

ABSTRACT
All major types of interferon (IFN) efficiently inhibit hepatitis C virus (HCV) replication in vitro and in vivo. Remarkably, HCV replication is not sensitive to IFN-γ in the hepatoma cell line Huh6, despite an intact signaling pathway. We performed transcriptome analyses between Huh6 and Huh-7 cells to identify effector genes of the IFN-γ response and thereby identified the DExD/H box helicase DEAD box polypeptide 60-like (DDX60L) as a restriction factor of HCV replication. DDX60L and its homolog DEAD box polypeptide 60 (DDX60) were both induced upon viral infection and IFN treatment in primary human hepatocytes. However, exclusively DDX60L knockdown increased HCV replication in Huh-7 cells and rescued HCV replication from type II IFN as well as type I and III IFN treatment, suggesting that DDX60L is an important effector protein of the innate immune response against HCV. In contrast, we found no impact of DDX60L on replication of hepatitis A virus. DDX60L protein was detectable only upon strong ectopic overexpression, displayed a broad cytoplasmic distribution, but caused cytopathic effects under these conditions. DDX60L knockdown did not alter interferon-stimulated gene (ISG) induction after IFN treatment but inhibited HCV replication upon ectopic expression, suggesting that it is a direct effector of the innate immune response. It most likely inhibits viral RNA replication, since we found neither impact of DDX60L on translation or stability of HCV subgenomic replicons nor additional impact on assembly of infectious virus. Similar to DDX60, DDX60L had a moderate impact on RIG-I dependent activation of innate immunity, suggesting additional functions in the sensing of viral RNA.
IMPORTANCE Interferons induce a plethora of interferon-stimulated genes (ISGs), which are our first line of defense against viral infections. In addition, IFNs have been used in antiviral therapy, in particular against the human pathogen hepatitis C virus (HCV); still, their mechanism of action is not well understood, since diverse, overlapping sets of antagonistic effector ISGs target viruses with different biologies. Our work identifies DDX60L as a novel factor that inhibits replication of HCV. DDX60L expression is regulated similarly to that of its homolog DDX60, but our data suggest that it has distinct functions, since we found no contribution of DDX60 in combatting HCV replication. The identification of novel components of the innate immune response contributes to a comprehensive understanding of the complex mechanisms governing antiviral defense.
INTRODUCTION
Hepatitis C virus (HCV), a positive-strand RNA virus grouped into the Flaviviridae family, is an important human pathogen affecting about 2 to 3% of the world's population (1). HCV infections are not cleared in most cases but become chronic in the livers of ca. 70% of infected individuals, often resulting in severe liver disease. A strong and multispecific adaptive immune response, especially CD8+ T cells, is one of the key determinants for clearance of the infection; however, the contribution of innate immunity to establishing and maintaining HCV persistence is still under debate (reviewed in reference 2). HCV induces a very potent interferon (IFN) response very early in infected hosts (3, 4), resulting in expression of interferon-stimulated genes (ISGs) as the first line of defense counteracting viral infection. ISG expression is driven by type I (IFN-α and IFN-β), II (IFN-γ), and III (IFN-λ) IFNs upon binding to their respective receptors and by activation of intracellular RNA sensors activating interferon regulatory factor 3 (IRF-3) in infected cells, inducing sets of partially overlapping genes (5–7). IFN-α is mainly produced by dendritic cells (8) and has been the backbone of anti-HCV therapy for decades (9). IFN-γ is the major cytokine of noncytolytic T cell actions against HCV (10). IFN-β and IFN-λ are mainly secreted upon sensing of viral RNA in HCV-infected cells (7, 11, 12) and result in autocrine and paracrine feedback activation of IFN responses. Although the viral protease NS3/4A cleaves mitochondrial antiviral signaling protein (MAVS), Riplet, and TRIF, which are important factors involved in IRF-3 responses (13), HCV seems to mount a strong innate immune response in infected cells, which is mainly mediated by IFN-λ (7, 12).
Several studies have already focused on the IFN response against HCV infection (5, 6, 14, 15) and identified ISGs directly affect HCV replication; among those are the genes for RSAD2/viperin, PLSCR1, IFIT3, IFITM1, IFITM3, and NOS2 (reviewed in reference 16). Still, no single ISG has been shown to be indispensable for effective IFN responses against HCV. Therefore, it is currently believed that IFNs induce overlapping and redundant sets of effector proteins tailored to interfere with replication of a wide set of viruses with various biologies (15, 17). Identifying novel factors contributing to the interferon response of particular virus groups and unraveling their mechanism of action are therefore important prerequisites for a better understanding of innate immune responses against viral infections.
Some ISG products belong to the large family of DExD/H-box helicases and contribute to antiviral defense by sensing and counteracting viral infection (reviewed in reference 18). Generally, DExD/H-box helicases share conserved domains and play a role in almost every step of RNA metabolism from transcription to degradation (19, 20). The most prominent ISG products among the DExD/H-box helicases family are the RIG-I-like helicases (RLH), which include RIG-I (DDX58) and melanoma differentiation-associated protein 5 (MDA5), two sensors of viral RNA molecules (21, 22). In addition, DEAD box polypeptide 60 (DDX60) and its highly similar homolog DEAD box polypeptide 60-like (DDX60L) have recently been described to be ISG products as well (23, 24). DDX60 and DDX60L are about 70% identical in their amino acid sequences, contain the same conserved DExD/H box domains, and likely have evolved from a gene duplication late in mammalian evolution (23). Their genes are neighbors on chromosome IV, and mice possess only DDX60 (23). DDX60 has been shown to contribute to RIG-I-dependent IRF-3 activation and viral RNA degradation (23, 25) and has also been described to be an inhibitor of HCV replication (15). In contrast, DDX60L has not been further characterized so far.
In this study, we aimed to identify novel factors that are part of the IFN response against HCV. HCV replication is highly sensitive to IFN-α and IFN-γ in the human hepatocellular carcinoma cell line Huh-7 and subclones thereof, which have been the most efficient and most widely used cellular model to study HCV replication (26). In contrast, HCV replication is not suppressed by IFN-γ treatment in the human hepatoblastoma cell line Huh6, while the virus is still sensitive to IFN-α treatment in these cells (27). This selective resistance to IFN-γ was neither due to mutations in the viral genome nor due to a general defect in IFN-γ signaling, since other viruses remained sensitive to IFN-γ in Huh6 cells (27). Therefore, we hypothesized that a specific component of the IFN-γ response against HCV was missing in Huh6 cells. By comparing the IFN-γ-induced gene expression profiles of Huh-7 and Huh6 cells and analyzing differentially expressed genes in a small interfering RNA (siRNA)-based screen, we identified DDX60L as a potent host restriction factor of HCV replication, acting independently of DDX60 and contributing to type I, II, and III IFN responses. Since DDX60L also strongly impaired production of lentiviral vectors, our results indicate a potential role as a restriction factor of retroviral replication.
MATERIALS AND METHODS
Cell lines.
All cell lines were cultured in Dulbecco's modified Eagle medium (DMEM; Life Technologies, Darmstadt, Germany) supplemented with 10% fetal bovine serum, nonessential amino acids (Life Technologies), 100 U/ml of penicillin, and 100 ng/ml of streptomycin (Life Technologies) and cultivated at 37°C and 5% CO2. Huh6 and Huh-7 cells harboring persistent replicons have been described before (27, 28), as have Huh-7 cells with persistent reporter replicons of genotypes 1b and 2a, LucubineoCon1 and LucubineoJFH1, respectively (10, 29); the cells were kept under selection with 1 mg/ml of G418 (Geneticin; Life Technologies). Two independent Huh6-based genotype 2a replicon cell lines were established (Huh6JFH), as was a second genotype 1b cell line (Huh6Con1), based on the selectable replicon I389neo-EI/NS3-3′_ET (27) to exclude clonal artifacts. Huh-7-Lunet is a cured replicon cell clone with high permissiveness for HCV replication (30). Huh-7-Lunet-T7 cells were described before (31). Huh7.5 cells were a kind gift of C. M. Rice, Rockefeller University, New York, NY. Cells transduced with lentiviral expression vectors were selected by adding 1 μg/ml of puromycin (Sigma-Aldrich, Steinheim, Germany) or 5 μg/ml of blasticidin (Sigma-Aldrich). Interferon treatment was performed either with IFN-γ (R&D Systems, Wiesbaden, Germany), IFN-α-2a (PBL Laboratories, Acris, Herfold, Germany), or IFN-λ1 (PeproTech, Hamburg, Germany). IFN-γ used for microarray experiments was obtained from a different source (Roche, Basel, Switzerland).
For long-term culture, stable replicon cell lines based on Huh-7-Lunet and Huh6 cells were seeded in 10-cm dishes and treated with 1 mg/ml of G418 and 10 ng/ml of IFN-γ. Cells were detached, counted, and reseeded every 3 to 4 days. At each time point, the same number of cells was reseeded for all cell lines and treated with 10 ng/ml of IFN-γ and 1 mg/ml of G418.
PHH.
Experiments with primary human hepatocytes (PHH; ordered from Biopredic, Rennes, France) are described in reference 6. RNA from these experiments was used for quantitative reverse transcription-PCR (qRT-PCR) as described below.
Plasmid constructs.
DDX60L was cloned from Huh-7-Lunet cell total RNA by long-template RT-PCR. Primer A_DDX60L_MluI (AACACGCGTTTATTCTAAATGAT) was used for synthesis of DDX60L-specific cDNA, which was then subjected to long-template PCR with primers S_DDX60L_5′UTR (GAGGTGCCATTCACATCAAAT) and A_DDX60L_MluI, followed by another round of long-template PCR with S_DDX60L_AsiSI (GATGCGATCGCGATGGGGTCAAAGGATCATG) and A_DDX60L_MluI (AACACGCGTTTATTCTAAATGAT). A consensus sequence was assembled based on 10 individual clones. This sequence has 6 amino acid exchanges compared to the DDX60L sequence present in the Refseq database (C336Y, V409L, C831R, E1222D, N1488D, and M1646V, compared to the sequence under GenBank accession number NM_001012967.2). However, all 6 deviations have been observed before, therefore reflecting polymorphisms in the human population (http://www.uniprot.org/uniprot/Q5H9U9). The coding sequence was then inserted into the pWPI backbone by digestion with AsiSI and MluI. To shuttle the DDX60L coding sequence into the pcDNA3.1 backbone, pWPI-DDX60Lpuro was digested with AscI and SpeI, blunted, and ligated into the pcDNA3.1 vector digested with EcoRV and XbaI. To transfer the DDX60L coding sequence into the pTM backbone, the 5′ part was amplified by PCR with primers S_NcoI_DDX60L (GTAGCCATGGGGTCAAAGGATCATGCAG) and A_DDX60L_4635 (CAATATTCTTTAACAATTGCTCATCATACAAG), followed by restriction digestion with NcoI and SpeI. The 3′ part was excised from the pWPI backbone by restriction digestion with SpeI and XhoI, and both parts were ligated into the pTM vector digested with NcoI and XhoI.
The two-plasmid doxycycline-inducible lentiviral expression system pLVX (Clontech, Mountain View, CA) was used for inducible expression of DDX60L. The DDX60L coding sequence was amplified by PCR using primers for_NotI_DDX60L (GTAAGCGGCCGCGTAGCCGCCACCATGGGGTCAAAGGATCATGCAG) and rev_NheI_DDX60L (GTAGCTAGCTGCTACTTATTCTAAATGATTTTGACTCATTTGAATTTG) and transferred into the pLVX expression vector by use of NotI and NheI restriction sites.
HCV and HAV reporter replicons used for transient-transfection assays were described before: pFKI389Luc-EI/NS3-3′_ET (LucCon1ET) (32), pFKI389Luc-EI/NS3-3′_JFH (LucJFH1) (33), or pT7-18f-Luc (LucHAV) (34, 35). The plasmid used for generation of reporter virus JcR2A was described before (36), as was the monocistronic HCV reporter replicon pFK-I389Luc-ubi/NS3-3′/JFH1 (33). This construct was modified to carry an additional internal ribosome entry site (IRES) derived from poliovirus, giving rise to pFK-I341PI-Luc-ubi/NS3-3′/JFH1, in which protein translation is independent of the HCV IRES. pRL-CMV (GenBank accession number AF025843) encodes Renilla luciferase under the transcriptional control of the T7 RNA polymerase promoter and was used to generate capped in vitro transcripts.
Plasmids encoding phosphatidyl inositol-4 kinase IIIα (PI4KIIIa) were described before in detail (31, 37). To generate stable knockdown cell lines, we cloned short hairpin RNA (shRNA) sequences into the pAPM backbone and prepared lentiviral particles according to a previously published protocol (38). The targeting sequences were shNT (ATCTCGCTTGGGCGAGAGTAAG), shDDX60L_1 (AACACTGAATGTCATAGTTATG), shDDX60L_3 (CGCCCTCAGTATCATACTTAAA), shRSAD2_1 (CTCTCGCTATCTCCTGTGACAG), and shRSAD2_2 (CAGGGATTATAGAGTCGCTTTC).
Transcriptome analysis.
Huh-7 and Huh6 cells were treated with 1,000 IU/ml of IFN-γ or 1,000 IU/ml of IFN-α or remained untreated for 24 h. Total RNA was prepared and analyzed using an Affymetrix HG U133 Plus 2.0 Genechip array.
Data were analyzed and normalized with the standard Affymetrix workflow of Chipster software (39). To select genes for further analyses, the log2 ratios of IFN-γ-treated and untreated cells were calculated separately for Huh6 and Huh-7 cells. To identify genes contributing to the IFN-γ response, we assumed that candidates should be strongly induced by IFN-γ in Huh-7 cells but not induced in Huh6 cells. We therefore subtracted the log2 ratios of Huh6 cells from Huh-7 cells and arbitrarily defined genes above a threshold of a 3-fold difference as candidates (Table 1). In the case that a signal was rated “absent” in the presence and absence of IFN-γ in the Huh6 samples, apparent induction was considered nonsignificant and the respective log2 ratio therefore defined as 0 (see Table S1 in the supplemental material). For the same reason, we excluded genes with apparently high IFN-γ induction but being rated as “absent” under all conditions. Genes upregulated more than 2-fold by IFN-γ in Huh6 cells were not considered candidates, even in the case of a much higher upregulation in Huh-7 cells (e.g., CXCL10; see Table S1).
TABLE 1.
Genes differentially induced by IFN-γ in Huh-7 and Huh6 cellsa
| Gene product | Fold difference between Huh-7 and Huh6 cells |
|---|---|
| CXCL92 | 13.8 |
| BTN3A31 | 10.5 |
| CASP11 | 9.4 |
| CARD161 | 8.5 |
| CXCL112 | 7.0 |
| MX13 | 6.8 |
| HLA-B4 | 6.1 |
| BTN3A21 | 5.6 |
| C1R2 | 5.5 |
| CFH2 | 5.5 |
| HERC61 | 4.3 |
| SERPINA71 | 4.0 |
| CTSS1 | 3.9 |
| IL-72 | 3.8 |
| HLA-A4 | 3.6 |
| TRIM311 | 3.5 |
| MT2A2 | 3.4 |
| NLRC51 | 3.2 |
| DDX60L1 | 3.2 |
| TLR33 | 3.1 |
| IL-322 | 3.1 |
| CFB2 | 3.1 |
| ACY31 | 3.0 |
IFN-γ-induced genes that are more strongly upregulated in Huh-7 compared to Huh6 identified by microarray gene expression analysis. Factors shown in bold were used for further analysis. Exclusion of factors in regular font was based on the following criteria, indicated by superscript numbers as follows: 1, candidate effector for further analysis; 2, secreted factor; 3, no anti-HCV activity; 4, unlikely direct effector.
Cell viability assay.
WST-1 cell proliferation reagent (Roche Life Sciences, Mannheim, Germany) was used to determine cell viability after siRNA or plasmid transfection and has been described earlier (40).
In vitro transcription, RNA transfection, and luciferase assay.
In vitro transcription and firefly luciferase assays (FLuc) as well as Renilla luciferase assays were performed as described elsewhere (reference 41 and references 36 and 41, respectively). In brief, plasmid DNA was linearized for 60 min at 37°C with AseI and ScaI (Con1), MluI (JFH1), and AgeI (HAV), purified by phenol-chloroform extraction, and subjected to in vitro transcription by T7 RNA polymerase. After overnight incubation at 37°C, DNase I (Promega, Mannheim, Germany) was added to digest template DNA. Another round of phenol-chloroform extraction was performed to purify in vitro-transcribed RNA. RNA concentration and integrity were determined by measurement of optical density (OD) at 260 nm on a NanoDrop Lite (Thermo Scientific, Braunschweig, Germany) and agarose gel electrophoresis, respectively. In the case of pRL-CMV, in vitro transcription reactions were performed with addition of G(5′)ppp(5′)G RNA cap structure analog (NEB, Frankfurt, Germany) according to the manufacturer's instructions to generate 5′-capped transcripts capable of direct translation.
For transient-replication assays, 1 × 107 Huh-7-Lunet cells or 1.75 × 107 Huh6 cells were resuspended in 1 ml of cytomix. A total of 2.5 μg of in vitro-transcribed RNA was used for the electroporation of a 100-μl single-cell suspension (Huh-7-Lunet) or 5 μg of in vitro-transcribed RNA for the electroporation of a 400-μl single-cell suspension (Huh-7-Lunet and Huh6) as described elsewhere (6). In the case of knockdown experiments, 100 pmol of siRNA was coelectroporated. For translation assays, 1 × 106 Huh-7-Lunet cells were transfected with siRNAs using the Lipofectamine RNAiMax (Life Technologies) reagent according to the manufacturer's instructions. Forty-eight hours later, cells were counted and 1 × 106 Huh-7-Lunet cells were transfected with 5 μg of replication-deficient monocistronic HCV FLuc reporter replicon RNA together with 5 μg of 5′-capped Renilla luciferase in vitro-generated RNA. For assays on stable FLuc reporter replicon cell lines, cells were seeded in 24-well plates and transfected with 6pmol of siRNA using RNAiMax (Life Technologies) according to the instructions of the manufacturer.
Predesigned FlexiTube siRNAs (Qiagen, Hilden, Germany) were used for candidate screening. Each gene was targeted with two different siRNAs as follows: for ACY3 (gene identifier [ID] 91703), Hs_ACY3_3 and Hs_ACY3_5; for BTN3A2 (gene ID 11118), Hs_BTN3A2_6 and Hs_BTN3A2_9; for BTN3A3 (gene ID 10384), Hs_BTN3A3_4 and Hs_BTN3A3_5; for CASP1 (gene ID 834), Hs_CASP1_14 and Hs_CASP1_15; for CARD16 (gene ID 114769), Hs_COPI_3 and Hs_COPI_4; for CTSS (gene ID 1520), Hs_CTSS_3 and Hs_CTSS_5; for DDX60L (gene ID 91351), Hs_FLJ31033_5 “siDDX60L_5” and Hs_FLJ31033_6 “siDDX60L_6”; for HERC6 (gene ID 550008), Hs_HERC6_4 and Hs_HERC6_5; for NLRC5 (gene ID 84166), Hs_NLRC5_3 and Hs_NLRC5_4; for SERPINA7 (gene ID 6906), Hs_SERPINA7_5 and Hs_SERPINA7_6; for TLR3 (gene ID 7098), Hs_TLR3_7 and Hs_TLR3_8); and for TRIM31 (gene ID 11074), Hs_TRIM31_6 and Hs_TRIM31_7. The other siRNAs used were siNT (ON-TARGETplus NTC; Dharmacon, Lafayette, CO), siIFNgR(CAUUAUCUCGUUUCCGGAA), siSTAT2 (GGCCGAUUAACUACCCUAA), siDDX60L_7 (GAACCUUAGUCCGGAUUCA), siDDX60L_8 (UGACAUCUGCUGUAAUUGA), and siDDX60 (GGCTAACAAACTTCGAAAA) (23). These siRNAs were ordered from Eurofins MWG Operon (Ebersberg, Germany) unless otherwise indicated.
Infection with reporter virus.
JcR2A reporter virus was produced as described before (42). Huh7.5 cells were transfected with siRNAs using Lipofectamine RNAiMax (Life Technologies). Twenty-four hours later, the cells were infected with JcR2A reporter virus at a multiplicity of infection (MOI) of 1 50% tissue culture infective dose (TCID50)/cell. Twenty-four hours after infection, IFN-α or IFN-γ was added. Forty-eight hours after infection, the medium was changed. Seventy-two hours after infection, the supernatant was used to reinfect untreated Huh7.5 cells, while the initially infected cells were subjected to Renilla luciferase assay to determine the replication of HCV. Seventy-two hours after reinfection, Renilla luciferase assay was performed to determine the infectivity of the newly formed viral particles during primary infection and IFN treatment and therefore used as a correlate of viral titers.
Production of lentiviral vectors and generation of stable cell lines.
A total of 5 × 106 293T cells were seeded into 10-cm cell culture dishes and transfected using polyethylenimine (PEI) and plasmid pWPI (encoding the gene of interest), pAPM (encoding shRNAs), or pLVX (encoding the gene of interest under the control of a doxycycline-inducible promoter) together with the packaging constructs pMD.G (vesicular stomatitis virus G protein) and pSPAX2 (HIV gag-pol). Virus containing supernatants were harvested 48 h after transfection and passed through a 0.45-μm filter. Supernatants were then stored at −80°C or directly added to target cells. Twenty-four hours after infection, the appropriate selection antibiotic was added. To determine lentivirus titers, 100-μl quantities of serial dilutions were added to 8,000 HeLa cells in each well of a 96-well plate. Four replicates were used per dilution step. Twelve hours after transduction, the appropriate selection antibiotic was added (blasticidin or puromycin). Four days after infection, cells were stained with crystal violet and vector titers were determined in CFU by counting resistant cell colonies per well.
Western blotting.
Cells were harvested from confluent 6-cm cell culture dishes by addition of NP-40 lysis buffer (0.1% NP-40, 20 mM Tris [pH 7.6], 100 mM NaCl, 50 mM NaF, protease inhibitor cocktail [Roche]). The protein concentration was determined by Bradford assay. SDS-PAGE was performed on a 10% polyacrylamide-SDS gel for HCV proteins, a 12.5% polyacrylamide-SDS gel for detection of HIV-1 p24 capsid protein, or an 8% polyacrylamide-SDS gel for detection of DDX60L. Proteins were blotted on nitrocellulose for detection of HCV or HIV-1 proteins (43) or polyvinylidene difluoride (PVDF) membranes for detection of DDX60L. Primary antibodies used for staining the membrane were NS3-specific polyclonal rabbit antiserum (44), a mouse monoclonal antibody against β-actin (A5441; Sigma-Aldrich), sheep anti-HIV-1 p24CA antiserum (kind gift from Barbara Müller), a mouse anti-transferrin receptor (Life Technologies), a mouse monoclonal antibody against the HA peptide (H3663; Sigma-Aldrich), or a polyclonal rabbit antibody against DDX60L (ARP68258_P050; Aviva Systems Biology, San Diego, CA). Another rabbit polyclonal antibody against DDX60L (Abcam; ab110772) did not give a signal. Secondary detection of HCV proteins was performed using IRDye-labeled (LI-COR Biosciences, Bad Homburg, Germany) secondary antibodies. Fluorescent signal was detected using an Odyssey infrared imaging system (LI-COR Biosciences), and ImageJ software was used for quantification. Detection of DDX60L and HIV-1 proteins was performed using horseradish peroxidase (HRP)-coupled secondary antibodies (Sigma-Aldrich).
IF analysis.
Immunofluorescence (IF) analysis was performed as described before (31). Primary antibodies used were rabbit anti-hemagglutinin (anti-HA; Ab9110; Abcam) and mouse anti-HCV-NS5A (9E10; C. M. Rice, Rockefeller University, New York, NY) at dilutions of 1:250 and 1:1,000, respectively. Two commercial rabbit polyclonal anti-DDX60L antibodies (ab110772 from Abcam and ARP68258_P050 from Aviva Systems Biology) were tested, but they did not detect DDX60L in cells overexpressing HA-DDX60L with a positive HA stain.
Northern blotting.
Northern blot analysis of full-length HCV RNA was performed as described elsewhere (33). Twenty micrograms of purified total RNA was denatured by glyoxal treatment and separated on a denaturing agarose gel, followed by Northern blotting, staining with methylene blue, and radiolabeling with 32P-riboprobes directed against β-actin or the neomycin phosphotransferase encoded in the replicon sequences. The membranes were exposed to a maximum-resolution autoradiography (MR) film (Sigma-Aldrich) at −70°C for 4 h to 4 days, depending on the signal intensity. For quantification, films were either scanned directly or exposed to phosphorimager plates and scanned with a phosphorimager. ImageJ software was used for quantification.
Determination of stability of genomic HCV RNA.
LucubineoJFH cells were transduced with pAPM-based lentivirus encoding different shRNAs at an MOI of 5 CFU/cell. Twenty-four hours later, the cells were treated with 100 μM 2′-C-methylcytidine (2′CMC; Sigma-Aldrich), which corresponds to ca. 5× the 90% inhibitory concentration (IC90). Twenty-four hours, 48 h, and 72 h later, total RNA was isolated, and qRT-PCR for HCV genomic RNA was performed as described below.
Overexpression of DDX60L.
For transient-overexpression experiments, 300-ng quantities of pcDNA vectors were transfected into Huh-7-Lunet cells using transIT-LT1 transfection reagent (Mirus Bio, Madison, WI) according to the manufacturer's instructions. pcDNA-GFP vector was used to keep the amount of transfected DNA at 300 ng and to control for transfection efficiency. Higher levels of overexpression can be achieved using the pTM plasmid system, based on T7 polymerase for transcription of RNA for the gene of interest downstream of an IRES sequence from encephalomyocarditis virus (EMCV). For transfection of pTM constructs, we transfected 2.5 μg of DNA into Huh-7-Lunet-T7 cells using transIT-LT1 transfection reagent (Mirus Bio) and analyzed the samples 24 h later.
Stable Huh-7-Lunet-derived cell lines containing the pLVX doxycycline-responsive transcriptional activator as well as the pLVX insert with the gene of interest were selected using blasticidin and puromycin. For HCV replication assays, selected cells were transfected with LucJFH reporter replicons as described above. Twenty-four hours after transfection, expression was induced by addition of 750 ng/ml of doxycycline (Sigma-Aldrich). Forty-eight hours later, HCV replication was determined by luciferase assay and total RNA was isolated; this was used to determine DDX60L and IFIT1 mRNA expression levels by qRT-PCR.
qRT-PCR.
For gene expression analysis, total RNA was isolated using the NucleoSpin RNA kit (Macherey-Nagel, Düren, Germany). Total RNA was used for qRT-PCR analysis with the 2× iTaq Universal SYBR green supermix (Bio-Rad, Munich, Germany). Reactions were performed on a CFX96 Touch real-time PCR detection system (Bio-Rad) as follows: 95°C for 3 min, 95°C for 10 s, and 60°C for 30 s. Primer sequences used were as follows: for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), S_GAPDH (GAAGGTGAAGGTCGGAGTC) and A_GAPDH (GAAGATGGTGATGGGATTTC); for DDX60L, S_DDX60L (TGAGGACCGCTTTAATTCTCCA) and A_DDX60L (ACAACATTGACTTTCATTCCCCA); for DDX60, S_DDX60 (CAGCTCCAATGAAATGGTGCC) and A_DDX60 (CTCAGGGGTTTATGAGAATGCC); for IFIT3, S_IFIT3 (GAACATGCTGACCAAGCAG) and A_IFIT3 (CAGTTGTGTCCACCCTTCC); for GBP1 S_GBP1, (CCAGTGCTCGTGAACTAAGGA) and A_GBP1 (TGTCATGTGGATCTCTGATGC); and for IFIT1, S_IFIT1 (GAAGCAGGCAATCACAGAAA) and A_IFIT1 (TGAAACCGACCATAGTGGAA). GAPDH was used as an internal reference gene; relative gene expression was determined using the threshold cycle (2−ΔΔCT) method (45).
For analysis of HCV genomic RNA, one-step qRT-PCR was performed using qScript XLT One-Step RT-qPCR ToughMix (Quanta Biosciences, Gaithersburg, MD) according to the manufacturer's instructions. In brief, 15 μl of reaction mixture contained 7.5 μl of 2× enzyme/buffer mix, a 1 μM concentration of each JFH1-specific primer (S-146 [GGGCATAGAGTGGGTTTATCCA] and A-219 [GGGCATAGAGTGGGTTTATCCA]), 0.27 μM JFH1-specific probe {A195 (6-carboxyfluorescein [FAM]-AAAGGACCCAGTCTTCCCGGCAATT-6-carboxytetramethylrhodamine [TAMRA])}, 3 μl of template RNA, and RNase-free water. To determine absolute RNA amounts, a serial dilution of an RNA standard (103 to 108 HCV RNA copies per reaction) was processed in parallel. Reactions were performed using the following program: 50°C for 10 min, 95°C for 1 min, and 40 cycles as follows: 95°C for 10 s and 60°C for 1 min.
Statistical analysis.
To test for significance, a two-tailed paired t test was performed using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA). A P value of <0.05 was considered statistically significant. In figures, statistical significance is indicated as follows: *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
Accession numbers.
The consensus sequence assembled based on 10 individual clones has been deposited in GenBank under accession number KR632542. Raw data from the transcriptome analysis were deposited in NCBI's Gene Expression Omnibus and are accessible through GEO series accession number GSE68927.
RESULTS
Impact of IFN-γ on HCV replication in Huh6 and Huh-7 cells.
Interestingly, HCV replication was resistant to treatment with IFN-γ in a Huh6 cell population harboring a persistent genotype 1b replicon, while other viruses remained sensitive and induction of ISGs upon IFN-γ treatment in Huh6 cells was comparable to that in Huh-7 cells (27). Therefore, we assumed that Huh6 cells might lack expression of distinct effector proteins mediating IFN-γ response against HCV.
We first reinvestigated the lack of suppression of HCV replication in response to IFN-γ in Huh6 cells by comparing IFN-γ sensitivity of persistent replicons in Huh6 cells compared to that in Huh-7-Lunet cells, a cell line highly permissive for HCV replication (30). We treated both genotype 1b (isolate Con1) and genotype 2a (isolate JFH1) Huh6 replicon cell lines with IFN-γ for 96 h and assessed HCV replication by quantification of NS3 protein expression by Western blotting (Fig. 1A and B) and HCV genomic RNA by Northern blotting (Fig. 1C and D). Con1 and JFH1 replication in Huh6 cells was moderately reduced, to a minimum of 40% compared to that in untreated cells, whereas in Huh-7 cells the RNA signal became undetectable within 48 h (Fig. 1C and D) and only residual amounts of protein were detectable at 96 h (Fig. 1A and B). To exclude the possibility that the apparent IFN-γ resistance in Huh6 cells was transient, we performed long-term culture experiments. The same numbers of Huh6 and Huh-7 cells harboring persistent replicons were seeded and treated with both IFN-γ and G418. On every third to fourth day, cells were detached and counted (Fig. 1E), and the same number of cells for each population was reseeded and retreated. We expected that IFN-γ-mediated inhibition of virus replication should result in decreased growth rates and even cell death upon longer treatment, since G418 resistance is dependent on active replication of the replicon. Indeed, at passage 5, Huh-7-Lunet cell numbers declined rapidly, while Huh6 cell numbers remained fairly stable throughout the experiment (Fig. 1E). Interestingly, the genotype 2a containing Huh-7 cell population was completely extinguished, while in the case of genotype 1b, at passage 8 an IFN-γ-resistant population emerged (Fig. 1E). This result demonstrated that HCV replication was indeed not sensitive to IFN-γ treatment in long-term Huh6 cultures and sensitive in Huh-7 cells. However, also in the case of Huh-7 cells, HCV can acquire IFN-γ resistance, albeit with very low efficiency (ca. 1 in 1 × 106 cells [K. Esser-Nobis and V. Lohmann, unpublished data]).
FIG 1.

IFN-γ sensitivity of HCV replicons in Huh-7 and Huh6 cells. (A) Western blot analysis of stable HCV replicon cell lines derived from Huh6 and Huh-7 cells. Cells were treated with 10 ng/ml of IFN-γ or remained untreated for the indicated times. HCV NS3 protein was detected by using secondary antibodies labeled with IRDye and the LI-COR Odyssey system. Actin was used as a loading control. (B) Quantification of Western blot results in panel A. Band intensity was determined by using ImageJ software. Background was subtracted, and NS3 band intensity was normalized (norm.) to actin band intensity. Shown is the ratio of treated to untreated samples in percent. (C) Northern blot analysis of stable HCV replicon cell lines derived from Huh6 and Huh-7 cells. Cells were treated with 10 ng/ml of IFN-γ or remained untreated for the indicated times. RNA was used for Northern blot analysis with 32P-labeled riboprobes specific for the neomycin resistance sequence, within the genomic HCV RNA or actin mRNA as a loading control. Two independent cell lines were analyzed for each genotype in the case of Huh6 cells and one was analyzed for Huh-7 cells. One representative blot is shown for each condition. (D) Quantification of Northern blot results in panel C. Band intensity was determined by using ImageJ software. Background was subtracted, and the HCV RNA band intensity was normalized to the actin band intensity. Two independent cell lines were analyzed for Huh6 cells and one was analyzed for Huh-7 cells. Shown is the ratio of treated to untreated samples in percent. (E) Long-term culture of stable Huh6 and Huh-7 replicon cell lines. Both replicon cell lines were seeded and then treated with G418 for selection of the HCV replicon. Additionally, 10 ng/ml of IFN-γ was added to select for IFN-γ-resistant cell populations. Cells were detached and counted, and the same amounts were reseeded every 3 to 4 days. Plotted is the ratio of cell numbers between G418- and IFN-γ-treated cells relative to that of cells treated with G418 alone. The experiment was performed twice with Huh6-derived cell lines and once with Huh-7-derived cell lines. Error bars indicate SDs. (F) Transient replication of HCV FLuc reporter replicons under treatment with IFN-γ. Naive Huh6 and Huh-7 cell lines were transfected with HCV FLuc reporter RNA (LucJFH1) and treated with IFN-γ at the given concentrations. Forty-eight hours after treatment, HCV replication was measured by luciferase assay. The experiment was performed 4 times; the results of one representative experiment are shown. Plotted are means between triplicate samples; error bars indicate SDs. rel, relative.
We furthermore assessed inhibition of HCV replication by IFN-γ in naive Huh6 and Huh-7-Lunet using genotype 2a firefly luciferase (FLuc) reporter replicons (LucJFH1 [40]), since only JFH1-based replicons replicated robustly enough in Huh6 cells to allow transient-replication assays (34). Indeed, HCV replication was completely abolished in Huh-7 cells (IC90 = 0.18 ng/ml) (Fig. 1F), whereas in Huh6 cells transient-replication levels were only moderately reduced, even at high concentrations of IFN-γ.
In summary, our results confirmed that HCV replication of genotypes 1 and 2 was sensitive to IFN-γ in Huh-7 cells and partially resistant in Huh6 cells, as has been described before (27, 29).
Screening for IFN-γ-induced candidate genes with potential effector functions in the IFN-γ response against HCV.
Resistance of HCV to IFN-γ appeared to be an intrinsic property of Huh6 cells. We hypothesized that this was due to factors involved in the IFN-γ response that were missing in Huh6 cells but present in Huh-7 cells. This should ultimately also include direct effectors mediating inhibition of viral replication. Since HCV replication was not entirely resistant to IFN-γ in Huh6 cells, we assumed that candidates might not be completely absent in Huh6 cells. However, we reasoned that candidate effector genes should be induced to a higher extent by IFN-γ in Huh-7 than in Huh6 cells. To identify such factors, we performed microarray-based gene expression analysis after IFN-γ treatment of naive Huh6 and Huh-7 cells (see Table S1 in the supplemental material), calculated the fold induction for each gene compared to that in the untreated cell population, and divided fold induction values in Huh-7 cells by those in Huh6 cells (Fig. 2A). Generally, levels of ISG induction upon IFN-γ treatment were similar in Huh6 and Huh-7 cells, resulting in a >2-fold upregulation of 115 and 140 genes, respectively (see Table S1). Genes which were less than 2-fold induced in Huh6 cells by IFN-γ and showing at least 3-fold-higher induction in Huh-7 cells than in Huh6 cells were considered candidates, in total resulting in a selection of 23 genes (Table 1). We focused on the identification of direct effectors implicated in the antiviral IFN-γ response, and therefore, we excluded several candidate genes. These included (i) genes for secreted proteins, such as interleukins, chemokines, and complement factors (CXCL9, CXCL11, CFH, IL-7, MT2A, IL-32, and CFB; although the lack of particular cytokines in Huh6 cells was striking and potentially contributed to the differences in the functional IFN-γ response in Huh6 and Huh-7 cells, e.g., by amplifying changes in gene expression in response to IFNs or by inducing additional subsets of genes, they were unlikely direct effectors), (ii) known ISGs lacking antiviral activity against HCV (Mx1 [46]), and (iii) genes unlikely having direct effector functions, like major histocompatibility complex (MHC) class I genes (HLA-A and HLA-B) and TLR3. Out of 23 genes, 11 were considered for further experimental validation: BTN3A3, CASP1, CARD16, BTN3A2, HERC6, SERPINA7, CTSS, TRIM31, NLRC5, DDX60L, and ACY3 (Table 1).
FIG 2.

Identification of effector proteins of the IFN-γ response against HCV. (A) Strategy to identify candidate effectors of the IFN-γ response against HCV by gene expression analysis. Huh-7 and Huh6 cells were treated with 1,000 IU/ml of IFN-γ or left untreated for 24 h. Total RNA was used for gene expression analysis by Affymetrix HGU133A Plus 2.0 Genechip microarray. Relative induction by IFN-γ treatment was calculated for each cell line, and relative induction values in Huh-7 cells were divided by relative induction values in Huh6 cells for each gene. Genes with more than ca. 3-fold (log2 > 1.58) higher induction in Huh-7 cells were considered candidate effectors of IFN-γ. (B) Experimental strategy used for screening of candidates. siRNAs directed against candidate genes listed in Table 1 were transfected together with HCV FLuc reporter replicon RNA (genotype 1b or genotype 2a) into naive Huh-7-Lunet cells. Twenty-four hours after transfection, cells were treated with 0.15 ng/ml of IFN-γ. HCV replication was determined 96 h after transfection by luciferase assay. (C) Results of the primary candidate screening. The experiment was performed as described for panel B, using genotype 1b (LucCon1ET) reporter replicons. Luciferase activity was normalized to that of the untreated siNT control. siIFNgR was used as a control for rescue of HCV replication. The experiment was performed twice; shown are means with SDs. (D) siRNAs against DDX60L were tested in the same setting as for panel B, but using a genotype 2a FLuc reporter replicon (LucJFH1) instead. Luciferase values were normalized to that of the untreated siNT control. Experiment was performed twice; shown are means with SDs. Note that in the case of siDDX60L_5, results did not reach statistical significance due to high variability among independent replicates. (E) Knockdown efficiency of siRNAs against DDX60L in the presence and absence of IFN-γ. DDX60L mRNA levels were determined by qRT-PCR 48 h after transfection. Results were normalized to those for untreated siNT.
To determine if any of these candidates played a role in IFN-γ-mediated inhibition of HCV replication, we silenced expression of each respective gene in Huh-7 cells to restrict the functional IFN-γ response and increase viral replication after IFN-γ treatment. This strategy previously allowed us to identify novel effector ISGs involved in IFN-α and IFN-γ responses against HCV (6).
In this study, we independently cotransfected two siRNAs per gene with HCV Con1 FLuc reporter replicons (LucCon1ET) into Huh-7-Lunet cells. Twenty-four hours after transfection, cells were treated with an IFN-γ concentration slightly below the IC90 (Fig. 1F), and 72 h later, HCV replication was assessed by measuring firefly luciferase activity (Fig. 2B). Knockdown of interferon gamma receptor 2 (IFN-γR) was used as a positive control for rescue of HCV replication in the presence of IFN-γ (6). Data were normalized to the luciferase activity obtained upon transfection of a nontargeting siRNA (siNT) in the absence of IFN-γ treatment, which was set to 100% (Fig. 2C). As expected, HCV replication was reduced to approximately 10% after IFN-γ treatment (and siNT transfection) and rescued by knockdown of IFN-γR to more than 50% (Fig. 2C). Most siRNAs did not show any or showed only minor effects on HCV replication under IFN-γ treatment, not reaching statistical significance. However, we did not test knockdown efficiencies in these experiments, so it cannot be excluded that these factors still play a role in the IFN-γ response against HCV. Interestingly, two siRNAs, both targeting DDX60L, mediated a significant rescue of HCV replication in the presence of IFN-γ, resulting in a 2- to 3-fold increase of HCV replication compared to siNT (Fig. 2C). We additionally tested the entire panel of candidate genes for their capability to rescue genotype 2a replication using the same experimental setup (replicon LucJFH1) (Fig. 2B). Again, only knockdown of DDX60L rescued HCV replication from IFN-γ treatment (Fig. 2D), as we had observed for genotype 1b, whereas knockdown of all other candidates had no significant effects (data not shown). We furthermore determined knockdown efficiency in the presence and absence of IFN-γ for the siRNAs targeting DDX60L (Fig. 2E). Indeed, both siRNAs reduced DDX60L mRNA levels, albeit to various extents, reflecting their efficiencies in rescuing HCV replication. Importantly, both siRNAs were capable of limiting induction of DDX60L mRNA expression upon IFN-γ treatment to the level of the untreated samples (Fig. 2E). In summary, by comparing genes differentially induced by IFN-γ in Huh-7 and Huh6 cells and screening for their effects on HCV replication, we identified DDX60L as a novel effector protein mediating the IFN-γ response against HCV in Huh-7 cells.
DDX60L acts independently from DDX60 and restricts HCV RNA replication in Huh-7 cells.
DDX60L is a member of the large family of DExD/H box helicases, but no distinct function has been assigned to it so far. The sequence of DDX60L is more than 70% identical to that of DEAD box polypeptide 60 (DDX60), which has been implicated in RIG-I signaling (23) and RNA stability (25) and has been identified as a candidate effector protein against HCV (15). Therefore, we next aimed to validate the specific role of DDX60L in the IFN-γ response against HCV using cell lines harboring persistent genotype 2a FLuc reporter replicons (LucubineoJFH1) (Fig. 3A) and to dissect the function of DDX60L and its homolog DDX60. We tested a broader set of siRNAs against DDX60L and an siRNA targeting DDX60 (23), all of which were specific for their respective targets (Fig. 3B and data not shown). The siRNA siDDX60L_6 was excluded from further experiments due to cytostatic effects (Fig. 3C), which were also apparent by microscopic analysis and might interfere with HCV replication (27). LucubineoJFH1 cells were transfected with siRNAs targeting DDX60L, DDX60, or IFN-γR, and HCV replication was assessed by measuring luciferase activity in the presence and absence of IFN-γ treatment (Fig. 3A, D, and E). Interestingly, HCV replication was already increased upon knockdown of DDX60L in the absence of IFN-γ (Fig. 3D), which was not the case in DDX60-silenced cells. This result indicated that DDX60L was a factor generally restricting HCV replication in Huh-7 cells. To assess the specific contribution of DDX60L to the IFN-γ response, we normalized the data to those for the untreated controls specific for the siRNAs (Fig. 3E). This again revealed a significant contribution of DDX60L but not of DDX60 to a functional IFN-γ response against HCV. Since transfection of siRNA into a cell line harboring persistent replicons is not efficient in all cells, limiting the overall measurement window, we confirmed rescue from IFN-γ response by lentiviral transduction with selectable vectors expressing DDX60L-specific shRNAs and selected for cell populations harboring the replicon and expressing shRNA. We used vectors encoding shRNAs against RSAD2 (viperin), a previously described direct inhibitor of HCV replication (47) which substantially contributes to type I and II IFN responses (6), as a positive control. Stable cell populations were treated with IFN-γ, and rescue of replication from IFN-γ was assessed by normalization to each individual untreated cell population. Stable knockdown of DDX60L was comparably efficient (Fig. 3G) and rescued HCV replication to an extent similar to that of RSAD2 (Fig. 3F), suggesting that DDX60L contributes to a functional IFN-γ response to a level comparable to those of previously described effectors.
FIG 3.

DDX60L knockdown increases HCV replication in the absence of IFN-γ and partially rescues HCV replication from IFN-γ treatment. (A) Experimental strategy using replicon cell lines with persistent reporter replicons. HCV FLuc reporter replicon cell lines derived from Huh-7-Lunet cells (LucubineoJFH1, genotype 2a) were transfected with siRNAs. Twenty-four hours after transfection, the cells were treated with 0.15 ng/ml of IFN-γ. Ninety-six hours after transfection, HCV replication was determined by measuring luciferase activity. (B) Knockdown efficiency of transfected siRNAs. Forty-eight hours after transfection of siRNAs, DDX60L mRNA levels were determined by qRT-PCR. Shown are mRNA levels relative to that of the siNT control. (C) Cell viability after siRNA transfection. LucubineoJFH1 cells were transfected with siRNAs as for panel B. Ninety-six hours after transfection, cell viability was determined by WST-1 assay and normalized to the siNT control. Shown are mean values with SDs (n = 3). (D) Replication of HCV after siRNA transfection in the absence of IFN-γ. LucubineoJFH1 cells were transfected with indicated siRNAs. Luciferase activity 96 h after transfection was normalized to that of the siNT control. (E) Rescue of HCV replication in the presence of IFN-γ. Light gray bars correspond to bars in panel D, but luciferase activity at 96 h after transfection was normalized to the value for the untreated control of each siRNA, eliminating differences in HCV replication by the siRNA. Given are mean values with SDs (n = 2). (F) Rescue of HCV replication in the presence of IFN-γ using shRNA-mediated knockdown. LucubineoJFH1 cells were transduced with selectable lentiviral vectors encoding shRNAs against DDX60L and RSAD2. After double selection of populations expressing shRNA and replicons, cells were treated with IFN-γ for 72 h and HCV replication was determined by luciferase assay. Results were normalized to those for the untreated control of each shRNA population. Shown are mean values with SDs (n = 4). (G) Knockdown efficiency of shRNAs. Cells were transduced with the corresponding vectors. Ninety-six hours after transduction, DDX6L mRNA levels were determined by qRT-PCR. mRNA levels are shown relative to that of the shNT control.
We next wanted to evaluate if DDX60L was an upstream component or a downstream effector of the IFN-γ pathway. Therefore, we measured the IFN-γ-induced upregulation of the mRNA expression of the IFIT3 and GBP1 genes, two signature genes of the IFN-γ response in Huh-7 cells (6, 27). However, knockdown of DDX60L, in contrast to IFN-γR, did not alter the IFN-γ-mediated induction of IFIT3 and GBP1 mRNA expression (Fig. 4), arguing against an impact of DDX60L on IFN-γ-mediated JAK/STAT signaling. Taken together, these experiments revealed that DDX60L is a general restriction factor of HCV replication in Huh-7 cells that contributes to functional IFN-γ responses to an extent similar to that of RSAD2/viperin. In contrast, silencing of the homolog DDX60 did not significantly increase HCV replication in Huh-7 cells in either the absence or presence of IFN-γ. Our results furthermore indicate that DDX60L does not affect IFN-γ-induced gene expression but most likely acts as a direct effector protein inhibiting HCV RNA replication.
FIG 4.
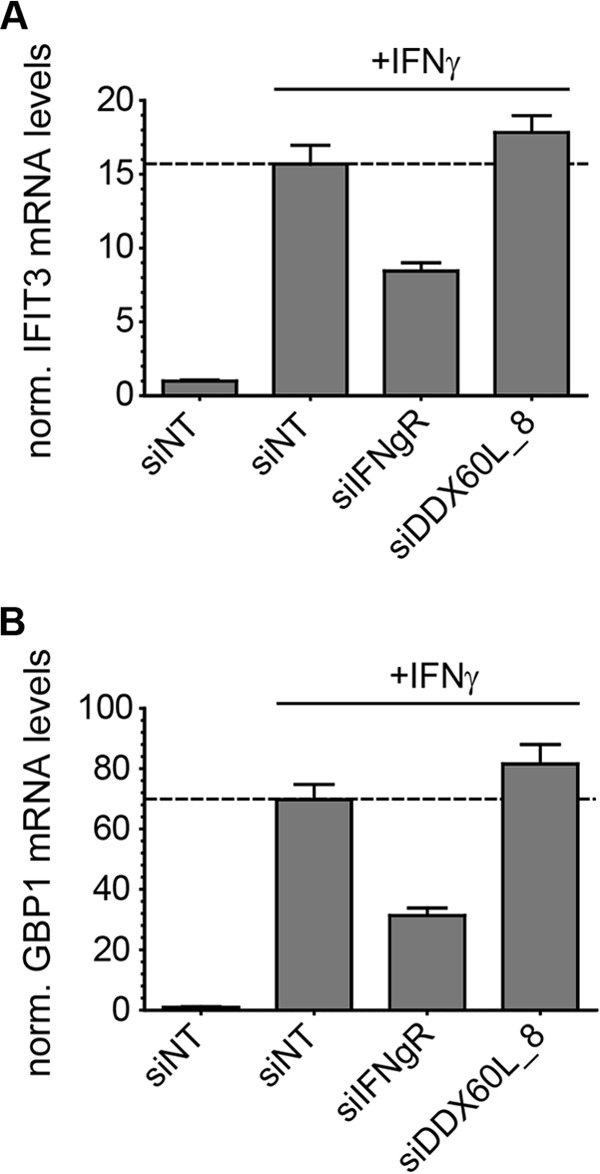
Absence of impact of DDX60L knockdown on ISG response to IFN-γ. Huh-7-Lunet cells cotransfected with LucJFH1 and siRNAs were treated with 0.1 ng/ml of IFN-γ. Twenty-four hours after treatment, IFIT3 (A) or GBP1 (B) mRNA levels were determined by qRT-PCR. Values were normalized to those for GAPDH and untreated siNT controls. Shown are mean values with SDs (n = 2).
HAV replication is not affected by DDX60L knockdown.
IFN-γ inhibits a broad range of viruses; however, previous studies suggested that different viruses and virus groups are targeted by various, partially overlapping sets of effector proteins. We therefore questioned whether DDX60L is involved in the IFN-γ response against HAV, a positive-strand RNA virus grouped into the Picornaviridae family. HAV and HCV share the same hepatotropisms, replicate with similar efficiencies in Huh-7 cells, and are comparably sensitive to IFN-γ (34). Although both viruses harbor a positive-strand genome and share distinct aspects of their biologies, they are also divergent in other respects; e.g., the HAV genome is linked to a 5′-terminal protein and contains a poly(A) tract at its 3′ end (48). HCV and HAV reporter replicons LucJFH1 and LucHAV (34) were cotransfected with siRNAs into Huh-7-Lunet cells, and viral replication and rescue from the IFN-γ response were determined by quantifying luciferase activity in the presence and absence of IFN-γ (Fig. 5A). However, knockdown of DDX60L did not significantly alter HAV replication in the absence of IFN-γ (Fig. 5B), nor did it rescue HAV replication from IFN-γ treatment, unlike replication of HCV (Fig. 5C). Still, these results did not formally rule out minor effector functions of DDX60L, since other ISG products might dominate the functional IFN response against HAV or HAV might be overall less sensitive to DDX60L.
FIG 5.
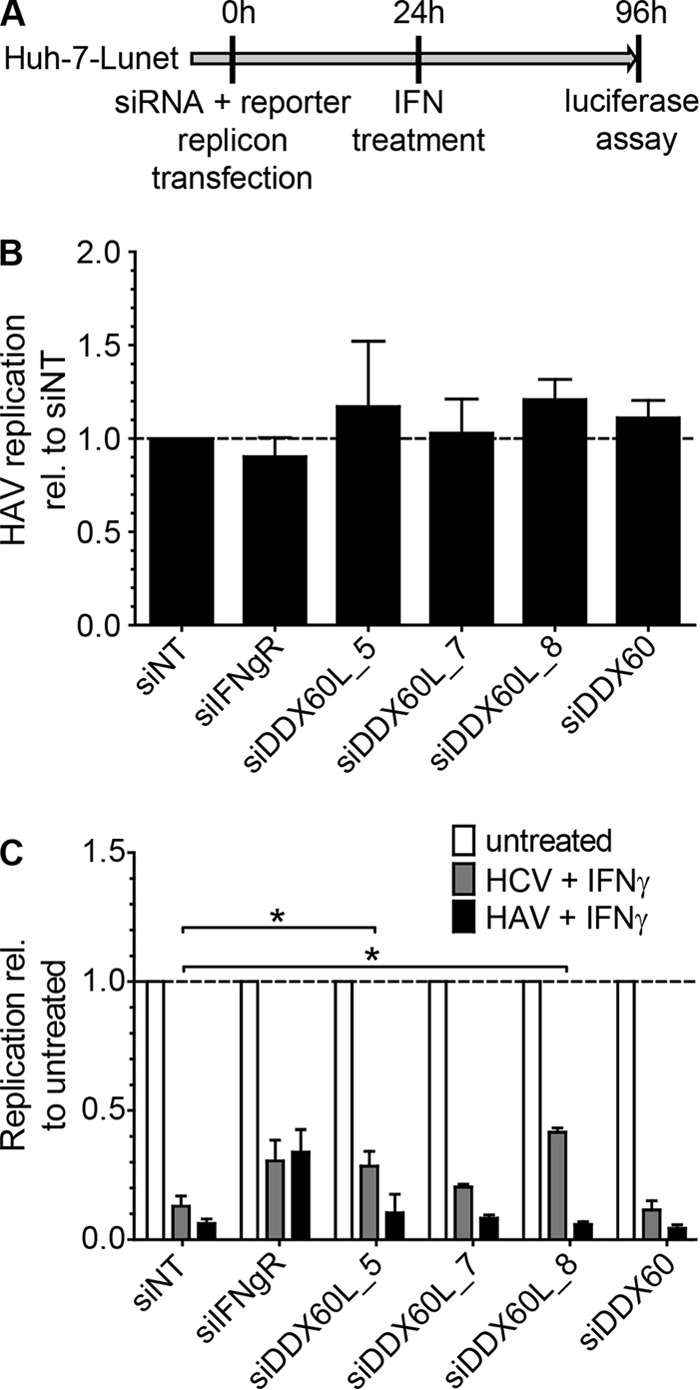
Impact of DDX60L knockdown on HAV replication and IFN-γ response to HAV. (A) Experimental procedure. Naive Huh-7-Lunet cells were cotransfected with HAV (LucHAV) or HCV FLuc reporter replicons (LucJFH1) and siRNAs. Twenty-four hours after transfection, cells were treated with 0.1 ng/ml of IFN-γ or left untreated. Ninety-six hours after transfection, replication was determined by measuring luciferase activity. (B) Impact of DD60XL knockdown on HAV replication. Luciferase activity 96 h after transfection was normalized to that of the siNT control. Shown are means with SDs (n = 2). (C) Absence of rescue of HAV replication from the IFN-γ response upon DDX60L knockdown. Luciferase activity 96 h after transfection was normalized to the untreated samples of each siRNA and reporter replicon, to determine rescue efficiency under IFN-γ treatment. Shown are means with SDs (n = 2).
DDX60L is involved in the type I and type III IFN responses against HCV.
Previous studies have pointed to partially overlapping sets of genes mediating type I, type II, and type III IFN responses (6). In addition, DDX60L had been previously described as a type I IFN response gene, similar to DDX60 (24). Therefore, we were interested in whether DDX60L also significantly contributes to type I and type III IFN responses against HCV. To study this, we first evaluated relative DDX60L induction levels by microarray gene expression analysis of Huh6 and Huh-7 cells treated with IFN-α (Fig. 2A). Interestingly, DDX60L was upregulated in both Huh6 and Huh-7 cells by treatment with IFN-α (Fig. 6A). DDX60L mRNA induction patterns therefore matched the IFN sensitivity pattern of HCV, which was sensitive to IFN-α in both cell types. To confirm DDX60L mRNA induction by IFN-α, we treated Huh-7-Lunet cells with different concentrations of IFN-α and analyzed DDX60L mRNA expression by qRT-PCR; we found it to be induced up to 20-fold upon IFN-α treatment (Fig. 6B). Basal expression of DDX60 mRNA was lower in Huh-7 cells than expression of DDX60L but reached levels comparable to those of DDX60L upon IFN-α treatment (data not shown). In primary human hepatocytes (PHH), DDX60L and DDX60 mRNA levels were strongly induced by IFN-α. They both reached comparable expression levels and displayed very similar induction kinetics, suggesting that both genes are part of the early ISG repertoire of the human liver (Fig. 6C).
FIG 6.

Role of DDX60L in the type I and type III IFN responses. (A) Induction of DDX60L mRNA in Huh-7 and Huh6 cells. Microarray gene expression analysis was performed as described for Fig. 2A. Shown are the relative induction levels of DDX60L in response to treatment with 1,000 IU/ml IFN-α or IFN-γ in Huh-7 and Huh6 cells. (B) Induction of DDX60L by IFN-α treatment in Huh-7-Lunet cells. Huh-7-Lunet cells were treated with the indicated concentrations of IFN-α for 24 h, and DDX60L mRNA was determined by qRT-PCR relative to the untreated control. Shown are mean values with SDs (n = 3). (C) Induction of DDX60L by IFN-α treatment in PHH. PHH were treated with 100 IU/ml of IFN-α, and DDX60L and DDX60 mRNA levels were determined by qRT-PCR and normalized to the value for the untreated control at each given time point. (D) DDX60L but not DDX60 knockdown partially rescues HCV replication upon IFN-α treatment. LucubineoJFH1 cells were transfected with siRNAs against DDX60L and treated with 2.5 IU/ml of IFN-α 24 h after transfection as shown in Fig. 3A. siSTAT2 was used as a control for rescue efficiency. Luciferase activity 96 h after transfection was used as a measure of HCV replication and normalized to that of the untreated control of each siRNA. Shown are means with SDs (n = 2). (E) Induction of DDX60L mRNA expression by IFN-λ treatment in Huh-7-Lunet cells. Huh-7-Lunet cells were treated with the indicated concentrations of IFN-λ for 24 h, and DDX60L mRNA was determined by qRT-PCR relative to that of the untreated control. Shown are mean values with SDs (n = 2). (F) DDX60L but not DDX60 knockdown partially rescues HCV replication upon IFN-λ treatment. LucubineoJFH1 cells were transfected with an siRNA against DDX60L and treated with 0.5 ng/ml of IFN-λ 24 h after transfection. siSTAT2 was used as a control for rescue efficiency. Luciferase activity 96 h after transfection was used as a measure of HCV replication and normalized to that of the untreated control of each siRNA. Shown are means with SDs (n = 2).
Since expression of DDX60L and DDX60 was upregulated by IFN-α in both Huh-7-Lunet cells and PHH, we next aimed to evaluate whether knockdown of these genes would also rescue HCV replication from IFN-α responses. We followed the same experimental strategy for IFN-α as for IFN-γ before (Fig. 3A), silenced DDX60L and DDX60 in HCV LucubineoJFH1 cells, and assessed HCV replication in the presence and absence of IFN-α. Silencing of STAT2 served as a positive control. Knockdown was specific for DDX60L and DDX60, dampening the IFN-α-mediated increase in mRNA expression with similar efficiencies for both genes (data not shown). Importantly, silencing of DDX60L but not of DDX60 substantially rescued HCV replication from IFN-α treatment (Fig. 6D), demonstrating that DDX60L indeed was a key effector protein not only of the type II but also of the type I IFN response against HCV replication in Huh-7 cells. Finally, using the same experimental model as for IFN-α, we assessed the contribution of DDX60L to the type III IFN response. Indeed, DDX60L mRNA expression was also upregulated dose dependently by IFN-λ (Fig. 6E), and HCV replication was partially rescued from the IFN-λ response upon knockdown of DDX60L but not DDX60 (Fig. 6F).
In summary, these results showed that DDX60L is induced upon IFN-α and IFN-λ treatment and contributes significantly to the suppression of HCV replication by type I and type III IFNs.
DDX60L is induced during HCV infection in PHH and contributes to RIG-I activation.
Since these results clearly established DDX60L as an important contributor to IFN responses against HCV in Huh-7 cells, we next aimed to clarify if DDX60L expression is directly induced by HCV. Recent studies have shown that HCV replication mounts a substantial ISG response in infected cells in vivo (3, 12) and in PHH (6, 49), despite its ability to cleave MAVS, an essential adaptor in the IRF-3 signaling pathway leading to suppressed type I and III IFN production (50). Since the direct activation of IRF-3-dependent responses is generally weak in hepatoma cells, like Huh-7 cells (51), we analyzed the ISG induction in PHH infected with HCV. To this end, we measured the expression levels of DDX60L, DDX60, and IFN-β mRNA at different time points after infection with cell culture-derived HCV particles at an MOI of 5 TCID50/ml. HCV infection resulted in strong induction of all genes tested starting as early as 18 h postinfection (Fig. 7A), at the onset of viral replication (Fig. 7B). Induction of DDX60L and DDX60 mRNA expression was comparable to that in IFN-α-treated cells and occurred in parallel to IFN-β mRNA induction but was much more sustained, similar to what was previously found for other ISGs (6). This fast and strong ISG response was most likely the reason for the limited replication of HCV in PHH, which only reached input RNA levels 48 h postinfection, before RNA levels declined again, as in a previous study (6) (Fig. 7B).
FIG 7.
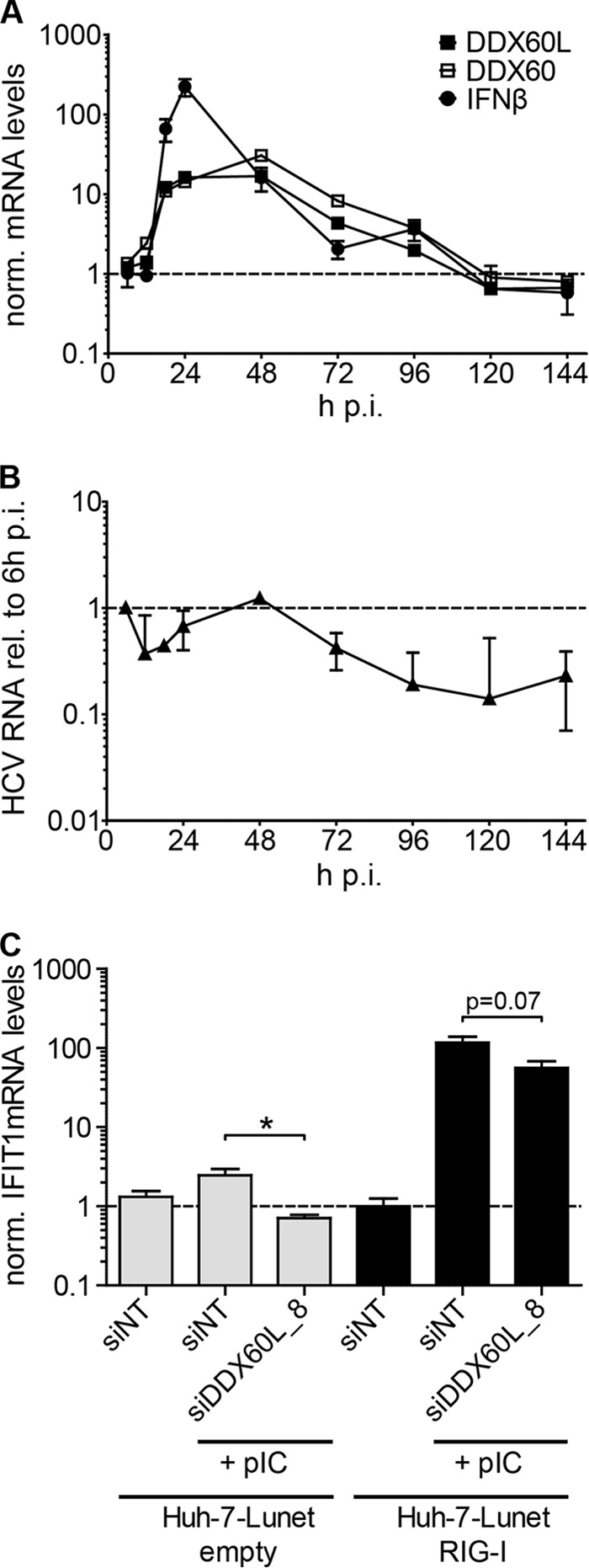
DDX60L and DDX60 expression during HCV infection and impact of DDX60L on RIG-I activation. (A and B) PHH were infected with cell culture-derived HCV with an MOI of 5 TCID50/cell for 6 h. mRNA levels of DDX60L, DDX60, and IFN-β as well as HCV RNA were determined by qRT-PCR at various time points after infection. The experiment was performed in triplicate with cells from one donor. (A) Expression of ISGs during HCV infection. DDX60L, DDX60, and IFN-β mRNA levels were determined by qRT-PCR and normalized to the mock-infected control at each time point. Note that baseline expressions of DDX60L and DDX60 were similar (data not shown). (B) Quantification of HCV RNA by qRT-PCR in samples shown in panel A. HCV RNA levels were normalized to the input RNA at 6 h postinfection. Relative values correspond to about 4 HCV strands per cell at 6 h postinfection (p.i.). (C) Impact of DDX60L on RIG-I activation. Huh-7-Lunet cells stably overexpressing RIG-I (Huh-7-Lunet-RIG-I) and empty control cells were transfected with an siRNA targeting DDX60L or a nontargeting control (siNT). Forty-eight hours later, cells were transfected with 100 ng of poly(I·C) and harvested 12 h later to quantify ISG induction. IFIT1 mRNA levels determined by qRT-PCR and normalized to GAPDH and unstimulated Huh-7-Lunet-RIG-I cells transfected with siNT control. Shown are mean values with SDs (n = 2).
DDX60 has been shown to enhance induction of the ISG response by stimulating RIG-I (23, 25). Since our results indicated that significant amounts of DDX60L are already present in the absence of IFN treatment, we reasoned that DDX60L also might enhance the RIG-I-dependent ISG response and thereby contribute to the onset of innate immune responses, similar to DDX60. To address a potential role for DDX60L in RIG-I-mediated IRF-3 activation, we silenced DDX60L in Huh-7-Lunet cells transduced with an empty vector or expressing RIG-I and stimulated the IRF-3 response by transfection of poly(I·C). Induction of IFIT1 mRNA expression was used as a readout for RIG-I activation (Fig. 7C). IFIT1 mRNA expression was increased only 2-fold in naive Huh-7-Lunet upon transfection of poly(I·C), as reported previously (42), but was dramatically increased upon overexpression of RIG-I (Fig. 7C). However, in both experimental settings, knockdown of DDX60L resulted in a decrease in IFIT1 mRNA induction levels (approximately 2-fold reduction), but this did not reach statistical significance in RIG-I-overexpressing cells (Fig. 7C). Nevertheless, these results implied that DDX60L might contribute to RIG-I-mediated activation of innate immune responses, as reported for DDX60 (23, 25).
These results revealed that DDX60L expression is induced early during acute HCV infection of PHH with a kinetics similar to that for the related DDX60. DDX60L furthermore enhances RIG-I-mediated IRF-3 activation, as previously shown for DDX60.
Ectopic expression of DDX60L inhibits HCV replication and hampers production of lentiviral vectors.
The above-described experiments defined a role for DDX60L in the interferon-mediated suppression of HCV replication by silencing of endogenous mRNA. We next tried to detect endogenous DDX60L protein by IF and Western blotting, using two commercially available antibodies. However, as judged by their responsiveness to interferon treatment or knockdown, we failed to detect signals specific for DDX60L in the presence and absence of IFN and proteasome inhibitors (data not shown). To overcome this problem, we aimed to overexpress DDX60L ectopically. Due to the limited availability of DDX60L cDNA from public libraries or commercial sources, which might be due to its large size (5.1 kb, encoding a 198-kDa protein), we decided to clone the cDNA from Huh-7-Lunet cells, thereby ensuring that we would obtain the specific coding sequence with a proven ability to inhibit HCV replication. We generated a consensus sequence based on 10 individual plasmid clones differing at 6 amino acid positions from the GenBank entry, reflecting documented polymorphisms in the human population.
Based on this Huh-7-specific consensus sequence, we next aimed to generate cell lines stably expressing DDX60L using a selectable lentiviral vector. However, we were not able to select a cell population that survived the selection process. In search of the underlying cause, we assessed which step of our vector production/transduction procedure was affected. Transduction of HeLa cells after serial dilution and subsequent selection to allow single colony formation revealed that virus stocks generated with empty vector controls contained approximately 1 × 107 CFU (Fig. 8A). Compared to this, titers of DDX60L-carrying lentiviruses were strongly reduced, by more than 4 orders of magnitude. A possible reason was the large insert size of DDX60L (5.1 kb, encoding a 198-kDa protein), which is known to reduce lentiviral vector titers (52). Therefore, we analyzed the lentiviral titers of a vector carrying the coding sequence for phosphatidylinositol-4-kinase IIIα (PI4KIIIα) (31), containing a 6.3-kb insert, encoding a 240-kDa protein. In fact, titers after PI4KIIIα transfection displayed a reduction of about 50-fold compared to the empty vector control (Fig. 8A). However, the DDX60L-encoding vectors were clearly impaired an additional 400-fold, suggesting a specific inhibitory effect of DDX60L on the vector titer. This defect did not manifest at the level of viral protein synthesis during vector production, since similar amounts of the Gag polyprotein p55 as well as its processing products p41, p31, and p24Gag were detected in the lysates of vector-producing 293T cells (Fig. 8B). The amounts of viral particles released in the cell culture supernatants were slightly (approximately 5-fold) reduced for DDX60L-containing vectors as assessed by reverse transcriptase activity or p24Gag protein levels (data not shown). However, this moderate reduction in the production of physical particles could not account for the dramatic drop in infectious titer observed.
FIG 8.

Ectopic expression of DDX60L using lentiviral vectors. (A) Titers of lentiviral vectors encoding DDX60L or PI4KIIIα compared to empty vectors. pWPI constructs encoding the indicated inserts and packaging plasmids were transfected into 293T cells to generate lentiviral vectors. Forty-eight hours after transfection, supernatants were harvested. All data represent mean values with SDs (n = 4). Infectious titers were determined by transduction of HeLa cells and selection for the appropriate antibiotic resistance (puro, puromycin; blr, blasticidin). CFU per milliliter were calculated by counting cell colonies 4 days posttransduction. (B) Western blotting for HIV-1 Ca protein in lentiviral vector-producing 293T cell lysates. Transferrin receptor was used as a loading control. Shown is a representative blot of three independent experiments. (C) Titers of lentiviral vectors encoding doxycycline-inducible DDX60L are not strongly reduced. Lentivirus was generated and titers were determined as described for panel A, with pLVX-based inducible expression vectors encoding DDX60L, HA- or eGFP-tagged DDX60L, or eGFP as a control. Data represent mean values with SDs (n = 2). (D) Expression of DDX60L by the inducible pLVX system. Huh-7-Lunet cells transduced with lentiviral vectors encoding the inducible DDX60L-HA or with an empty vector were transfected with HCV Fluc reporter replicons. Twenty-four hours after transfection, cells were treated with doxycycline (Dox) to induce expression of DDX60L. Seventy-two hours after transfection, total RNA was isolated and DDX60L mRNA levels were determined by qRT-PCR. DDX60L mRNA levels were normalized to that of the empty vector control (−Dox). Shown are mean values with SDs (n = 2). (E) Inducible overexpression of DDX60L reduces HCV replication. Cells were treated as described for panel D. Seventy-two hours after transfection, HCV replication was determined by luciferase assay. Shown are mean values normalized to that of the empty vector control (−Dox) with SDs (n = 4). (F) Absence of impact of inducible DDX60L overexpression on ISG response. IFIT1 mRNA levels were determined in total RNA samples from panel D. Shown are mean values with SDs (n = 2).
Since the infectivity of DDX60L containing lentiviral vectors was markedly reduced, suggesting the DDX60L protein as a potent restriction factor of lentiviral infection, we next generated lentiviral vectors with doxycycline-inducible DDX60L expression because in this setting DDX60L should not be expressed during production of the lentiviral particles. In this case, vector titers were only slightly reduced by the presence of a DDX60L insert compared to controls (Fig. 8C), and we were able to select for stably transduced cells. However, we were not able to detect DDX60L protein either by IF or by Western blotting, using antibodies specific for DDX60L or the HA tag or autofluorescence of green fluorescent protein (GFP)-tagged DDX60L in the presence and absence of doxycycline. Therefore, we confirmed DDX60L mRNA expression by qRT-PCR and found already enhanced mRNA levels prior to induction, which were further increased by addition of doxycycline (Fig. 8D). Indeed, HCV replication was repressed in cells transduced with inducible vectors encoding DDX60L compared to cells transduced with an empty vector (Fig. 8E), arguing for a direct antiviral activity of DDX60L. Importantly, we found no evidence for induction of other ISGs upon ectopic expression of DDX60L (Fig. 8F), suggesting that DDX60L indeed is a direct effector counteracting viral replication and does not act by enhancing expression of other ISGs.
DDX60L protein shows a broad cytoplasmic distribution, forms aggregates, and causes cytopathic effects upon high expression levels.
To further analyze the impact of DDX60L overexpression on HCV replication and to finally detect DDX60L protein, we next tried transient transfection of plasmids encoding DDX60L under transcriptional control of the strong cytomegalovirus (CMV) promoter. We transfected increasing amounts of plasmid DNA encoding DDX60L into LucubineoJFH1 cells. The total amount of transfected DNA was kept constant by addition of vector DNA encoding GFP, which also served as a negative control (Fig. 9A). As expected, HCV replication decreased upon transfection of increasing doses of DDX60L-encoding plasmid DNA (Fig. 9A). However, cell viability was dose dependently affected by transfection of DDX60L DNA (Fig. 9B), which was not observed using inducible lentiviral vectors (data not shown). Therefore, we could not distinguish unequivocally whether decreased HCV replication was due to direct DDX60L-mediated inhibition or due to cell death induced by DDX60L overexpression. Again, neither untagged DDX60L nor HA- and GFP-tagged DDX60L proteins could be detected by Western blotting or IF. To finally visualize DDX60L protein, we next implicated an even stronger expression system based on transient transfection of genes under the transcriptional control of the T7 RNA polymerase promoter into cell lines constitutively expressing T7 RNA polymerase, which allowed in previous studies efficient expression even of huge open reading frames, like those of HCV NS3-5B (ca. 290 kDa) and PI4KIIIα (240 kDa) (31). Indeed, using HA-tagged DDX60L, we could detect the protein by Western blotting (Fig. 9D), but under standard denaturing conditions (5 min at 95°C), the entire protein species ran at extremely high molecular masses, suggesting that it forms aggregates. Using milder denaturing conditions, some of the protein could be resolved, but it remained a smear covering an extremely broad range of mainly higher molecular masses (Fig. 9D). Very similar results were obtained upon in vitro translation of radiolabeled DDX60L, again giving rise not to a distinct band at the expected size of 200 kDa but rather to a broad smear (data not shown). Neither molecular mass distribution nor abundance of DDX60L could be changed by treatment with proteasome or protease inhibitors (data not shown). These results suggested that DDX60L tends to form aggregates. However, by being able to detect HA-tagged DDX60L, we could confirm that one of the commercially available antibodies was specific for DDX60L (Fig. 9D), suggesting that the amount of endogenous DDX60L was indeed beyond the limit of detection by Western blotting. With the ability to detect ectopically expressed HA-tagged DDX60L protein, we next analyzed the impact of DDX60L expression on HCV replication (Fig. 9E) and the localization of DDX60L relative to HCV proteins (Fig. 9F). We transfected subgenomic replicons into Huh7-T7 cells and transfected plasmids encoding DDX60L, enhanced GFP (eGFP), or empty vectors 48 h later, to first allow the establishment of robust HCV replication (Fig. 9C). Indeed, only plasmids encoding DDX60L substantially suppressed HCV replication (Fig. 9E), confirming our previous data using other expression models (Fig. 8E and 9A) but now allowing study of the subcellular localization of DDX60L. However, most cells with strong HA-DDX60L expression clearly displayed cytopathic effects in the presence (Fig. 9F, marked with an asterisk) and absence (data not shown) of HCV, indicating that high expression levels of DDX60L indeed might cause cell death. DDX60L was widely distributed throughout the cytoplasm (Fig. 9F), similar to DDX60 (23, 25). Strong DDX60L expression was rarely found in the presence of HCV replication (Fig. 9F). However, in these cases we found no indication for a generally altered localization of DDX60L (Fig. 9F, right). DDX60L and NS5A only sometimes colocalized in individual spots, but not to a statistically significant level (data not shown).
FIG 9.

Ectopic expression of DDX60L using transient transfection. (A and B) DDX60L overexpression under transcriptional control of the CMV promoter by transient transfection of plasmids. LucubineoJFH1 cells were transfected with 300 ng of plasmid, either pcDNA-GFP or 10, 100, or 300 ng of pcDNA-DDX60L, adjusted with pcDNA-GFP to 300 ng or mock transfected, and analyzed 48 h later. Shown are mean values with SDs normalized to values for mock-transfected samples (n = 2). (A) HCV replication was determined by luciferase assay. (B) Cell viability after DDX60L overexpression was determined by WST-1 cell proliferation assay. (C) Experimental strategy for analyzing the impact of DDX60L overexpression on HCV replication using a T7 RNA polymerase-based expression system. Huh-7-Lunet cells expressing T7-RNA polymerase were transfected with HCV FLuc reporter replicons (genotype 2a). After 48 h, the cells were transfected with pTM vectors encoding DDX60L variants, eGFP, or an empty vector control. Twenty-four hours later, cells were harvested and analyzed by luciferase assay for HCV replication (E) or by IF (F). (D) DDX60L protein detection by Western blotting. Protein lysates from Huh-7-Lunet-T7 cells transfected with the indicated pTM constructs were denatured in SDS sample buffer at different temperatures. Denatured lysates were analyzed by SDS-PAGE followed by Western blotting using antibodies detecting HA or DDX60L, as indicated. Note that a lower denaturation temperature led to a slightly better resolution of protein aggregates. β-Actin was used as a loading control. (E) DDX60L overexpression decreases HCV replication. Huh-7-Lunet-T7 cells transfected with HCV Fluc reporter replicon RNA were transfected with pTM vectors encoding DDX60L or eGFP as a control as indicated in panel C. HCV replication was determined by luciferase assay 72 h after transfection of HCV RNA and 24 h after transfection of plasmid DNA. Values are normalized to that of the empty vector control. Shown are mean values with SDs (n = 2). (F) Detection of DDX60L and HCV NS5A by immunofluorescence (IF) microscopy. Cells transfected with HCV replicon RNA and pTM plasmids encoding HA-tagged DDX60L as indicated in panel C or mock-transfected cells were fixed, DDX60L was visualized by IF staining for the HA tag, and HCV replication complexes were detected by using 9E10 antibody directed against NS5A. The HA signal is shown in green, the NS5A signal in red. An asterisk indicates a dying cell.
Taken together, the results showed that ectopic expression of DDX60L in three different models inhibited HCV replication but did not induce expression of other ISG products, further confirming that DDX60L is a direct effector protein inhibiting viral RNA replication. DDX60L protein was detectable only upon massive overexpression, which was associated with cytopathic effects. The protein is widely distributed throughout the cytoplasm, and we found only minor colocalization with HCV NS5A.
DDX60L affects viral RNA replication independently from IRES-dependent translation and RNA stability.
The lack of impact of DDX60L on IFN-γ-induced gene expression as well as the inhibition of HCV replication upon DDX60L expression suggested that DDX60L acts as a direct effector protein inhibiting HCV replication, although we did not find a strong colocalization of DDX60L and HCV proteins. We therefore aimed to narrow down which step of the replication cycle was affected by DDX60L. All our experiments so far were based on subgenomic replicons lacking the viral structural proteins, suggesting that viral RNA replication was the most plausible target of DDX60L. Still, we aimed to exclude a further inhibitory role for DDX60L in viral assembly and release and therefore analyzed the impact of DDX60L knockdown on the rescue of HCV infection and particle production using infectious virus. To this end, we used a monocistronic reporter virus (JcR2A [36]) to infect Huh7.5 cells after transfection of siRNA (Fig. 10A). Twenty-four hours after infection, cells were pulse-treated with IFN for 24 h to assess the impact of DDX60L on rescue of viral infection by determining Renilla luciferase activity in cell lysates (Fig. 10B). The IFN-free supernatant at the time point of cell harvest was used for reinfection of naive Huh7.5 cells to determine additional effects of DDX60L knockdown on viral assembly and particle release (Fig. 10C). DDX60L knockdown indeed rescued JcR2A replication after infection and treatment with IFN-α and IFN-γ (Fig. 10B), demonstrating that DDX60L also is an important effector in the IFN response against HCV infection. We also obtained more infectious virus from cells transfected with siDDX60L compared to siNT (Fig. 10C). However, the relative increase in viral titers simply mirrored the rescue of replication after infection (compare Fig. 10C and B), suggesting that DDX60L mainly targets viral RNA replication and has no further impact on viral assembly or release.
FIG 10.

Impact of DDX60L on HCVcc infection, IRES-dependent translation, and stability of HCV RNA. (A) Experimental strategy to determine the impact of DDX60L knockdown on replication and assembly and release of full-length HCV reporter virus JcR2A, schematically shown at the top. Huh7.5 cells were transfected with siRNAs 24 h before being infected with cell culture-derived JcR2A reporter virus at an MOI of 1 TCID50/cell. Twenty-four hours later, cells were treated with 0.3 ng/ml of IFN-γ or 5 IU/ml of IFN-α. Forty-eight hours after infection, the medium was replaced by interferon-free medium. Seventy-two hours after infection, cells were harvested and analyzed for luciferase activity to assess the contribution of DDX60L to the IFN response against HCV infection. Supernatants were transferred to naive Huh7.5 cells for quantification of HCV particle production (“reinfection”). Luciferase activity in the reinfection experiment was used as a correlate for titers of infectious virus. (B) JcR2A infection is rescued from IFN treatment by DDX60L knockdown. Huh7.5 cells transfected with the indicated siRNA and infected with JcR2A reporter virus were treated with IFN-γ or IFN-α and analyzed for Renilla luciferase activity as indicated in panel A. Luciferase values were normalized to the respective untreated controls to determine rescue of HCV infection from the IFN response by DDX60L knockdown. Shown are mean values with SDs (n = 2). (C) JcR2A particle production is rescued from IFN treatment by DDX60L knockdown. Huh7.5 cells were infected with supernatants from the samples in panel B. Seventy-two hours after reinfection, cell lysates were subjected to Renilla luciferase assay to analyze the rescue of HCV production from the IFN response upon silencing of DDX60L. Luciferase activity was normalized to the respective untreated controls. Shown are mean values with SDs (n = 2). (D) DDX60L knockdown does not influence translation of the HCV IRES. Huh-7-Lunet cells were transfected with siRNA targeting DDX60L and 48 h later electroporated with replication-deficient reporter replicons, relying on either the HCV or the unrelated poliovirus IRES for translation of FLuc. Capped in vitro transcripts encoding Renilla luciferase (RLuc) were cotransfected to normalize for transfection efficiency. FLuc and RLuc activity was determined in cell lysates 2 h after transfection. Shown are mean values with SDs (n = 2). (E) Stability of HCV RNA is not altered by DDX60L knockdown. LucubineoJFH1 cells were transduced with lentiviruses encoding shRNAs against DDX60L at an MOI of 5. Twenty-four hours later, the cells were treated with 100 μM 2′CMC to inhibit the de novo synthesis of viral RNA. At the indicated time points, total RNA was isolated to determine the amount of HCV genomes by qRT-PCR. Values were normalized to the amount of total RNA. Shown are mean values with SDs (n = 2).
Translation of HCV proteins depends on the IRES in its 5′ untranslated region (UTR) (53), and indeed, several previous studies have shown that IFN results in inhibition of HCV IRES-dependent translation (15, 54). To address whether DDX60L might target activity of the HCV IRES or other unrelated IRES elements, we silenced DDX60L in Huh-7-Lunet cells and transfected monocistronic replication-deficient reporter replicons encoding firefly luciferase driven either by the HCV IRES or by the poliovirus IRES, which differ significantly in their mechanisms of ribosome recruitment (55) (Fig. 10D). To normalize for transfection efficiency, we added a capped in vitro transcript encoding Renilla luciferase (RLuc). However, knockdown of DDX60L had no impact on the efficiency of IRES-mediated translation, either from the HCV IRES or from the poliovirus IRES (Fig. 10D). The bicistronic replicons used in most experiments furthermore contain a heterologous EMCV-IRES element (28), which could be a target of DDX60L. Therefore, we confirmed that knockdown of DDX60L also increased replication of monocistronic replicons in Huh-7-Lunet cells and rescued them from the IFN-γ response to an extent similar to those of bicistronic replicons (data not shown).
Finally, we aimed to determine whether DDX60L acts by actively degrading HCV RNA, as has been recently proposed for DDX60 (25). Therefore, we transduced Huh-7 cells harboring a subgenomic JFH1 replicon with lentiviral vectors encoding nontargeting or DDX60L-specific shRNA, inhibited viral RNA synthesis by addition of 2′CMC, a nucleosidic inhibitor of HCV replication, and quantified HCV copy numbers to assess a direct contribution of DDX60L to degradation of viral RNA (Fig. 10E). However, the HCV genome copy number declined with the same kinetics in cells transduced with nontargeting shRNA as with knockdown of DDX60L. Therefore, DDX60L, in contrast to DDX60, seems not to be involved in the degradation of HCV RNA but affects a different step in viral RNA replication.
Overall, we found no evidence for an impact of DDX60L on HCV assembly and release, on IRES-mediated translation, or on RNA stability. We therefore assume that DDX60L most likely affects viral RNA replication. Still, further in-depth analyses are required to gain deeper insights into the mechanisms governing suppression of HCV replication by DDX60L.
DISCUSSION
Even though efficient IFN-γ responses are induced in Huh6 as well as Huh-7 cells, Huh6 cells fail to induce an antiviral state, most likely due to the lack of particular effector proteins (27). In this study, we used comparative transcriptome analyses of Huh-7 and Huh6 cells followed by an siRNA-based screening approach to identify genes contributing to functional IFN-γ responses against HCV. We identified the yet-uncharacterized putative DExD/H-box helicase DDX60L as an important effector protein of the type I, type II, and type III interferon responses against HCV, also restricting viral replication in the absence of IFN.
To screen for effectors involved in the IFN-γ response, we silenced individual candidate genes and analyzed their impact on HCV replication in the presence of IFN-γ. In the case of substantial contribution of a factor to the antiviral effect of interferons, viral replication will be rescued to a significant level, as previously shown on a larger scale (6). Comparison with the complementing strategy employing overexpression of a candidate gene and directly testing its effect on viral replication (15, 17) shows that both approaches have certain strengths and weaknesses. A certain advantage of the knockdown is that each single gene is tested in the context of the full-blown IFN-induced antiviral state with authentic gene expression levels, thereby avoiding potential artifacts from overexpression, e.g., due to cytotoxic effects inhibiting viral replication. However, the knockdown approach will identify only candidates with a substantial contribution to the IFN response, since a complementing and overlapping set of ISGs targets each virus (6, 15); therefore, minor candidates might be missed. For example, one siRNA slightly but reproducibly increased HCV replication in the presence of IFN-γ in the case of ACY3 and NLRC5, a protein previously described to increase ISG levels after IFN-γ treatment (56). However, our study focused on the identification of factors substantially contributing to IFN responses; therefore, these potential candidates were not further validated. By following the knockdown approach, we identified DDX60L as a novel inhibitor of HCV genotype 1 and 2 replication and as a main effector of the type I, type II, and type III IFN responses against HCV. The contribution of DDX60L was comparable to those other major ISG products targeting HCV, like RSAD2/viperin (47), IFITM3, and NOS2 (6) (data not shown).
Surprisingly, it was extremely difficult to detect DDX60L protein, and we succeeded only by using a strong transient-overexpression model. Thereby we could verify the specificity of one commercially available antibody toward DDX60L, allowing in principle the detection of endogenous protein by Western blotting but not by IF. However, even after IFN treatment, endogenous DDX60L stayed below the limit of detection; therefore, it remained difficult to assess protein functions in a physiological context. Still, working with the overexpressed protein, we could define some principal properties. (i) DDX60L forms SDS-resistant aggregates, similar to amyloids (57), since in Western blotting and in vitro translation, no distinct band of the expected size was found, but rather a smear covering a very broad range of mainly higher molecular masses than expected. Proteasome inhibitors had no impact on protein abundance or size distribution. At the current state it is therefore difficult to draw firm conclusions on DDX60L protein stability. (ii) DDX60L was widely distributed throughout the cytoplasm but was not found in the nucleus, similar to DDX60 (23, 25). We furthermore found no striking colocalization with HCV NS5A. (iii) DDX60L caused cytotoxicity at high expression levels. Indeed, other DExD/H box helicases have been shown to induce apoptosis or to arrest the cell cycle (reviewed in reference 19), and expression of other ISGs can inhibit cell proliferation or promote cell death (reviewed in reference 58). Still, it is currently hard to judge whether this is also a physiological role of DDX60L or rather an artifact of overexpression. Overall, our study thereby set the ground and defined reagents for a more in-depth analysis of properties and functions of DDX60L. However, additional studies, going beyond the scope of this work, will be required to clarify the mechanism of action of DDX60L in greater detail, e.g., by reverse genetic analysis of mutants or by seeking for physical interactions with viral RNA or proteins.
DDX60L had a strong impact on infectivity of lentiviral vectors, demonstrating that its activity is not restricted to HCV. While our studies did not address the mechanism underlying this restriction, they define that the effect of DDX60L is determined during lentivirus production but ultimately impacts target cell infection. Our result is reminiscent of a recent study, which failed to generate lentiviral vectors encoding DDX60, DDX3X, and MOV10 (15), which are also RNA helicases, like DDX60L. The magnitude of the antilentiviral effect of DDX60L warrants further investigation of the underlying mechanism, particularly regarding its relevance for the spread of replication-competent lentiviruses such as HIV-1, and the involvement of DDX60L in the protective effects of interferons in physiological target cells.
Little is known so far about the function of DDX60L. It has yet only been part of some genome-wide siRNA screens on HIV host factors (59), nonalcoholic fatty liver disease (60), and childhood obesity (61) and was identified in a genome-wide association study as a biomarker for the antidepressant responses of selective serotonin reuptake inhibitors (62). DDX60L has also been recognized as an ISG product before in a study searching for genes induced by IFN-β in Epstein-Barr virus-infected multiple sclerosis patients (24). However, it was not included in two previous comprehensive functional analyses of potential effector proteins involved in the interferon response against HCV (6, 15). Our results now indicate that DDX60L is an important ISG product involved in the type I, type II, and type III antiviral responses against HCV and probably also other viruses.
DDX60L and DDX60 are 70% identical at the amino acid level. Interestingly, DDX60 has been identified before as a potential inhibitor of HCV replication upon overexpression of the protein, and the same study also pointed toward an interference of DDX60 with the production of lentiviral vectors (15), suggesting similar and overlapping functions of DDX60 and DDX60L. In addition, our data revealed that mRNAs of both proteins are upregulated to the same extent by HCV infection and IFN-α treatment of PHH, emphasizing their potential physiological significance. However, our results did not point to a functional role of DDX60 in the interferon response against HCV, at least in Huh-7 cells, since silencing did not rescue HCV replication in the presence of IFNs, nor did it increase replication in the absence of IFNs. Although we cannot entirely exclude a function of DDX60 due to low baseline expression in Huh-7 cells, our data clearly show that DDX60L has a distinct and specific role in the ISG response against HCV. In addition, DDX60L is a host factor generally restricting HCV replication in Huh-7 cells.
Many DExD/H box helicases have been found to directly sense viral RNA (RIG-I, Mda5, and DHX9 [21, 22, 63]) or to contribute to the RIG-I response (e.g., DDX3 [64]). Previous studies revealed that DDX60 also has a stimulating role in the induction of RIG-I responses (23), and a very recent paper showed that DDX60L is also involved in RIG-I activation, with a pathogen-associated molecular pattern (PAMP) profile slightly differing from that of DDX60, also including single-stranded, triphosphorylated RNAs (25). The contribution of DDX60 to RIG-I responses to viral infections in vivo has furthermore been demonstrated in DDX60 knockout mice (25). Unfortunately, this is not possible for DDX60L, since mice generally lack the gene for this protein (25). Indeed, DDX60L knockdown also in our hands slightly reduced the induction of the RIG-I pathway upon poly(I·C) transfection, thereby confirming these findings and arguing for conserved functions of DDX60 and DDX60L in sensing of viral RNA. However, it seems unlikely that this mechanism restricted HCV replication in our experiments, since several studies have shown that the RIG-I pathway is barely activated upon HCV infection in Huh-7 cells (42, 51, 65). In addition, HCV has developed potent means to block the RIG-I-mediated response by cleavage of MAVS once replication is established (50). Furthermore, blocking of the RIG-I pathway did not increase permissivity of Huh-7 cells in our hands (51). Therefore, more in-depth studies will be required to ultimately clarify the physiological relevance of RNA sensing by DDX60 and DDX60L and their contribution to induction of IFN responses in the case of HCV.
Our data strongly suggest that DDX60L most likely acts as a direct antiviral effector protein against HCV, targeting the step of RNA replication. First, we show that DDX60L does not generally stimulate ISG expression induced by IFN-γ, suggesting that it rather acts as a direct effector. Still, although we think this is unlikely, we cannot formally exclude that DDX60L transcriptionally regulates expression of a certain subset of genes with effector functions against HCV. Second, expression of DDX60L inhibited HCV replication without inducing expression of other ISGs. Third, our candidates were solely derived from HCV subgenomic replicon models; therefore, putative restriction factors can target only viral translation or RNA replication and not viral entry or particle production. Fourth, we excluded that DDX60L acts on IRES-mediated translation. In contrast to the case with DDX60, which has recently been shown to contribute to the degradation of HCV RNA (25), we could not find evidence for an impact of DDX60L silencing on the half-life of HCV RNA after inhibition of viral replication. Therefore, we think it unlikely that DDX60L acts by directly degrading HCV RNA. Currently, the precise mechanism of action of DDX60L remains elusive. Besides their already-discussed role in sensing viral RNA, DExD/H box helicases have been described to be involved in viral replication as both dependency factors and restriction factors (reviewed in reference 18). In the case of HCV, proviral functions were found for DDX3 (66–68) and DDX6 (69), as well as for helicases involved in the generation of miR-122, which is essential for HCV translation and replication (66). However, very little is known so far about the mechanism underlying direct antiviral activities of RNA helicases, which could, e.g., pry apart stretches of double-stranded RNA, remove proteins bound to nucleic acids (70), or even act by mechanisms completely independent of helicase activity (71). Taken together, our results therefore argue for inhibition of HCV RNA replication by DDX60L; however, the distinct mechanism needs to be further defined in future studies, e.g., investigating colocalization of DDX60L with viral RNA or interaction studies with viral proteins.
Interestingly, DDX60L had no impact on HAV replication, although HAV and HCV share similar biologies, the same tropism, and an identical sensitivity to IFNs in Huh-7 cells (34). Two recent large-scale screens of ISGs identified other genes that act specifically on HCV but not on either of the picornaviruses poliovirus or coxsackie B virus (15, 17), stressing the need for a specific yet complementary set of genes required for effective defense against different viruses. It will be interesting to evaluate in future studies which other virus families might be targeted by the antiviral action of DDX60L.
In conclusion, we have identified DDX60L as a novel ISG product with an important function in the interferon-mediated antiviral response against HCV and with potential antiretroviral activity. Although this protein shares a high degree of similarity with its homolog DDX60, our results indicate that it has clearly distinct functions. Still, further in-depth studies will be required to shed light on the mechanism of action of this new player in the innate immune response.
Supplementary Material
ACKNOWLEDGMENTS
This project was funded by grants from the Deutsche Forschungsgemeinschaft (FOR1202, TP3 to V.L., FOR1202, TP1 to R.B., and SFB1129 project 8 to O.F.). O.F. acknowledges funding from the BMBF consortium ImmunoQuant (Teilprojekt Q 0316170C).
We especially thank Rahel Klein and Ulrike Herian for excellent technical assistance. Anti-HIV-1 p24CA serum was a kind gift of Barbara Müller, and HCV isolate JFH1 was provided by T. Wakita. We thank Charles M. Rice for 9E10 monoclonal antibody as well as for Huh7.5 cells.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01297-15.
REFERENCES
- 1.Hajarizadeh B, Grebely J, Dore GJ. 2013. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol 10:553–562. doi: 10.1038/nrgastro.2013.107. [DOI] [PubMed] [Google Scholar]
- 2.Thimme R, Binder M, Bartenschlager R. 2012. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol Rev 36:663–683. doi: 10.1111/j.1574-6976.2011.00319.x. [DOI] [PubMed] [Google Scholar]
- 3.Bigger CB, Brasky KM, Lanford RE. 2001. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infection. J Virol 75:7059–7066. doi: 10.1128/JVI.75.15.7059-7066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, Chisari FV. 2002. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A 99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. 2012. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A 109:4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Metz P, Dazert E, Ruggieri A, Mazur J, Kaderali L, Kaul A, Zeuge U, Windisch MP, Trippler M, Lohmann V, Binder M, Frese M, Bartenschlager R. 2012. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology 56:2082–2093. doi: 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- 7.Thomas E, Gonzalez VD, Li Q, Modi AA, Chen W, Noureddin M, Rotman Y, Liang TJ. 2012. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 142:978–988. doi: 10.1053/j.gastro.2011.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dreux M, Garaigorta U, Boyd B, Decembre E, Chung J, Whitten-Bauer C, Wieland S, Chisari FV. 2012. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 12:558–570. doi: 10.1016/j.chom.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heim MH. 2013. 25 years of interferon-based treatment of chronic hepatitis C: an epoch coming to an end. Nat Rev Immunol 13:535–542. doi: 10.1038/nri3463. [DOI] [PubMed] [Google Scholar]
- 10.Jo J, Aichele U, Kersting N, Klein R, Aichele P, Bisse E, Sewell AK, Blum HE, Bartenschlager R, Lohmann V, Thimme R. 2009. Analysis of CD8+ T-cell-mediated inhibition of hepatitis C virus replication using a novel immunological model. Gastroenterology 136:1391–1401. doi: 10.1053/j.gastro.2008.12.034. [DOI] [PubMed] [Google Scholar]
- 11.Wieland S, Makowska Z, Campana B, Calabrese D, Dill MT, Chung J, Chisari FV, Heim MH. 2014. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology 59:2121–2130. doi: 10.1002/hep.26770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheahan T, Imanaka N, Marukian S, Dorner M, Liu P, Ploss A, Rice CM. 2014. Interferon lambda alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe 15:190–202. doi: 10.1016/j.chom.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gokhale NS, Vazquez C, Horner SM. 2014. Hepatitis C virus. Strategies to evade antiviral responses. Future Virol 9:1061–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 15.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Metz P, Reuter A, Bender S, Bartenschlager R. 2013. Interferon-stimulated genes and their role in controlling hepatitis C virus. J Hepatol 59:1331–1341. doi: 10.1016/j.jhep.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 17.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fullam A, Schroder M. 2013. DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim Biophys Acta 1829:854–865. doi: 10.1016/j.bbagrm.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuller-Pace FV. 2006. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res 34:4206–4215. doi: 10.1093/nar/gkl460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linder P, Jankowsky E. 2011. From unwinding to clamping—the DEAD box RNA helicase family. Nat Rev Mol Cell Biol 12:505–516. doi: 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- 21.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr, Akira S, Yonehara S, Kato A, Fujita T. 2005. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 22.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 23.Miyashita M, Oshiumi H, Matsumoto M, Seya T. 2011. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol Cell Biol 31:3802–3819. doi: 10.1128/MCB.01368-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khsheibun R, Paperna T, Volkowich A, Lejbkowicz I, Avidan N, Miller A. 2014. Gene expression profiling of the response to interferon beta in Epstein-Barr-transformed and primary B cells of patients with multiple sclerosis. PLoS One 9:e102331. doi: 10.1371/journal.pone.0102331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oshiumi H, Miyashita M, Okamoto M, Morioka Y, Okabe M, Matsumoto M, Seya T. 2015. DDX60 is involved in RIG-I-dependent and independent antiviral responses, and its function is attenuated by virus-induced EGFR activation. Cell Rep 11:1193–1207. doi: 10.1016/j.celrep.2015.04.047. [DOI] [PubMed] [Google Scholar]
- 26.Lohmann V, Bartenschlager R. 2014. On the history of hepatitis C virus cell culture systems. J Med Chem 57:1627–1642. doi: 10.1021/jm401401n. [DOI] [PubMed] [Google Scholar]
- 27.Windisch MP, Frese M, Kaul A, Trippler M, Lohmann V, Bartenschlager R. 2005. Dissecting the interferon-induced inhibition of hepatitis C virus replication by using a novel host cell line. J Virol 79:13778–13793. doi: 10.1128/JVI.79.21.13778-13793.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 29.Frese M, Schwarzle V, Barth K, Krieger N, Lohmann V, Mihm S, Haller O, Bartenschlager R. 2002. Interferon-gamma inhibits replication of subgenomic and genomic hepatitis C virus RNAs. Hepatology 35:694–703. doi: 10.1053/jhep.2002.31770. [DOI] [PubMed] [Google Scholar]
- 30.Friebe P, Boudet J, Simorre JP, Bartenschlager R. 2005. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J Virol 79:380–392. doi: 10.1128/JVI.79.1.380-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harak C, Radujkovic D, Taveneau C, Reiss S, Klein R, Bressanelli S, Lohmann V. 2014. Mapping of functional domains of the lipid kinase phosphatidylinositol 4-kinase type III alpha involved in enzymatic activity and hepatitis C virus replication. J Virol 88:9909–9926. doi: 10.1128/JVI.01063-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J Virol 77:3007–3019. doi: 10.1128/JVI.77.5.3007-3019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Binder M, Quinkert D, Bochkarova O, Klein R, Kezmic N, Bartenschlager R, Lohmann V. 2007. Identification of determinants involved in initiation of hepatitis C virus RNA synthesis by using intergenotypic replicase chimeras. J Virol 81:5270–5283. doi: 10.1128/JVI.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esser-Nobis K, Harak C, Schult P, Kusov Y, Lohmann V. 2015. Novel perspectives for hepatitis A virus therapy revealed by comparative analysis of hepatitis C virus and hepatitis A virus RNA replication. Hepatology 62:397–408. doi: 10.1002/hep.27847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gauss-Müller V, Kusov YY. 2002. Replication of a hepatitis A virus replicon detected by genetic recombination in vivo. J Gen Virol 83:2183–2192. doi: 10.1099/0022-1317-83-9-2183. [DOI] [PubMed] [Google Scholar]
- 36.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Buhler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reiss S, Harak C, Romero-Brey I, Radujkovic D, Klein R, Ruggieri A, Rebhan I, Bartenschlager R, Lohmann V. 2013. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog 9:e1003359. doi: 10.1371/journal.ppat.1003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaul A, Stauffer S, Berger C, Pertel T, Schmitt J, Kallis S, Zayas M, Lohmann V, Luban J, Bartenschlager R. 2009. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog 5:e1000546. doi: 10.1371/journal.ppat.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kallio MA, Tuimala JT, Hupponen T, Klemela P, Gentile M, Scheinin I, Koski M, Kaki J, Korpelainen EI. 2011. Chipster: user-friendly analysis software for microarray and other high-throughput data. BMC Genomics 12:507. doi: 10.1186/1471-2164-12-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esser-Nobis K, Romero-Brey I, Ganten TM, Gouttenoire J, Harak C, Klein R, Schemmer P, Binder M, Schnitzler P, Moradpour D, Bartenschlager R, Polyak SJ, Stremmel W, Penin F, Eisenbach C, Lohmann V. 2013. Analysis of hepatitis C virus resistance to silibinin in vitro and in vivo points to a novel mechanism involving nonstructural protein 4B. Hepatology 57:953–963. doi: 10.1002/hep.26260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmitt M, Scrima N, Radujkovic D, Caillet-Saguy C, Simister PC, Friebe P, Wicht O, Klein R, Bartenschlager R, Lohmann V, Bressanelli S. 2011. A comprehensive structure-function comparison of hepatitis C virus strain JFH1 and J6 polymerases reveals a key residue stimulating replication in cell culture across genotypes. J Virol 85:2565–2581. doi: 10.1128/JVI.02177-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiet MS, Bauhofer O, Zayas M, Roth H, Tanaka Y, Schirmacher P, Willemsen J, Grunvogel O, Bender S, Binder M, Lohmann V, Lotteau V, Ruggieri A, Bartenschlager R. 2015. Control of temporal activation of hepatitis C virus-induced interferon response by domain 2 of nonstructural protein 5A. J Hepatol doi: 10.1016/j.jhep.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 43.Haller C, Muller B, Fritz JV, Lamas-Murua M, Stolp B, Pujol FM, Keppler OT, Fackler OT. 2014. HIV-1 Nef and Vpu are functionally redundant broad-spectrum modulators of cell surface receptors, including tetraspanins. J Virol 88:14241–14257. doi: 10.1128/JVI.02333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Backes P, Quinkert D, Reiss S, Binder M, Zayas M, Rescher U, Gerke V, Bartenschlager R, Lohmann V. 2010. Role of annexin A2 in the production of infectious hepatitis C virus particles. J Virol 84:5775–5789. doi: 10.1128/JVI.02343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 46.Frese M, Pietschmann T, Moradpour D, Haller O, Bartenschlager R. 2001. Interferon-alpha inhibits hepatitis C virus subgenomic RNA replication by an MxA-independent pathway. J Gen Virol 82:723–733. doi: 10.1099/0022-1317-82-4-723. [DOI] [PubMed] [Google Scholar]
- 47.Helbig KJ, Eyre NS, Yip E, Narayana S, Li K, Fiches G, McCartney EM, Jangra RK, Lemon SM, Beard MR. 2011. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology 54:1506–1517. doi: 10.1002/hep.24542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin A, Lemon SM. 2006. Hepatitis A virus: from discovery to vaccines. Hepatology 43:164–172. doi: 10.1002/hep.21052. [DOI] [PubMed] [Google Scholar]
- 49.Marukian S, Andrus L, Sheahan TP, Jones CT, Charles ED, Ploss A, Rice CM, Dustin LB. 2011. Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology 54:1913–1923. doi: 10.1002/hep.24580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 51.Binder M, Kochs G, Bartenschlager R, Lohmann V. 2007. Hepatitis C virus escape from the interferon regulatory factor 3 pathway by a passive and active evasion strategy. Hepatology 46:1365–1374. doi: 10.1002/hep.21829. [DOI] [PubMed] [Google Scholar]
- 52.al Yacoub N, Romanowska M, Haritonova N, Foerster J. 2007. Optimized production and concentration of lentiviral vectors containing large inserts. J Gene Med 9:579–584. doi: 10.1002/jgm.1052. [DOI] [PubMed] [Google Scholar]
- 53.Tsukiyama-Kohara K, Iizuka N, Kohara M, Nomoto A. 1992. Internal ribosome entry site within hepatitis C virus RNA. J Virol 66:1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang C, Pflugheber J, Sumpter R Jr, Sodora DL, Hui D, Sen GC, Gale M Jr. 2003. Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J Virol 77:3898–3912. doi: 10.1128/JVI.77.7.3898-3912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lozano G, Martinez-Salas E. 2015. Structural insights into viral IRES-dependent translation mechanisms. Curr Opin Virol 12:113–120. doi: 10.1016/j.coviro.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 56.Kuenzel S, Till A, Winkler M, Hasler R, Lipinski S, Jung S, Grotzinger J, Fickenscher H, Schreiber S, Rosenstiel P. 2010. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J Immunol 184:1990–2000. doi: 10.4049/jimmunol.0900557. [DOI] [PubMed] [Google Scholar]
- 57.Chiti F, Dobson CM. 2006. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 58.Barber GN. 2000. The interferons and cell death: guardians of the cell or accomplices of apoptosis? Semin Cancer Biol 10:103–111. doi: 10.1006/scbi.2000.0313. [DOI] [PubMed] [Google Scholar]
- 59.Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS. 2008. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 60.Chalasani N, Guo X, Loomba R, Goodarzi MO, Haritunians T, Kwon S, Cui J, Taylor KD, Wilson L, Cummings OW, Chen YD, Rotter JI, Nonalcoholic Steatohepatitis Clinical Research Network. 2010. Genome-wide association study identifies variants associated with histologic features of nonalcoholic Fatty liver disease. Gastroenterology 139:1567–1576. doi: 10.1053/j.gastro.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Comuzzie AG, Cole SA, Laston SL, Voruganti VS, Haack K, Gibbs RA, Butte NF. 2012. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS One 7:e51954. doi: 10.1371/journal.pone.0051954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morag A, Pasmanik-Chor M, Oron-Karni V, Rehavi M, Stingl JC, Gurwitz D. 2011. Genome-wide expression profiling of human lymphoblastoid cell lines identifies CHL1 as a putative SSRI antidepressant response biomarker. Pharmacogenomics 12:171–184. doi: 10.2217/pgs.10.185. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Z, Yuan B, Lu N, Facchinetti V, Liu YJ. 2011. DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J Immunol 187:4501–4508. doi: 10.4049/jimmunol.1101307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schröder M, Baran M, Bowie AG. 2008. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J 27:2147–2157. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keskinen P, Nyqvist M, Sareneva T, Pirhonen J, Melen K, Julkunen I. 1999. Impaired antiviral response in human hepatoma cells. Virology 263:364–375. doi: 10.1006/viro.1999.9983. [DOI] [PubMed] [Google Scholar]
- 66.Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, Chien M, Weir DB, Russo JJ, Ju J, Brownstein MJ, Sheridan R, Sander C, Zavolan M, Tuschl T, Rice CM. 2007. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci U S A 104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ariumi Y, Kuroki M, Abe K, Dansako H, Ikeda M, Wakita T, Kato N. 2007. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J Virol 81:13922–13926. doi: 10.1128/JVI.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Owsianka AM, Patel AH. 1999. Hepatitis C virus core protein interacts with a human DEAD box protein DDX3. Virology 257:330–340. doi: 10.1006/viro.1999.9659. [DOI] [PubMed] [Google Scholar]
- 69.Jangra RK, Yi M, Lemon SM. 2010. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J Virol 84:6810–6824. doi: 10.1128/JVI.00397-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jankowsky E. 2011. RNA helicases at work: binding and rearranging. Trends Biochem Sci 36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fuller-Pace FV. 2013. The DEAD box proteins DDX5 (p68) and DDX17 (p72): multi-tasking transcriptional regulators. Biochim Biophys Acta 1829:756–763. doi: 10.1016/j.bbagrm.2013.03.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.