

ABSTRACT
Influenza virus hemagglutinin (HA) mediates virus entry by binding to cell surface receptors and fusing the viral and endosomal membranes following uptake by endocytosis. The acidic environment of endosomes triggers a large-scale conformational change in the transmembrane subunit of HA (HA2) involving a loop (B loop)-to-helix transition, which releases the fusion peptide at the HA2 N terminus from an interior pocket within the HA trimer. Subsequent insertion of the fusion peptide into the endosomal membrane initiates fusion. The acid stability of HA is influenced by residues in the fusion peptide, fusion peptide pocket, coiled-coil regions of HA2, and interactions between the surface (HA1) and HA2 subunits, but details are not fully understood and vary among strains. Current evidence suggests that the HA from the circulating pandemic 2009 H1N1 influenza A virus [A(H1N1)pdm09] is less stable than the HAs from other seasonal influenza virus strains. Here we show that residue 205 in HA1 and residue 399 in the B loop of HA2 (residue 72, HA2 numbering) in different monomers of the trimeric A(H1N1)pdm09 HA are involved in functionally important intermolecular interactions and that a conserved histidine in this pair helps regulate HA stability. An arginine-lysine pair at this location destabilizes HA at acidic pH and mediates fusion at a higher pH, while a glutamate-lysine pair enhances HA stability and requires a lower pH to induce fusion. Our findings identify key residues in HA1 and HA2 that interact to help regulate H1N1 HA stability and virus infectivity.
IMPORTANCE Influenza virus hemagglutinin (HA) is the principal antigen in inactivated influenza vaccines and the target of protective antibodies. However, the influenza A virus HA is highly variable, necessitating frequent vaccine changes to match circulating strains. Sequence changes in HA affect not only antigenicity but also HA stability, which has important implications for vaccine production, as well as viral adaptation to hosts. HA from the pandemic 2009 H1N1 influenza A virus is less stable than other recent seasonal influenza virus HAs, but the molecular interactions that contribute to HA stability are not fully understood. Here we identify molecular interactions between specific residues in the surface and transmembrane subunits of HA that help regulate the HA conformational changes needed for HA stability and virus entry. These findings contribute to our understanding of the molecular mechanisms controlling HA function and antigen stability.
INTRODUCTION
The influenza virus envelope protein, hemagglutinin (HA), is organized as a noncovalently associated homotrimer on the viral surface. Each monomer of HA is posttranslationally cleaved into HA1 and HA2 subunits that are disulfide linked. The HA trimer consists of a large membrane-distal, globular domain formed only by HA1 and an elongated membrane-proximal stem domain comprised of HA2 and the N- and C-terminal segments of HA1. HA1 mediates virus binding to cell surface sialic acid receptors to initiate viral entry through endocytosis. The acidic pH in endosomes induces an irreversible, large-scale conformational change in HA2 that mediates the fusion of viral and endosomal membranes and then uncoating (1, 2).
High-resolution structural information is currently available for multiple HA subtypes (3–9). A hinge region referred to as the B loop, which connects two antiparallel α-helical segments of HA2 in the neutral pH conformation, has a high propensity for a helical conformation (10). At the pH of fusion, the B loop adopts a helical conformation connecting the adjoining helices to form a single long α helix in the postfusion conformation of HA. Transition to the postfusion conformation repositions the fusion peptide at the N terminus of HA2 approximately 100 Å closer to the target membrane (2, 11–14).
In the prefusion state, the B loop is trapped in a metastable conformation during HA expression and transport through the endoplasmic reticulum and Golgi compartments to the cell surface (15, 16). Some rearrangement of HA1 is needed to release the B loop for the conformational change (17–19). A large body of literature has suggested that the acid stability of HA is influenced by the residues in the fusion peptide, the fusion peptide pocket, coiled-coil regions of HA2, and local interactions between the HA1 and HA2 subunits (20–25). Molecular modeling studies have shown a strong electrostatic attraction between the HA1 subunits (positively charged) and the HA2 subunits (negatively charged) at neutral pH (26, 27). However, the interactions between the HA1 and HA2 subunits and the molecular mechanism for the B-loop release from its metastable state are not fully understood.
The acid stability of HA varies among different strains and is an important factor affecting virus infectivity and adaptation to host cells. The HA from the pandemic 2009 H1N1 influenza A virus [A(H1N1)pdm09] has been reported to be less stable than the HAs of other seasonal influenza A virus strains (28–31), a feature that likely contributes to challenges in the production of A(H1N1)pdm09 vaccines (28, 29). Moreover, it was recently reported that the currently circulating A(H1N1)pdm09 virus is acquiring mutations that improve HA stability (31, 32) and may therefore improve viral fitness.
In our prior studies aimed at mapping the neutralizing epitopes in HA, we noticed that certain combinations of chimeric HA involving HA1 and HA2 subunits from A(H1N1)pdm09 and other seasonal H1N1 strains abolished HA fusion activity (33). Inspection of the HA structure and sequence differences between the HAs from these strains led us to identify residues in HA1 and the B loop of HA2 that could potentially interact to regulate HA function. In this report, we show that interactions between these residues help to regulate B-loop conformational changes. These studies identify a key pair of residues affecting HA stability and influenza virus infectivity.
MATERIALS AND METHODS
Plasmids and cell lines.
pCMV/R constructs expressing the full-length HA open reading frame (ORF) with the Q223R mutation from A/Mexico/4108/2009 (Mex; GenBank accession number GQ223112) and the full-length wild-type HA ORFs from A/New Caledonia/20/1999 (NCD; GenBank accession number AY289929) and A/Brisbane/59/2007 (Bris; GenBank accession number CY058487) as well as the full-length wild type neuraminidase (NA) ORF from A/California/04/2009 (GenBank accession number FJ966084) were described previously (33). In addition, with mutations at residue 205 in HA1 and residue 72 in HA2 were introduced into HA using standard molecular biology protocols. The codon-optimized human airway trypsin (HAT)-like protease gene expression construct (pCAGGS-HATcop) was described before (34). The HIV gag/pol (pCMVΔR8.2) and luciferase (Luc) reporter (pHR′CMV-Luc) constructs were described previously (35, 36) and obtained from Gary J. Nabel (NIH, Bethesda, MD). A plasmid expressing the β-galactosidase (β-Gal) α subunit and 293T cells stably expressing the β-Gal ω subunit (37) were generously provided by Nathaniel Landau (New York University, New York, NY). MDCK cells, 293T cells, and 293T cells expressing the β-Gal ω subunit were cultured in Dulbecco's modified Eagle medium (DMEM) with high glucose, l-glutamine, minimal essential medium nonessential amino acids, penicillin-streptomycin, and 10% fetal calf serum.
Antibodies.
Rabbit antisera against the H1N1 HA2 C helix were produced via immunization with the A/New Caledonia/20/1999 HA2 C-helix peptide conjugated to keyhole limpet hemocyanin (KLH; Pierce) as described previously (38).
Production of HA pseudoviruses.
HA pseudoviruses carrying a Luc reporter gene were produced in 293T cells as described previously (39). HA pseudoviruses were collected at 48 h posttransfection, filtered through a 0.45-μm-pore-size low-protein-binding filter, and used immediately or stored at −80°C. HA and gag in pseudoviruses were measured by immunoblot analysis and a p24 enzyme-linked immunosorbent assay, respectively, as described previously (39). HA pseudovirus titers were measured by infecting 293T cells with HA pseudoviruses for 48 h prior to measuring luciferase activity in infected cells using a luciferase assay reagent (Promega) as described previously (39). HA pseudovirus infectivity titers were expressed as the number of relative luminescence units (RLU) of HA pseudovirus supernatants per nanogram of p24.
Generation of influenza viruses by reverse genetics.
The HA gene segment of A/Brisbane/59/2007 and the engineered mutations were amplified by PCR with universal primers (40) and inserted into the pHW2000 plasmid, provided by Maryna Eichelberger (FDA, Center for Biologics Evaluation and Research). Reassortant viruses bearing the HA of A/Brisbane/59/2007 and the complementary seven gene products of A/Puerto Rico/8/1934 were rescued by 8-plasmid reverse genetics, as previously described (41). All viruses were passaged once on MDCK cells.
Protease sensitivity and Western blotting.
The whole viral particles and purified HA proteins have successfully been used to demonstrate the acidic pH-induced HA conformational changes and HA sensitivity to proteolysis (15, 38, 42–46). Following the acidic pH-induced conformational change of HA, l-(tosylamido-2-phenyl ethyl) chloromethyl ketone (TPCK)-treated trypsin digests the HA1 subunit but not the HA2 subunit (42). The HA pseudoviruses, made in serum-free medium in the absence of HAT, were used for limited proteolysis by trypsin. In brief, HA pseudovirus supernatant samples were mixed with 1 M citrate buffers of different pHs and 10% n-dodecyl β-d-maltoside (DDM) to have final concentrations of 0.1 M citrate and 1% DDM, and the mixtures were incubated at 37°C for 1 h. The samples were neutralized with 1 M Tris (pH 8) and then digested with TPCK-treated trypsin (Pierce) at a final concentration of 100 μg/ml at room temperature for 20 h. The trypsin-digested samples were resolved on nonreducing SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Invitrogen) for Western blot analysis. The blots were probed with rabbit antisera against the HA2 C helix and horseradish peroxidase-linked anti-rabbit goat immunoglobulin antibodies and detected by use of the LumiGLO Reserve substrate (KPL). The amount of undigested HA was calculated according to the band intensity and band area.
Cell-cell fusion assay.
The HA conformational change was assessed in an HA-mediated cell-cell fusion assay on the basis of β-Gal complementation (47). As described previously (38), 293T cells were transfected with plasmids expressing HA, HAT, and the β-Gal α subunit using the Fugene 6 reagent. At 48 h after transfection, the transfected 293T cells were detached using a nonenzymatic cell dissociation solution (Sigma) and washed with DMEM. A total of 6 × 104 cells per well were then added to β-Gal ω subunit-expressing 293T target cells that had been seeded the night before at 3 × 104 cells per well on a 96-well plate. The cells were cocultivated for 3 h at 37°C. The culture supernatants were then removed and replaced with Dulbecco's phosphate-buffered saline (DPBS) previously adjusted to the desired pH with 0.1 M citric acid. The cells were treated for 4 min in the DPBS-citrate buffer and then cultured with DMEM. Sixteen hours later, cell-cell fusion was scored by determination of the β-Gal activity in cocultured cell lysates using a Galacto-Star kit (Applied Biosystems) according to the manufacturer's instructions.
Agglutination of erythrocytes.
Turkey, sheep, and horse erythrocytes (red blood cells; Lampire Biological Laboratories) were washed three times with phosphate-buffered saline (PBS) and diluted to 0.5% for hemagglutination assay with HA pseudoviruses and viruses generated by reverse genetics using standard protocols (48).
Computational analysis.
The interactions between the HA1 and HA2 subunits were modeled by use of the UCSF Chimera program (http://www.cgl.ucsf.edu/chimera/) and the PyMOL molecular graphics system (version 1.7.4; Schrödinger, LLC) using the protein structures with Protein Data Bank (PDB) accession numbers 3M6S (9), 3LZG (49), 1RU7 (3), 3QQB (46), 2HMG (50), 2IBX (51), and 4XKF (52).
Data analysis.
The infectivity data reported were from at least three independent experiments. A t test for comparison of paired data and the corresponding P value were analyzed using GraphPad Prism software. P values of <0.05 were considered statistically significant.
RESULTS
Potential molecular interactions between HA1 and HA2 subunits.
In our prior studies mapping neutralizing determinants in the HA of the A(H1N1)pdm09 virus, we generated several chimeric HAs by combining the HA1 subunit from one strain with the HA2 subunit from another strain (33). The function of the chimeric HA was assessed in infectivity studies involving lentiviral pseudotype particles bearing the chimeric HA on their surface, as previously described (34). Using the HA1 and HA2 subunits from the pandemic strain A/Mexico/4108/2009 (Mex) and two recent seasonal H1N1 strains, A/New Caledonia/20/1999 (NCD) and A/Brisbane/59/2007 (Bris), we found that all chimeras between these strains were functional except chimeras involving the Mex HA1 subunit combined with the HA2 subunit from the seasonal strains (Mex.HA1-Bris.HA2 and Mex.HA1-NCD.HA2) (33). Thus, we analyzed the nonconserved residues among the NCD, Bris, and Mex HA1 and HA2 subunits that could potentially contact each other using available H1 HAs with PDB accession numbers 3M6S, 3LZG, and 1RU7.
We noted that the residue pair consisting of residue 205 in HA1 and residue 399 in the B loop (hinge) of HA2 (residue 72, HA2 numbering) in Mex differs from that in NCD or Bris (Fig. 1A). For all three structures from PDB it was found that residue 205 in HA1 and residue 72 in HA2 are in close proximity, suggesting that they may be involved in intermonomer, intersubunit interactions (Fig. 1B). The interactions between the arginine at residue 205 in HA1 and the lysine at residue 72 in HA2 (R-K pair) in the chimeric HAs involving Mex.HA1-NCD.HA2 or Mex.HA1-Bris.HA2 may be different from the interactions seen in the other pairs involving (i) the arginine at residue 205 in HA1 and the histidine at residue 72 (R-H pair) in the wild-type Mex HA, (ii) the histidine at residue 205 and the lysine at residue 72 (H-K pair) in wild-type NCD and Bris HAs or the chimeric HA involving NCD.HA1-Bris.HA2 and Bris.HA1-NCD.HA2, and (iii) the histidine at residue 205 and the histidine at residue 72 (H-H pair) in the chimeric HA involving Bris.HA1-Mex.HA2 and NCD.HA1-Mex.HA2. We therefore asked whether the R-K pair alters the HA function.
FIG 1.

Intermonomer interactions between residue 205 in HA1 and residue 72 in HA2. (A) Alignment of NCD, Bris, and Mex HA1 amino acid residues 200 to 250 and HA2 amino acid residues 350 to 400 (HA2 residues 23 to 73). The residues in the 205-72 pair are marked with blue and red ovals, respectively. (B) (Left) The 205-72 pair position in the HA trimer complex based on the structure with PDB accession number 3M6S; (right) proposed residue interactions of R-H, R-K, H-H, and H-K in the 205-72 pair.
The R-K pair reduces HA pseudovirus infectivity.
To investigate the effect of R-K interactions on HA function, we introduced mutations at residue 205 in HA1 and residue 72 in HA2 in different HAs, as well as created a chimeric HA1-HA2 with these mutations. As shown in Fig. 2, compared to the effect of the wild-type HAs (the R-H pair in Mex, the H-K pair in NCD and Bris), the H-H pair had little effect on HA pseudoviruses infectivity, while the R-K pair in Mex, Bris, and NCD HA pseudoviruses greatly reduced infectivity (Fig. 2A). As expected, in the chimeric Mex.HA1-NCD.HA2 and Mex.HA1-Bris.HA2 HA, the R-K pair also resulted in the low infectivity of HA pseudoviruses (Fig. 2B). However, these HA pseudoviruses gained infectivity when the R-K pair was converted to H-K, R-H, and H-H pairs. The levels of mature HA in all pseudoviruses were similar, indicating that the R-K pair directly alters HA function.
FIG 2.
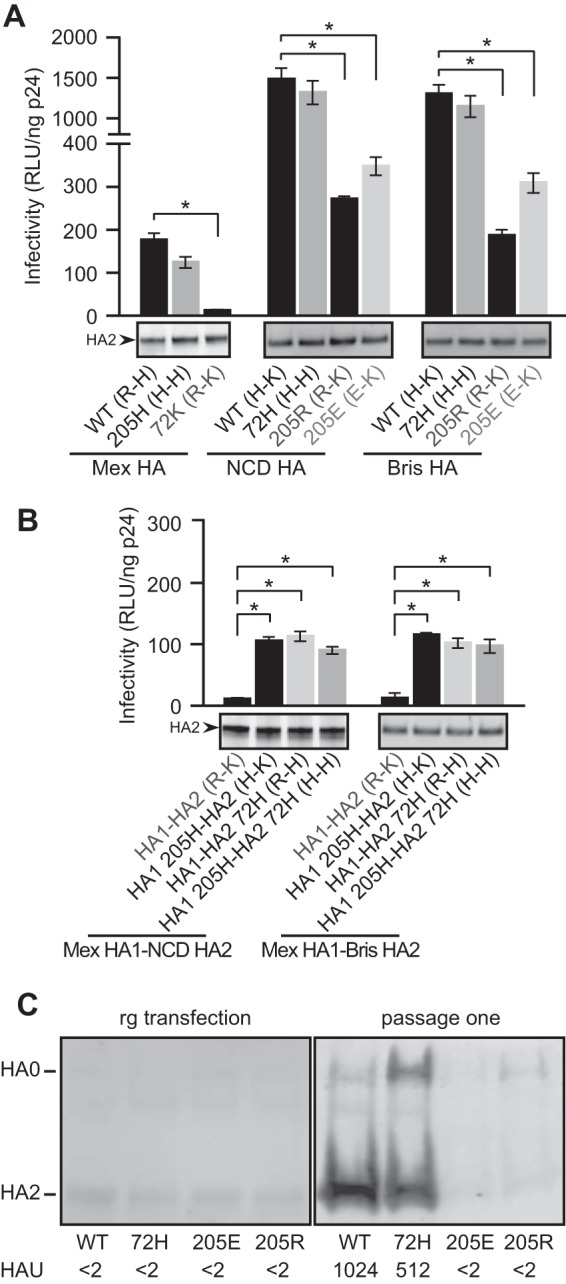
R-K and E-K pairs at the 205-72 pair in HA decrease viral infectivity. (A and B) The HA pseudovirus infectivities of wild-type (WT), NCD, Bris, and Mex HAs containing R-H, H-H, H-K, R-K, and E-K pairs at the 205-72 pair (A) and of chimeric Mex-NCD and Mex-Bris containing the R-H, H-H, H-K, R-K, and E-K pairs at the 205-72 pair (B) are compared. Data are shown as the mean and standard deviation from three independent experiments. *, t test, P < 0.05. (Bottom) The levels of mature HA (HA2) in pseudoviruses shown by Western blotting were detected with rabbit antisera against the HA2 C helix. (C) Influenza viruses with Bris HA generated by reverse genetics (rg) and recovered from the supernatants of transfected cells (left) or after a single passage on MDCK cells (right). Infectivity was scored by quantifying the HAU on turkey red blood cells. The relative levels of HA in viruses recovered from the supernatants shown by Western blotting were detected with rabbit antisera against the HA2 C helix. The data shown are representative of those from two independent experiments.
We also assessed the effect of residues in the residue 205-residue 72 pair (the 205-72 pair) on the function of Bris HA in the context of replicating virus. Construction of viruses with the H-to-E (205E) or H-to-R (205R) substitution at residue 205 to create an E-K pair or an R-K pair in HA using reverse genetics yielded viruses with extremely poor replication capacities, while the viruses with a K-to-H substitution at position 72 (72H) to create an H-H pair retained infectivity, as indicated by increasing numbers of hemagglutination units (HAU) after passage (Fig. 2C). These findings are consistent with the infectivity results of the corresponding HA pseudotypes. The levels of mature HA in all viruses generated by reverse genetic from the transfection supernatants were similar (Fig. 2C, left), confirming that the E-K and R-K pairs directly alter HA function and the ability of the virus to replicate.
The R-K pair destabilizes HA at acidic pH.
Since the Bris, NCD, and Mex 205-72 pairs contain a histidine, it seemed likely that histidine is needed to regulate HA conformational changes at acidic pH. We therefore investigated the effect of the R-K pair on acidic pH-induced conformational changes. Following acidic pH treatment, TPCK-treated trypsin digests the HA1 subunit but not the HA2 subunit, and the undigested HA2 is associated with incompletely digested HA1 under nonreducing condition (42). After acidic pH and TPCK-treated trypsin treatment, the Bris HA R-K pair underwent a structural transition when the pH dropped below 6.0, while wild-type Bris HA containing the H-K pair underwent a structural transition at a pH of less than 5.5 (Fig. 3A). The dose-response curve of trypsin digestion of wild-type Bris HA showed a 50% conversion to the postfusion conformation at pH 5.2 and 37°C for 1 h. However, the HA with the R-K pair underwent a conformation conversion at pH 5.6 (Fig. 3A). The destabilization of HA conferred by the R-K pair at acidic pH was also observed in the Mex.HA1-Bris.HA2 chimeric HA (Fig. 3B). Similar results were obtained with Mex HA, NCD HA, and the Mex.HA1-NCD.HA2 chimeric HA (data not shown).
FIG 3.

The R-K pair at the 205-72 pair destabilizes the HA prefusion structure at fusogenic pH. HA pseudoviruses were treated at acidic pH and digested with TPCK-trypsin. Nonreducing Western blots of TPCK-trypsin-digested Bris (A) and chimeric Mex.HA1-Bris.HA2 (B) HA pseudoviruses are shown. (Bottom) The undigested HA contents shown in the Western blots at different pHs were quantified. Full-length undigested HA (HA0) and HA fragments (disulfide-bonded HA1 and HA2 proteolytic fragments) were detected with rabbit antisera against the HA2 C helix.
The R-K pair confers HA-mediated cell-cell fusion at a higher pH.
The effect of the R-K pair on acidic pH-induced HA-mediated fusion was also assessed in a cell-cell fusion assay using complementation of β-galactosidase subunits in effector and target cells (37, 47). Cell numbers and surface expression levels of HA were controlled and similar within each set of experiments (data not shown). As expected, the R-K pair greatly influenced HA sensitivity to acidic pH (Fig. 4A). Wild-type Bris HA (H-K pair) induced half-maximal membrane fusion at pH 5.2, while the HA containing the R-K pair mediated half-maximal membrane fusion at pH 5.5 and 95% maximal membrane fusion at pH 5.2. These results indicate that HA containing the R-K pair can undergo conformational changes and remain capable of membrane fusion but that the membrane fusion of HA containing the R-K pair is sensitive to conformational changes at a higher pH. The results of these functional assays are consistent with the conformational transitions of HA observed in the trypsin digestion experiments (Fig. 3). Similar results were obtained when Mex, NCD, and chimeric HA were used (data not shown).
FIG 4.
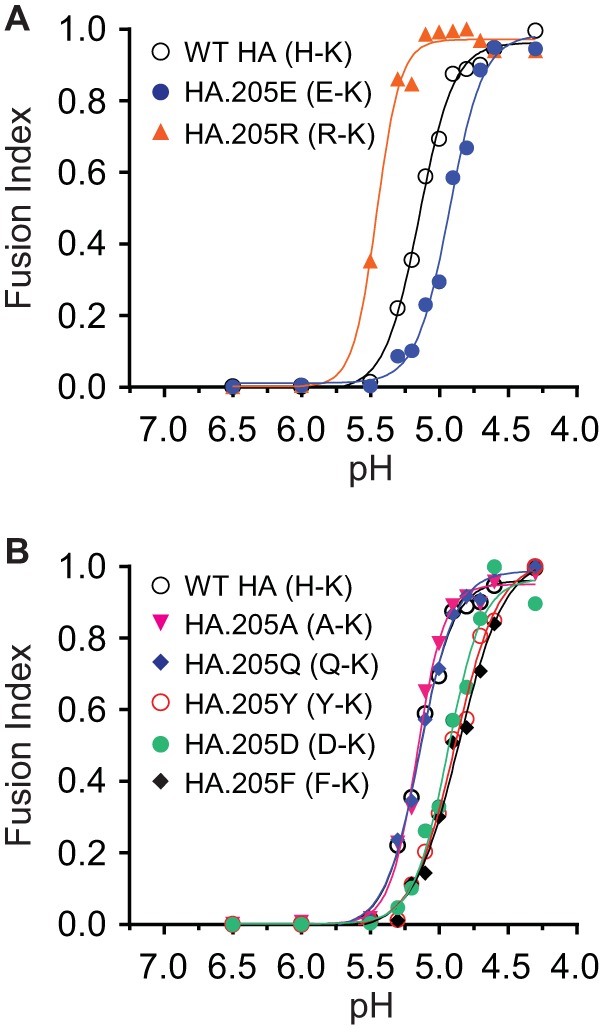
Effects of the 205-72 pair on HA sensitivity to acidic pH in HA-mediated cell-cell fusion. The fusion indexes of each HA containing different residues in 205-72 pair-mediated cell-cell fusion are compared. To obtain the fusion index, the fusions in each fusion curve (response to pH) were normalized to the maximum fusion for that curve. (A) Comparison of wild-type Bris H-K, R-K, and E-K HA sensitivity to pH. (B) Comparison of wild-type Bris H-K, A-K, Q-K, Y-K, D-K, and F-K HA sensitivity to pH. Fusion data are shown as the means from three independent experiments.
A salt bridge between residue 205 in HA1 and residue 72 in HA2 stabilizes HA.
To further confirm the intersubunit interactions between residue 205 in HA1 and residue 72 in HA2, we mutated the histidine at residue 205 in Bris and NCD HA1 to glutamate. This mutation changes the H-K pair to an E-K pair at residues 205 and 72 in the Bris and NCD HAs, which potentially introduces a salt bridge (E-K pair). We then tested the effect of the E-K pair on HA pseudovirus infectivity, HA stability at acidic pH, and the pH of HA-mediated membrane fusion, as described above. Consistent with the E-K pair forming a salt bridge, we found that the HA with this pair was much more stable than wild-type HA. The HA with the E-K pair underwent a structural transition at a much lower pH than the wild type (pH < 5.0) (Fig. 3A). The dose-response curve of trypsin digestion showed a 50% conversion to the postfusion conformation at pH 4.9 (Fig. 3A).
In the cell-cell fusion assay, the HA with the E-K pair likewise displayed fusion activity at a lower pH than the wild type (Fig. 4A). While wild-type Bris HA (H-K pair) induced half-maximal membrane fusion at pH 5.2, the HA with the E-K pair mediated half-maximal fusion at pH 5.0 and 90% maximal fusion at pH 4.5. These results suggest that the HA containing the E-K pair also undergoes pH-induced conformational changes for membrane fusion at a pH analogous to that for wild-type HA, but the E-K pair requires a lower pH. Curiously, the increased stability conferred by the E-K pair greatly impaired virus infectivity (Fig. 2), suggesting that the E-K pair in HA is too stable for fusion in the endosome during infection.
A histidine in the 205-72 pair is important for HA function.
The infectivity, cell-cell fusion, and protease digestion data of HAs containing the R-K and E-K pairs suggested that a histidine in the 205-72 pair is important for HA function. To investigate this possibility, we first asked whether histidine is conserved in the 205-72 pair in HA. After reviewing the H1 influenza virus HA sequences (www.fludb.org/brc/home.do?decorator=influenza) available as of 19 December 2014, we noted that there are no R-K and E-K pairs in H1 HAs. Most human seasonal influenza H1 HAs (∼91%) have an H-K pair, while avian H1 HAs mostly have a K-N pair (∼97%). Swine H1 HAs display more diversity in the 205-72 pairs, but the R-H and K-H pairs are dominant (∼50%) (Fig. 5A). For A(H1N1)pdm09 virus HAs, 99.7% of the HAs contain R-H or K-H pairs (Fig. 5B). Altogether, these finding suggest that histidine may be critical to optimal HA function in human cells.
FIG 5.
The 205-72 pair is conserved in H1 HAs. (A) Different 205-72 pair contents in seasonal human, swine, and avian influenza virus HAs. All available seasonal H1 virus HA sequences in the database were counted. (B) Different 205-72 pair contents in the HAs of A(H1N1)pdm09 influenza virus isolated from human, swine, and avian sources.
Next, we mutated the histidine in the H-K pair of Bris HA with various residues. As shown in Fig. 4B, the HA containing an alanine at residue 205 (H205A, A-K pair) or a glutamine at residue 205 (H205Q, Q-K pair) displayed a fusion curve similar to that of wild-type HA, while an HA containing a tyrosine at residue 205 (H205Y, Y-K pair), a glutamic acid at position 205 (H205D, D-K pair), or a phenylalanine at position 205 (H205F, F-K pair) required a more acidic pH for fusion. The HAs containing the last three pairs mediated cell-cell fusion similarly to the E-K pair (Fig. 4A). Consistent with these observations, we found that HAs containing A-K and Q-K pairs underwent a conformational transition at a pH of less than 5.5, similar to the pH required for wild-type HA, but the HAs containing the Y-K, D-K, and F-K pairs underwent a conformational transition at a pH lower than that for wild-type HA (Fig. 6A). The dose-response curve of trypsin digestion of HA showed that the pH for 50% conversion to the postfusion conformation of the HAs containing A-K and Q-K pairs was the same as the pH 5.2 for wild-type HA, while pH 5.0 is needed for the HA containing the Y-K pair and pH 4.9 is needed for the HAs containing the D-K and F-K pairs (Fig. 6B), which is the same as that for the HA containing the E-K pair (Fig. 3A). These results suggest that the A-K and Q-K pairs do not alter the HA stability according to the pH but that the Y-K, D-K, and F-K pairs make the HA more stable. However, all these pairs reduced HA pseudovirus infectivity relative to that of the wild type (Fig. 6C). The pseudoviruses bearing more stable HAs (Y-K, D-K, and F-K pairs) exhibited even less infectivity, suggesting that these HAs are too stable and do not effectively undergo pH-induced conformational changes in endosomes prior to transport to lysosomes. Similar results were observed with the NCD HA (data not shown).
FIG 6.

A histidine in the 205-72 pair is important for HA stability and virus infectivity. (A) HA pseudoviruses were treated with acidic pH and digested with TPCK-trypsin. Nonreducing Western blots were used to compare TPCK-trypsin-digested Bris HA pseudoviruses containing A-K, Q-K, Y-K, D-K, and F-K pairs. Full-length HA (HA0) and HA fragments (disulfide-bonded HA1 and HA2 proteolytic fragments) were detected with rabbit antisera against the HA2 C helix. (B) The contents of the undigested HA in panel A were quantified at different pHs. (C) Infectivities of HA pseudoviruses containing the A-K, Q-K, Y-K, D-K, and F-K pairs. (Bottom) The levels of mature HA (HA2) in pseudoviruses shown by Western blotting were detected with rabbit antisera against the HA2 C helix. Data are shown as the mean and standard deviation from three independent experiments. *, t test, P < 0.05.
Intermolecular interactions between residue 205 in HA1 and residue 72 in HA2 have no effects on receptor binding of HA.
In addition to the change of the pH of fusion, the R-K pair that destabilized HA may also change HA binding to receptors to decrease HA pseudovirus infectivity. To investigate this possibility, we performed a hemagglutination assay with HA pseudoviruses using turkey, sheep, and horse erythrocytes. Turkey erythrocytes are known to contain a mixture of α2,3- and α2,6-linked sialic acids as HA receptors, whereas sheep and horse erythrocytes primarily have only α2,3-linked sialic acids (53, 54). Using these erythrocytes, we could thus also identify the difference of HA binding to α2,3- and α2,6-linked sialic acids. However, compared to wild-type Bris and NCD HAs, neither the R-K nor E-K pair changed the ability of HA to bind to receptors or the profile of HA binding to receptors (Table 1).
TABLE 1.
Summary of hemagglutination by HA pseudoviruses
| HA (amino acids at 205-72 pair) | Agglutinationa (no. of HAU/50 ml) |
||
|---|---|---|---|
| Turkey RBCs | Sheep RBCs | Horse RBCs | |
| Wild-type Bris HA (H-K) | 16 | <2 | <2 |
| Bris HA.205R (R-K) | 16 | <2 | <2 |
| Bris HA.205E (E-K) | 16 | <2 | <2 |
| Wild-type NCD HA (H-K) | 32 | <2 | <2 |
| NCD HA.205R (R-K) | 32 | <2 | <2 |
| NCD HA.205E (E-K) | 32 | <2 | <2 |
Hemagglutination assays were performed using standard techniques. Red blood cell (RBC) numbers and the HA levels of HA pseudoviruses were controlled and similar within each set of experiments. Turkey red blood cells contain a mixture of α2,3- and α2,6-linked sialic acids as HA receptors, whereas sheep and horse red blood cells primarily have only α2,3-linked sialic acids.
DISCUSSION
The acidic environment of the endosome triggers a large-scale conformational change in HA2, including transition of the B loop in HA2 to a helix. This transition creates an extended coiled-coil that repositions the fusion peptide at the N terminus of HA2 approximately 100 Å toward the endosomal membrane to initiate fusion. The acid stability of HA is influenced by the residues in the fusion peptide, the fusion peptide pocket, the coiled-coil regions of HA2, and local interactions between the HA1 and HA2 subunits (20–27, 31, 32, 55). It therefore seems likely that different combinations of interactions among residues in these regions act together in various ways among different influenza virus strains to regulate the fusogenic conformational changes needed for virus entry. Such a network of interacting residues across HA subunits and between monomers may provide the plasticity needed for the virus to adapt to various host conditions. Here, using chimeric H1N1 HA pseudoviruses, we found that intermonomer interactions between residue 72 in the B loop of HA2 and residue 205 in HA1 play an important role in the acid stability of HA and that a histidine residue appears to finely tune HA stability at acidic pH for the infectivity for H1N1 viruses.
Histidine residues have been reported to be important molecular switches in regulating the conformational changes of viral fusion proteins (46, 56–59). Histidines can be protonated in the acidic milieu of the endosome. However, nearby residues influence the susceptibility of histidine to protonation. Here, we have identified for the first time that the 205-72 residue pair in HAs from influenza A H1N1 viruses can regulate pH-induced conformational changes. Significantly, the residues in the 205-72 pair are conserved among H1N1 viruses. More than 90% of human seasonal H1N1 virus HAs contain a histidine in this pair, and the A(H1N1)pdm09 virus HA contains only the R-H or K-H pair at residues 205 and 72. In contrast, the K-N pair is favored in avian viruses. The K-Q pair, which is structurally similar to the K-N pair, reduced the infectivity of human Bris and NCD HA pseudoviruses, further suggesting the importance of the histidine residue. The HAs from swine influenza viruses, however, display more diversity in this residue pair. While histidines are predominant, the K-N and K-Q pairs are present in a small percentage of virus isolates from swine. We note that the codons for H, Q, and N have only 1 base pair difference, which may facilitate adaptive changes in swine.
The influence of the R-K pair on reducing HA stability may explain why Mex.HA1-Bris.HA2 (or Mex.HA1-NCD.HA2) HA pseudoviruses have low infectivity. A less stable HA may be more prone to premature inactivation. An unstable HA might lead to a lower level of fusion due to a lack of sufficient numbers of fusion-competent HAs to efficiently support cooperative conformational changes within an HA trimer or between the multiple HA trimers that are likely needed for fusion. Alternatively, it is conceivable that fusion may need to take place in a particular endosomal compartment to proceed to the next step of the infection. In this case, the pH of HA activation may need to be closely matched to the pH of the proper (infection-permissive) endosomal compartment.
The importance of interactions between residues in the 205-72 pair is confirmed by the demonstration that an E-K pair in Bris, Mex, and NCD HAs, which has the potential to form a salt bridge, conferred greater acid stability to HA and reduced the infectivity of the respective pseudoviruses. Similarly, the low infectivity of HA pseudoviruses with a K-D pair indicates an interaction between the pair of amino acids at residues 205 and 72. In addition, structural analysis indicates that the A-K and Q-K pairs likely have little interference (Fig. 7A), which may explain why they do not obviously change HA stability. On the other hand, the Y and F residues have a benzene ring that could form H bonds with K (Fig. 7A), thus increasing the stability of the HAs with Y-K and F-K pairs and reducing infectivity. HAs that are too stable may not efficiently undergo fusion-inducing conformational changes before delivery to the lysosome for degradation.
FIG 7.

Intermonomer interactions between HA1 and HA2. (A) The intermonomer interactions between HA1 residue 205 and HA2 residue 72 are modeled for the A-K, Q-K, Y-K, and F-K pairs using the structure with PDB accession number 3M6S. The HA1 residues (A, Q, Y, and F) at position 205 are shown in green. The K residue at residue 72 of HA2 is shown in red. (B and C) Different rotational perspectives of the local environment surrounding the intermonomer interface between HA1 residue 205 and HA2 residue 72 of different HA subtypes (H1, PDB accession number 1RU7; H2, PDB accession number 3QQB; H3, PDB accession number 2HMG; H5, PDB accession number 2IBX; H6, PDB accession number 4XKF). For clarity, only residues 198 to 213 in HA1 are shown and illustrated as purple ribbons. Positions 72 and 205 from the H1 subtype are shown in yellow. For other subtypes, residue 72 of HA2 is shown in red, while residue 205 of HA1 is shown in blue. The blue dotted lines indicate the direction between C alpha carbons for the respective residue pairs. (D and E) Alternate interfaces between HA1 and HA2 are shown for subtype H3 (PDB accession number 2HMG) (D), and alternate interfaces between two adjacent HA2 monomers and HA1 are shown for subtype H6 (PDB accession number 4XKF) (E). Residue positions are indicated. Magenta, HA1; green, HA2.
We also looked for potential interactions between the corresponding amino acids at residues 205 and 72 in other subtypes using coordinates available for high-resolution HA structures for H1-H3 and H5-H6 subtypes (Fig. 7B and C). We first noted that the general orientation of the side chain of residue 72 in these other structures and their distance to nearby HA1 residues resembles that seen in HAs from the H1 subtype. However, these distances are variable; sometimes approach a residue further C terminal to residue 205, as in the case of H3 (Fig. 7D); and are often just outside the range needed to make polar contacts. However, because these residues reside in a loop structure, there is potential for interactions in solution due to thermal conformational sampling. Second, in some structures, for example, HA from subtype H6 (Fig. 7E), there is often a histidine near but more N terminal in the loop to position 72 that is in the vicinity of positively charged residues in the neighboring HA1 and HA2 subunits. These observations underscore the structural complementarity between residues in the HA2 B loop and residues in the HA1 subunit among different subtypes. Further experiments are needed to confirm the functional interactions in these other subtypes that may be analogous to those that we demonstrated for H1N1 under physiological conditions. Altogether, these finding suggest that combinations of pairs of amino acids involving pH-sensitive intermononer interactions between HA1 and B-loop residues in HA2 serve as important regulators of HA conformational changes.
In summary, we identified two key residues in the HA1 and HA2 subunits of influenza A H1N1 viruses involved in intermonomer interactions that facilitate the acid pH-induced release of the HA2 B loop needed for virus entry. Our data further suggest that there may be a window of pH activation of HA that needs to match the timing of fusion-inducing conformational changes with the timing of virus transport in the endocytic pathway.
ACKNOWLEDGMENTS
This work was funded by the U.S. Food and Drug Administration.
We thank Xianghong Jing and Hongquan Wan (U.S. Food and Drug Administration, Silver Spring, MD) for critical readings of the manuscript.
W.W., C.J.D., and C.D.W. conceived of and designed the experiments. W.W., C.J.D., E.A-F., and R.V. performed the experiments. W.W., C.J.D., and C.D.W. analyzed the data and wrote the manuscript.
We do not have commercial or other associations that might pose a conflict of interest and have no competing interests to disclose.
REFERENCES
- 1.Huang RT, Rott R, Klenk HD. 1981. Influenza viruses cause hemolysis and fusion of cells. Virology 110:243–247. doi: 10.1016/0042-6822(81)90030-1. [DOI] [PubMed] [Google Scholar]
- 2.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 3.Gamblin SJ, Haire LF, Russell RJ, Stevens DJ, Xiao B, Ha Y, Vasisht N, Steinhauer DA, Daniels RS, Elliot A, Wiley DC, Skehel JJ. 2004. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 303:1838–1842. doi: 10.1126/science.1093155. [DOI] [PubMed] [Google Scholar]
- 4.Ha Y, Stevens DJ, Skehel JJ, Wiley DC. 2002. H5 avian and H9 swine influenza virus haemagglutinin structures: possible origin of influenza subtypes. EMBO J 21:865–875. doi: 10.1093/emboj/21.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stevens J, Blixt O, Tumpey TM, Taubenberger JK, Paulson JC, Wilson IA. 2006. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science 312:404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- 6.Stevens J, Corper AL, Basler CF, Taubenberger JK, Palese P, Wilson IA. 2004. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 303:1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- 7.Wilson IA, Skehel JJ, Wiley DC. 1981. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 8.Russell RJ, Gamblin SJ, Haire LF, Stevens DJ, Xiao B, Ha Y, Skehel JJ. 2004. H1 and H7 influenza haemagglutinin structures extend a structural classification of haemagglutinin subtypes. Virology 325:287–296. doi: 10.1016/j.virol.2004.04.040. [DOI] [PubMed] [Google Scholar]
- 9.Yang H, Carney P, Stevens J. 2010. Structure and receptor binding properties of a pandemic H1N1 virus hemagglutinin. PLoS Curr 2:RRN1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward CW, Dopheide TA. 1980. Influenza virus haemagglutinin. Structural predictions suggest that the fibrillar appearance is due to the presence of a coiled-coil. Aust J Biol Sci 33:441–447. [DOI] [PubMed] [Google Scholar]
- 11.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 12.Carr CM, Kim PS. 1993. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 73:823–832. doi: 10.1016/0092-8674(93)90260-W. [DOI] [PubMed] [Google Scholar]
- 13.Harrison SC. 2008. Viral membrane fusion. Nat Struct Mol Biol 15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carr CM, Chaudhry C, Kim PS. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc Natl Acad Sci U S A 94:14306–14313. doi: 10.1073/pnas.94.26.14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J, Skehel JJ, Wiley DC. 1999. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc Natl Acad Sci U S A 96:8967–8972. doi: 10.1073/pnas.96.16.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbey-Martin C, Gigant B, Bizebard T, Calder LJ, Wharton SA, Skehel JJ, Knossow M. 2002. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology 294:70–74. doi: 10.1006/viro.2001.1320. [DOI] [PubMed] [Google Scholar]
- 18.Godley L, Pfeifer J, Steinhauer D, Ely B, Shaw G, Kaufmann R, Suchanek E, Pabo C, Skehel JJ, Wiley DC, Wharton S. 1992. Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell 68:635–645. doi: 10.1016/0092-8674(92)90140-8. [DOI] [PubMed] [Google Scholar]
- 19.Kemble GW, Bodian DL, Rose J, Wilson IA, White JM. 1992. Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J Virol 66:4940–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cross KJ, Wharton SA, Skehel JJ, Wiley DC, Steinhauer DA. 2001. Studies on influenza haemagglutinin fusion peptide mutants generated by reverse genetics. EMBO J 20:4432–4442. doi: 10.1093/emboj/20.16.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daniels RS, Downie JC, Hay AJ, Knossow M, Skehel JJ, Wang ML, Wiley DC. 1985. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 40:431–439. doi: 10.1016/0092-8674(85)90157-6. [DOI] [PubMed] [Google Scholar]
- 22.Ilyushina NA, Govorkova EA, Russell CJ, Hoffmann E, Webster RG. 2007. Contribution of H7 haemagglutinin to amantadine resistance and infectivity of influenza virus. J Gen Virol 88:1266–1274. doi: 10.1099/vir.0.82256-0. [DOI] [PubMed] [Google Scholar]
- 23.Steinhauer DA, Martin J, Lin YP, Wharton SA, Oldstone MB, Skehel JJ, Wiley DC. 1996. Studies using double mutants of the conformational transitions in influenza hemagglutinin required for its membrane fusion activity. Proc Natl Acad Sci U S A 93:12873–12878. doi: 10.1073/pnas.93.23.12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinhauer DA, Wharton SA, Skehel JJ, Wiley DC. 1995. Studies of the membrane fusion activities of fusion peptide mutants of influenza virus hemagglutinin. J Virol 69:6643–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thoennes S, Li ZN, Lee BJ, Langley WA, Skehel JJ, Russell RJ, Steinhauer DA. 2008. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology 370:403–414. doi: 10.1016/j.virol.2007.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Q, Opitz R, Knapp EW, Herrmann A. 2002. Protonation and stability of the globular domain of influenza virus hemagglutinin. Biophys J 82:1050–1058. doi: 10.1016/S0006-3495(02)75464-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Q, Sivaramakrishna RP, Ludwig K, Korte T, Bottcher C, Herrmann A. 2003. Early steps of the conformational change of influenza virus hemagglutinin to a fusion active state: stability and energetics of the hemagglutinin. Biochim Biophys Acta 1614:3–13. doi: 10.1016/S0005-2736(03)00158-5. [DOI] [PubMed] [Google Scholar]
- 28.Farnsworth A, Cyr TD, Li C, Wang J, Li X. 2011. Antigenic stability of H1N1 pandemic vaccines correlates with vaccine strain. Vaccine 29:1529–1533. doi: 10.1016/j.vaccine.2010.12.120. [DOI] [PubMed] [Google Scholar]
- 29.Feshchenko E, Rhodes DG, Felberbaum R, McPherson C, Rininger JA, Post P, Cox MM. 2012. Pandemic influenza vaccine: characterization of A/California/07/2009 (H1N1) recombinant hemagglutinin protein and insights into H1N1 antigen stability. BMC Biotechnol 12:77. doi: 10.1186/1472-6750-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galloway SE, Reed ML, Russell CJ, Steinhauer DA. 2013. Influenza HA subtypes demonstrate divergent phenotypes for cleavage activation and pH of fusion: implications for host range and adaptation. PLoS Pathog 9:e1003151. doi: 10.1371/journal.ppat.1003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang H, Chang JC, Guo Z, Carney PJ, Shore DA, Donis RO, Cox NJ, Villanueva JM, Klimov AI, Stevens J. 2014. Structural stability of influenza A(H1N1)pdm09 virus hemagglutinins. J Virol 88:4828–4838. doi: 10.1128/JVI.02278-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cotter CR, Jin H, Chen Z. 2014. A single amino acid in the stalk region of the H1N1pdm influenza virus HA protein affects viral fusion, stability and infectivity. PLoS Pathog 10:e1003831. doi: 10.1371/journal.ppat.1003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang W, Anderson CM, De Feo CJ, Zhuang M, Yang H, Vassell R, Xie H, Ye Z, Scott D, Weiss CD. 2011. Cross-neutralizing antibodies to pandemic 2009 H1N1 and recent seasonal H1N1 influenza A strains influenced by a mutation in hemagglutinin subunit 2. PLoS Pathog 7:e1002081. doi: 10.1371/journal.ppat.1002081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W, Xie H, Ye Z, Vassell R, Weiss CD. 2010. Characterization of lentiviral pseudotypes with influenza H5N1 hemagglutinin and their performance in neutralization assays. J Virol Methods 165:305–310. doi: 10.1016/j.jviromet.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 36.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 37.Holland AU, Munk C, Lucero GR, Nguyen LD, Landau NR. 2004. Alpha-complementation assay for HIV envelope glycoprotein-mediated fusion. Virology 319:343–352. doi: 10.1016/j.virol.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Alvarado-Facundo E, Gao Y, Ribas-Aparicio RM, Jimenez-Alberto A, Weiss CD, Wang W. 2015. Influenza M2 protein ion channel activity helps maintain the pandemic 2009 H1N1 hemagglutinin fusion competence during transport to the cell surface. J Virol 89:1975–1985. doi: 10.1128/JVI.03253-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang W, Butler EN, Veguilla V, Vassell R, Thomas JT, Moos M Jr, Ye Z, Hancock K, Weiss CD. 2008. Establishment of retroviral pseudotypes with influenza hemagglutinins from H1, H3, and H5 subtypes for sensitive and specific detection of neutralizing antibodies. J Virol Methods 153:111–119. doi: 10.1016/j.jviromet.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 40.Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. 2001. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol 146:2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- 41.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A 97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skehel JJ, Bayley PM, Brown EB, Martin SR, Waterfield MD, White JM, Wilson IA, Wiley DC. 1982. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc Natl Acad Sci U S A 79:968–972. doi: 10.1073/pnas.79.4.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruigrok RW, Cremers AF, Beyer WE, de Ronde-Verloop FM. 1984. Changes in the morphology of influenza particles induced at low pH. Arch Virol 82:181–194. doi: 10.1007/BF01311162. [DOI] [PubMed] [Google Scholar]
- 44.Boulay F, Doms RW, Wilson I, Helenius A. 1987. The influenza hemagglutinin precursor as an acid-sensitive probe of the biosynthetic pathway. EMBO J 6:2643–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vanderlinden E, Goktas F, Cesur Z, Froeyen M, Reed ML, Russell CJ, Cesur N, Naesens L. 2010. Novel inhibitors of influenza virus fusion: structure-activity relationship and interaction with the viral hemagglutinin. J Virol 84:4277–4288. doi: 10.1128/JVI.02325-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu R, Wilson IA. 2011. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J Virol 85:5172–5182. doi: 10.1128/JVI.02430-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossi F, Charlton CA, Blau HM. 1997. Monitoring protein-protein interactions in intact eukaryotic cells by beta-galactosidase complementation. Proc Natl Acad Sci U S A 94:8405–8410. doi: 10.1073/pnas.94.16.8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kendal AP, Pereira MS, Skehel JJ. 1982. Concepts and procedures for laboratory-based influenza surveillance. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control, Atlanta, GA. [Google Scholar]
- 49.Xu R, Ekiert DC, Krause JC, Hai R, Crowe JE Jr, Wilson IA. 2010. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 328:357–360. doi: 10.1126/science.1186430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weis WI, Brunger AT, Skehel JJ, Wiley DC. 1990. Refinement of the influenza virus hemagglutinin by simulated annealing. J Mol Biol 212:737–761. doi: 10.1016/0022-2836(90)90234-D. [DOI] [PubMed] [Google Scholar]
- 51.Yamada S, Suzuki Y, Suzuki T, Le MQ, Nidom CA, Sakai-Tagawa Y, Muramoto Y, Ito M, Kiso M, Horimoto T, Shinya K, Sawada T, Usui T, Murata T, Lin Y, Hay A, Haire LF, Stevens DJ, Russell RJ, Gamblin SJ, Skehel JJ, Kawaoka Y. 2006. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature 444:378–382. doi: 10.1038/nature05264. [DOI] [PubMed] [Google Scholar]
- 52.Tzarum N, de Vries RP, Zhu X, Yu W, McBride R, Paulson JC, Wilson IA. 2015. Structure and receptor binding of the hemagglutinin from a human H6N1 influenza virus. Cell Host Microbe 17:369–376. doi: 10.1016/j.chom.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ito T, Suzuki Y, Mitnaul L, Vines A, Kida H, Kawaoka Y. 1997. Receptor specificity of influenza A viruses correlates with the agglutination of erythrocytes from different animal species. Virology 227:493–499. doi: 10.1006/viro.1996.8323. [DOI] [PubMed] [Google Scholar]
- 54.Medeiros R, Escriou N, Naffakh N, Manuguerra JC, van der Werf S. 2001. Hemagglutinin residues of recent human A(H3N2) influenza viruses that contribute to the inability to agglutinate chicken erythrocytes. Virology 289:74–85. doi: 10.1006/viro.2001.1121. [DOI] [PubMed] [Google Scholar]
- 55.Byrd-Leotis L, Galloway SE, Agbogu E, Steinhauer DA. 2015. Influenza hemagglutinin (HA) stem region mutations that stabilize or destabilize the structure of multiple HA subtypes. J Virol 89:4504–4516. doi: 10.1128/JVI.00057-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Z, Blissard GW. 2011. Autographa californica multiple nucleopolyhedrovirus GP64 protein: roles of histidine residues in triggering membrane fusion and fusion pore expansion. J Virol 85:12492–12504. doi: 10.1128/JVI.05153-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mair CM, Meyer T, Schneider K, Huang Q, Veit M, Herrmann A. 2014. A histidine residue of the influenza virus hemagglutinin controls the pH dependence of the conformational change mediating membrane fusion. J Virol 88:13189–13200. doi: 10.1128/JVI.01704-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kalani MR, Moradi A, Moradi M, Tajkhorshid E. 2013. Characterizing a histidine switch controlling pH-dependent conformational changes of the influenza virus hemagglutinin. Biophys J 105:993–1003. doi: 10.1016/j.bpj.2013.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kampmann T, Mueller DS, Mark AE, Young PR, Kobe B. 2006. The role of histidine residues in low-pH-mediated viral membrane fusion. Structure 14:1481–1487. doi: 10.1016/j.str.2006.07.011. [DOI] [PubMed] [Google Scholar]