

ABSTRACT
To understand the interplay between host cytotoxic T-lymphocyte (CTL) responses and the mechanisms by which HIV-1 evades them, we studied viral evolutionary patterns associated with host CTL responses in six linked transmission pairs. HIV-1 sequences corresponding to full-length p17 and p24 gag were generated by 454 pyrosequencing for all pairs near the time of transmission, and seroconverting partners were followed for a median of 847 days postinfection. T-cell responses were screened by gamma interferon/interleukin-2 (IFN-γ/IL-2) FluoroSpot using autologous peptide sets reflecting any Gag variant present in at least 5% of sequence reads in the individual's viral population. While we found little evidence for the occurrence of CTL reversions, CTL escape processes were found to be highly dynamic, with multiple epitope variants emerging simultaneously. We found a correlation between epitope entropy and the number of epitope variants per response (r = 0.43; P = 0.05). In cases in which multiple escape mutations developed within a targeted epitope, a variant with no fitness cost became fixed in the viral population. When multiple mutations within an epitope achieved fitness-balanced escape, these escape mutants were each maintained in the viral population. Additional mutations found to confer escape but undetected in viral populations incurred high fitness costs, suggesting that functional constraints limit the available sites tolerable to escape mutations. These results further our understanding of the impact of CTL escape and reversion from the founder virus in HIV infection and contribute to the identification of immunogenic Gag regions most vulnerable to a targeted T-cell attack.
IMPORTANCE Rapid diversification of the viral population is a hallmark of HIV-1 infection, and understanding the selective forces driving the emergence of viral variants can provide critical insight into the interplay between host immune responses and viral evolution. We used deep sequencing to comprehensively follow viral evolution over time in six linked HIV transmission pairs. We then mapped T-cell responses to explore if mutations arose due to adaption to the host and found that escape processes were often highly dynamic, with multiple mutations arising within targeted epitopes. When we explored the impact of these mutations on replicative capacity, we found that dynamic escape processes only resolve with the selection of mutations that conferred escape with no fitness cost to the virus. These results provide further understanding of the complicated viral-host interactions that occur during early HIV-1 infection and may help inform the design of future vaccine immunogens.
INTRODUCTION
Exploring the dynamic interplay between human immune responses and the mechanisms by which HIV-1 evades these responses can help inform which types and specificity of immunity a vaccine should elicit. At peak viral load, the viral population is usually homogenous (1–3). In approximately 75% of HIV transmissions, a single variant (the “founder” virus) establishes fulminant infection with expansion throughout the body (3–8). The appearance of the first HIV-specific cytotoxic T-lymphocyte (CTL) responses is temporally associated with the initial decline in peak viremia (9), likely due to the ability of these early CTLs to eliminate HIV-infected cells and thus curtail viral replication. As a result, founder-specific CTL responses impose selective pressure on the viral population, and consequently, mutations both within and flanking targeted epitopes are selected from within the virus population that confer escape from the immune response (5, 10–13). Furthermore, if the founder virus carries mutations previously selected to confer CTL escape in the transmitting partner, these mutations sometimes revert to the more sensitive form upon transmission to a new host not sharing that HLA allele (14). On the basis of these findings, CTL pressure in the form of selective escape and, to a lesser extent, reversion has been suggested to be a major driving force of early HIV evolution.
Preferential CTL targeting of the Gag protein has been associated with lower viral loads during chronic infection and thus is largely considered to be the most critical target for CTL-based vaccines (15–18). Escape and potential reversion mutations within Gag epitopes during both acute and chronic HIV infections have been widely reported. One factor that is believed to influence the likelihood of escape and reversion is the impact of the mutation on the replicative fitness of the virus (13, 19–22). Given the differential structural and functional constraints across the Gag protein, some amino acid sites exhibit high replicative fitness costs upon mutation, whereas others can readily tolerate multiple different amino acid substitutions (23–26). Understanding CTL escape and reversion pathways during acute infection can help identify vulnerable viral regions that elicit the greatest fitness consequences if changed. The most well-defined CTL escape and reversion pathways are from responses targeted by HLA alleles associated with control of HIV replication (notably B*27 and B*57). These responses consistently promote dominant escape and reversion pathways in which only a select few mutations are required (27–29). However, in many responses detected during primary HIV-1 infection, clear evidence of reversions is difficult to determine and escape pathways are less consistent (5, 11, 30). Interestingly, frequent sampling of viral populations during acute infection revealed that escape processes can be highly dynamic, with multiple simultaneous or rapidly sequential and mutually exclusive escape mutations arising within a targeted epitope (5). It is unclear which selective forces promote these dynamic escape processes and why some variants are selected over others. Understanding fitness-balanced escape processes in complex escape pathways is likely to help in the design of HIV vaccines that are widely immunogenic and encompass the most vulnerable viral regions (31).
To gain critical insight into the early CTL response, escape, and reversion processes and linked fitness consequences, we studied founder-specific Gag immune responses and ensuing viral evolution in six phylogenetically and epidemiologically linked HIV transmission pairs. We performed massively parallel viral sequencing at frequent intervals, permitting a detailed view of Gag evolution during the first few years of untreated HIV-1 infection. Furthermore, we generated autologous peptide sets for each individual, which permitted the detection of Gag-specific T-cell responses in both the transmitting partner near the time of transmission and the seroconverter's responses to both the founder sequence and unique viral variants that arose over time. We found little evidence for CTL reversion occurring during primary infection. In contrast, the vast majority of CTL escape processes detected were highly dynamic, with often multiple epitope variants present in the viral population. Overall, these dynamic escape processes were found to resolve with the selection of fitness-balanced escape mutations. Our results provide further definition and understanding of the complicated viral-host interactions that occur during early HIV-1 infection and highlight the value of frequent sampling of the viral population to better capture these highly dynamic host-pathogen interactions.
MATERIALS AND METHODS
Study subjects.
All subjects were recruited through the University of Washington Primary Infection Clinic (PIC) (32–34). The relevant institutional review boards approved all human subject protocols, and all subjects provided written informed consent before enrollment. Seroconverting partners (SP) were recruited during primary HIV infection (Fiebig stages I to V) and followed at frequent intervals. Transmitting partners (TPs) were recruited for a single visit around the time of transmission. Duration of infection was recorded as days post onset of symptoms (dps), which refers to the onset of symptoms of primary infection, a median of 12 days after acquisition (32). All SPs were antiretroviral therapy naive during the study period, and no TPs were on therapy at the time point analyzed. HLA typing was performed using sequence-based typing as previously described (35).
Peptides and gamma interferon/interleukin-2 (IFN-γ/IL-2) FluoroSpot.
For each subject, a set of autologous 15-mer peptides overlapping by 11 amino acids (aa) was synthesized (Sigma-Aldrich, St. Louis, MO). Peptide sets were designed to reflect all Gag p17 and p24 variants present in >5% of corrected sequence reads from each individual's viral population. Peptides from all subjects were pooled based on 15-mer windows, resulting in a total of 85 peptide pools used to screen all pairs.
The IFN-γ/IL-2 FluoroSpot assay was used for detection of Gag-specific T-cell responses in all SPs and TPs as previously described (36). Briefly, cryopreserved peripheral blood mononuclear cells (PBMC) were thawed and incubated in R10 medium overnight before stimulation. Between 70,000 and 100,000 PBMC/well were plated in 96-well IPFL plates and stimulated overnight with peptide pools or individual 11-mers added at a final concentration of 2 μg/ml. Each plate contained two wells stimulated with 1.8 μg/ml of phytohemagglutinin (PHA) as a positive control and six negative-control wells of cells incubated with medium alone. Spots were counted using the AID iSpot FluoroSpot reader system.
All TPs and SPs with sufficient PBMC samples (n = 10) were first screened for responses to the 85 Gag peptide pools. SPs were screened at one or two time points during the first year of infection, and TPs were screened at one time point following transmission. PBMC time points were selected based on sample availability and closest correspondence to time points from which viral sequences were obtained. For each positive pool, the autologous peptides from each partner pair (i.e., from both the TP and SP) were tested in duplicate in a confirmatory IFN-γ/IL-2 FluoroSpot. Positivity for both pool and single variant responses was defined as (i) >55 spot-forming cells (SFC)/million PBMC, (ii) >4 times the average of at least six negative-control wells, (iii) >3 standard deviations above average negative-control wells, and (iv) at least 5 spots per well.
Functional avidity.
Optimal epitopes (the shortest and most immunogenic sequence) were determined for all positive responses detected in SPs to the 15-mer peptides. Optimal epitopes were first predicted based on well-known epitopes in the Los Alamos National Laboratory HIV sequence database (HIVDB) as well as on HLA binding motifs (37). Single amino acid truncations and extensions of the predicted optimal epitope were designed as previously described (38). All potential optimal epitopes were synthesized (Sigma-Aldrich) and tested over 8 log10 dilutions using IFN-γ/IL-2 FluoroSpot. The peptide concentration needed to induce the half-maximal response rate (half-maximal effective concentration [EC50]) was determined for each peptide. The sequence with the lowest EC50 (i.e., highest avidity) was designated the optimal epitope. If an individual recognized variants in a particular region as determined by the 15-mer peptide analysis, then additional peptides were generated to reflect the optimal epitope containing these variants. Variant peptides were also titrated over 8 log10 dilutions using IFN-γ/IL-2 FluoroSpot, and the EC50 was calculated. The majority of responses were IFN-γ secretions, and therefore, all EC50s were calculated based on IFN-γ.
PIC38417 immunogenicity assays.
For PIC38417, a total of 93 HIV-1 whole-genome sequences were obtained from 9 time points starting at 25 dps through 247 dps (5). Autologous 15-mer peptides were designed to cover amino acid sites undergoing positive selection and displaying mutually exclusive substitution patterns (n = 14 amino acid sites, corresponding to five distinct peptide regions). Additional 15-mer peptides were generated to reflect all variants detected over 247 days of follow-up at these sites. IFN-γ ELISpot assays were performed as previously described (39) on cryopreserved PBMC from 53 dps and 247 dps. Optimal epitopes were determined as described above.
Pyrosequencing.
DNA sequencing was performed as described in reference 40. Briefly, HIV-1 RNA was extracted from the plasma of infected individuals using the QIAamp viral RNA minikit (Qiagen, Valencia, CA) according to the manufacturer's protocol. cDNA was synthesized using the TaKaRa BluePrint first-strand synthesis kit (Clontech 6115A) according to the manufacturer's protocol, using gene-specific primers, F683 (5′CTCTCGACGCAGGACTCGGCTTG-3′) and R3337-1 (5′-TTTCCYACTAAYTTYTGTATRTCATTGAC-3′) for gag-pol, at a final concentration of 400 nM. First- and second-round PCRs were performed using Kapa HiFi HS (Kapa Biosystems, Boston, MA). Emulsion PCR and sequencing were performed according to the manufacturer's GS FLX Titanium protocols (Roche). PCR products were added to the emulsion PCR at a ratio of 1 or 2 molecules per bead. The picotiter sequencing plate was prepared with a single gasket, creating two regions. Four million enriched beads were distributed equally across the two regions.
To process output from 454 sequencing, raw sequences were screened to remove any reads that were less than 100 bp or contained the ambiguous base N. Sequences generated for each sample were then aligned to a participant-specific consensus using BLAST with the following parameters: match, 1; mismatch, −1; gap existence, 1; and gap extension, 2. Reads were corrected using the Indel-Carryforward Correction (ICC) method (41).
In cases where material and/or viral load was limiting, 15 to 20 single-template-derived Sanger sequences were generated as previously described (5).
Generation of mutant NL4-3 viral stocks.
Mutations identified during epitope evolution in PIC64236 RK9 (T28, E28, R28, and Q28), PIC68008 TW10 (E248 and N242), and PIC38417 NL9 (D640, T641, and A644) and FF9 (R11 and E12) were introduced into the HIV-1NL4-3 genome. Site-directed mutagenesis was performed using restriction-free cloning off synthesized gblocks (synthetic gene products up to 500 bp) containing the mutation of interest (Integrated DNA Technologies, Coralville, IA). The gblock containing the mutated site of interest was amplified in a first round of PCR using primers listed in Table S1 in the supplemental material (42). The first round of PCR used Phusion Hot Start Flex polymerase (New England BioLabs, Ipswich, MA) and the following conditions: 98°C for 30 s; 25 cycles of 98°C for 8 s, 55°C for 20 s, and 72°C for 15 s; and a final extension at 72°C for 5 min. Amplified gblock PCR fragments were purified using a PCR cleanup kit (Clontech). One hundred nanograms of purified amplified double-stranded gblock DNA was used as a primer in the second-round PCR using the following conditions: 98°C for 30 s; 25 cycles of 98°C for 8 s, 60°C for 20 s, and 72°C for 1 min 15 s; and a final extension step at 72°C for 5 min (42).
Second-round PCR products were digested with DpnI and transformed into One Shot TOP10 E. coli (Invitrogen). Single colonies were picked and grown overnight, followed by plasmid DNA extraction using QIAprep Spin Miniprep kit (Qiagen); desired mutations were confirmed by Sanger sequencing.
Plasmid preps were prepared using the QIAPrep HiSpeed Plasmid Midi kit (Qiagen) with an additional endotoxin removal step for use in mammalian cell transfection experiments (43). HEK 293T cells were transfected with 2 μg of plasmid DNA using the XtremeGENE 9 DNA transfection reagent (Roche, San Francisco, CA). Following 48 h at 37°C, cell-free supernatants were collected and cleaned by double centrifugation. Viral aliquots were stored at −80°C until use. The titer for each stock was calculated by determining endpoints in a serial dilution assay (44) on PBMC from a single uninfected donor. Positive wells were identified by p24 enzyme-linked immunosorbent assay (ELISA) (45).
Pairwise growth competition assay.
PBMC from the same uninfected donor as used for virus titer determination were infected with NL4-3 and variant virus 72 h after PHA stimulation (43). Cells were cultured in Iscove's modified Dulbecco's medium (IMDM) supplemented with 20U/ml of human IL-2 (hIL-2; Roche), 10% fetal bovine serum (Sigma-Aldrich), and 1% penicillin-streptomycin. Viruses were added at a multiplicity of infection (MOI) of 0.005 to 3 × 105 PBMC/well and were washed 24 h postinfection (43). All dual infections were done in triplicate at a minimum. Cell-free supernatant (400 μl) was collected from each replicate on days 0, 3, 5, 7, and 9 and stored at −80°C in 200-μl aliquots.
Viral RNA was extracted from supernatant aliquots with a QIAxtractor robot (Qiagen) as described in the manufacturer's protocol. cDNA was synthesized using SuperScript III (Invitrogen) with primer RT2 or ED5 (see Table S1 in the supplemental material) with the following conditions: 50°C for 1.5 h followed by 70°C for 15 min. Depending on the competition, the PIC64236_RK9, PIC68008_TW10, PIC38417_FF9, or PIC38417_NL9 region was PCR amplified using epitope-specific gblock primers (see Table S1 in the supplemental material) and Phusion Hot Start Flex polymerase under the following conditions: 98°C for 30 s; 25 cycles of 98°C for 8 s, 70°C for 20 s, and 72°C for 30 to 60 s; and a final extension step at 72°C for 5 min. Purified PCR products containing the gene of interest were submitted directly for Sanger sequencing. Chromatograms were assembled and edited using Geneious 5.6.5 (Biomatters Ltd., Auckland, New Zealand).
The relative proportion of the two viruses in culture over time was determined by measuring the area under the curve on chromatograms at the mutated nucleotide of interest using the Chromatquant web tool (http://indra.mullins.microbiol.washington.edu/cgi-bin/chromatquant.cgi). The relative fitness (1+s) was estimated using a web-based linear regression algorithm (http://bis.urmc.rochester.edu/vFitness/FitnessMulti.aspx) that allows for the assessment of the rate of change in mutant frequency over multiple data points within exponential virus growth phase from days 3 to 7 (43).
Data analysis.
Breadth of responses in SPs and PIC38417 was calculated as the number of optimal epitopes defined during the first year of infection. In other TPs, breadth was calculated based on the number of targeted 15-mer peptides. Responses to two consecutive overlapping 15-mers were counted as a single epitope. Escape mutations were defined as variant amino acids that did not elicit a positive response in the 15-mer peptide screen.
Statistical analysis.
Nonlinear regressions were performed to determine the EC50 in the functional-avidity analyses. Relative fitness differences between viruses were determined using Student's t test. P values were not corrected for multiple comparisons. GraphPad Prism X was used for all statistical analyses.
RESULTS
Detection of primarily IFN-γ-secreting Gag-specific T-cell responses in transmitting and seroconverting partners.
Six epidemiologically and phylogenetically linked HIV-1 transmission pairs from the Seattle Primary Infection Clinic (PIC) were included in this study. HIV RNA derived from plasma from seroconverting partners (SPs) were first sequenced a median of 25 days post onset of symptoms (dps) and then followed for a median of 827 dps, whereas transmitting partner (TP) plasma RNA was sequenced at one time point at a median of 28.5 dps in the SP (Table 1). Viral sequences corresponding to Gag p17 and p24 coding regions were obtained by 454 pyrosequencing of a median of 245 viral templates or by generating 15 to 20 single-template derived Sanger sequences when material and/or viral load was limiting (40). Phylogenetic analysis supported a single viral variant establishing infection in 5/6 SPs, whereas PIC51861 had at least two founding variants.
TABLE 1.
PIC partner pair characteristicsa
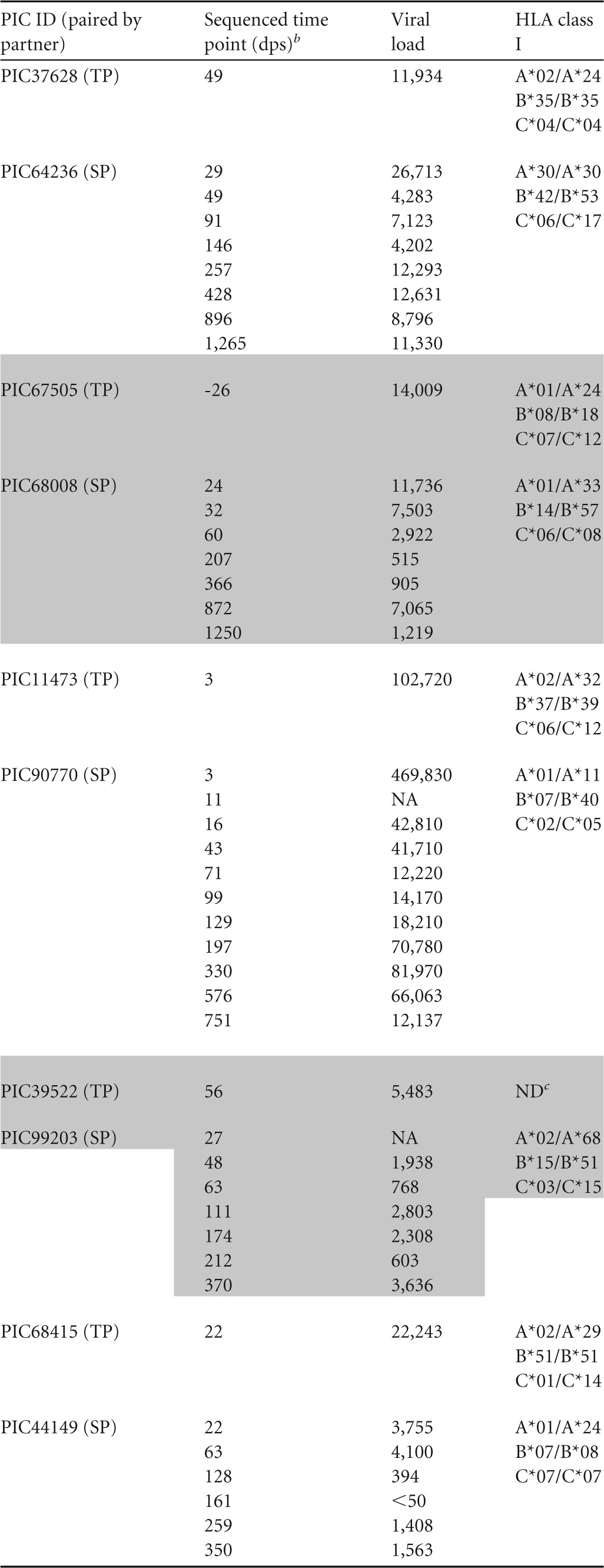
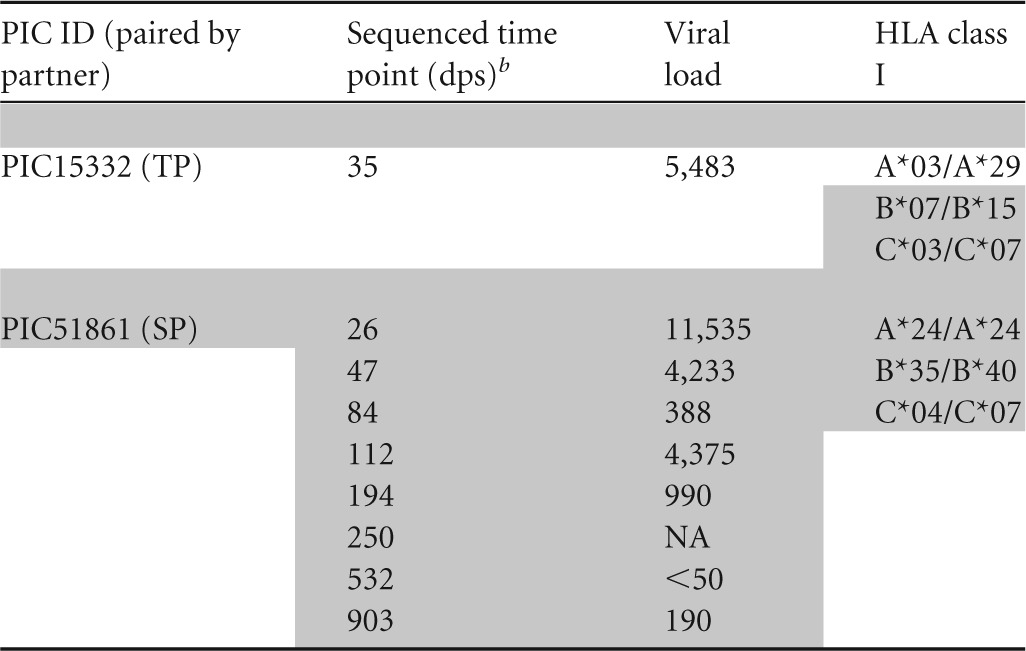
Days after onset of symptoms. Data for transmitting partners (TPs) and seroconverting partners (SPs) are grouped vertically, as indicated by the presence and absence of shading. ID, identifier; NA, not available.
Days postonset of symptoms.
ND, not determined due to lack of available sample.
For each partner pair, we generated autologous 15-mer peptides overlapping by 11 amino acids (aa), corresponding to full-length Gag p17 and p24 sequences. These peptides reflected the founder sequence and any amino acid variant found in ≥5% of sequence reads obtained from either the TP or SP at any time point. Each subject was screened for responses by IFN-γ/IL-2 FluoroSpot using both partners' autologous peptide sets. TPs were screened for responses at the same visit date as used for sequencing. SPs were screened for responses at 1 to 3 visit dates during the first year of HIV-1 infection depending on sample availability. One TP (PIC39522) and one SP (PIC90770) could not be screened due to insufficient PBMC. We also determined the optimal epitopes for responses detected in SPs. All SPs were antiretroviral therapy naive, and no TPs were on treatment on or near the visit used for immunological assays.
In the tested TPs, we detected a median of two Gag-specific T-cell responses with a median magnitude of 1,552 SFC/million (Fig. 1) at the single time point tested. Within the SPs, we compared breadth and magnitude of Gag-specific T-cell responses detected before and after 100 dps. We found an increase in breadth over time, with a median of 1.25 Gag responses before 100 dps increasing to a median of 4.5 responses following 100 dps (Fig. 1A). Similarly, the median magnitude increased from 456 SFC/million prior to 100 dps to a median of 1,199 SFC/million after 100 dps (Fig. 1B). Cytokine responses were primarily IFN-γ+, with an IL-2 response detected in only one TP (PIC11473 [data not shown]). In a previous study of progressive HIV infection, 95% of IFN-γ ELISpot responses were found to be CTL mediated (46). However, given the use of bulk PBMC in our assays, we cannot rule out the possibility that we were also detecting CD4+ T-cell responses.
FIG 1.

Breadth and magnitude of responses in transmission pairs. Partner pairs are listed on the x axis. N/A, sample not available. (A) Number of targeted epitopes. (B) Total magnitude of responses. White bars, transmitting partner; gray bars, seroconverting partner at <100 dps; black bars, seroconverting partner at >100 dps.
Little evidence for reversion in the evolution of TP-targeted epitopes in SPs.
We defined a reversion pattern as the transmission of a sequence containing an amino acid of lower frequency in the Los Alamos National Laboratory HIV sequence database (HIVDB) that confers escape from a TP T-cell response, followed by a reversion to a higher-frequency sensitive amino acid (i.e., pre-escape amino acid) in the SP (5). Although we found no evidence for this pattern in the six TP epitopes that evolved over time in SP, we did observe two unique cases of viral evolution in epitopes targeted by TPs (Fig. 2).
FIG 2.

Epitopes targeted in TP with corresponding sequence evolution in the SP. (A) PIC67505/68008; (B) PIC11473/90770; (C) PIC37628/64236. Values for SFC/million are for VEEKAFSPEVIPMFS and AFSPEVIPMFSALAE. (D) PIC15332/51861; (E) PIC68415/44149. Red labels indicate epitope variant that reached >80% frequency in the viral population (i.e., the replacement variant). Immunological assays were performed at the same time point as used for sequencing.
Phylogenetic analyses identified at least two founder variants in SP PIC51861 (data not shown). Two variants within 5ASVLSGGELDRWEKI19, E12, and A12 (positions refer to Gag residues within the HXB2 reference strain) were identified in the SP by 35 dps, whereas only the E12 variant was detected in the TP's (PIC15332) plasma. Although the A12 variant was dominant early in the SP's infection, the E12 variant rose in frequency and was the dominant variant by 532 dps (Fig. 2A). The TP had T-cell responses to 15-mer peptides containing E12 but not the A12 variant, indicating that the A12 mutation could confer escape from T-cell recognition (Fig. 2A). Thus, rather than being absent in the TP and arising de novo in the SP, the A12 variant could have been emerging in the TP and present at levels below detection at the time of transmission. The fact that the E12 variant eventually became dominant in the SP suggests that the escape mutation (A12) carried fitness costs that prevented fixation over the sensitive amino acid form (E12).
In TP PIC37628, the dominant variant in 159VEEKAFSPEVIPMFSALAE177 around the time of transmission contained V159/A176. A minor variant, V159/S176, was also detected at a frequency of 5.3%, and this emerged as the founder sequence in SP PIC64236 (Fig. 2B). Over time, an I159 mutation, reflecting a dramatic decrease in HIVDB frequency (V, 98%, I, 1%), arose in the SP and mostly replaced the founder sequence by 428 dps (Fig. 2B). Both the V159 and I159 variants and the A176 and S176 variants were recognized by the TP (Fig. 2B), suggesting that these did not represent classical TCR or binding escape variants in the TP. Interestingly, A176 is predicted to have a higher human proteasome cleavage score (0.7676) using a proteasomal cleavage prediction algorithm (http://www.cbs.dtu.dk/services/NetChop/) compared to S176 (0.3168), suggesting that A176 may have dominated in the TP viral population as a result of disrupted processing of the targeted epitope (47, 48).
We found two additional examples of viral evolution within the SP viral population in regions previously targeted by the TP; however, these did not suggest occurrence of CTL reversions (Fig. 2C and D).
Highly dynamic T-cell epitopic escape processes are common during HIV-1 infection.
We identified a total of 20 Gag-specific responses in SP during the first year of infection. Over the course of follow-up, subsequent viral evolution occurred in 13 epitopes, while seven epitopes remained unchanged (Fig. 3 and 4A). Dynamic epitope evolution was common, with multiple epitope variants detected in 9/13 evolving responses and a median of 2 variants per response (Fig. 4A). Interestingly, we found a moderate correlation between the number of epitope variants detected and epitope entropy (Spearman ρ = 0.43; P = 0.05) (Fig. 4B).
FIG 3.

Epitopes targeted in SP and their sequence variants. (A) PIC51861; (B) PIC64236; (C) PIC68008; (D) PIC44149; (E) PIC99203. Optimal epitopes were experimentally defined except where noted and are represented by shaded areas within 15-mer sequences. Red labels indicate epitope variants that reached >80% frequency in the viral population (i.e., the replacement variant), and yellow highlighting represents de novo epitopes containing escape mutations to founder-specific responses. Time points used for immunological assays are noted for each SP.
FIG 4.

Multiple variants often found within targeted epitopes. (A) Number of epitope variants detected in each of the 20 responses detected in the SPs. The absence of detection of epitope variants indicates a persistent epitope. (B) Epitope entropy is correlated to the number of epitope variants detected per response. (C) The nine epitopes in which a replacement variant was selected in the viral population are listed on the x axis. For each epitope, the duration (in dps) is shown between the time the response was first detected and the time in which the replacement variant achieved >80% frequency in the viral population.
At the end of follow-up, we found that in nine epitopes, a single epitope variant became fixed in the viral population (i.e., the replacement variant). The time to fixation was long, with a median of 575 days between the time point in which the response was first detected and that at which the replacement variant achieved >80% frequency in the viral population (Fig. 4C). Over this long course of virus-host interactions, we found limited similarities in the mutational patterns of T-cell escape leading to the selection of a replacement variant, highlighting the remarkable plasticity of T-cell escape processes. The following distinct patterns of CTL escape in cases were observed.
(i). Classical escape.
In PIC68008, the p17 founder sequence 65QPSLQTGSEELKSLY79 began to decline following 366 dps and F79 became the major variant (Fig. 3C). Only the 15-mer reflecting the founder sequence was recognized at both 207 dps and 1453 dps, indicating that the F79 mutation conferred escape (Fig. 3C). The optimal epitope was determined to be 71GSEELKSLY79 (GY9) and was predicted to be restricted by A*01. Interestingly, his transmitting partner (PIC67505) also had responses to overlapping 15-mer peptides containing the Y79 variant (65QPSLQTGSEELKSLYNTVA83) but not to peptides containing the F79 variant (Fig. 2C). Both variants were detected in his virus population, suggesting that F79 was an escape mutation at the time of transmission. TP PIC67505 also expresses HLA A*01, and although we did not determine the optimal epitope in him due to lack of sample availability, the same GY9 epitope may have also been targeted. Taken together, these observations show that it is possible that because the sensitive form of the epitope was in the founder, PIC68008 was able to target and promote the same escape pathway as the one predicted in the linked TP.
(ii) Cross-reactive responses.
In the PIC51861-targeted p17 peptide 17EKIRLRPGGKKKYKL31, an R28 variant arose by 112 dps and was predominant by 532 dps (Fig. 3A). Peptides spanning the founder and R28 variant were recognized with comparable magnitude responses at both 54 and 1,322 dps (Fig. 3A). However, the optimal epitope in this case was 18KIRLRPGGKKKY29 (KY12), which was recognized with ∼8-fold higher avidity than the emerging variant sequence (see Fig. S1A in the supplemental material), suggesting that the R28 variant likely conferred escape through loss of avidity.
(iii) Sequential responses.
In PIC68008, the p24 founder sequence 143VHQAISPRTLNAWVK157 declined following 60 dps and was undetectable by 366 dps, replaced by the P146 variant (Fig. 3C). Neither variant was recognized at 47 dps (Fig. 3C), both variants were recognized by 207 dps, and by 1,453 dps, only a high-magnitude response to the P146 variant was detected (Fig. 3A). At 207 dps, the optimal epitope was 147ISPRTLNAW155 (IW9 [see Fig. S1B in the supplemental material]), a B*57 targeted epitope. The P146 mutation is a well-characterized escape mutation that disrupts processing of the IW9 epitope (49), suggesting that it was selected to confer escape from IW9-specific T cells. However, the emerging strong and specific response to the overlapping P146-containing epitope, 144HQPISPRTL152 (HL9), suggests that the P146 mutation was selected because it conferred escape to IW9 but subsequently became targeted as a result of the creation of a new epitope, HL9 (see Fig. S1C in the supplemental material). Despite CTL targeting for over 3 years, however, no escape mutations were observed in the HL9 epitope. We found similar patterns of de novo mutations broadening Gag-specific responses over time in PIC68008 (DE10/DR11 [Fig. 3C]) and PIC64236 (LY12/LI10 [Fig. 3B]).
(iv) Simultaneous escape and reversion.
One of nine epitopes showed evidence for simultaneous escape and reversion processes. In SP PIC44149, the p24 founder sequence 77SLFNTIAVLFCVHQR91 at 22 dps (V82/F86 [Fig. 3D]) was not detected in his TP PIC68415, who instead had I82/F86 as the major variant (Fig. 2E).
Closely following transmission, TP PIC68415 responded to a 15-mer peptide in Gag77-91, which contains the immunodominant A*02 SL9 epitope (77SLFNTIAVL85 [Fig. 2E]). Although we did not fine map the response due to limited sample availability, it is highly likely that this epitope is being targeted since PIC68415 expresses HLA-A*02. While I82 is present in approximately 24% of sequences from the HIVDB, F86 is found in only <3%. Five of six variants tested, all except S86, were recognized by TP PIC68415 (Fig. 2E), indicating that the majority of variation occurring at site 86 does not disrupt recognition, potentially because it is outside the SL9 epitope. It is possible that F86 disrupts processing of the SL9 epitope and thus was maintained in the TP viral population as an escape mutation.
Multiple mutations arose in SP PIC44149 at position 86 (L86, S86, C86, and Y86) with the eventual fixation of Y86 (Fig. 2E and 3D). Notably, Y86 is found in 97% of HIVDB sequences, whereas all other observed mutations are rare. At the single time point tested (169 dps), responses to 15-mers corresponding to the founder and the L86, C86, and Y86 variants were detected. As in the TP, no response was detected to the S86 variant (Fig. 3D). Interestingly, the optimal epitope contained the L86 variant, 81TVAVLLCV88 (TV8), which arose before 63 dps, suggesting that it was the L86 variant that primed the TV8-specific response. The F86, C86 and Y86 variants were all recognized with lower avidity (see Fig. S1D in the supplemental material). Collectively, these results suggest that following a rapid selection for mutations to replace the F86 variant transmitted by the TP, one of these variants (L86) became the target of the SP host immune response. These dual selective pressures may have promoted a complicated mutational process that eventually resulted in the fixation of the Y86 variant, which was the most frequently occurring amino acid at this site in the HIV database (putative reversion) and was also less well recognized by the TV8-specific response (escape).
Fixation of replacement variants reflects fitness-balanced escape processes.
Selection of CTL escape mutations is believed to be the result of a balance between the replicative fitness costs of a particular mutation and its ability to confer immune escape (13, 22). We hypothesized that while some transient mutations could permit escape, the fitness costs of these mutations allowed for further selection of more fit escape mutants. Hence, fitness competition assays were performed between parental HIV-1 NL4-3 virus and NL4-3 variants with mutated sites corresponding to the variants observed in PIC64236 RK9 (Fig. 3B) and PIC68008 TW10 (Fig. 3C). These epitopes displayed substitution patterns that included an intermediate, transient variant(s) before fixation of a replacement variant. Pairwise fitness competitions were performed with PBMC cultures from a single healthy donor, and relative fitness was calculated by measuring the proportion of parental NL4-3 versus mutant virus at different time points following infection, as previously described (43).
In PIC64236, the p17 founder sequence 17EKIRLRPGGKKKYKL31 began to decline after 91 dps until it was undetectable by 428 dps. Multiple independent variants (T28, E28, Q23, R28, Q28, and R30) arose in the viral population by 146 dps, and Q28 became fixed by the end of the study period at 1,265 dps (Fig. 5A). At 41 dps, 15-mers representing the founder sequence and the R30 variant (mutation C-terminal to epitope) were recognized with comparable magnitudes. By 321 dps, these two variants plus R28 were recognized. Peptides corresponding to the four other observed variants did not elicit a response at either visit date, indicating that those substitutions conferred escape (Fig. 5B). The optimal epitope, 17RLRPGGKKK28 (RK9 [Fig. 5C]), was found in the 15-mers representing the founder sequence and the R30 variant, accounting for their similar levels of IFN-γ induction. The R28 mutation was previously reported to cause a decrease in avidity when RK9 was restricted by HLA A*03 (50). In PIC64236, who does not possess HLA A*03, this epitope was predicted to be restricted by HLA A*30, as also reported in a study describing the promiscuity of CTL epitopes (51), possibly explaining why the R28 variant was recognized with comparable avidity.
FIG 5.

Early escape mutations confer fitness costs compared to replacement variants. (A) Frequency of variants at amino acid positions 23, 28, and 30 within Gag17-31 in PIC64236. (B) Magnitude of IFN-γ T-cell response by PIC64236 to 15-mers reflecting variation in Gag17-31 at 41 dps (open bars) and 321 dps (filled bars). (C) RK9 epitope (K26/K27/K28 EC50 of 468 ng/ml, black; R28 EC50 of 631 ng/ml, blue; Q26 not recognized, dark gray; Q27 not recognized, light gray) in PIC64236 at 41dps. (D) Highly basic region located in the N-terminal domain of p17. Blue indicates basic residues. The diagram was adapted from reference 53. (E) Fitness costs of observed RK9 epitope variants relative to K28 within an otherwise homologous HIV-1NL4-3 background.
The RK9 epitope is located within the highly basic region (HBR) of Gag p17 (Fig. 5D), which plays an important role following Gag targeting to the plasma membrane, where basic amino acid residues (R and K) in the HBR interact with negatively charged phospholipids (52). We engineered mutations in NL4-3 to reflect escape mutations occurring at aa 28 (K28 in the founder sequence) within the RK9 epitope, including a positively charged amino acid (R28), a negatively charged amino acid (E28), and polar uncharged amino acids (T28 and Q28). Neither the transient R28 variant, which did not confer escape, nor the eventual dominant escape Q28 variant conferred a fitness cost, whereas the E28 and T28 transient escape mutations had lower fitness than did the Q28 eventual replacement (P = 0.02 for each [Fig. 5E]). Hence, the eventual dominant mutant was unique in that it conferred both escape and no loss of fitness.
Next we addressed why the majority of mutations occurred at site 28, hypothesizing that this site was more tolerant to change than other positively charged amino acid sites within the epitope. Mutations were engineered into NL4-3 that mimicked the replacement variant (K to Q) at aa 26 and 27. Both of these mutations conferred escape within the RK9 epitope (Fig. 5C); hence, these mutations could have been selected as alternative escape pathways. However, the Q27 mutation proved to be lethal, whereas the Q26 mutation had no significant fitness cost (Fig. 5E).
Between 32 dps and 207 dps in PIC68008, the p24 founder sequence, 239TTSTLQEQVGWMTSN253, was first replaced by an E248 variant and then rapidly replaced by an N242 variant that became fixed by 366 dps. We also detected an N242/I247 variant at low levels in the viral population at both 366 dps and 1250 dps (7.1% and 6.6% of sequence reads, respectively) (Fig. 6A). At 47 dps, PIC68008 recognized all 15-mer variants tested, but by 207 dps only responses targeted against the founder sequence and the E248 variant were detectable, and no responses were detected at 1453 dps (Fig. 6B). The optimal epitope, 240TSTLQEQVGW249 (TW10), is an immunodominant epitope targeted by B*57 individuals during acute infection (27). While the founder sequence and E248 were recognized with similar avidities at 47 dps, the N242 and N242/I247 variants had reduced avidity (Fig. 6C). Interestingly, the E248 mutation has been reported to increase susceptibility to the NK cell response by abrogating KIR3DL1 binding (53). We did not KIR type our study subjects, and therefore, it is unknown if NK-mediated selection pressures may have influenced the removal of the E248 mutation from the viral population.
FIG 6.

Sequential selection of epitope variants due to fitness costs of multiple mutations within the TW10 epitope. (A) Frequency of variants at amino acid positions 242, 247, and 248 within Gag239-253 over 1,250 dps of follow-up in PIC68008. (B) Magnitude of IFN-γ T-cell response by PIC68008 to 15-mers reflecting variation in Gag17-31 at 47 dps (open bars), 207 dps (filled bars), and 1,453 dps (patterned bars, no responses detected). (C) TW10 epitope (founder EC50 of 575 ng/ml, black; E248 EC50 of 100 ng/ml, green; N242 EC50 of 19,953 ng/ml, red; N242/I247 EC50 of 25,118,864 ng/ml, purple; N242/E248 not recognized, gray) in PIC68008 at 47dps. (D) Host protein cyclophilin A interacts with p24 residues as described in references 27 and 54. (E) Fitness costs of single or double amino acid substitutions relative to T242/G248 within an otherwise homologous HIV-1NL4-3 background.
The TW10 epitope lies within a region of Gag p24 that interacts with host protein cyclophilin A (Fig. 6D), which is crucial for a number of viral processes (27, 54). In PIC68008, a mutually exclusive substitution pattern occurred within TW10 in which a transient E248 variant first emerged and was subsequently replaced by N242 (Fig. 6A). Neither mutation resulted in significant fitness costs in the HIVNL4-3 background (Fig. 6E), consistent with other recent studies with PBMC (43, 55). As this epitope displayed a mutually exclusive substitution pattern, we hypothesized that multiple simultaneous mutations could incur a fitness cost, possibility due to greater disruption of the cyclophilin A interaction. To explore this, an N242/E248 double mutant was tested. In contrast to the single mutants, peptides, including both mutations did not elicit immune responses, suggesting that the double mutation permitted more efficient escape (Fig. 6C). However, the double mutation conferred significant fitness costs compared to E248 (P = 0.001) or N242 (P = 0.003) alone (Fig. 6E). Our results agree with previous studies that reported significant fitness costs of simultaneous mutations occurring at sites 242 and 248 and others within the TW10 epitope, which highlights the functional constraints of this epitope (28, 56).
Maintenance of multiple escape mutants in the absence of fitness costs.
In a previous study (5), we reported a number of mutually exclusive substitution patterns occurring throughout the viral genome of PIC38417. Given the results found in PIC64236 and PIC68008, we assessed whether the observed mutually exclusive substitution patterns in PIC38417 were the result of fitness-balanced escape processes in viral proteins other than Gag p17 and p24. To this end, we tested for responses by IFN-γ enzyme-linked immunosorbent spot (ELISpot) assay using an autologous peptide set corresponding to sites within the viral proteome displaying mutually exclusive substitution patterns. We found evidence for two cases of CTL response and subsequent viral escape in Gag-Pol and Env gp41.
In Gag-Pol, the founder sequence 7AFPQGKARKFSSEQTR22 began to decline following 50 dps, as two mutually exclusive mutations (R11 and E12) arose in the Pol sequence, without impact on the Gag amino acid sequence (Fig. 7A). All three variants were present in the viral population at the end of the study period. Only the founder sequence elicited a T-cell response at 53 dps, indicating that R11 and E12 conferred escape (Fig. 7B). The optimal epitope 9FPQGKARKF16 was predicted to be restricted by B*35 (Fig. 7C). Interestingly, G11 and K12 are encoded within the Gag-Pol frameshift stimulator loop (Fig. 7D), which promotes a −1 translational frameshift that results in translation of the Gag-Pol protein (57). The R11 and E12 substitutions each involve a single nucleotide change, which is likely to affect the RNA base pairing of the stimulator loop.
FIG 7.

Single escape mutations do not confer fitness costs in FF9 epitope. (A) Frequency of variants at sites 11 and 12 within Gag-Pol7-22 over 247 dps of follow-up. (B) Magnitude of IFN-γ response to 15-mers reflecting variation in Gag-Pol7-22 at 53 dps (open bars) and 247 dps (filled bars). (C) FF9 epitope (founder EC50 of 513 ng/ml, black, left axis; R11/E12 EC50 of 33,113 ng/ml, gray, right axis) in PIC38417 at 53 dps. (D) Nucleotide changes corresponding to G11R or K12E amino acid substitutions within the frameshift stimulator stem-loop (57). (E) Fitness costs of single or double amino acid substitutions relative to G11/E12 within an otherwise homologous HIV-1NL4-3 background.
Despite changes imposed on the stimulator loop, neither the R11 or E12 mutations impaired fitness (Fig. 7E). However, an R11/E12 double mutant had a 2-fold reduction in avidity compared to the founder peptide, although it elicited a higher-magnitude response (Fig. 7C). The double mutant also had significantly reduced fitness compared to the R11 (P = 0.0009) or E12 (P = 0.002) mutant alone (Fig. 7E), in agreement with a previous report that multiple simultaneous mutations within the stimulator loop negatively affect viral viability (57).
In Env gp41, the founder env sequence 635ISNYTNIIYTLIENS649 declined after 40 dps and was undetectable by 116 dps, while multiple mutations arose in a mutually exclusive substitution pattern at sites 640, 641, and 644 (Fig. 8A). The IFN-γ response to the founder sequence was robust at 53 dps but declined by 247. One cross-reactive variant (H640) was also recognized at 53 dps but not at 247 dps. All other variants tested (D640, S640, T641, L641, and A644) failed to elicit a response at either visit date, indicating that they conferred escape (Fig. 8B). The optimal epitope, 637NYTNIIYTL645, was predicted to be restricted by A*24 (Fig. 8C). This epitope is located within the gp41 C-terminal heptad repeat (CHR) (Fig. 8D), which binds to the N-terminal heptad repeat (NHR) to facilitate viral fusion with the host cell membrane (58). Interestingly, the residues mutated during the escape process are not involved in the interaction between the CHR and NHR (58). In contrast, the HLA anchor residues in the epitope (638 and 645) are directly involved in binding with the NHR (58).
FIG 8.

Fitness costs of escape variants vary across sites in the C-terminal heptad repeat of gp41. (A) Frequency of variants at sites 640, 641, and 644 within Env635-649 over 247 dps of follow-up. (B) Magnitude of IFN-γ response to 15-mers reflecting variation in Env635-649 at 53 dps (open bars) and 247 dps (filled bars). All variants are color coordinated between graphs A and B. Optimal epitopes are boxed. (C) NL9 epitope (founder EC50 of 51 ng/ml, black; A638 EC50 of 4,786 ng/ml, gray squares; R645 EC50 of 15,849 ng/ml, gray triangles) in PIC38417 at 53dps. (D) The gp41 C-terminal and N-terminal heptad repeats bind to facilitate viral fusion. Interacting amino acids are circled and linked. The NL9 epitope is circled in purple (58). (E) Fitness costs of observed and hypothetical NL9 epitope variants relative to S640/L641/T644 within an otherwise homologous HIV-1NL4-3 background.
Whereas sites involved with binding to NHR are highly conserved among HIV-1 clade B sequences, other sites within this region are highly variable. Consequently, there were amino acid differences in sites 640, 641, and 644 between the PIC38417 founder sequence and NL4-3. Several of the observed escape mutations were engineered into NL4-3 at the three evolving sites (D640, T641, and A644), as well as at the anchor residues involved in NHR binding (A638 and R645), the latter with mutations predicted to disrupt binding to HLA-A*24 (59). The D640 and A644 mutations did not incur significant fitness costs relative to parental NL4-3, while T641 had a minor fitness deficit (Fig. 8E). Interestingly, both mutations at anchor residues were lethal (Fig. 8E), highlighting the functional constraints of the CHR-NHR binding for virus viability.
DISCUSSION
In this study, massively parallel sequencing of viral populations was combined with detection of T-cell responses using autologous peptides in six pairs of linked HIV transmissions to provide a unique perspective into T-cell escape and reversion processes during HIV infection.
Reports have been variable on the extent of CTL reversions during primary HIV infection (5, 11, 60). As our study included transmission pairs, we were able to screen TPs for T-cell responses near the time of transmission, allowing us to determine potential reverting sites, but only those associated with contemporaneously targeted TP epitopes, as responses often wane following fixation of escape mutations (22). With peptides reflecting only a snapshot of the TP infection, this may explain why we found a lower breadth of responses in the TP than at later time points in the SP, in which the responses detected corresponded to the current viral population. We found a low frequency of potentially reverting sites, and the ongoing evolution observed at these sites over the first 1 to 3+ years of infection did not reflect expected reversion patterns. The relative paucity of reversions in our experimental study compared to reversions determined using HLA associations in large cohort studies is in line with the finding that escape mutations identified as HLA-associated systematically favor those that escape and revert rapidly (61). Interestingly, several of the SPs had relatively low viral loads during early infection. As transmitted polymorphisms have been associated with lower viral loads, the lack of reversions in these subjects may have contributed to the maintenance of low viremia (62).
We identified Gag-specific T-cell responses to 20 epitopes within the first year of infection in the six SPs, with 13 evolving over the course of the study period. The majority of evolving epitopes displayed complex escape processes, which were frequently dynamic, with the emergence of multiple near-simultaneous mutations. Our study revealed a correlation between epitope entropy and quantity of epitope variants, which suggests a relationship between the tolerance for variability within the epitope and selection for multiple escape mutations. Thus, epitope entropy may serve as a useful predictor of the number of potential escape pathways available following a targeted response.
Previously reported escape mutations were found in several targeted epitopes (e.g., RK9, TW10, and IW9), confirming that common escape mutations often occur across different individuals (27, 28, 49, 63). However, frequent sampling, as done in this study, revealed that even though a single common escape mutation typically became fixed in the viral population, the evolutionary processes leading to that selection were complex, with the emergence and loss of various transient escape mutations. In some cases, these processes can take years to resolve. In this study, we found that it took a median of 575 days from detection of response to selection of a replacement variant. Importantly, the precision of these calculations is greatly influenced by the time points available for analysis. Therefore, these findings further emphasize the need for frequent sampling from acute through chronic infections in order to accurately define these timelines and afford a more comprehensive understanding of the complexity of viral adaptation to cellular immune responses.
The majority of T-cell responses to evolving epitopes demonstrated some level of cross-reactivity to the variants that arose over time. In some cases, variants were recognized with a lower functional avidity (e.g., N242 in PIC68008), which may reflect a decrease in cytotoxic potential (64–66), and consequently, these variants may persist in the viral population due to weakened CTL-mediated pressure. Recognition of epitope variants can also result from de novo responses to the same peptides as previously selected to confer escape (30, 67). We found that escape mutations can also promote responses to new epitopes located in close proximity to the originally targeted epitope. Therefore, escape processes during primary HIV infection have the potential to increase the breadth and depth of Gag-specific CTL responses. We recently reported that during chronic HIV infection, progressors maintain higher levels of epitope variant recognition than viremic controllers (36). Our current results demonstrate that cross-reactivity and induction of de novo responses to epitope variants over time may lead to the high levels of variant recognition observed during chronic progressive infection.
To explore the impact of epitopic mutations on viral replicative capacity, we performed viral fitness assays using NL4-3 viruses modified to reflect mutations observed during the dynamic escape processes observed. In targeted epitopes in which a replacement variant was selected (PIC64236 RK9 and PIC68008 TW10), we found that compared to the replacement variant, transient mutations either carried replicative fitness costs or did not confer escape. The fitness costs we observed, while significant, were not very pronounced; however, we may be underestimating this effect due to engineering the mutations into the backbone of the fast-growing lab strain NL4-3 rather than within the subjects' autologous virus. Nonetheless, our data suggest that even minor fitness defects can prevent fixation of a particular escape variant. These results indicate that eventual selection of a replacement variant and resolution of dynamic escape processes are the result of an optimal balance between immune escape and replicative fitness costs. Interestingly, in epitopes that remained variable over the course of the study period (PIC64236 NL9 and FF9), the majority of detectable mutations conferred escape without incurring replicative fitness costs.
We found that escape processes involve mutations occurring at one site within an epitope or a complex mutually exclusive substitution pattern, suggesting that functional constraints limit the number of sites tolerant to mutation(s) in order to facilitate escape. We explored this directly in our viral fitness assays by engineering NL4-3 viruses to express mutations that reflect alternative escape pathways (i.e., mutations at different sites in the epitope or simultaneous mutations). While all of these mutations conferred escape through loss of recognition or decreased avidity, dramatic replicative fitness costs were observed, and in some cases, these mutations were lethal. These observations help explain why mutations are limited to particular sites or to single mutations during dynamic escape processes. Importantly, by using complementary sequencing and epitope mapping techniques, these types of escape patterns can be revealed and can help identify viral regions that are immunogenic yet functionally constrained.
In summary, we have shown that the principle of fitness-balanced immune escape guides the selection of mutations in complicated dynamic escape processes. Several vaccine immunogens have been designed to induce CTL responses that have the potential to circumvent escape pathways either by only including regions that are under such high functional constraints that they cannot mutate without loss of function (24) or by inducing immune responses to commonly occurring variants (68, 69). The complex escape pathways identified in this study highlight the difficulty of blocking all viable variants representing escape routes for the virus. However, our results indicate that some viral regions are limited in the number of sites available to facilitate escape. Potentially, having potent cross-reactive or overlapping T-cell responses directed against low entropy regions may allow for sufficient coverage of the sites tolerant to mutation. This may exert sufficient selective pressure on the virus to mutate the intolerable sites or select for simultaneous mutations, which could have greater fitness consequences and thus significantly contribute to viral control. Further investigations into the identification of these vulnerable viral regions may aid in the design of improved immunogens based on HIV Gag.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers P01AI057005 and T32AI007140 and by the Seattle Centers for AIDS Research Molecular Profiling and Computational Biology Core (NIH P30AI027757).
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The opinions expressed herein are those of the authors and should not be construed as official or representing the views of the U.S. Department of Defense or the Department of the Army.
We thank all study participants for their participation in this research study as well as the clinic staff for their critical work and dedication. We thank Stephen Voght for editorial assistance.
We declare no conflicts of interest.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01876-15.
REFERENCES
- 1.Delwart EL, Sheppard HW, Walker BD, Goudsmit J, Mullins JI. 1994. Human immunodeficiency virus type 1 evolution in vivo tracked by DNA heteroduplex mobility assays. J Virol 68:6672–6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang LQ, MacKenzie P, Cleland A, Holmes EC, Brown AJ, Simmonds P. 1993. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J Virol 67:3345–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu T, Mo H, Wang N, Nam DS, Cao Y, Koup RA, Ho DD. 1993. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 261:1179–1181. doi: 10.1126/science.8356453. [DOI] [PubMed] [Google Scholar]
- 4.Abrahams MR, Anderson JA, Giorgi EE, Seoighe C, Mlisana K, Ping LH, Athreya GS, Treurnicht FK, Keele BF, Wood N, Salazar-Gonzalez JF, Bhattacharya T, Chu H, Hoffman I, Galvin S, Mapanje C, Kazembe P, Thebus R, Fiscus S, Hide W, Cohen MS, Karim SA, Haynes BF, Shaw GM, Hahn BH, Korber BT, Swanstrom R, Williamson C. 2009. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-Poisson distribution of transmitted variants. J Virol 83:3556–3567. doi: 10.1128/JVI.02132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herbeck JT, Rolland M, Liu Y, McLaughlin S, McNevin J, Zhao H, Wong K, Stoddard JN, Raugi D, Sorensen S, Genowati I, Birditt B, McKay A, Diem K, Maust BS, Deng W, Collier AC, Stekler JD, McElrath MJ, Mullins JI. 2011. Demographic processes affect HIV-1 evolution in primary infection before the onset of selective processes. J Virol 85:7523–7534. doi: 10.1128/JVI.02697-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A 105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol 73:10489–10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gottlieb GS, Heath L, Nickle DC, Wong KG, Leach SE, Jacobs B, Gezahegne S, van 't Wout AB, Jacobson LP, Margolick JB, Mullins JI. 2008. HIV-1 variation before seroconversion in men who have sex with men: analysis of acute/early HIV infection in the multicenter AIDS cohort study. J Infect Dis 197:1011–1015. doi: 10.1086/529206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol 68:4650–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, McNevin J, Cao J, Zhao H, Genowati I, Wong K, McLaughlin S, McSweyn MD, Diem K, Stevens CE, Maenza J, He H, Nickle DC, Shriner D, Holte SE, Collier AC, Corey L, McElrath MJ, Mullins JI. 2006. Selection on the human immunodeficiency virus type 1 proteome following primary infection. J Virol 80:9519–9529. doi: 10.1128/JVI.00575-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, Keele BF, Learn GH, Turnbull EL, Salazar MG, Weinhold KJ, Moore S, Letvin N, Haynes BF, Cohen MS, Hraber P, Bhattacharya T, Borrow P, Perelson AS, Hahn BH, Shaw GM, Korber BT, McMichael AJ. 2009. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med 206:1253–1272. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, Zody MC, Erlich RL, Green LM, Berical A, Wang Y, Casali M, Streeck H, Bloom AK, Dudek T, Tully D, Newman R, Axten KL, Gladden AD, Battis L, Kemper M, Zeng Q, Shea TP, Gujja S, Zedlack C, Gasser O, Brander C, Hess C, Gunthard HF, Brumme ZL, Brumme CJ, Bazner S, Rychert J, Tinsley JP, Mayer KH, Rosenberg E, Pereyra F, Levin JZ, Young SK, Jessen H, Altfeld M, Birren BW, Walker BD, Allen TM. 2012. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog 8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Troyer RM, McNevin J, Liu Y, Zhang SC, Krizan RW, Abraha A, Tebit DM, Zhao H, Avila S, Lobritz MA, McElrath MJ, Le Gall S, Mullins JI, Arts EJ. 2009. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog 5:e1000365. doi: 10.1371/journal.ppat.1000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJ. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med 10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 15.Rolland M, Heckerman D, Deng W, Rousseau CM, Coovadia H, Bishop K, Goulder PJ, Walker BD, Brander C, Mullins JI. 2008. Broad and Gag-biased HIV-1 epitope repertoires are associated with lower viral loads. PLoS One 3:e1424. doi: 10.1371/journal.pone.0001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med 13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 17.Masemola A, Mashishi T, Khoury G, Mohube P, Mokgotho P, Vardas E, Colvin M, Zijenah L, Katzenstein D, Musonda R, Allen S, Kumwenda N, Taha T, Gray G, McIntyre J, Karim SA, Sheppard HW, Gray CM. 2004. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: correlation with viral load. J Virol 78:3233–3243. doi: 10.1128/JVI.78.7.3233-3243.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuñiga R, Lucchetti A, Galvan P, Sanchez S, Sanchez C, Hernandez A, Sanchez H, Frahm N, Linde CH, Hewitt HS, Hildebrand W, Altfeld M, Allen TM, Walker BD, Korber BT, Leitner T, Sanchez J, Brander C. 2006. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J Virol 80:3122–3125. doi: 10.1128/JVI.80.6.3122-3125.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganusov VV, Goonetilleke N, Liu MK, Ferrari G, Shaw GM, McMichael AJ, Borrow P, Korber BT, Perelson AS. 2011. Fitness costs and diversity of the cytotoxic T lymphocyte (CTL) response determine the rate of CTL escape during acute and chronic phases of HIV infection. J Virol 85:10518–10528. doi: 10.1128/JVI.00655-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, Pillay T, Rousseau C, Mullins JI, Brander C, Walker BD, Stuart DI, Kiepiela P, Goulder P. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol 80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedrich TC, Dodds EJ, Yant LJ, Vojnov L, Rudersdorf R, Cullen C, Evans DT, Desrosiers RC, Mothe BR, Sidney J, Sette A, Kunstman K, Wolinsky S, Piatak M, Lifson J, Hughes AL, Wilson N, O'Connor DH, Watkins DI. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat Med 10:275–281. doi: 10.1038/nm998. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, McNevin J, Zhao H, Tebit DM, Troyer RM, McSweyn M, Ghosh AK, Shriner D, Arts EJ, McElrath MJ, Mullins JI. 2007. Evolution of human immunodeficiency virus type 1 cytotoxic T-lymphocyte epitopes: fitness-balanced escape. J Virol 81:12179–12188. doi: 10.1128/JVI.01277-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boutwell CL, Carlson JM, Lin TH, Seese A, Power KA, Peng J, Tang Y, Brumme ZL, Heckerman D, Schneidewind A, Allen TM. 2013. Frequent and variable cytotoxic-T-lymphocyte escape-associated fitness costs in the human immunodeficiency virus type 1 subtype B Gag proteins. J Virol 87:3952–3965. doi: 10.1128/JVI.03233-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rolland MMS, Swain JV, Lanxon-Cookson EC, Kim M, Westfall DH, Larsen BB, Gilbert PB, Mullins JI. 2013. HIV-1 conserved-element vaccines: relationship between sequence conservation and replicative capacity. J Virol 87:5461–5467. doi: 10.1128/JVI.03033-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manocheewa S, Swain JV, Lanxon-Cookson E, Rolland M, Mullins JI. 2013. Fitness costs of mutations at the HIV-1 capsid hexamerization interface. PLoS One 8:e66065. doi: 10.1371/journal.pone.0066065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rihn SJ, Wilson SJ, Loman NJ, Alim M, Bakker SE, Bhella D, Gifford RJ, Rixon FJ, Bieniasz PD. 2013. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog 9:e1003461. doi: 10.1371/journal.ppat.1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brockman MA, Schneidewind A, Lahaie M, Schmidt A, Miura T, Desouza I, Ryvkin F, Derdeyn CA, Allen S, Hunter E, Mulenga J, Goepfert PA, Walker BD, Allen TM. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A J Virol 81:12608–12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miura T, Brockman MA, Schneidewind A, Lobritz M, Pereyra F, Rathod A, Block BL, Brumme ZL, Brumme CJ, Baker B, Rothchild AC, Li B, Trocha A, Cutrell E, Frahm N, Brander C, Toth I, Arts EJ, Allen TM, Walker BD. 2009. HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare gag variants associated with reduced viral replication capacity and strong cytotoxic T-lymphocyte [corrected] recognition. J Virol 83:2743–2755. doi: 10.1128/JVI.02265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneidewind A, Brockman MA, Sidney J, Wang YE, Chen H, Suscovich TJ, Li B, Adam RI, Allgaier RL, Mothe BR, Kuntzen T, Oniangue-Ndza C, Trocha A, Yu XG, Brander C, Sette A, Walker BD, Allen TM. 2008. Structural and functional constraints limit options for cytotoxic T-lymphocyte escape in the immunodominant HLA-B27-restricted epitope in human immunodeficiency virus type 1 capsid. J Virol 82:5594–5605. doi: 10.1128/JVI.02356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, McNevin JP, Holte S, McElrath MJ, Mullins JI. 2011. Dynamics of viral evolution and CTL responses in HIV-1 infection. PLoS One 6:e15639. doi: 10.1371/journal.pone.0015639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rolland M, Nickle DC, Mullins JI. 2007. HIV-1 group M conserved elements vaccine. PLoS Pathog 3:e157. doi: 10.1371/journal.ppat.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schacker T, Collier AC, Hughes J, Shea T, Corey L. 1996. Clinical and epidemiologic features of primary HIV infection. Ann Intern Med 125:257–264. doi: 10.7326/0003-4819-125-4-199608150-00001. [DOI] [PubMed] [Google Scholar]
- 33.Schacker TW, Hughes JP, Shea T, Coombs RW, Corey L. 1998. Biological and virologic characteristics of primary HIV infection. Ann Intern Med 128:613–620. doi: 10.7326/0003-4819-128-8-199804150-00001. [DOI] [PubMed] [Google Scholar]
- 34.Stekler J, Sycks BJ, Holte S, Maenza J, Stevens CE, Dragavon J, Collier AC, Coombs RW. 2008. HIV dynamics in seminal plasma during primary HIV infection. AIDS Res Hum Retroviruses 24:1269–1274. doi: 10.1089/aid.2008.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janes H, Friedrich DP, Krambrink A, Smith RJ, Kallas EG, Horton H, Casimiro DR, Carrington M, Geraghty DE, Gilbert PB, McElrath MJ, Frahm N. 2013. Vaccine-induced gag-specific T cells are associated with reduced viremia after HIV-1 infection. J Infect Dis 208:1231–1239. doi: 10.1093/infdis/jit322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sunshine J, Kim M, Carlson JM, Heckerman D, Czartoski J, Migueles SA, Maenza J, McElrath MJ, Mullins JI, Frahm N. 2014. Increased sequence coverage through combined targeting of variant and conserved epitopes correlates with control of HIV replication. J Virol 88:1354–1365. doi: 10.1128/JVI.02361-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Llano AF, Brander NC. 2009. How to optimally define optimal cytotoxic T lymphocyte epitopes in HIV infection? p 3–24. In Yusim K, Brander C, Haynes BF, Koup R, Moore JP, Watkins DI (ed), HIV molecular immunology. Los Alamos National Laboratory, Theoretical Biology and Biophysics Group, Los Alamos, NM. [Google Scholar]
- 38.Frahm N, Adams S, Kiepiela P, Linde CH, Hewitt HS, Lichterfeld M, Sango K, Brown NV, Pae E, Wurcel AG, Altfeld M, Feeney ME, Allen TM, Roach T, St John MA, Daar ES, Rosenberg E, Korber B, Marincola F, Walker BD, Goulder PJ, Brander C. 2005. HLA-B63 presents HLA-B57/B58-restricted cytotoxic T-lymphocyte epitopes and is associated with low human immunodeficiency virus load. J Virol 79:10218–10225. doi: 10.1128/JVI.79.16.10218-10225.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frahm N, DeCamp AC, Friedrich DP, Carter DK, Defawe OD, Kublin JG, Casimiro DR, Duerr A, Robertson MN, Buchbinder SP, Huang Y, Spies GA, De Rosa SC, McElrath MJ. 2012. Human adenovirus-specific T cells modulate HIV-specific T cell responses to an Ad5-vectored HIV-1 vaccine. J Clin Invest 122:359–367. doi: 10.1172/JCI60202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larsen BB, Chen L, Maust BS, Kim M, Zhao H, Deng W, Westfall D, Beck I, Frenkel LM, Mullins JI. 2013. Improved detection of rare HIV-1 variants using 454 pyrosequencing. PLoS One 8:e76502. doi: 10.1371/journal.pone.0076502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng W, Maust BS, Westfall DH, Chen L, Zhao H, Larsen B, Iyer S, Liu Y, Mullins JI. 2013. Indel and Carryforward Correction (ICC): a new analysis approach for processing 454 pyrosequencing data. Bioinformatics doi: 10.1093/bioinformatics/btt434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van den Ent F, Lowe J. 2006. RF cloning: a restriction-free method for inserting target genes into plasmids. J Biochem Biophys Methods 67:67–74. doi: 10.1016/j.jbbm.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 43.Lanxon-Cookson EC, Swain JV, Manocheewa S, Smith RA, Maust B, Kim M, Westfall D, Rolland M, Mullins JI. 2013. Factors affecting relative fitness measurements in pairwise competition assays of human immunodeficiency viruses. J Virol Methods 194:7–13. doi: 10.1016/j.jviromet.2013.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reed LJM, H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg 27:493–497. [Google Scholar]
- 45.McClure J, van't Wout AB, Tran T, Mittler JE. 2007. Granulocyte-monocyte colony-stimulating factor upregulates HIV-1 replication in monocyte-derived macrophages cultured at low density. J Acquir Immune Defic Syndr 44:254–261. doi: 10.1097/QAI.0b013e318030f5c5. [DOI] [PubMed] [Google Scholar]
- 46.Addo MM, Yu XG, Rathod A, Cohen D, Eldridge RL, Strick D, Johnston MN, Corcoran C, Wurcel AG, Fitzpatrick CA, Feeney ME, Rodriguez WR, Basgoz N, Draenert R, Stone DR, Brander C, Goulder PJR, Rosenberg ES, Altfeld M, Walker BD. 2003. Comprehensive epitope analysis of HIV-1-specific T cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J Virol 77:2081–2092. doi: 10.1128/JVI.77.3.2081-2092.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nielsen M, Lundegaard C, Lund O, Kesmir C. 2005. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 57:33–41. doi: 10.1007/s00251-005-0781-7. [DOI] [PubMed] [Google Scholar]
- 48.Kesmir C, Nussbaum AK, Schild H, Detours V, Brunak S. 2002. Prediction of proteasome cleavage motifs by neural networks. Protein Eng 15:287–296. doi: 10.1093/protein/15.4.287. [DOI] [PubMed] [Google Scholar]
- 49.Draenert R, Le Gall S, Pfafferott KJ, Leslie AJ, Chetty P, Brander C, Holmes EC, Chang SC, Feeney ME, Addo MM, Ruiz L, Ramduth D, Jeena P, Altfeld M, Thomas S, Tang Y, Verrill CL, Dixon C, Prado JG, Kiepiela P, Martinez-Picado J, Walker BD, Goulder PJ. 2004. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J Exp Med 199:905–915. doi: 10.1084/jem.20031982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Streeck H, Brumme ZL, Anastario M, Cohen KW, Jolin JS, Meier A, Brumme CJ, Rosenberg ES, Alter G, Allen TM, Walker BD, Altfeld M. 2008. Antigen load and viral sequence diversification determine the functional profile of HIV-1-specific CD8+ T cells. PLoS Med 5:e100. doi: 10.1371/journal.pmed.0050100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frahm N, Yusim K, Suscovich TJ, Adams S, Sidney J, Hraber P, Hewitt HS, Linde CH, Kavanagh DG, Woodberry T, Henry LM, Faircloth K, Listgarten J, Kadie C, Jojic N, Sango K, Brown NV, Pae E, Zaman MT, Bihl F, Khatri A, John M, Mallal S, Marincola FM, Walker BD, Sette A, Heckerman D, Korber BT, Brander C. 2007. Extensive HLA class I allele promiscuity among viral CTL epitopes. Eur J Immunol 37:2419–2433. doi: 10.1002/eji.200737365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chukkapalli V, Ono A. 2011. Molecular determinants that regulate plasma membrane association of HIV-1 Gag. J Mol Biol 410:512–524. doi: 10.1016/j.jmb.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brackenridge S, Evans EJ, Toebes M, Goonetilleke N, Liu MK, di Gleria K, Schumacher TN, Davis SJ, McMichael AJ, Gillespie GM. 2011. An early HIV mutation within an HLA-B*57-restricted T cell epitope abrogates binding to the killer inhibitory receptor 3DL1. J Virol 85:5415–5422. doi: 10.1128/JVI.00238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hue S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ. 2011. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog 7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boutwell CL, Rowley CF, Essex M. 2009. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J Virol 83:2460–2468. doi: 10.1128/JVI.01970-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song H, Pavlicek JW, Cai F, Bhattacharya T, Li H, Iyer SS, Bar KJ, Decker JM, Goonetilleke N, Liu MK, Berg A, Hora B, Drinker MS, Eudailey J, Pickeral J, Moody MA, Ferrari G, McMichael A, Perelson AS, Shaw GM, Hahn BH, Haynes BF, Gao F. 2012. Impact of immune escape mutations on HIV-1 fitness in the context of the cognate transmitted/founder genome. Retrovirology 9:89. doi: 10.1186/1742-4690-9-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dulude D, Berchiche YA, Gendron K, Brakier-Gingras L, Heveker N. 2006. Decreasing the frameshift efficiency translates into an equivalent reduction of the replication of the human immunodeficiency virus type 1. Virology 345:127–136. doi: 10.1016/j.virol.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 58.Liu S, Jing W, Cheung B, Lu H, Sun J, Yan X, Niu J, Farmar J, Wu S, Jiang S. 2007. HIV gp41 C-terminal heptad repeat contains multifunctional domains. Relation to mechanisms of action of anti-HIV peptides. J Biol Chem 282:9612–9620. [DOI] [PubMed] [Google Scholar]
- 59.Sidney J, Peters B, Frahm N, Brander C, Sette A. 2008. HLA class I supertypes: a revised and updated classification. BMC Immunol 9:1. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li B, Gladden AD, Altfeld M, Kaldor JM, Cooper DA, Kelleher AD, Allen TM. 2007. Rapid reversion of sequence polymorphisms dominates early human immunodeficiency virus type 1 evolution. J Virol 81:193–201. doi: 10.1128/JVI.01231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fryer HR, Frater J, Duda A, Palmer D, Phillips RE, McLean AR. 2012. Cytotoxic T-lymphocyte escape mutations identified by HLA association favor those which escape and revert rapidly. J Virol 86:8568–8580. doi: 10.1128/JVI.07020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goepfert PA, Lumm W, Farmer P, Matthews P, Prendergast A, Carlson JM, Derdeyn CA, Tang J, Kaslow RA, Bansal A, Yusim K, Heckerman D, Mulenga J, Allen S, Goulder PJ, Hunter E. 2008. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J Exp Med 205:1009–1017. doi: 10.1084/jem.20072457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, Swenson LC, Tao I, Szeto S, Rosato P, Sela J, Kadie CM, Frahm N, Brander C, Haas DW, Riddler SA, Haubrich R, Walker BD, Harrigan PR, Heckerman D, Mallal S. 2009. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One 4:e6687. doi: 10.1371/journal.pone.0006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alexander-Miller MA, Leggatt GR, Berzofsky JA. 1996. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl Acad Sci U S A 93:4102–4107. doi: 10.1073/pnas.93.9.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Derby M, Alexander-Miller M, Tse R, Berzofsky J. 2001. High-avidity CTL exploit two complementary mechanisms to provide better protection against viral infection than low-avidity CTL. J Immunol 166:1690–1697. doi: 10.4049/jimmunol.166.3.1690. [DOI] [PubMed] [Google Scholar]
- 66.Mailliard RB, Smith KN, Fecek RJ, Rappocciolo G, Nascimento EJ, Marques ET, Watkins SC, Mullins JI, Rinaldo CR. 2013. Selective induction of CTL helper rather than killer activity by natural epitope variants promotes dendritic cell-mediated HIV-1 dissemination. J Immunol 191:2570–2580. doi: 10.4049/jimmunol.1300373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Allen TM, Yu XG, Kalife ET, Reyor LL, Lichterfeld M, John M, Cheng M, Allgaier RL, Mui S, Frahm N, Alter G, Brown NV, Johnston MN, Rosenberg ES, Mallal SA, Brander C, Walker BD, Altfeld M. 2005. De novo generation of escape variant-specific CD8+ T-cell responses following cytotoxic T-lymphocyte escape in chronic human immunodeficiency virus type 1 infection. J Virol 79:12952–12960. doi: 10.1128/JVI.79.20.12952-12960.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fischer W, Perkins S, Theiler J, Bhattacharya T, Yusim K, Funkhouser R, Kuiken C, Haynes B, Letvin NL, Walker BD, Hahn BH, Korber BT. 2007. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat Med 13:100–106. doi: 10.1038/nm1461. [DOI] [PubMed] [Google Scholar]
- 69.Nickle DC, Rolland M, Jensen MA, Pond SL, Deng W, Seligman M, Heckerman D, Mullins JI, Jojic N. 2007. Coping with viral diversity in HIV vaccine design. PLoS Comput Biol 3:e75. doi: 10.1371/journal.pcbi.0030075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.