone-activated nonenveloped virus perforates the physiologically relevant endoplasmic reticulum membrane. J Virol 81:12996–13004. doi: 10.1128/JVI.01037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]