

ABSTRACT
The polyprotein Gag is the primary structural component of retroviruses. Gag consists of independently folded domains connected by flexible linkers. Interactions between the conserved capsid (CA) domains of Gag mediate formation of hexameric protein lattices that drive assembly of immature virus particles. Proteolytic cleavage of Gag by the viral protease (PR) is required for maturation of retroviruses from an immature form into an infectious form. Within the assembled Gag lattices of HIV-1 and Mason-Pfizer monkey virus (M-PMV), the C-terminal domain of CA adopts similar quaternary arrangements, while the N-terminal domain of CA is packed in very different manners. Here, we have used cryo-electron tomography and subtomogram averaging to study in vitro-assembled, immature virus-like Rous sarcoma virus (RSV) Gag particles and have determined the structure of CA and the surrounding regions to a resolution of ∼8 Å. We found that the C-terminal domain of RSV CA is arranged similarly to HIV-1 and M-PMV, whereas the N-terminal domain of CA adopts a novel arrangement in which the upstream p10 domain folds back into the CA lattice. In this position the cleavage site between CA and p10 appears to be inaccessible to PR. Below CA, an extended density is consistent with the presence of a six-helix bundle formed by the spacer-peptide region. We have also assessed the affect of lattice assembly on proteolytic processing by exogenous PR. The cleavage between p10 and CA is indeed inhibited in the assembled lattice, a finding consistent with structural regulation of proteolytic maturation.
IMPORTANCE Retroviruses first assemble into immature virus particles, requiring interactions between Gag proteins that form a protein layer under the viral membrane. Subsequently, Gag is cleaved by the viral protease enzyme into separate domains, leading to rearrangement of the virus into its infectious form. It is important to understand how Gag is arranged within immature retroviruses, in order to understand how virus assembly occurs, and how maturation takes place. We used the techniques cryo-electron tomography and subtomogram averaging to obtain a detailed structural picture of the CA domains in immature assembled Rous sarcoma virus Gag particles. We found that part of Gag next to CA, called p10, folds back and interacts with CA when Gag assembles. This arrangement is different from that seen in HIV-1 and Mason-Pfizer monkey virus, illustrating further structural diversity of retroviral structures. The structure provides new information on how the virus assembles and undergoes maturation.
INTRODUCTION
The major structural component of retroviruses is the polyprotein Gag. The expression of Gag alone is sufficient to mediate the assembly and release of virus-like particles (VLPs). Gag proteins from all retroviruses contain an N-terminal membrane-binding matrix (MA) domain, two capsid (CA) domains, and a nucleocapsid (NC) domain (1). These domains are structurally similar across retroviral genera but differ greatly in sequence. Outside these core domains, Gag proteins vary among retroviruses, and other linkers and domains may be present.
The canonical assembly pathway of Gag into immature particles in the infected cell is mediated by interactions between MA and the plasma membrane (PM), between NC and RNA, and between CA domains, including short amino acid sequences immediately C-terminal to CA (2, 3). For most retrovirus genera assembly takes place on the plasma membrane, but for betaretroviruses the particles are assembled in the cytoplasm and then transported to the PM. Depending on the retrovirus, interactions that include other nonconserved domains may also contribute to assembly.
Concomitant with, or shortly after, particle release, cleavage of Gag by the viral protease (PR) gives rise to separate MA, CA, and NC proteins. This cleavage induces a rearrangement of the internal viral structure, with CA forming the shell of the mature viral core. Full proteolytic cleavage of Gag into its individual domains is necessary for virus infectivity (3).
The tertiary structures of retroviral CA domains are well conserved (4–8). Within the immature virus particle they are assembled into a hexameric protein lattice (9–11). The C-terminal domain of CA (CA-CTD), and the first amino acids downstream of CA (in some retroviruses called the spacer region) form a key assembly determinant (12, 13). Low-resolution cryo-electron tomography data indicated that the overall arrangement of the CA domains within the immature hexameric protein lattice is similar in Rous sarcoma virus (RSV), Mason-Pfizer monkey virus (M-PMV), and HIV-1 (11). However, recent higher-resolution cryo-electron tomography structures revealed that while the arrangement of the CA-CTD is well conserved, the arrangement of the N-terminal domain of CA (CA-NTD) within immature HIV-1 and M-PMV differs (14). It remains unclear whether in other retroviruses one of these two CA-NTD arrangements invariably occurs or, alternatively, whether other CA-NTD arrangements also have evolved to help build the hexameric Gag lattice.
RSV is an alpharetrovirus endemic in chickens that is long established as a robust retroviral model system. The same as for HIV-1 Gag, addition of nucleic acid to purified variants of the RSV Gag protein leads to the spontaneous formation of spherical VLPs that closely resemble immature virus particles that are released from cells. These in vitro-assembled RSV VLPs have been studied extensively (11, 15–17). Unlike the Gag proteins of other retroviruses, RSV Gag contains a further immature assembly determinant N-terminal to CA, the C-terminal portion of the 62-amino-acid residue p10 domain. Mutation or omission of this domain prevents assembly of spherical VLPs in vitro (16, 18) and in vivo (19). X-ray crystallography and cysteine cross-linking data suggest that p10 forms a trans-interaction with neighboring CA-NTD domains in the assembled lattice, thereby stabilizing CA hexamers (19, 20).
Also unlike HIV-1 Gag, which has a single PR cleavage site separating the MA and CA domains, RSV Gag has four cleavage sites in that region, which delimit the small mature proteins or peptides named p2A, p2B, and p10, in addition to MA and CA (Fig. 1a). The order of Gag cleavages in infectious RSV is not well defined, although the cleavage between CA and SP occurs late (21, 22). As for all retrovirus Gag proteins, to what degree amino acid sequence on the one hand, and three dimensional structure on the other, determine cleavage rates is not well understood (23).
FIG 1.

In vitro assembly and purification of VLPs. (a) Diagram of RSV Gag (top) and the extent of the GagΔMBD construct (black rectangle, bottom). GagΔMBD starts at residue 84 of MA. (b) Sedimentation purification of RSV GagΔMBD VLPs. VLPs banded to 40 to 50% (wt/wt) sucrose were separated from unassembled and aggregated protein on a 30 to 60% sucrose step gradient. (c) Negative stain EM image of purified VLPs at low (left) and high (right) magnification (the scale bars are 500 and 100 nm, respectively).
We have determined the structure of in vitro-assembled immature RSV Gag VLPs by cryo-electron tomography and subtomogram averaging to a resolution of ∼8 Å. We also compared PR-mediated proteolytic processing of Gag in solution with processing of Gag assembled into VLPs.
MATERIALS AND METHODS
Preparation of RSV Gag particles.
Purification of ΔMBDΔPR (referred to as GagΔMBD) was performed as previously reviewed in reference 24. SUMO-tagged (25) GagΔMBD was induced in, and purified from, BL21 bacterial cultures. Cells were lysed in buffer (20 mM Tris-HCl [pH 8], 500 mM NaCl, 5% glycerol, 2 μM ZnCl2, 2 mM Tris(2-carboxyethyl)phosphine) (TCEP), 1 mM phenylmethylsulfonyl fluoride) by sonication, and the lysate cleared by centrifugation in a TLA-110 Beckman rotor at 90,000 rpm for 45 min. Nucleic acid was removed by addition of polyethyleneimine to cleared lysate to 0.3%, followed by centrifugation of the formed precipitate. Ammonium sulfate was added to 20% to precipitate protein followed by centrifugation. Pelleted protein was resuspended in buffer (20 mM Tris-HCl [pH 8], 50 mM NaCl, 5% glycerol, 2 μM ZnCl2, 2 mM TCEP), and purified by cation-exchange chromatography (HiTrap SP FF; GE Healthcare), followed by Ni+2 affinity chromatography (His Trap HP; GE Healthcare). ULP1 protease was added to the eluted protein to cleave off the SUMO tag. The cleavage reaction was dialyzed overnight at 4°C against buffer (20 mM Tris-HCl [pH 8], 500 mM NaCl, 5% glycerol, 2 μM ZnCl2, 10 mM dithiothreitol). The SUMO tag and ULP1 protease was removed by Ni+2 affinity chromatography. Purified protein at ∼5 mg/ml was flash frozen and stored at −80°C. GagΔMBD used in protease cleavage experiments was further purified by size-exclusion chromatography (HiPrep 16/60, Sephacryl S-100; Amersham Pharmacia).
GagΔMBD VLP assembly was performed as previously described (19). Briefly, Protein at 5 mg/ml was mixed with 1:10 by mass GT25 (an oligonucleotide with 25 repeats of GT) and diluted 1:5 with buffer (MES [morpholineethanesulfonic acid; pH 6.5], 2 μM ZnCl2, 2 mM TCEP). To separate VLPs from unassembled protein and protein aggregates, assembly reactions were overlaid onto a 30 to 60% (wt/wt) sucrose step gradient and centrifuged in a Beckman TLS55 swinging bucket rotor at 35,000 rpm for 3.5 h. Gradients were fractionated and analyzed by polyacrylamide gel electrophoresis to identify the fractions containing VLPs. These fractions were diluted with buffer (MES [pH 6.5], 100 mM NaCl, 2 μM ZnCl2, 2 mM TCEP), and VLPs were pelleted by ultracentrifugation. Purified VLPs were screened by negative stain (2% uranyl acetate) electron microscopy (EM) on a Morgagni 268 transmission electron microscope. Purified VLPs were stored on ice.
Proteolytic cleavage with RSV PR.
RSV protease was purified as previously described (26), and stored at 1 mg/ml in buffer (20 mM MES [pH 6.5], 100 mM NaCl, 2 μM ZnCl2) at −80°C until use. For digestions of protein with GT25 oligonucleotide, protein was mixed with oligonucleotide as described for VLP assembly, and the mixture was incubated for 1 h prior to the addition of protease (a method similar to one used to analyze HIV Gag cleavage [27]). PR digestions were performed in buffer (20 mM MES [pH 6.5], 100 mM NaCl) at a 1:1 mass ratio of PR to GagΔMBD protein for 10 min. Cleavage was stopped by addition of 5× sodium dodecyl sulfate (SDS) loading buffer (0.3 M Tris-HCl [pH 6.8], 10% SDS, 50% glycerol, 25% 2-mercaptoethanol, 0.1% bromophenol blue). After 10 min of incubation, the cleavage is not yet complete, and differences in cleavage rate between assembled and unassembled Gag can be assessed by monitoring intermediate products.
Cleavage products were identified by immunoblot analysis using primary antibodies against Escherichia coli-expressed and purified MAp2, p10, and CA. In the context of GagΔMBD, cleavage at the MA-p2 site and the SP-NC cleavage site results in products that differ in size by only three amino acids, while in full-length Gag, these products differ significantly in size. We therefore also performed protease tests on full-length Gag to determine that this cleavage product represents MA-SP.
Cryo-electron tomography.
Degassed C-Flat 2/2-3C grids were glow discharged for 30 s at 20 mA. The VLP solution was diluted with phosphate-buffered saline containing 10-nm colloidal gold. Then, 2 μl of this mixture was applied to grids and plunge frozen in liquid ethane using an FEI Vitrobot Mark2. The grids were stored in liquid nitrogen until imaging.
Cryo-electron tomography data acquisition was performed similarly, as described previously (14, 28). In brief, tilt series were imaged on a FEI Titan Krios electron microscope operated at 200 keV, with a GIF2002 post-column energy filter (using a slit width of 20 eV) and a 2×2 K Gatan Multiscan 795 charge-coupled device camera. Low-magnification montages for navigation were acquired using serial EM (29), and tilt series were acquired at appropriate positions using FEI tomography software, version 4. The nominal magnification was ×81,000, giving a calibrated pixel size of 1.035 Å. The tilt range was from −45° to +45° or from −30° to +30° in 3° steps, collecting first from 0° to negative tilt angles and then from 3° to positive tilt angles. Tilt series were collected at a range of nominal defoci between −1.5 and −5 μm. The total dose applied to each tilt series ranged from approximately 24 to 34 e/Å2. Tomograms were reconstructed using the IMOD software suite (30).
Image processing.
Image processing was performed essentially as described previously (14, 28) with minor modifications. Subtomogram averaging calculations were performed using Matlab scripts derived from the AV3 (31), TOM packages (32), and the Dynamo software package (33). Visualization of tomograms was performed in either IMOD (30) or Amira (FEI Visualization Sciences Group) using the EM toolbox (34).
Initial processing was performed on 8× binned data (pixel size, 8.28 Å). To obtain a starting reference, one tomogram, acquired at a defocus of −5 μm and containing two VLPs, was chosen. Subvolumes were extracted from the surface of a sphere with the radius corresponding to the diameter of the VLPs with initial angles corresponding to the geometry of the sphere and averaged. The volumes were aligned iteratively in six dimensions against this reference until the structure stabilized. Now all subtomograms extracted from the entire data set were aligned for two iterations against the initial reference, low-pass filtered to 60 Å. After the first two iterations a cross-correlation-based cleaning was performed to remove subtomograms that contained no density corresponding to the GagΔMBD protein layer. Subsequently, subtomograms were split into even/odd data sets, storing the determined x, y, and z positions and psi/theta angles of the previous iterations but randomizing all phi-angles to avoid overcorrelation between the data sets. The now split data sets were aligned for another nine iterations against the same starting reference using a dynamic low-pass filter. At the end of the 8× binned alignments, subvolumes that converged onto the same positions in both data sets were removed.
The defocus of each tomogram was measured using Matlab. Theoretical contrast transfer function (CTF)-curves were fitted to averaged power spectra from 512 square pixel tiles generated from all images in a tilt series. CTF correction was done using the program “ctf phase flip” implemented in IMOD (35). Subvolumes with a size of 248 Å3 were extracted from 2× binned, CTF-corrected tomograms (pixel size, 2.07 Å) at the positions determined in the 8× binned alignments. References for both data sets were generated independently by averaging of subvolumes using the angles and positions determined in the binned alignments. The subvolumes were subjected to four further rounds of alignment. Sixfold symmetry was applied during all alignment steps. Comparison of the averages of all subtomograms after alignment (45,492 and 45,498 asymmetric units in each of the half-data sets, respectively) by Fourier shell correlation (FSC) indicated a resolution of 8.2 Å. After the removal of subtomograms with lower cross-correlation values, final averages were generated from 25,044 and 25,206 asymmetric units in each of the half-data sets, giving a slightly improved resolution of 7.7 Å. Subsequently, we averaged the two half-data set maps and sharpened them with an empirically determined negative B-factor of −700, while filtering to the resolution determined at the 0.143 FSC threshold (36).
Structure fitting and analysis.
Visualization of structures was performed in UCSF Chimera (37). Published structures of immature HIV (PDB 4usn and EMDB-2706) and M-PMV (EMDB-2707) were used for comparison (14).
For generation of the immature RSV capsid model, PDB 3g1i (38) (one CA-CTD monomer) and PDB 1p7n (20) (p10/CA-NTD) were rigid body docked into the structure using the “Fit in Map” option in Chimera. Redundant residues of the antiparallel dimer of PDB 1p7n were removed. Missing residues in the helix 7/helix 8 linker region of CA, the connection loop between p10 and CA-NTD and residues upstream of the helix in p10 were manually modeled using Coot (39).
Subsequently, 18 copies of the p10-CA-NTD/CTD model were rigid body docked into the EM density map. Flexible fitting was performed using the molecular dynamics flexible fitting program (40) and NAMD version 2.9 (41). Simulations were performed until the fit stabilized and backbone atoms stopped moving.
Cryo-electron microscopy structures and a representative tomogram have been deposited in the Electron Microscopy Data Bank (EMDB) under accession numbers EMD-3101 and EMD-3102, the fitted RSV atomic model has been deposited in the Protein Data Bank (PDB) under accession number PDB 5a9e.
RESULTS AND DISCUSSION
RSV GagΔMBD was purified and assembled in vitro into VLPs as described in Materials and Methods (Fig. 1). The assembled particles were imaged by cryo-electron tomography. As previously reported (11), they appeared to be approximately spherical, with a multilayered density (Fig. 2a).
FIG 2.

Structure of the immature RSV p10-CA-SP lattice. (a) Sum of 10 computational slices through a Gaussian-filtered tomogram containing one RSV GagΔMBD VLP. The protein density is black. The red box marks the approximate radial position of the capsid density. Scale bar, 25 nm. (b to d) Isosurface representations of the final subtomogram average of the immature RSV p10-CA-SP lattice as derived from subtomogram averaging. High-resolution structures for p10 (yellow), CA-NTD (blue), and CA-CTD (red) have been flexibly fitted into the density. The density is shown as a side view in panel b, from the outside of the VLP in panel c, and from the inside of the VLP in panel d. Scale bars, 25 Å. See also Movie S1 in the supplemental material. (e) Views of the final structural model of the immature RSV p10-CA-SP lattice from outside the virus showing the indicated CA domains. The coloring is as described for panels b to d. Six- and three-fold symmetry axes are indicated by black hexagons and triangles, respectively.
To determine the structure of the protein lattice we extracted subvolumes from the surface of the spheres and performed subtomogram averaging as described in Materials and Methods. The overall arrangement of the protein density in the final structure is consistent with that previously described at low resolution (11). The density corresponding to the N-terminal CA domain is arranged in hexameric rings around large holes, below which the C-terminal domain of CA links through to the spacer region which appears as an extended column-like density on the 6-fold symmetry axis (Fig. 2b to d). The hexamer-hexamer spacing at the CA-CTD is ∼8 nm, an observation consistent with the dimensions reported for other retroviruses (42).
We observed no ordered densities for regions upstream of p10 and downstream of the spacer peptide, being an indication that these domains are not ordered with respect to the hexameric assembly of p10-CA-SP. This observation is in agreement with cryo-electron tomography and subtomogram averaging results of immature HIV-1 and M-PMV particles (14).
The resolution of the final structure in the CA region was determined by Fourier shell correlation to be 7.7 Å. At this resolution all the alpha-helices in CA are clearly resolved, as well as some of the connecting loops, permitting automated docking of available crystal structures as rigid bodies into the electron density. We subsequently applied molecular dynamics-based flexible fitting to generate a complete structural model (see Materials and Methods for details) (Fig. 2e; see also Movie S1 in the supplemental material). Only minor movements were seen upon flexible fitting, indicating that the available crystal structures were largely consistent with the electron density.
During review of the manuscript Schulten and coworkers published a model of the immature RSV lattice derived using available crystal structures, molecular dynamics simulations and mutagenesis studies (43). Although the relative positions of individual alpha-helices in their model deviate by up to 1 nm from those observed in our structure, the overall arrangement of the CA domains is in excellent agreement.
We compared the arrangement of the CA-CTD in our structure to available structures of RSV CA-CTD dimers, and found that the CA dimers that had been crystallized at mildly alkaline conditions (PDB 3g1i and PDB 3g1g) (38) most closely mimic the CA arrangement in the assembled particles. These capsid-dimer conformations were previously predicted to possibly be involved in the formation of the immature particle (38). The major interaction across the dimeric interface between adjacent hexamers at the CA-CTD is in the region of hydrophobic residues Pro426 and Val427 (Fig. 3a) in helix 9. Within the dimer, the monomers are slightly closer together and helices 9 cross with a slightly steeper angle than in HIV-1, perhaps reflecting smaller hydrophobic residues in the interface and the slightly tighter curvature of the lattice. RSV has a cysteine residue (Cys431) at the base of helix 9 that has been speculated to participate in the interaction across the CA-CTD dimer interface (5). In our model the distance between Cys431 residues across the dimer interface is too large to permit disulfide bridge formation in the immature particle.
FIG 3.
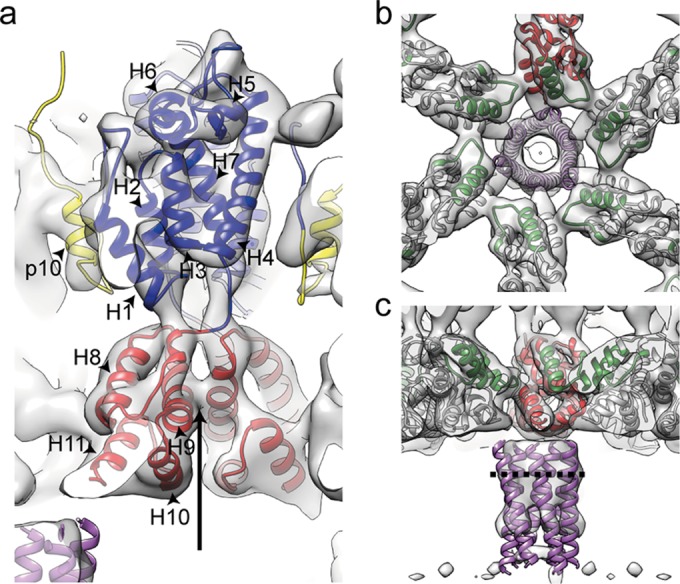
Interactions and structural details in the immature RSV lattice. (a) Flexible fit of RSV-CA into the EM density. Helices are numbered. An arrow indicates the position at which contacts are formed between the helices 9 in the vicinity of Pro426/Val427. The density for the helix-7/helix-8 linker is visible. (b) The CA-CTD forms interactions around the hexameric ring. Residues in the MHR (green) contribute to these interfaces. The putative 6-helix bundle (45) (purple) has been fitted into the density. (c) Isosurface representation of the density corresponding to RSV-SP shown in a side view of the VLP, with the six-helix bundle model generated for RSV-SP (45) fitted into the density. The dashed line indicates the approximate position of the cleavage site between CA and SP.
Residues in the highly conserved major homology region (MHR; I395-V414) within CA-CTD are in positions where they could establish contacts around the hexameric ring (Fig. 3b). In general, the arrangement of the C-terminal CA domain is similar to that previously described for HIV-1 and M-PMV (14), indicating that the interactions that stabilize the immature retroviral lattice at the CA-CTD are well conserved among retroviruses.
Below CA is an extended, hollow cylinder of density corresponding to the spacer region (SP). The spacer peptide itself, as defined by cleavage sites, is 12 amino acid residues in length, but by mutagenesis the inferred SP assembly domain extends upstream 8 residues and downstream 4 to 6 residues (44, 45). The entire SP assembly domain is predicted to form a helix, and a 29-residue peptide comprising this segment of Gag forms hexamers in vitro (45). The electron density in this region is not as well defined as in the CA region and is connected to CA-CTD only by weak densities. Individual alpha-helices cannot be resolved. These observations suggest that the linkers between CA-CTD and SP are flexible, providing freedom for SP to move relative to CA, a finding consistent with NMR studies of monomeric RSV CA-SP that identified flexibility in the CA-SP hinge (8). The electron density becomes weaker at the C-terminal end of SP, suggesting further increasing disorder. We fitted the six-helix bundle model for the RSV SP helix (45) into the map and found that its dimensions are approximately consistent with the observed hollow cylinder (Fig. 3b and c). The approximate position of the cleavage site between CA and SP in the assembled immature virus would leave it accessible for RSV PR (Fig. 3c).
The EM density map allows modeling of the trajectory of the linker connecting CA-CTD with the upstream CA-NTD. The linker seems to be stabilized by contacting the base of helix 1 and by interactions with residues in the loop connecting helix 8 and 9 (Fig. 3a). Similar to HIV-1, we do not observe an extensive CA-NTD/CA-CTD interface.
When fitting the CA-NTD into the electron density, all of the alpha-helices of the N-terminal domain are accommodated within appropriately shaped densities, but one rod-like density is not occupied by the fitted crystal structure. We hypothesized that this density is derived from p10. A protein comprising the C-terminal 25 residues of p10 and CA-NTD crystallizes as an antiparallel dimer with an extended interface between the NTD and p10 from the neighboring molecule (20). We therefore fitted the structure of the NTD and associated p10 (PDB 1p7n [20]) into the EM map. The associated p10 helix perfectly fits into the unoccupied electron density (Fig. 4). p10 forms an integral part of the assembled lattice, linking NTDs around the hexamer. The N-terminal p10-helix folds back into the CA-lattice, forming interactions in trans with the neighboring CA molecule around the hexamer that have been predicted correctly in the crystal-structure and also by mutational and modeling analyses (19, 20). It is possible that residues in the linker between p10 and CA are stabilized by residues at the top of helix 4 of an adjacent capsid monomer.
FIG 4.

p10 is an integral part of the assembled lattice. Representations of p10-CA-NTD interactions are shown. Flexible fits alone and with the corresponding EM density are shown in the left and right panels, respectively. Arrowheads mark the proteolytic cleavage site between p10 and CA. (a) Three p10-CA-NTD monomers are shown from the outside of the VLP with one monomer highlighted in yellow (p10) and blue (CA-NTD). The hole on the 6-fold axis is indicated by black lines. (b and c) Side view of two p10-CA-NTD monomers as seen from the hexamer center (b) and rotated by 180° around the vertical axis (c). For clarity, one p10-CA-NTD monomer has been removed from the side views in panels b and c.
Surprisingly, the N-terminal domain is arranged to form a lattice that differs both from that seen in HIV-1 and that seen in M-PMV (Fig. 2e; Fig. 5). The overall arrangement of CA-NTDs is more similar to M-PMV, with a dimeric interface linking across hexamers, including helices 2 and 7 (Fig. 2e). The distance across the dimeric interface is larger, presumably caused by the presence of a flexible loop region between helices 4 and 5 in RSV (Fig. 2e and Fig. 5).
FIG 5.
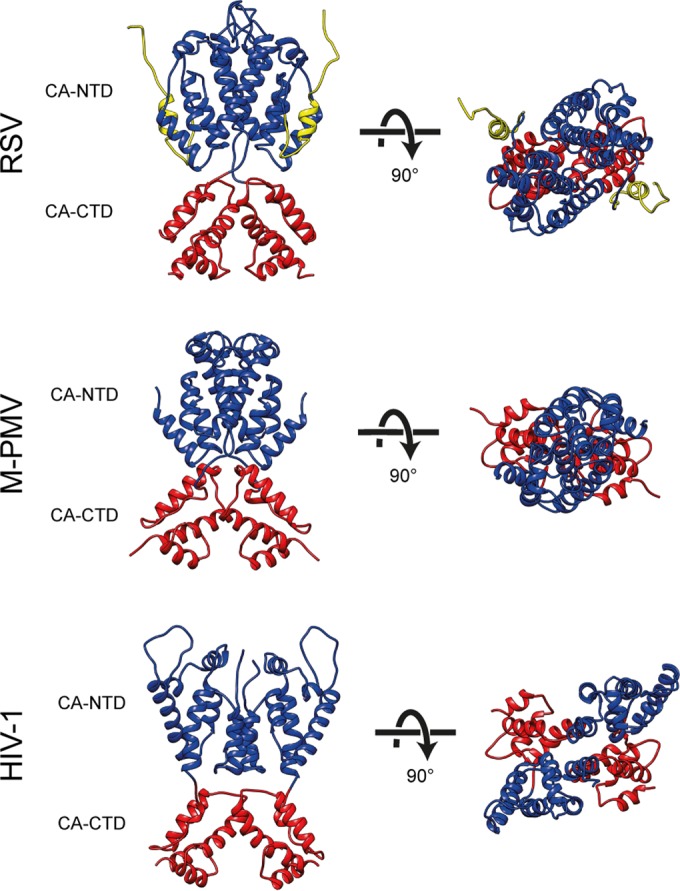
Comparison of immature capsid assembly in different retroviruses. A comparison of immature retroviral capsid dimers (RSV, M-PMV, and HIV-1) is presented. Dimers are shown in orthogonal and top views. CA-CTD interactions are largely conserved between the retroviruses, while the arrangement of the CA-NTD varies. The coloring is as in Fig. 2.
Within the assembled lattice the cleavage site between p10 and CA is located in the region between the CA-CTD and CA-NTD (Fig. 4c), where it would not be accessible to the RSV protease. Cleavage at this site could occur only at the edges of the lattice or after induced disordering of the NTD arrangement or removal of p10, perhaps caused by an upstream cleavage between MA and p2 or between p2 and p10. The interactions between p10 and CA cannot be retained in the lattice after maturation since p10 would block interactions formed by helix 2 in the mature capsid assembly (46, 47). Thus, it is tempting to speculate that p10 acts as a maturation regulator or switch.
We wished to test the structural prediction that the p10-CA cleavage site is not easily accessible to the protease within the assembled Gag lattice. To do this we performed cleavage of RSV Gag constructs in vitro using purified recombinant RSV PR (26). We found that cleavage of GagΔMBD protein in the presence of GT25 oligonucleotide, under conditions where ca. 50% of the protein is assembled into VLPs (15), was slower than cleavage of GagΔMBD in solution in the unassembled state (Fig. 6A lanes 1 to 3, see Fig. 6b for the correspondence between band numbers and cleavage products). In particular, there was a buildup of intermediate products MA-SP (band 2), and p2-NC (band 3), suggesting a reduced rate of cleavage at the sites between p2 and CA. RSV GagΔMBD particles, as in other retroviral particles (11), have incomplete Gag shells, making it unlikely that this effect results from limited protease access to the center of the VLP. Instead, accessibility of a cleavage site to the protease is presumably determined locally by the structure of the lattice.
FIG 6.

Cleavage of RSV Gag protein and VLPs. (a) Coomassie blue-stained, 5 to 15% gradient gel showing cleavage of unassembled GagΔMBD, unassembled GagΔMBD-11C (E227C and T259C), and purified GagΔMBD-11C (E227C and T259C) VLPs. The lanes represent protein with or without GT25 oligonucleotide (+/−) and with or without PR (+/−). The numbers in lanes 5 and 6 correspond to identification of products in panel b. (b) Lanes 5 and 6 were copied from panel a, with each band annotated to indicate the corresponding cleavage product (see Materials and Methods). Band 9 is protease.
To further test the hypothesis that the p10-CA cleavage site is protected in the assembled VLPs, we used GagΔMBD-11C (E227C, T259C) (referred to as GagΔMBD-11C+CC). Upon oxidation, both in vitro and in Gag particles collected from transfected cells, this mutant protein is known to form hexameric Gag complexes due to covalent cross-linking between CA-NTD and p10 from the neighboring Gag molecule via formation of a disulfide bond between the ectopic cysteines (19). Compared to cleavage of GagΔMBD, cleavage of GagΔMBD-11C+CC was slower (Fig. 6a, lanes 4 to 6). In the presence of the GT25 oligonucleotide, the accumulation of CA (band 5) from GagΔMBD-11C+CC was significantly reduced. Cleavage of GagΔMBD-11C+CC in VLPs that had been purified by sedimentation showed that no CA accumulated (Fig. 6a, lanes 9 to 10, purified VLPs). Instead, full-length Gag, p2-NC, and p2-SP were observed, strongly supporting the hypothesis that the cleavages between p2 and CA are hindered in the VLP Gag lattice.
The structure of the immature-like RSV CA-NTD lattice determined here differs from that in both HIV-1 and M-PMV, suggesting that despite the conserved tertiary structure of the CA-NTD domain, its quaternary arrangement within immature retroviruses is highly variable. In the case of RSV, the upstream p10 region contributes to the assembled CA-NTD lattice. Together with the observed effect of assembly on proteolytic cleavage, we speculate that the mechanism by which structural maturation of CA is regulated, in particular at the N-terminal end of CA, is also variable between retroviruses. In contrast, the arrangement of CA-CTD is well conserved in RSV (an alpharetrovirus), M-PMV (a betaretrovirus), and HIV-1 (a lentivirus), suggesting conserved function during assembly.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a grant from the Deutsche Forschungsgemeinschaft to J.A.G.B. (BR 3635/2-1) and by USPHS grant GM-107013 to V.M.V.
We thank members of the Briggs group for helpful discussions. The Briggs laboratory acknowledges financial support from the European Molecular Biology Laboratory and the Chica und Heinz Schaller Stiftung. This study was technically supported by EMBL IT services.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01502-15.
REFERENCES
- 1.Bell NM, Lever AM. 2013. HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol 21:136–144. doi: 10.1016/j.tim.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Lingappa JR, Reed JC, Tanaka M, Chutiraka K, Robinson BA. 2014. How HIV-1 Gag assembles in cells: putting together pieces of the puzzle. Virus Res 193:89–107. doi: 10.1016/j.virusres.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sundquist WI, Krausslich HG. 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med 2:a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macek P, Chmelik J, Krizova I, Kaderavek P, Padrta P, Zidek L, Wildova M, Hadravova R, Chaloupkova R, Pichova I, Ruml T, Rumlova M, Sklenar V. 2009. NMR structure of the N-terminal domain of capsid protein from the Mason-Pfizer monkey virus. J Mol Biol 392:100–114. doi: 10.1016/j.jmb.2009.06.029. [DOI] [PubMed] [Google Scholar]
- 5.Campos-Olivas R, Newman JL, Summers MF. 2000. Solution structure and dynamics of the Rous sarcoma virus capsid protein and comparison with capsid proteins of other retroviruses. J Mol Biol 296:633–649. doi: 10.1006/jmbi.1999.3475. [DOI] [PubMed] [Google Scholar]
- 6.Tang C, Ndassa Y, Summers MF. 2002. Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nat Struct Biol 9:537–543. [DOI] [PubMed] [Google Scholar]
- 7.Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278:849–853. doi: 10.1126/science.278.5339.849. [DOI] [PubMed] [Google Scholar]
- 8.Kingston RL, Fitzon-Ostendorp T, Eisenmesser EZ, Schatz GW, Vogt VM, Post CB, Rossmann MG. 2000. Structure and self-association of the Rous sarcoma virus capsid protein. Structure 8:617–628. doi: 10.1016/S0969-2126(00)00148-9. [DOI] [PubMed] [Google Scholar]
- 9.Wright ER, Schooler JB, Ding JH, Kieffer C, Fillmore C, Sundquist WI, Grant JJ. 2007. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J 26:2218–2226. doi: 10.1038/sj.emboj.7601664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG. 2009. Structure and assembly of immature HIV. Proc Natl Acad Sci U S A 106:11090–11095. doi: 10.1073/pnas.0903535106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Marco A, Davey NE, Ulbrich P, Phillips JM, Lux V, Riches JD, Fuzik T, Ruml T, Krausslich HG, Vogt VM, Briggs JA. 2010. Conserved and variable features of Gag structure and arrangement in immature retrovirus particles. J Virol 84:11729–11736. doi: 10.1128/JVI.01423-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Accola MA, Höglund S, Göttlinger HG. 1998. A putative α-helical structure which overlaps the capsid-p2 boundary in the human immunodeficiency virus type 1 Gag precursor is crucial for viral particle assembly. J Virol 72:2072–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Datta SA, Temeselew LG, Crist RM, Soheilian F, Kamata A, Mirro J, Harvin D, Nagashima K, Cachau RE, Rein A. 2011. On the role of the SP1 domain in HIV-1 particle assembly: a molecular switch? J Virol 85:4111–4121. doi: 10.1128/JVI.00006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schur FK, Hagen WJ, Rumlova M, Ruml T, Muller B, Krausslich HG, Briggs JA. 2015. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 A resolution. Nature 517:505–508. doi: 10.1038/nature13838. [DOI] [PubMed] [Google Scholar]
- 15.Yu F, Joshi SM, Ma YM, Kingston RL, Simon MN, Vogt VM. 2001. Characterization of Rous sarcoma virus Gag particles assembled in vitro. J Virol 75:2753–2764. doi: 10.1128/JVI.75.6.2753-2764.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campbell S, Vogt VM. 1997. In vitro assembly of virus-like particles with Rous sarcoma virus Gag deletion mutants: identification of the p10 domain as a morphological determinant in the formation of spherical particles. J Virol 71:4425–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Briggs JAG, Johnson MC, Simon MN, Fuller SD, Vogt VM. 2006. Cryo-electron microscopy reveals conserved and divergent features of Gag packing in immature particles of Rous sarcoma virus and human immunodeficiency virus. J Mol Biol 355:157–168. doi: 10.1016/j.jmb.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 18.Joshi SM, Vogt VM. 2000. Role of the Rous sarcoma virus p10 domain in shape determination of Gag virus-like particles assembled in vitro and within Escherichia coli. J Virol 74:10260–10268. doi: 10.1128/JVI.74.21.10260-10268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phillips JM, Murray PS, Murray D, Vogt VM. 2008. A molecular switch required for retrovirus assembly participates in the hexagonal immature lattice. EMBO J 27:1411–1420. doi: 10.1038/emboj.2008.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nandhagopal N, Simpson AA, Johnson MC, Francisco AB, Schatz GW, Rossmann MG, Vogt VM. 2004. Dimeric Rous sarcoma virus capsid protein structure relevant to immature Gag assembly. J Mol Biol 335:275–282. doi: 10.1016/j.jmb.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 21.Bennett RP, Rhee S, Craven RC, Hunter E, Wills JW. 1991. Amino acids encoded downstream of gag are not required by Rous sarcoma virus protease during gag-mediated assembly. J Virol 65:272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pepinsky RB, Papayannopoulos IA, Chow EP, Krishna NK, Craven RC, Vogt VM. 1995. Differential proteolytic processing leads to multiple forms of the CA protein in avian sarcoma and leukemia viruses. J Virol 69:6430–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogt VM. 1996. Proteolytic processing and particle maturation, p 95–131. In Kr̈ausslich H-G. (ed), Morphogenesis and maturation of retroviruses, vol 214 Springer, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 24.Bush DL, Vogt VM. 2014. In Vitro Assembly of Retroviruses. Annu Rev Virol 1:561–580. doi: 10.1146/annurev-virology-031413-085427. [DOI] [PubMed] [Google Scholar]
- 25.Malakhov MP, Mattern MR, Malakhova OA, Drinker M, Weeks SD, Butt TR. 2004. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics 5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- 26.Schatz GW, Reinking J, Zippin J, Nicholson LK, Vogt VM. 2001. Importance of the N terminus of Rous sarcoma virus protease for structure and enzymatic function. J Virol 75:4761–4770. doi: 10.1128/JVI.75.10.4761-4770.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deshmukh L, Ghirlando R, Clore GM. 2015. Conformation and dynamics of the Gag polyprotein of the human immunodeficiency virus 1 studied by NMR spectroscopy. Proc Natl Acad Sci U S A 112:3374–3379. doi: 10.1073/pnas.1501985112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schur FK, Hagen WJ, de Marco A, Briggs JA. 2013. Determination of protein structure at 8.5A resolution using cryo-electron tomography and sub-tomogram averaging. J Struct Biol 184:394–400. doi: 10.1016/j.jsb.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 29.Mastronarde DN. 2005. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 30.Kremer JR, Mastronarde DN, McIntosh JR. 1996. Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 31.Forster F, Medalia O, Zauberman N, Baumeister W, Fass D. 2005. Retrovirus envelope protein complex structure in situ studied by cryo-electron tomography. Proc Natl Acad Sci U S A 102:4729–4734. doi: 10.1073/pnas.0409178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nickell S, Forster F, Linaroudis A, Net WD, Beck F, Hegerl R, Baumeister W, Plitzko JM. 2005. TOM software toolbox: acquisition and analysis for electron tomography. J Struct Biol 149:227–234. doi: 10.1016/j.jsb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Castano-Diez D, Kudryashev M, Arheit M, Stahlberg H. 2012. Dynamo: a flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments. J Struct Biol 178:139–151. doi: 10.1016/j.jsb.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 34.Pruggnaller S, Mayr M, Frangakis AS. 2008. A visualization and segmentation toolbox for electron microscopy. J Struct Biol 164:161–165. doi: 10.1016/j.jsb.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 35.Xiong Q, Morphew MK, Schwartz CL, Hoenger AH, Mastronarde DN. 2009. CTF determination and correction for low dose tomographic tilt series. J Struct Biol 168:378–387. doi: 10.1016/j.jsb.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenthal PB, Henderson R. 2003. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 37.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 38.Bailey GD, Hyun JK, Mitra AK, Kingston RL. 2009. Proton-linked dimerization of a retroviral capsid protein initiates capsid assembly. Structure 17:737–748. doi: 10.1016/j.str.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Trabuco LG, Villa E, Schreiner E, Harrison CB, Schulten K. 2009. Molecular dynamics flexible fitting: a practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods 49:174–180. doi: 10.1016/j.ymeth.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Briggs JA, Krausslich HG. 2011. The molecular architecture of HIV. J Mol Biol 410:491–500. doi: 10.1016/j.jmb.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 43.Goh BC, Perilla JR, England MR, Heyrana KJ, Craven RC, Schulten K. 2015. Atomic modeling of an immature retroviral lattice using molecular dynamics and mutagenesis. Structure doi: 10.1016/j.str.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keller PW, Johnson MC, Vogt VM. 2008. Mutations in the spacer peptide and adjoining sequences in Rous sarcoma virus Gag lead to tubular budding. J Virol 82:6788–6797. doi: 10.1128/JVI.00213-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bush DL, Monroe EB, Bedwell GJ, Prevelige PE Jr, Phillips JM, Vogt VM. 2014. Higher-order structure of the Rous sarcoma virus SP assembly domain. J Virol 88:5617–5629. doi: 10.1128/JVI.02659-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bailey GD, Hyun JK, Mitra AK, Kingston RL. 2012. A structural model for the generation of continuous curvature on the surface of a retroviral capsid. J Mol Biol 417:212–223. doi: 10.1016/j.jmb.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 47.Cardone G, Purdy JG, Cheng N, Craven RC, Steven AC. 2009. Visualization of a missing link in retrovirus capsid assembly. Nature 457:694–698. doi: 10.1038/nature07724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.