

ABSTRACT
Phenotypic diversity in prion diseases can be specified by prion strains in which biological traits are propagated through an epigenetic mechanism mediated by distinct PrPSc conformations. We investigated the role of host-dependent factors on phenotypic diversity of chronic wasting disease (CWD) in different host species that express the same prion protein gene (Prnp). Two CWD strains that have distinct biological, biochemical, and pathological features were identified in transgenic mice that express the Syrian golden hamster (SGH) Prnp. The CKY strain of CWD had a shorter incubation period than the WST strain of CWD, but after transmission to SGH, the incubation period of CKY CWD was ∼150 days longer than WST CWD. Limited proteinase K digestion revealed strain-specific PrPSc polypeptide patterns that were maintained in both hosts, but the solubility and conformational stability of PrPSc differed for the CWD strains in a host-dependent manner. WST CWD produced PrPSc amyloid plaques in the brain of the SGH that were partially insoluble and stable at a high concentration of protein denaturant. However, in transgenic mice, PrPSc from WST CWD did not assemble into plaques, was highly soluble, and had low conformational stability. Similar studies using the HY and DY strains of transmissible mink encephalopathy resulted in minor differences in prion biological and PrPSc properties between transgenic mice and SGH. These findings indicate that host-specific pathways that are independent of Prnp can alter the PrPSc conformation of certain prion strains, leading to changes in the biophysical properties of PrPSc, neuropathology, and clinical prion disease.
IMPORTANCE Prions are misfolded pathogenic proteins that cause neurodegeneration in humans and animals. Transmissible prion diseases exhibit a spectrum of disease phenotypes and the basis of this diversity is encoded in the structure of the pathogenic prion protein and propagated by an epigenetic mechanism. In the present study, we investigated prion diversity in two hosts species that express the same prion protein gene. While prior reports have demonstrated that prion strain properties are stable upon infection of the same host species and prion protein genotype, our findings indicate that certain prion strains can undergo dramatic changes in biological properties that are not dependent on the prion protein. Therefore, host factors independent of the prion protein can affect prion diversity. Understanding how host pathways can modify prion disease phenotypes may provide clues on how to alter prion formation and lead to treatments for prion, and other, human neurodegenerative diseases of protein misfolding.
INTRODUCTION
Prion diseases are transmissible protein misfolding diseases that cause fatal neurodegeneration in humans and animals. The mechanism of prion formation has been proposed to proceed by a template-dependent, or seeded, protein polymerization (1, 2). In seeded polymerization epigenetic information transfer is mediated by the pathogenic prion protein (PrPSc), which self-assembles into multimers and/or amyloid by a conformation-dependent mechanism that involves the misfolding and incorporation of the cellular isoform of the prion protein (PrPC) into the elongating amyloid. In prion diseases, phenotypic diversity within a host species has been attributed to both the primary structure of PrPC and the tertiary or quaternary structure of the PrPSc multimer. Mutations and polymorphisms in the prion protein gene (Prnp) have been linked to phenotypic diversity among the inherited and sporadic human prion diseases (3–9) and to experimental scrapie prion infection in mice (10, 11). However, phenotypic diversity also is observed for the infectious forms of disease during intraspecies prion transmission in the absence of Prnp mutations and polymorphisms. Here, prion diversity is propagated by distinct prion strains, which can be stably maintained within the same host species to produce characteristic incubation periods, clinical symptoms, and neuropathology (12, 13). The molecular basis of prion strain diversity is encoded in the structure of PrPSc multimers and/or amyloid, such that distinct PrPSc conformations can propagate from a similar PrPC resulting in different disease outcomes (14–17). This mechanism accounts for strain diversity not only for mammalian prion diseases but also in yeast prions and prion-like neurodegenerative diseases of humans (18–22).
Identification of a new prion phenotype is often described upon experimental transmission of prions into rodents from a human or ruminant host with prion disease. Interspecies prion transmission results in a reduction in the kinetics of new PrPSc formation by seeded polymerization due to mismatches in the amino acid sequence between the infectious PrPSc and host-encoded PrPC (23–25). Consistent with these findings is that interspecies transmission leads to an inefficient disease process that results in very long prion incubation periods or, in several cases, no disease transmission (26, 27). After this initial prion adaptation in a new rodent host, there is an increase in the efficiency of PrPSc propagation and a selection for fast replicating prions upon additional serial passages in the new host species. This is partially due to homotypic PrPC-PrPSc interactions in which both isoforms of the prion protein now have the same amino acid sequence (25, 28). After several passages, the prion phenotype will acquire stable and highly reproducible biological and neuropathological features in the new host species. In some studies, two prion phenotypes, or strains, with different disease properties have been identified following interspecies transmission of a natural prion isolate (29). The origin of these phenotypes is often difficult to assess because there are only a few examples in which the newly identified prion strains are inoculated back into the original host species (29, 30). In these studies, it is possible to distinguish between the prion strain present in the original host and was isolated upon interspecies transmission into rodents, versus prion strains that arise in rodents due to heterotypic PrPC-PrPSc interactions and adaptation to the new host species. Furthermore, the role of cellular factors that can influence adaptation and selection of prion strains in a new host species is unclear because Prnp is the major determinant for prion replication; in the absence of Prnp, there is no PrPSc formation or prion transmission (31, 32). Genetic analyses suggest that additional host genes can influence prion incubation periods, but no candidate genes have been implicated to have a significant role (33–35).
In the present study, we investigated interspecies transmission of prions to different rodent hosts that express the same Prnp in order to investigate the effect of Prnp-independent, host-specific factors on prion strain diversity. An isolate of chronic wasting disease (CWD) from white-tailed deer was transmitted to transgenic mice expressing Prnp from Syrian golden hamsters (SGH) and two CWD strains were identified that had distinct biological and PrPSc biophysical properties. Upon interspecies transmission to SGH there were dramatic changes to the phenotype of the CWD strains, but similar rodent transmission studies using the HY and DY strains of transmissible mink encephalopathy (TME) only led to minor differences in the properties of the TME strains. These findings indicate that host-dependent cellular pathways that are independent of Prnp can have a profound impact on the biophysical properties of PrPSc and the phenotypic diversity of certain prion strains.
MATERIALS AND METHODS
Animal inoculations, body weight measurements, and tissue collection.
Transgenic mice were generated from PrP-null mice and were engineered to express the SGH prion protein under the control of the rat neuron-enolase specific promoter, as previously described (and kindly provided by B. Chesebro and R. Race, NIH Rocky Mountain Laboratories, Hamilton, MT) (36, 37). These transgenic mice are designated HPrP7752KO mice and are referred to as transgenic mice here. HPrP7752KO mice were estimated to have an ∼4-fold-higher level of PrPC in the brain than found in SGH (38). HPrP7752KO mice were intracerebrally (i.c.) inoculated with 30 μl of a 10% (wt/vol) brain homogenate from a free-ranging white-tailed deer with CWD (kindly provided by E. Hoover, Colorado State University, Fort Collins, CO). Brains from these CWD-infected transgenic mice were serially passaged into additional transgenic mice by intracerebral inoculation of brain homogenates at a dilution of 10−2 or 10−4. In addition, brain homogenates from transgenic mice were serially passaged into weanling, Syrian golden hamsters (Harlan Laboratories, Indianapolis, IN) by i.c. inoculation with 50 μl of a 1 or 10% (wt/vol) brain homogenate. Age-matched SGH were also inoculated with brain homogenates from normal transgenic mice or SGH (i.e., mock-infected group). After inoculation, individual hamster body weights were measured on a weekly basis. Transgenic mice and SGH were observed at least three times per week for the onset of clinical symptoms and were euthanized during the early to middle stages of neurological disease. For biochemical tissue analysis, tissues were collected, immediately frozen over dry ice, and stored at −80°C until use. For collection of tissues for immunohistochemistry, hamsters were intracardially perfused with periodate-lysine-paraformaldehyde fixative, and tissues were dissected and processed for embedding in paraffin wax as previously described (39–41).
PrPSc enrichment and Western blotting.
Frozen brain samples were homogenized in lysis buffer (10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.5% sodium deoxycholate, and 0.5% ipegal) containing 1× Complete protease inhibitor (Roche Diagnostics, Indianapolis, IN) to 10% (wt/vol). Brain samples were homogenized using a Bullet Blender (Next Advance, Averill Park, NY) and 0.5-mm-diameter glass beads. The protein concentration in tissue homogenates was determined using a micro-BCA assay (Pierce Protein Research, Rockford, IL). To detect PrPSc in brain, 100 μg of protein from clinically ill mice and SGH was digested with proteinase K (PK; Roche Diagnostics Corp., Indianapolis, IN) at 10 and 100 μg/ml at a protein sample concentration of 1 mg/ml. Enzymatic reactions were performed at 37°C for 1 h with constant agitation, followed by the addition of 1 mM Pefabloc (Roche Diagnostics Corp., Indianapolis, IN). For studies involving the removal of N-linked carbohydrates from proteins, enrichment for PrPSc in brain samples was performed by extraction in buffer containing 10% Sarkosyl, differential ultracentrifugation, and proteinase K digestion as previously described (39, 42). Deglycosylation was performed using N-endoglycosidase F (PNGase F) enzymatic removal of carbohydrates (New England BioLabs, Beverly, MA) as previously described according to the manufacturer's instructions (43).
SDS-PAGE was performed in 12% morpholinepropanesulfonic acid NuPAGE gels (Invitrogen, Carlsbad, CA), and proteins were transferred to polyvinylidene difluoride membranes for Western blot as previously described (44). Detection of PrPC and PrPSc on membranes was performed using mouse (12B2; kindly provided by J. P. M. Langeveld, Central Veterinary Institute of Wageningen, Lelystad, The Netherlands) (45) and mouse-human chimeric monoclonal antibodies to the prion protein (D13 and D18; kindly provided by D. Burton, The Scripps Research Institute, La Jolla, CA) (46) as previously described (43, 47) or by using LI-COR IRDye680LT and IRDye800LT secondary antibodies directed to mouse or human IgG and a LI-COR Odyssey CLx Imager (LI-COR Biosciences, Lincoln, NE).
Prion CSSA.
A prion conformation solubility and stability assay (CSSA) was performed as previously described with the following modifications (48). Brain homogenates were adjusted to 2.5 mg of protein/ml in 50 mM Tris (pH 7.4) and 2% N-lauroylsarcosine. After agitation for 1 h at 37°C, 50 μg of protein was removed and added to an equal volume of GdnHCl solution so that the final GdnHCl concentration ([GdnHCl]) was 0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, or 4.0 M. Samples were incubated for 1 h at 37°C with constant agitation. Ultracentrifugation was performed in an Optima MAX and TLA-55 rotor at 21,000 rpm (20,000 × g) for 1 h (Beckman Coulter Instruments, Fullerton, CA). The supernatant was carefully removed, and the pellet and supernatant were analyzed by SDS-PAGE and Western blotting with anti-PrP D18 antibody. The amount of prion protein signal (LI-COR Image Studio software) in the supernatant and pellet fractions at each [GdnHCl] was combined and given a value of 1.0. The percentage of prion protein that remained in the pellet fraction represents insoluble PrP, and this was plotted versus the [GdnHCl] to generate a prion protein solubility and stability curve. A best-fit line was produced using the four-parameter nonlinear regression analysis in Prism software (GraphPad). The median prion denaturation dose (DD50) was defined as the [GdnHCl] that resulted in a 50% reduction in the amount of the prion protein in the insoluble fraction compared to the starting amount of prion protein that was insoluble at 0 M GdnHCl. Brain from three different animals was analyzed for each prion strain and rodent host combination.
PrPSc IHC.
PrPSc immunohistochemistry (IHC) was performed on paraffin embedded tissue as previously described (39–41). Tissue sections were subjected to antigen retrieval by treatment with 99% (wt/vol) formic acid for 30 min and successively incubated with anti-PrP monoclonal 3F4 antibody overnight at 4°C, horse anti-mouse IgG biotinylated antibody (1:400; Vector Laboratories, Burlingame, CA) at room temperature for 30 min, and streptavidin-horseradish peroxidase (HRP) at room temperature for 20 min. PrPSc was visualized by localization of the HRP activity with DAB+ (Dako Cytomation, Carpinteria, CA). Tissue sections were counterstained with hematoxylin and coverslip mounted with mounting medium (Richard-Allen Scientific, Kalamazoo, MI) for viewing with a Nikon Eclipse E600 microscope. Controls for PrPSc IHC included the use of mock-infected tissues and substituting a similar concentration of murine IgG isotype control for the anti-PrP 3F4 monoclonal antibody.
RT-QuIC assay.
A real-time quaking-induced-conversion (RT-QuIC) analysis was performed as previously described with minor modifications (42, 49). Briefly, brain was homogenized to 10% (wt/vol) in buffer (10 mM phosphate buffer [pH 7.4], 300 mM NaCl, 0.5% Triton X-100, and Complete protease inhibitor) using a BioSpec Mini-BeadBeater and 1-mm-diameter glass beads in 1.5-ml tubes (BioSpec Products, Inc., Bartlesville, OK). After low-speed centrifugation (2,000 × g for 2 min), the supernatant was collected and frozen at −80°C until use. For RT-QuIC reactions, brain supernatants were serially diluted in diluent (phosphate-buffered saline, 0.1% SDS, and N2 medium supplement), and 2 μl of each was added to 98 μl containing reaction buffer (10 mM phosphate buffer [pH 7.4], 300 mM NaCl, 1 mM EDTA tetrasodium salt), 10 μM Thioflavin T (ThT; Sigma-Aldrich, Inc., Atlanta, GA), and hamster recombinant PrP 90-231 (0.1 mg/ml) (49, 50). Quadruplicate reactions were assayed in a 96-well microtiter plate (a black plate with a clear bottom) using a FLUOstar Omega plate reader (BMG Labtech, Cary, NC) at 50°C for 1,500 cycles (one cycle consists of shaking at 700 rpm for 1 min and resting for 1 min), and ThT fluorescence measurements were made every 45 min. Spearman-Kärber analysis was used to calculate the median prion seeding dose (SD50) per mg of brain, and the SD50 concentration was defined as the sample amount giving a positive response in 50% of replicate RT-QuIC reactions (51). Positive RT-QuIC reactions were designated as reactions with ThT fluorescence that was >200% of the average of ThT fluorescence signal for control samples (PrPSc negative).
Ethics statement.
All procedures involving animals were approved by the Montana State University and Colorado State University IACUC and were in compliance with the Guide for the Care and Use of Laboratory Animals; these guidelines were established by the Institute of Laboratory Animal Resources and approved by the Governing Board of the U.S. National Research Council.
RESULTS
Clinical features of WST and CKY CWD in rodents expressing hamster Prnp.
To investigate the role of host species-specific factors on prion strain diversity, an isolate of chronic wasting disease from white-tailed deer was serially passaged in transgenic mice expressing the Syrian golden hamster prion protein gene (HPrP7752KO mice) and, subsequently, it was serially passaged into SGH. The rationale for this approach was that the higher expression level of hamster Prnp in HPrP7752KO mice would result in a more efficient CWD infection and adaptation upon interspecies transmission than direct inoculation of SGH. Interspecies transmission from HPrP7752KO mice to SGH does not result in a change in the Prnp, so any observed changes to the disease phenotype must be mediated by host factors other than Prnp or the PrPSc conformation. Table 1 summarizes the transmission studies of CWD into HPrP7752KO mice and SGH. A portion of this table appeared in a previous publication and is included here in order to present the transmission history of the wasted (WST) strain of CWD (previously called CWD line A) and CWD line B, now identified as the cheeky (CKY) strain of CWD (40). The new animal transmission data includes second and third serial passages of CKY CWD and WST CWD in SGH, respectively, and serial backpassage of each into HPrP7752KO mice.
TABLE 1.
Incubation period after serial passage of CWD isolates into transgenic mice and Syrian golden hamsters
| Recipient groupa | Inoculumb (incubation period in days) | HPrP7752KO mice |
Syrian golden hamsters |
Total no. of serial passages | ||||
|---|---|---|---|---|---|---|---|---|
| Incubation period (days) ± SEM | A/Ic | Pass no.d | Incubation period (days) ± SEM | A/Ic | Passage | |||
| Control (M6148) | CWD (NA) | 417 ± 23.6 | 4/4 | First | 1 | |||
| WST CWD passage | ||||||||
| M6299 | M6148.1 (369) | 198 ± 3.1 | 5/5 | Second | 2 | |||
| M6339 | M6299.1 (192) | 189 ± 5.7 | 4/4 | Third | 3 | |||
| M6386 | M6339.2 (189) | 187 ± 1.8 | 4/4 | Fourth | 4 | |||
| H1471 | M6299.1 (192) | 379 ± 3.0 | 4/4 | First | 3 | |||
| H1572 | H1471.4 (382) | 343 ± 5.0 | 8/8 | Second | 4 | |||
| H1733 | H1572.1 (340) | 323 ± 2.1 | 19/19 | Third | 5 | |||
| M6429 | H1572.1 (340) | 180 ± 8.3 | 3/3 | First back pass | 5 | |||
| M6595 | M6429.3 (197) | 218 ± 9.1 | 5/5 | Second back pass | 6 | |||
| CKY CWD passage | ||||||||
| M6332 | M6148.3 (473) | 360 ± 16 | 3/3 | Second | 2 | |||
| M6374 | M6332.1 (343) | 160 ± 4.6 | 4/4 | Third | 3 | |||
| M6388 | M6374.4 (151) | 157 ± 0.0 | 3/3 | Fourth | 4 | |||
| H1604 | M6374.4 (151) | 477 ± 15.4 | 3/3 | First | 4 | |||
| H1727 | H1604.2 (468) | 479 ± 6.4 | 20/20 | Second | 5 | |||
| M6448 | H1604.2 (468) | 156 ± 2.5 | 5/5 | First back pass | 5 | |||
| M6596 | M6448.1 (151) | 178 ± 2.5 | 6/6 | Second back pass | 6 | |||
Recipient rodents were either HPrP7752KO mice (indicated by an “M” preceding the recipient group number) or Syrian golden hamsters (indicated by an “H” preceding the recipient group number).
The inoculum was a 1% brain homogenate, a 10% brain homogenate (for initial passage of CWD isolate into HPrP7752KO mice and for inoculation of CKY CWD into Syrian golden hamsters), or a 0.01% brain homogenate (M6148.3). All inoculations were administered i.c. using the inoculum from an individual animal in the recipient group (indicated by the suffixes .1, .2, .3, and .4). The incubation period for the transgenic mouse or Syrian golden hamster used with the inoculum is indicated in parentheses. NA, not applicable.
A/I, number of animals affected by prion disease versus the total number inoculated.
Number of serial passages in either HPrP7752KO mice or Syrian golden hamsters.
Serial transmission of WST CWD into HPrP7752KO mice resulted in incubation periods that stabilized at ∼190 days on the third and fourth passages (Table 1, recipient groups M6339 and M6386). The clinical symptoms presented as reduced activity and an oily appearance to the fur coat, and this was followed by kyphosis, a shortened trunk, and somnolence. CWD-adapted to HPrP7752KO mice were transmitted into SGH, and the precise onset of symptoms was difficult to assess due to the subtle nature of the neurological changes. On the third serial passage in SGH, we estimate the incubation period to be 323 ± 2.1 days (Table 1, recipient group H1733), which in retrospect is likely to be representative of first and second serial passage in SGH even though longer incubation periods were initially reported. At around 41 weeks postinfection, the hamsters exhibited an increase in physical activity and a progressive loss of body weight over the remainder of the disease (Fig. 1A). Additional symptoms were progressive and included rearing, head tremor, craning of the neck backward, loss of balance, jerky movements when walking, circling behavior, low profiling when walking, and altered burrowing behavior.
FIG 1.
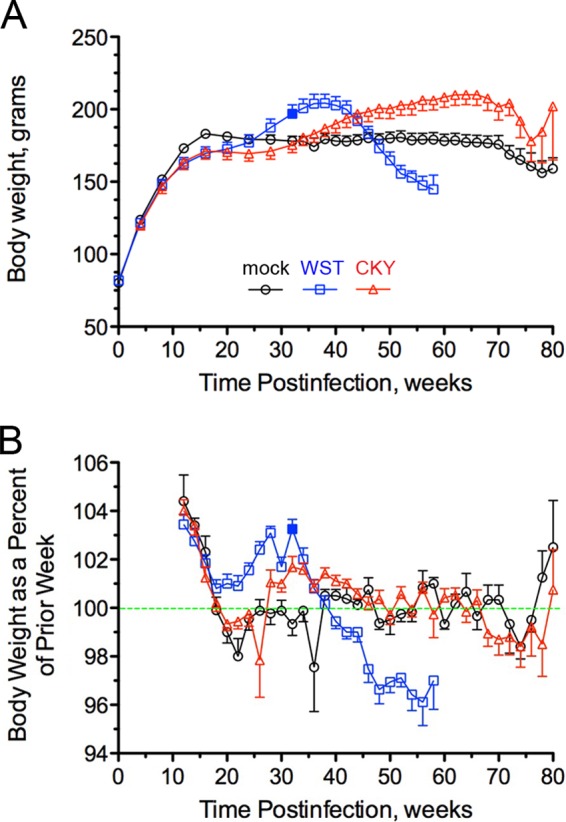
Body weight of SGH during prion infection with chronic wasting disease. Mock-infected (○) and CWD-infected hamsters (□ and △) (see Table 1, recipient groups H1733 for WST CWD and H1727 for CKY CWD) were monitored weekly over the course of infection for mean body weight (A) and the percent change in body weight (BW) from the prior week or BW replacement value (B). The green dashed line in panel B indicates no change in the BW replacement value, whereas values above or below the line indicate a net gain or loss, respectively, in body weight. A solid blue box at 32 weeks postinfection is indicated for WST CWD. The error bars indicate the standard errors of the mean and values changed when hamsters were sacrificed due to illness and for collection of age-matched controls (e.g., at 50 to 60 weeks and at 70 to 80 weeks postinfection). For illustration purposes, data points are plotted for every other week.
An examination of body weight as a percentage of the body weight from the previous week, or the BW replacement value, reveals a steady decline starting at 32 weeks postinfection in SGH (Fig. 1B). However, there was not a net loss in body weight until 40 weeks postinfection (Fig. 1A) because the BW replacement value was >100% until 38 weeks postinfection even though it was on the decline (Fig. 1B). It was not until after 44 weeks postinfection that the BW replacement value dropped below 99%, and for the remainder of the disease the BW replacement value declined 2.5 to 4% per week in SGH (Fig. 1B). The mean body weight in WST CWD-infected SGH reached a peak of 206 ± 5.7 g at 39 weeks postinfection, which corresponded to a 10% increase in peak body weight compared to mock-infected SGH (Table 2). The mean body weight at the terminal stage of the disease was 135 ± 4.3 g at 55 weeks postinfection, or a 34% reduction from peak body weight. The biological properties of CWD in hamsters led us to call this the “wasted” (WST) strain of CWD. It is the only prion strain that has been described in SGH that causes a progressive, long-term reduction in body weight.
TABLE 2.
Body weights of Syrian golden hamsters with WST and CKY CWD
| Prion strain | Peak (mean ± SEM) |
Terminal (mean ± SEM) |
Mean terminal/peak body wt % change ± SEMb | ||
|---|---|---|---|---|---|
| Body wt (g) | Time (wpi)a | Body wt (g) | Time (wpi) | ||
| Mock | 186 ± 3.2 | 48 ± 6.5 | 172 ± 5.8 | 73 ± 4.9 | –0.07 ± 0.01 |
| WST CWD | 206 ± 5.7 | 39 ± 0.9 | 135 ± 4.3 | 55 ± 0.6 | –0.34 ± 0.01 |
| CKY CWD | 219 ± 6.8 | 63 ± 1.2 | 188 ± 9.4 | 73 ± 1.0 | –0.14 ± 0.02 |
wpi, weeks postinfection.
That is, the percent change in body weight from peak to terminal stages.
The CKY CWD passage line was established using first passage HPrP7752KO mouse brain at a 10,000-fold dilution for the second passage, compared to a 100-fold dilution used to establish the WST CWD passage line (see recipient group M6148 and inoculation numbers M6148.1 and M6148.3 in Table 1). This approach previously has been used to identify slower replicating prions that can be concealed by faster replicating prions in a mixture of prion strains, especially when the latter is present at lower doses (52). At the second serial passage into HPrP7752KO mice, the incubation period was long due to the high dilution of inoculum (360 ± 16 days, see recipient group M6332 in Table 1). At the third and fourth serial transmission, now using a 100-fold dilution of the brain inoculum, the incubation period in HPrP7752KO mice stabilized around 160 days postinfection, and clinical symptoms were characterized by ptosis, torticollis, and ultimately somnolence and kyphosis (Table 1, see recipient groups M6374 and M6388). These findings indicate that the CWD strains were present as a mixture in the CWD inocula and/or brain of transgenic mice after interspecies transmission of CWD, but we would have predicted different results, i.e., that the shorter incubation period CKY CWD would cause clinical disease prior to WST CWD upon serial passage and not require high dilution of the brain inocula in order to emerge.
Subsequent transmission of fourth-passage transgenic mouse brain from the CKY CWD passage line into SGH resulted in incubation periods of ∼475 days on the first and second serial passages (Table 1, see recipient groups H1604 and H1727). The onset of clinical symptoms was insidious, and it was difficult to assign a definitive start date. At the second serial passage in SGH, there was an irreversible and progressive closing of the eyelids in the absence of microbial infection at 355 ± 13 days (range, 263 to 465 days). The incubation period was estimated to be 479 ± 6.4 days (range, 438 to 522 days) and was characterized by a ruffling and loss of fur, lack of coordination, and labored or erratic breathing. A significant number of SGH exhibited periods of incessant hiccup-like behavior that was often accompanied by inflation of one or both cheek pouches. In these cases, for each single hiccup, inhalation of air resulted in a stepwise inflation of the cheek pouch until it was full like a balloon. Eventually, the cheek pouch would rapidly deflate when the trapped air was released all at once. Based on the clinical phenotype of these hamsters, we termed this the “cheeky” (CKY) strain of CWD. SGH infected with CKY CWD exhibited a steady increase in body weight that peaked at 219 ± 6.8 g, which was a 15% increase compared to mock-infected SGH (Table 2). For CKY CWD, the peak body weight was reached at 63 weeks postinfection, and this was 24 and 15 weeks later than peak body weight in WST CWD-infected and mock-infected SGH, respectively (Fig. 1A and Table 2). The BW replacement value began to decline in CKY CWD beginning 66 weeks postinfection, and terminal disease was observed at 73 weeks postinfection. The body weight at terminal disease was reduced 14% compared to the peak body weight (Fig. 1B and Table 2).
WST CWD and CKY CWD exhibited distinct biological features in both HPrP7752KO mice and SGH expressing a similar hamster Prnp genotype. What was unusual about the interspecies transmission history of these two CWD strains was that CKY CWD had a shorter incubation period than WST CWD in HPrP7752KO mice at the third serial passage (160 ± 4.6 days versus 189 ± 8.3 days), but upon transmission to SGH, CKY CWD had a significantly longer incubation period than WST CWD at the second serial passage (479 ± 6.4 days versus 323 ± 2.1 days) (Table 1). This reversal in the length of incubation periods for the CWD strains upon interspecies transmission was Prnp independent and was therefore controlled by a host-mediated pathway.
Prnp-independent changes in disease onset of TME.
To determine whether the reversal in the length of the incubation period for the WST and CKY CWD strains upon transmission between HPrP7752KO mice and SGH can occur with additional prion strains, we inoculated the short-incubation “hyperexcitable” (HY) and long-incubation “drowsy” (DY) TME strains adapted to SGH into HPrP7752KO mice (26, 29). The incubation periods of HY and DY TME in SGH were 63 ± 2.1 days and 161 ± 2.0 days, respectively. Upon serial transmission to HPrP7752KO mice, HY TME established an approximately 70- to 90-day shorter incubation period than DY TME during four serial passages (Table 3). For the HY and DY TME strains, there were 20 and 17% increases, respectively, in the lengths of the incubation period from first passage to the fourth serial passage in HPrP7752KO mice, even though interspecies transmission did not result in a change of the Prnp genotype. This feature was also unusual since incubation periods typically shorten upon serial passage into a new host, especially since HPrP7752KO mice express higher levels of PrPC than do SGH. Therefore, there also appear to be host-specific changes to the biological properties of the TME strains following interspecies transmission that were independent of Prnp. These changes could be related to differences in TME brain titers in HPrP7752KO mice and SGH, as described below. Interspecies transmission of the TME strains did not follow a similar trend as the CWD strains. For the TME strains, the incubation periods were shorter for HY TME in both rodents, and differences in the lengths of the incubation period between hosts for each strain were slightly reduced in transgenic mice and considerably less than those found for the CWD strains.
TABLE 3.
Adaptation of HY and DY TME from SGH to HPrP7752KO mice
| Serial passage | HY TME |
DY TME |
||
|---|---|---|---|---|
| Mean incubation period (days) ± SEM | A/Ia | Mean incubation period (days) ± SEM | A/Ia | |
| 1 | 46 ± 0.2 | 5/5 | 116 ± 2.6 | 6/6 |
| 2 | 51 ± 0.4 | 8/8 | 119 ± 1.5 | 10/10 |
| 3 | 58 ± 1.7 | 7/7 | 146 ± 5.1 | 7/7 |
| 4 | 57 ± 1.8 | 6/6 | 140 ± 0.8 | 9/9 |
A/I, number of animals affected by prion disease versus the total number inoculated.
Another explanation for the reversal in the lengths of the incubation periods for the WST and CKY CWD strains upon interspecies transmission from HPrP7752KO mice to SGH is that there was a difference in brain titer between the CWD strains that correlates with disease onset. The standard method to measure prion titer is to calculate the median lethal dose (LD50) concentration by endpoint dilution of samples and animal bioassay (51, 53). In the present study, we used the RT-QuIC assay to measure the median seeding dose (SD50) concentration because this correlates with the LD50 concentration in SGH brains infected with scrapie (49). We previously reported a 100-fold-higher LD50 concentration in the brains of SGH with HY TME compared to DY TME (29), and the SD50 concentration for HY TME was also 10- to 100-fold higher than with DY TME in SGH, illustrating that the RT-QuIC assay is a valid in vitro assay to compare prion levels between these prion strains (Table 4). In HPrP7752KO mice, the brain SD50 concentration was at most 10-fold higher in HY TME than in DY TME, and the SD50 concentration was 10- to 100-fold lower in HPrP7752KO than in SGH for each strain. For the CWD strains, the SD50 concentration was 10- to 100-fold higher in the brains of CKY CWD compared to WST CWD for both HPrP7752KO mice and SGH, even though the overall SD50 concentrations were lower in HPrP7752KO mice for the CWD strains (Table 4). These findings indicate that the pattern of relative prion levels in the brain among strains is maintained in both rodent species. There was a higher SD50 concentration for HY TME compared to DY TME and for CKY CWD compared to WST CWD in transgenic mice and SGH. In addition, there was no correlation between the amount of prion in the brain, or SD50 concentration, and the length of the prion incubation period (Table 1, compare HY TME and CKY CWD), nor was there a significant change in SD50 concentration between CKY CWD and WST CWD that can explain the reversal in the order of the incubation periods upon interspecies transmission from HPrP7752KO mice to SGH.
TABLE 4.
Incubation periods and median seeding titers of TME and CWD strains in HPrP7752KO mice and SGH
| Prion strain | HPrP7752KO mice |
Syrian golden hamsters |
||
|---|---|---|---|---|
| Mean incubation period (days) ± SEM | SD50 per mg of braina | Mean incubation period (days) ± SEM | SD50 per mg of brainb | |
| HY TME | 58 ± 1.6 | 107.2, 107.2 | 63 ± 2.1 | 109.2, 108.9, 108.9 |
| DY TME | 146 ± 5.1 | 106.2, 106.9 | 161 ± 2.0 | 107.2, 107.2, 108.7 |
| CKY CWD | 160 ± 4.6 | 108.7, 108.7 | 479 ± 6.4 | 109.9, 1010.4, 108.7 |
| WST CWD | 189 ± 5.7 | 106.9, 106.7 | 323 ± 2.1 | 107.7, 108.4, 107.7 |
| Mock | >250 | <102, <102 | >600 | <102, <102, <102 |
Tenfold serial dilutions of brain were used in an RT-QUiC assay to calculate the median seeding dose (SD50).
A one-way analysis of variance and Tukey's multiple-comparison test indicated a difference (P < 0.05) between CKY CWD and WST CWD and between CKY CWD and DY TME. Statistical analysis in transgenic mice was not performed due to the limited n value.
PrPSc distribution in brain of WST and CKY CWD-infected SGH and HPrP7752KO mice.
PrPSc IHC was performed on the brains of SGH infected with WST and CKY CWD during the later stages of disease. PrPSc was widely distributed throughout the brain, and there were many brain structures in which PrPSc deposition was found in both of the CWD strains (Fig. 2). Overall, there was a lower PrPSc signal intensity for WST CWD in SGH at third serial passage compared to CKY CWD, especially in the hippocampus. At the second serial passage, WST CWD had a stronger PrPSc signal in the hippocampus and more closely resembled the brain distribution of CKY CWD at second serial passage. In the mesencephalon, PrPSc staining was very intense in the superior colliculus for both CWD strains, and moderate deposition was found in the CA1 region of the hippocampus, medial geniculate nucleus, substania nigra, interpeduncular nucleus, and middle cortical layers of the cerebrum (Fig. 2A to C). Further rostral in the brain, the strongest PrPSc deposition was found in layers CA1 to CA3 of the hippocampus, thalamic nuclei (e.g., the lateral geniculate nucleus and the lateral, ventral posteromedial, and posterior thalamic nuclei), and middle layers of the cerebral cortex (Fig. 2D to F). More intense PrPSc staining was consistently found in the hypothalamic nuclei in WST CWD-infected SGH. Even further rostral, strong PrPSc deposition was found in the laterodorsal thalamic nucleus, and moderate staining was noted in the caudate putamen, reticular formation, and middle layers of the cerebral cortex. More intense PrPSc staining was consistently found in the globus pallidus in CKY CWD-infected SGH (Fig. 2G to I). In the telencephalon, moderate PrPSc deposition was found in the caudate putamen, septal nuclei, cingulate cortex, and middle cortical layers of the cerebrum (Fig. 2J to L). The brain distribution of PrPSc was more similar between WST and CKY CWD than it was different.
FIG 2.

PrPSc deposition pattern in the brains of Syrian golden hamsters (SGH) infected with WST and CKY CWD. Brain sections from CKY CWD (A, D, G, B, E, H, and K), WST CWD (C, F, I, J, and L), and mock (M, N, and O) infections in SGH were stained for PrPSc by IHC (brown deposit) or stained with hematoxylin and eosin (A, D, G, J, M, N, and O). Brain tissue was from the second (CKY CWD) and third (WST CWD) serial passages in SGH. VC, visual cortex; M, motor cortex; Cg, cingulate cortex; S1, somatosensory cortex; Pir, piriform cortex; HC, hippocampus; SC, superior colliculus; SNR, substania nigra; RN, red nucleus; MG, medial geniculate nucleus; VPM, ventral posterior thalamic nucleus; IPR, interpedunclar nucleus; PN, pretectal nucleus; ZI, zona incerta; H, hypothalamic nuclei; AM, amygdala; Rt, reticular formation; CPu, caudate-putamen; GP, globus pallidum; LS, lateral septal nucleus; MS, medial septal nucleus; ic, internal capsule; cc, corpus callous; LV, lateral ventricle; 3, third ventricle.
Another feature of prion infection in the brain of SGH infected with WST and CKY CWD was large PrPSc deposits and PrPSc amyloid plaques. PrPSc plaques were most often associated with the ventricles, either in the subependymal layer and/or extending into the ventricle, and the vascular associated with pia arterioles and likely the ascending arterioles in the brain parenchyma (Fig. 3). It was common to observe strong PrPSc deposition along the entire length of the third and lateral ventricles (Fig. 3A and B), as well as in narrow ventricular passages, especially along the lateral ventricle between the alveus of the hippocampus and the cerebral cortex. PrPSc plaques were both unicentric and multicentric as evident by hematoxylin and eosin staining and were associated intense PrPSc deposition (Fig. 3C and F). PrPSc plaques were birefringent when stained with Congo red and often had a feathery appearance that emanated from the center of the plaque when stained with hematoxylin and eosin (Fig. 3E). PrPSc plaques were more frequent and more widely distributed in WST CWD than in CKY CWD. Cerebrovascular PrPSc deposition was also observed in the brain parenchyma, often in the hypothalamus, cerebellar cortex, and cerebral cortex; this pattern of PrPSc staining also was more common in WST than in CKY CWD (not shown). These findings indicate that PrPSc plaques was prominent in both WST and CKY CWD infections of SGH and is consistent with PrPSc deposition in CWD infection of transgenic mice that express cervid Prnp (54, 55).
FIG 3.
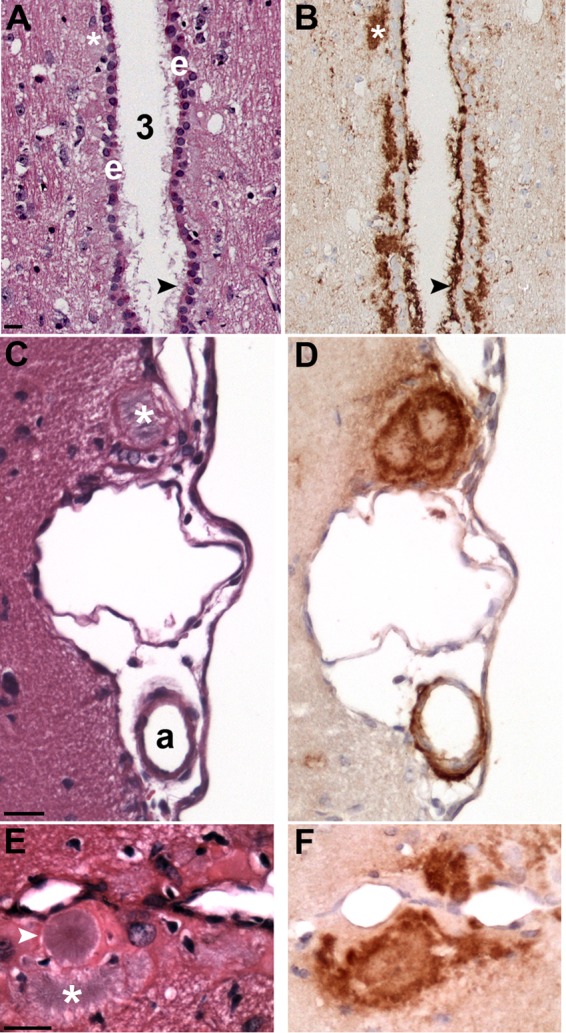
PrPSc deposition associated with brain ventricles and vasculature. PrPSc IHC (B, D, and F) and hematoxylin and eosin stain (A, C, and E) in adjacent tissue sections of CKY CWD (A and B)- and WST CWD (C to F)-infected SGH. (A and B) Heavy PrPSc deposition associated with the ependymal cell layer (e) of the third ventricle (3). PrPSc plaques (white asterisks and black arrowheads) were located below the ependymal cells, PrPSc extended through this cell layer, and PrPSc projected into the third ventricle. (C and D) A large PrPSc plaque (white asterisk) at the brain surface was found adjacent to PrPSc deposition surrounding an arteriole (a) in the pia. (E and F) Several PrPSc plaques were observed along the midline of the cerebral cortex. Large unicentric and multicentric hematoxylin-positive plaques (white arrowhead and asterisk, respectively) were often observed in WST CWD but were less common in CKY CWD infection of SGH. Brain tissue was from the second (CKY CWD) and third (WST CWD) serial passages in SGH. Scale bar, 20 μm.
The brain distributions of PrPSc in WST and CKY CWD infections of HPrP7752KO mice were both similar to and distinct from CWD infections in SGH. First, the level of PrPSc deposition was much lower in transgenic mice than in SGH, especially for WST CWD (data not shown). Both CWD strains had moderate levels of PrPSc in the thalamus in a distribution pattern similar to that found in SGH, particularly in the medial geniculate nucleus, laterodorsal thalamic nucleus, and ventral posterior thalamic nucleus. For CKY CWD, lower levels of PrPSc deposition were also found in the mesencephalon, telencephalon, hypothalamus, and rhombencephalon in similar brain structures as in SGH, although in WST CWD these brain regions were mostly devoid of PrPSc, except for an occasional PrPSc deposit. However, several brain regions with strong and moderate PrPSc deposition in the SGH were either lacking or had low levels of PrPSc deposition in transgenic mice, including the superior colliculus, the hippocampus, and the cerebral cortex. Based on the brain distribution of PrPSc during CWD infection, moderate changes to CKY CWD and significant changes for WST CWD were observed upon transmission from HPrP7752KO mice to SGH. The other notable difference between CWD infections in the two rodent species was the absence of PrPSc plaques in the brain of transgenic mice and little evidence for periventricular and perivasculature PrPSc deposition (data not shown). In contrast to CWD infection of SGH, intracellular PrPSc deposits were frequently observed in transgenic mice, likely in the soma of neurons, during both WST and CKY CWD infections.
Antigenic mapping of PrPSc from TME and CWD strains in HPrP7752KO mice and SGH.
To investigate strain-specific biochemical properties of PrPSc among the TME and CWD strains, we performed proteinase K (PK) digestion of brain homogenates from HPrP7752KO mice and SGH, followed by Western blotting and antigenic mapping. Limited PK digestion results in the truncation of ∼90 amino acids from the N terminus of PrPSc, and anti-PrP antibodies directed at the truncated and ragged N terminus of PrPSc can distinguish molecular weight differences between PrPSc polypeptides (14, 56). Prior studies that describe a difference in the molecular weight of PrPSc polypeptides between HY TME and DY TME in SGH also found that anti-PrP antibody directed to the PK truncated N terminus reacted poorly with PrPSc from DY TME but not HY TME (56, 57). In the present study, anti-PrP 12B2 antibody, which recognizes amino acids 88 to 92 of the prion protein, did not immunoreact with PrPSc from DY TME but did react strongly with PrPSc from HY TME in both HPrP7752KO mice and SGH (Fig. 4). Anti-PrP 12B2 immunoreacts with PrPSc in the absence of PK digestion, indicating that PK cleavage removed the epitope at the truncated N terminus of PrPSc from DY TME. PrPSc immunoreactivity was partially restored for DY TME in both host species using anti-PrP D13 antibody, which recognizes amino acid sequences that are adjacent to, or partly within, the N terminus after PK digestion (Fig. 4). Strong PrPSc immunoreactivity was observed for both HY and DY TME with anti-PrP D18 antibody that recognizes an internal PrP domain that is distant from the N terminus. PrPSc immunoreactivity with anti-PrP D18 antibody also reveals that the 1- to 2-kDa molecular mass difference between the TME strains and the faster migration of PrPSc polypeptides in DY TME is consistent with additional PK truncation at the N terminus. This molecular mass difference is most pronounced for the aglycosylated, or lowest-molecular-mass, PrPSc polypeptide in the 20-kDa range. The strain-specific PrPSc polypeptide migration pattern of HY and DY TME was also evident after PrPSc enrichment, PK digestion, and deglycosylation with PNGase F (Fig. 5A and B). These results illustrate that the strain-specific PK cleavage of PrPSc for the TME strains was maintained across different rodent species that express hamster Prnp.
FIG 4.

Antigenic mapping of PrPSc from TME and CWD infection of transgenic mice and Syrian golden hamsters. (A) Prion-infected brain homogenates from HPrP7752KO mice and Syrian golden hamsters were digested with or without proteinase K (PK) and analyzed by SDS-PAGE and Western blotting with the anti-PrP antibodies 12B2, D13, and D18. The hamster amino acid (aa) sequence that is recognized (12B2) or used as a peptide antigen (D13 and D18) is indicated below each antibody. Brain tissues were from the second (CKY CWD) and third (WST CWD) serial passages in SGH. The three short horizontal bars between the PK-treated samples from HPrP7752KO mice and Syrian golden hamsters correspond to molecular masses of 20, 30, and 40 kDa. The bracket indicates the doublet PrPSc polypeptide found in CKY CWD. (B) Schematic of the linear amino acid map of the mature prion protein, including the octapeptide repeat (OR) region, two N-linked glycosylation sites, and carbohydrate structures (the Y-like branches). Limited PK digestion of PrPSc results in the degradation of the N-terminal region. This results in a ragged N terminus with new N termini around amino acid 90. The locations of the peptides used to generate the anti-PrP antibodies 12B2, D13, and D18 are indicated with horizontal bars below the prion protein map. Enzymatic deglycosylation of PK-treated PrPSc removes the two N-linked carbohydrate structures and results in an ∼25% reduction in molecular weight and a single major PrPSc polypeptide (see Fig. 5B).
FIG 5.
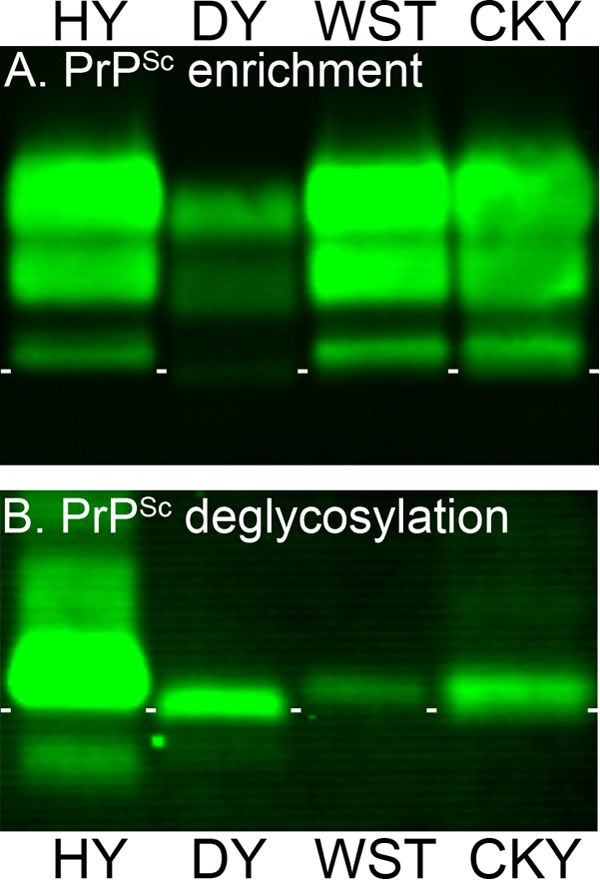
Enrichment and deglycosylation of PrPSc from TME and CWD strains. Brain homogenates from SGH infected with HY TME, DY TME, WST CWD, and CKY CWD were enriched for PrPSc by detergent extraction, ultracentrifugation, and proteinase K digestion (A), and N-linked carbohydrates were removed after enzymatic deglycosylation with PNGase F (B). Brain tissue was from second (CKY CWD) and third (WST CWD) serial passages in SGH. Samples were analyzed by SDS-PAGE and Western blotting with anti-PrP D18 antibody. Short white horizontal lines are located below the aglycosylated polypeptide band of HY TME and placed between each lane for reference purposes.
Antigenic mapping of PrPSc from WST and CKY CWD in HPrP7752KO mice and SGH also revealed strain-specific properties after PK digestion, but these differences were subtle. The immunoreactivity of anti-PrP 12B2 antibody indicated that PrPSc from both CWD strains had polypeptide migration patterns similar to that of PrPSc from HY TME (Fig. 4). When we used anti-PrP D13 antibody, PrPSc polypeptides from WST CWD appeared to migrate slightly faster than those from HY TME, especially in HPrP7752KO mice, but more slowly than PrPSc polypeptides from CKY CWD. These differences were also evident for CWD infection of SGH and PrPSc immunoreactivity with anti-PrP D18 antibody. In this case, the PrPSc polypeptides from WST CWD migrate similarly to those of HY TME, whereas for CKY CWD there were two aglycosylated PrPSc polypeptides that appeared as a doublet band in addition to the glycosylated PrPSc polypeptides (Fig. 4). The higher-molecular-weight aglycosylated PrPSc polypeptide from CKY CWD in SGH was immunoreactive with anti-PrP 12B2 antibody, whereas the lower-molecular-weight aglycosylated PrPSc polypeptide was not immunoreactive with 12B2 antibody (Fig. 4). This lower-molecular-weight PrPSc polypeptide had a migration similar to that of the aglycosylated PrPSc polypeptide in DY TME with anti-PrP D18 antibody. These subtle differences in PrPSc polypeptides between WST CWD and CKY CWD were more evident in brain homogenates than after PrPSc enrichment and PK digestion, but they were observed after deglycosylation of PrPSc polypeptides (Fig. 5A and B). These results demonstrate that strain-specific antigenic properties of PrPSc from the CWD strains were also maintained across different rodent species that express hamster Prnp.
Conformational analysis of PrPSc from TME and CWD strains in SGH and HPrP7752KO mice.
In addition to PK digestion and antigenic mapping of PrPSc, the biophysical properties of PrPSc from the TME and CWD strains in the two rodent species were investigated by a combination of detergent extraction, protein denaturation, and ultracentrifugation. A PrPSc CSSA was used to measure the median denaturation dose, which was defined as the concentration of GdnHCl that results in a 50% increase in PrPSc solubility compared to in the absence of GdnHCl. Western blot was used to visualize and quantify the percentage of PrP in the supernatant (i.e., soluble) and pellet (i.e., insoluble) at each concentration of GdnHCl between 0 and 4 M and to generate PrPSc denaturation curves. Figure 6 illustrates the results from a single SGH brain infected with each of the TME and CWD strains, and a gradual shift in the PrPSc distribution was observed from the pellet to the supernatant with increasing concentrations of GdnHCl. The CSSA was used to generate solubility and denaturation curves for each prion strain in SGH (Fig. 7A). The percentage of insoluble PrP in N-laurylsarcosine and 0 M GdnHCl distinguishes the prion CSSA curves into two patterns. HY TME and CKY CWD have a high percentage of insoluble PrP in 0 M GdnHCl, while DY TME and WST CWD have a moderate amount of insoluble PrP under these conditions (Fig. 7A). Although the PrPSc denaturation curves appear to slightly deviate between HY TME and CKY CWD at ≥2.0 M GdnHCl, the median denaturation doses (DD50) were similar, i.e., 2.08 and 2.15 M GdnHCl, respectively, between the prion strains. There was a more pronounced departure between DY TME and WST CWD, and this is reflected in DD50s of 1.57 and 2.40 M GdnHCl, respectively. These findings provide additional support for distinct features among the TME and CWD strains based on the biophysical properties of PrPSc.
FIG 6.

Western blot of prion protein from the TME and CWD strains in Syrian golden hamsters after a conformation solubility and stability assay (CSSA). SGH brain homogenates from HY TME (A), DY TME (B), WST CWD (C), and CKY CWD (D) were extracted in Sarkosyl, incubated with GdnHCl from 0 to 4.0 M, and subjected to ultracentrifugation as described for the CSSA. For each GdnHCl treatment, the supernatant (S) and pellet (P) fraction after ultracentrifugation were analyzed in adjacent lanes by SDS-PAGE and Western blotting with anti-PrP D18 antibody. Brain tissue was from the second (CKY CWD) and third (WST CWD) serial passages in SGH. For each panel, two separate Western blots prepared for strain analysis, and for illustrative purposes, they were merged between the 2.0 and 2.5 M GdnHCl values.
FIG 7.
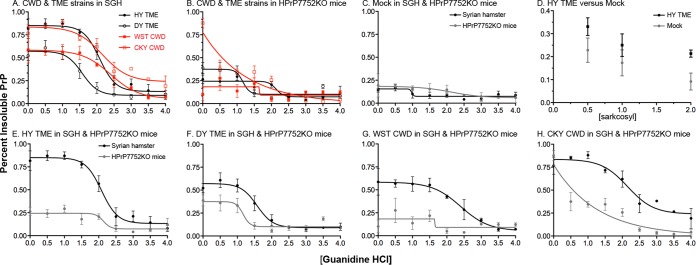
Conformational stability and solubility of prion protein in transgenic mice and Syrian golden hamsters infected with TME and CWD strains. The CSSA was performed using three individual brains from the TME and CWD strains in transgenic mice and SGH. The percentage of insoluble prion protein (PrP) indicates the percentage in the pellet fraction when 100% is equal to the sum of the soluble and pellet fraction for a given [GdnHCl]. The CSSA results for the prion strains in SGH (A) and HPrP7752KO mice (B) are plotted on the same graph. The CSSA results for each individual prion strain in both host species are also plotted on a single panel (E through H). The CSSA results for uninfected SGH and HPrP7752KO mice (C), and the solubility of PrP in N-laurylsarcosine and 0 M GdnHCl from mock-infected and HY TME-infected HrP7752KO brain (D) are also illustrated. Brain tissue was obtained from the second (CKY CWD) and third (WST CWD) serial passages in SGH and from the fourth (CKY CWD) and third (WST CWD) serial passages in HPrP7752KO mice.
A direct comparison of the CSSA curves for each prion strain in HPrP7752KO mice and SGH revealed that the biophysical features of PrPSc were not maintained upon interspecies transmission. There was a dramatic increase in brain PrP solubility in 2% N-laurylsarcosine in the absence of GdnHCl in HPrP7752KO mice compared to SGH for three of the four prion strains (Fig. 7B and Fig. 7E to H; data not shown). For HY TME, the percent insoluble PrP in 0 M GdnHCl was 81% ± 10% for SGH versus 26% ± 4.6% in HPrP7752KO mice (Fig. 7E). This was not due to a significant difference in the solubility of PrPC in uninfected brains from HPrP7752KO mice and SGH (Fig. 7C) or to the detergent conditions to separate PrPC and PrPSc in brain extracts of HPrP7752KO mice (Fig. 7D). The solubility of brain PrP in 0 M GdnHCl in SGH and HPrP7752KO mice was also distinct for DY TME (52% ± 5.1% versus 38% ± 5.9%), WST CWD (58% ± 14.4% versus 10% ± 7%), and CKY CWD (76% ± 9.3% versus 87% ± 5.3%) (Fig. 7F to H; data not shown). Despite these differences in PrPSc solubility, the DD50s for HY TME were 2.08 and 2.13 M GdnHCl for HPrP7752KO mice and SGH, respectively, and 1.57 and 1.16 M GdnHCl, respectively, in DY TME. The median denaturation doses for WST CWD and CKY CWD were 2.40 and 2.15 M GdnHCl in SGH, respectively, but ambiguous in HPrP7752KO mice using a four-parameter nonlinear regression analysis (Fig. 7B). For WST CWD in HPrP7752KO mice, the percentage of insoluble PrP appears to be unchanged with increasing concentrations of GdnHCl (Fig. 7B and G), suggesting that the majority of PrPSc was solubilized at low concentrations of GdnHCl. Additional support for this conclusion were the similarities in denaturation curves for WST CWD in HPrP7752KO mice and in mock-infected HPrP7752KO mice; both exhibited a flat-line fitted curve (Fig. 7B and C). These results indicate that the biophysical properties of PrPSc were not maintained for the CWD strains upon interspecies passage to a host with the same Prnp genotype, but these properties were largely maintained for the TME strains.
Passage of CWD strains into SGH can modify strain properties in HPrP7752KO mice.
Our findings provide evidence for Prnp-independent modification of both prion strain biological properties and PrPSc biophysical properties upon transmission of CWD strains from HPrP7752KO mice to SGH. We next investigated whether the strain-specific properties of WST CWD and CKY CWD in SGH can be maintained upon passage back into HPrP7752KO mice. Brain samples from SGH at the first and second serial passages of CKY CWD and WST CWD, respectively, were inoculated into HPrP7752KO mice for two serial passages. For CKY CWD, on the first and second serial backpassages the incubation periods in HPrP7752KO mice were 156 ± 2.5 days and 178 ± 2.5 days, respectively (Table 1, recipient groups M6448 and M6596) compared to 160 ± 4.6 days prior to interspecies transmission to SGH. For WST CWD in SGH, backpassage into HPrP7752KO mice resulted in incubation periods of 180 ± 8.3 days and 218 ± 9.1 days on the first and second serial passages, respectively (Table 1, recipient groups M6429 and M6595). The incubation period of WST CWD prior to interspecies transmission to SGH was 187 to 198 days. These findings indicate that the incubation period of CKY CWD was able to revert to a shorter length than for WST CWD upon backpassage into HPrP7752KO mice, as was observed in HPrP7752KO mice prior to interspecies transmission to SGH. However, the incubation periods for both CWD strains after serial backpassage were now longer than before interspecies transmission to SGH indicating that there was additional Prnp-independent modification of CWD strain properties.
To investigate the PrPSc polypeptide pattern of WST CWD and CKY CWD after backpassage into HPrP7752KO mice, PK digestion of brain homogenates was performed on SGH and transgenic mouse tissues. Western blot analysis with anti-PrP D18 antibody revealed that the PrPSc polypeptide migration pattern was similar between the SGH brain inoculum and HPrP7752KO mice for each of the CWD strains upon serial backpassage (Fig. 8). PrPSc polypeptides from WST CWD had an equal or slower migration than those from CKY CWD, and PrPSc from CKY CWD had the characteristic doublet PrPSc for the aglycosylated polypeptides. These results indicate that the strain-specific PrPSc polypeptide pattern for the CWD strains is maintained upon transmission from HPrP7752KO mice to SGH and backpassage into HPrP7752KO mice.
FIG 8.
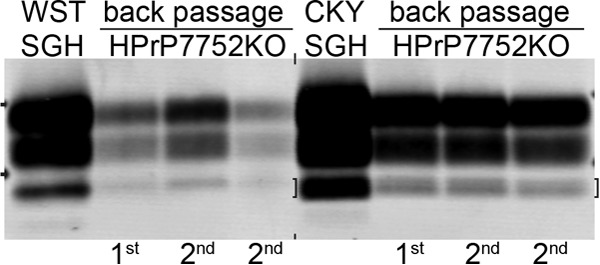
Western blot analysis of PrPSc following backpassage of CWD strains from Syrian golden hamsters (SGH) into transgenic mice. SGH brain samples from the second (CKY CWD) and third (WST CWD) serial passages were subsequently passaged back into transgenic mice (Mo) two times (e.g., lanes “1st” and “2nd”) as presented in Table 1. Brain homogenates were digested with proteinase K and analyzed by SDS-PAGE and Western blotting with anti-PrP D18 antibody. The two short horizontal bars at both ends of the membrane correspond to molecular masses of 20 and 30 kDa. The bracket indicates the doublet PrPSc polypeptide found in CKY CWD.
DISCUSSION
This study investigated interspecies prion transmission into two rodent species with the same Prnp genotype in order to determine whether there are host-dependent cellular pathways that influence prion strain properties. Our findings indicate that serial passage of prion strains into a new host species, but expressing a similar Prnp, can result in either minor or significant changes to the biophysical and biological properties of the prion agent. CWD transmission from HPrP7752KO mice to SGH resulted in a reversal in the length of the prion incubation period between the WST and CKY CWD strains, an increase in the conformational stability of PrPSc, and the formation of PrPSc amyloid plaques. These results indicate that the cellular environment for epigenetic PrPSc propagation and/or manifestation of prion-induced changes can vary between host species, and these differences can play a role in influencing prion phenotypic diversity. Although a strong host species effect was observed for WST and CKY CWD, the HY and DY TME strains were relatively unchanged with respect to incubation period and conformational stability of PrPSc when adapted to both HPrP7752KO mice and SGH. Prior reports also indicate the maintenance of prion phenotypes upon interspecies transmission into different host species that express the same Prnp. Transmission of sporadic Creutzfeldt-Jakob disease (sCJD), bovine spongiform encephalopathy, and CWD to transgenic mice that overexpress the human, bovine, and cervid Prnp, respectively, resulted in brain disease phenotypes that were similar to the disease in the original host species (54, 55, 58–61). Interestingly, in one study the transmission of sCJD was more efficient in transgenic mice that overexpress a chimeric human-mouse PRNP that was different from the human PrPC at six amino acid positions. This was interpreted to indicate that murine-specific factors can interact with either mouse PrPC or PrPSc more effectively than with the human prion protein and promote prion formation of the chimeric PrPC (58). Therefore, we propose that adaptation of the WST and CKY CWD strains to the hamster Prnp genotype can be used to determine how changes to the biophysical properties of PrPSc can translate to specific disease outcomes.
Several unusual biological features of prion disease were discovered during the isolation of WST and CKY CWD strains on the hamster Prnp background. One was the unexpected identification of the longer incubation period WST CWD strain upon serial passage at a low dilution of inocula (i.e., a high concentration of prion strains) and the subsequent identification of the shorter incubation period CKY CWD strain after a high dilution of CWD inocula (i.e., a low concentration of prion strains). Since a similar strategy was previously used during the early passages of Stetsonville TME to identify DY TME in SGH, we would expect CKY CWD to emerge after serial passage of CWD at low dilutions of inocula in transgenic mice (52). Based on prion competition experiments after coinoculation of short- and long-incubation-period prion strains, there is no precedence to explain the emergence of CKY CWD from a mixture also containing WST CWD only after the dilution of inocula (52). Another unusual feature of the CWD strains isolated in transgenic mice was the reversal in the length of the prion incubation periods upon transmission to SGH. CKY CWD had an incubation period in transgenic mice that was 30 days shorter than WST CWD, but the incubation periods for both CWD strains were two to three times longer in SGH and CKY CWD had an incubation period that was 150 days longer than WST CWD. This was not observed for HY and DY TME, which had comparable, but slightly shorter incubation periods upon transmission from SGH to HPrP7752KO mice. This difference in incubation periods between the CWD strains in the two rodent hosts was probably not due to a reduction in prion levels in SGH because the relative prion brain titer, as estimated by median seeding dose, was higher for CKY CWD than WST CWD in both rodent species. A reversal in the lengths of the incubation periods also has been reported for the Sc237 and 139H scrapie strains in SGH upon transmission to Chinese hamsters, but this interspecies passage is to a host with a nonhomologous Prnp, indicating that these changes to the prion phenotype are likely to be Prnp dependent (62). Lastly, the clinical symptoms of CKY CWD were unusual, and the long-term eyelid closure, erratic and labored breathing, persistent hiccups, and inflation of the cheek pouch have not been described in prion infection of SGH. These clinical features are consistent with prion-induced alterations to the autonomic nervous system and suggest that there is an altered function of the sphincter muscles of the cheek pouch. It is noteworthy that a previous study used a similar strategy to adapt CWD from deer and elk to both HPrP7752KO mice and SGH (63). Although these earlier studies reported prion incubation periods that are consistent with some of our results, it is difficult to compare the findings from the two studies with respect to identification of similar prion phenotypes based on their description of clinical signs and PrPSc brain distribution.
Our analyses indicate that the biophysical properties of PrPSc were distinct among the TME and CWD strains and suggest that the higher-order structure of multimeric PrPSc is different for each prion strain. In addition, for WST and CKY CWD the conformational stability of PrPSc was different for each strain in the two rodent hosts despite a similar hamster Prnp genotype. Based on detergent solubility in the absence of GdnHCl denaturant, the majority of the PrPSc in the brains of transgenic mice was soluble in three of the four prion strains, but in SGH under the same conditions, insoluble PrPSc average levels ranged from 55 to 92% among the TME and CWD strains. This indicates that the host species can affect the PrPSc biochemical properties independent of the prion strain. Differences in PrPSc solubility have been previously described for several prion strains, including HY and DY TME in SGH (57, 64). For the TME strains in HPrP7752KO mice and SGH there were minor differences to the stability of the PrPSc conformation, but for the CWD strains the biophysical properties of PrPSc undergo dramatic changes between rodent species. In transgenic mice the PrPSc denaturation curve for WST CWD was indistinguishable from that of PrPC, but upon transmission of WST CWD to SGH it had the highest conformational stability among the prion strains. The high detergent solubility of PrPSc from WST CWD in transgenic mice is a likely explanation for the negligible effect of GdnHCl in the CSSA. Another unusual finding was the undefined DD50 for PrPSc from CKY CWD in HPrP7752KO mice but not in SGH. The PrPSc denaturation curve for CKY CWD in transgenic mice was heterogeneous, especially at the lower concentrations of GdnHCl, even though most PrP was insoluble under these conditions. One possible explanation for this finding is that CKY CWD contains a mixture of prion strains and therefore does not produce a single-slope denaturation curve. Consistent with this explanation is the wide range for the onset of incubation periods for CKY CWD in SGH (from 438 to 522 days), which could indicate the presence of multiple prion phenotypes. Furthermore, in CKY CWD the aglycosylated PrPSc polypeptide migrated as a doublet, and this may also indicate a mixture of two distinct PrPSc types.
The expression level of Prnp in HPrP7752KO mice and SGH could provide an explanation for the changes in prion phenotype upon interspecies transmission. It has been reported that HPrP7752KO mice have four times the level of brain PrPC than SGH (38), and this can explain the shorter incubation periods for the TME and CWD strains in transgenic mice compared to SGH. Previous studies also report shorter incubation times for scrapie strains in transgenic mice that overexpress SGH Prnp. For the Sc237 and 139H scrapie strains, shorter incubation periods and a smaller difference in disease onset between these strains was found in Tg81 mice (81 days versus 106 days, respectively) and Tg7 mice (54 days versus 59 days, respectively) compared to SGH (77 days versus 167 days, respectively) (62). Serial passage of HY and DY TME in HPrP7752KO mice resulted in incubation periods that were only 5 to 15 days, respectively, shorter than the same TME strain in SGH, suggesting that overexpression of PrPC in HPrP7752KO mice had only a small effect on incubation periods compared to SGH. However, for WST and CKY CWD the incubation periods were 140 to 320 days, respectively, longer in SGH than in transgenic mice. These findings suggest that there is a strong host species effect on the CWD strains, but that it is likely to be independent of Prnp expression level based on the minor effect on the TME strains. We cannot exclude the possibility that there exist differences in the amount of PrPC expression at the cellular level and/or in specific neuronal subsets between these rodent host species that can influence incubation period and other disease related phenotypes. Other studies indicate that transmission of mouse-adapted scrapie strains between wild-type mice and transgenic mice that overexpress murine Prnp can also have unpredictable outcomes on incubation periods (65).
Another variable that can influence prion strain properties in a host-dependent manner are the cellular sites of Prnp expression. In HPrP7752KO mice the SGH Prnp is driven by the rat neuron-specific enolase promoter, which restricts brain PrPC expression to neurons (66). NSE promoter activity can vary among different neuronal subtypes, as can the activity of the Prnp promoter. PrPC expression from the endogenous Prnp promoter in the brains of mice and SGH is primarily found in neurons (67, 68). Both the NSE and the Prnp promoters are widely expressed in the mouse brain at high levels, and expression is found in many similar brain regions. There is one report of PrPC expression in glial cells (69), but other studies report no PrPC expression in astrocytes and a lack of detectable Prnp mRNA in glia (68, 70, 71). In transgenic mice that express SGH Prnp driven by the glial fibrillary acidic protein promoter (GFAP), prion incubation periods are very long and disease penetrance is incomplete (72). These studies suggest that astrocytes are not likely to be a prominent source of either PrPC or PrPSc propagation, especially in wild-type rodents, because Prnp expression is lower in astrocytes from the endogenous Prnp promoter compared to transgenic GFAP-Prnp mice. Therefore, differences in the cellular sites of Prnp expression between HPrP7752KO mice and SGH do not appear to provide an explanation for the changes that we observed for the CWD strains upon transmission between these host species. However, in another study, neuron-specific Prnp expression was removed using a conditional-inducible knockout mouse during the course of prion infection and, afterward, PrPSc was found to accumulate in astrocytes (73). This suggests that PrPSc formation can occur in astrocytes. An alternate interpretation is that since PrPSc accumulation was not observed in the astrocytes of these prion-infected mice when Prnp expression was not perturbed, PrPSc localization to astrocytes in the conditional Prnp knockout mice could represent cellular redistribution of PrPSc to astrocytes. The brain sites of PrPSc deposition could also influence the disease course during CWD infection, and PrPSc accumulation in the soma of neurons was more common in transgenic mice than in SGH, whereas PrPSc amyloid plaques were prominent in SGH. These differences may be influenced by the rate of PrPSc formation and turnover, as well as the duration of prion infection at a particular site. For the CWD strains there were minor differences in the pattern of PrPSc deposition in the brain of SGH, but there were many brain regions in which PrPSc was found for both strains at terminal disease. This may reflect the long disease duration and widespread dissemination of PrPSc more than the role of prion targeting to specific brain structures in defining prion strain phenotypes (74). The observation that the incubation periods and PrPSc biophysical properties of HY and DY TME were only slightly modified upon transmission from SGH to HPrP7752KO mice indicates that interspecies transmission between these rodents is not sufficient to significantly alter prion strain properties. In order to explain how prion phenotypes were altered for the CWD strains upon interspecies transmission, we propose that host-specific cellular factors, other than or in addition to the levels and cellular sites of PrPC expression, contribute to the disease process. WST and CKY CWD provide an experimental model to identify putative host-dependent factors that modify prion strain diversity.
ACKNOWLEDGMENTS
This study was supported by Public Health Service grant AI05543 from the National Institutes of Health. This study was supported in part by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases.
We thank Glenn Telling, Bian Jifeng, Lindsay Parrie, and Terry Spraker for helpful discussions and the staff at Laboratory Animal Resources for excellent animal care.
REFERENCES
- 1.Jarrett JT, Lansbury PT Jr. 1993. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 2.Groveman BR, Dolan MA, Taubner LM, Kraus A, Wickner RB, Caughey B. 2014. Parallel in-register intermolecular beta-sheet architectures for prion-seeded prion protein (PrP) amyloids. J Biol Chem 289:24129–24142. doi: 10.1074/jbc.M114.578344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldgaber D, Goldfarb LG, Brown P, Asher DM, Brown WT, Lin S, Teener JW, Feinstone SM, Rubenstein R, Kascsak RJ, et al. 1989. Mutations in familial Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker's syndrome. Exp Neurol 106:204–206. doi: 10.1016/0014-4886(89)90095-2. [DOI] [PubMed] [Google Scholar]
- 4.Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, Westaway D, Ott J, Prusiner SB. 1989. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature 338:342–345. doi: 10.1038/338342a0. [DOI] [PubMed] [Google Scholar]
- 5.Owen F, Poulter M, Lofthouse R, Collinge J, Crow TJ, Risby D, Baker HF, Ridley RM, Hsiao K, Prusiner SB. 1989. Insertion in prion protein gene in familial Creutzfeldt-Jakob disease. Lancet i:51–52. [DOI] [PubMed] [Google Scholar]
- 6.Doh-ura K, Tateishi J, Sasaki H, Kitamoto T, Sakaki Y. 1989. Pro→Leu change at position 102 of prion protein is the most common but not the sole mutation related to Gerstmann-Straussler syndrome. Biochem Biophys Res Commun 163:974–979. doi: 10.1016/0006-291X(89)92317-6. [DOI] [PubMed] [Google Scholar]
- 7.Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, Cortelli P, Julien J, Vital C, Pendelbury WW. 1992. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science 258:806–808. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- 8.Medori R, Tritschler HJ, LeBlanc A, Villare F, Manetto V, Chen HY, Xue R, Leal S, Montagna P, Cortelli P. 1992. Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N Engl J Med 326:444–449. doi: 10.1056/NEJM199202133260704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speer MC, Goldgaber D, Goldfarb LG, Roses AD, Pericak-Vance MA. 1991. Support of linkage of Gerstmann-Straussler-Scheinker syndrome to the prion protein gene on chromosome 20p12-pter. Genomics 9:366–368. doi: 10.1016/0888-7543(91)90266-H. [DOI] [PubMed] [Google Scholar]
- 10.Dickinson AG, Meikle VM. 1971. Host-genotype and agent effects in scrapie incubation: change in allelic interaction with different strains of agent. Mol Gen Genet 112:73–79. doi: 10.1007/BF00266934. [DOI] [PubMed] [Google Scholar]
- 11.Westaway D, Goodman PA, Mirenda CA, McKinley MP, Carlson GA, Prusiner SB. 1987. Distinct prion proteins in short and long scrapie incubation period mice. Cell 51:651–662. doi: 10.1016/0092-8674(87)90134-6. [DOI] [PubMed] [Google Scholar]
- 12.Dickinson AG, Fraser H. 1977. Scrapie pathogenesis in inbred mice: an assessment of host control and response involving many strains of agent, p 3–14. In ter Meulen V, Katz M (ed), Slow viral infections of the central nervous system. Springer-Verlag, New York, NY. [Google Scholar]
- 13.Fraser H. 1976. The pathology of natural and experimental scrapie, p 267–323. In Kimberlin RH. (ed), Slow virus diseases of man and animals. Elsevier, New York, NY. [Google Scholar]
- 14.Bessen RA, Marsh RF. 1994. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68:7859–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. 1995. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 375:698–700. doi: 10.1038/375698a0. [DOI] [PubMed] [Google Scholar]
- 16.Caughey B, Raymond GJ, Bessen RA. 1998. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem 273:32230–32235. doi: 10.1074/jbc.273.48.32230. [DOI] [PubMed] [Google Scholar]
- 17.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. 1996. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 18.Wickner RB, Shewmaker F, Edskes H, Kryndushkin D, Nemecek J, McGlinchey R, Bateman D, Winchester CL. 2010. Prion amyloid structure explains templating: how proteins can be genes. FEMS Yeast Res 10:980–991. doi: 10.1111/j.1567-1364.2010.00666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wickner RB, Edskes HK, Kryndushkin D, McGlinchey R, Bateman D, Kelly A. 2011. Prion diseases of yeast: amyloid structure and biology. Semin Cell Dev Biol 22:469–475. doi: 10.1016/j.semcdb.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VM. 2013. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, Diamond MI. 2014. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82:1271–1288. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watts JC, Condello C, Stohr J, Oehler A, Lee J, DeArmond SJ, Lannfelt L, Ingelsson M, Giles K, Prusiner SB. 2014. Serial propagation of distinct strains of Aβ prions from Alzheimer's disease patients. Proc Natl Acad Sci U S A 111:10323–10328. doi: 10.1073/pnas.1408900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kocisko DA, Priola SA, Raymond GJ, Chesebro B, Lansbury PT Jr, Caughey B. 1995. Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc Natl Acad Sci U S A 92:3923–3927. doi: 10.1073/pnas.92.9.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Come JH, Fraser PE, Lansbury PT Jr. 1993. A kinetic model for amyloid formation in the prion diseases: importance of seeding. Proc Natl Acad Sci U S A 90:5959–5963. doi: 10.1073/pnas.90.13.5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scott M, Foster D, Mirenda C, Serban D, Coufal F, Walchli M, Torchia M, Groth D, Carlson G, DeArmond SJ, Westaway D, Prusiner SB. 1989. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 59:847–857. doi: 10.1016/0092-8674(89)90608-9. [DOI] [PubMed] [Google Scholar]
- 26.Marsh RF, Bessen RA, Lehmann S, Hartsough GR. 1991. Epidemiological and experimental studies on a new incident of transmissible mink encephalopathy. J Gen Virol 72(Pt 3):589–594. doi: 10.1099/0022-1317-72-3-589. [DOI] [PubMed] [Google Scholar]
- 27.Taylor DM, Dickinson AG, Fraser H, Marsh RF. 1986. Evidence that transmissible mink encephalopathy agent is biologically inactive in mice. Neuropathol Appl Neurobiol 12:207–215. doi: 10.1111/j.1365-2990.1986.tb00051.x. [DOI] [PubMed] [Google Scholar]
- 28.Prusiner SB, Scott M, Foster D, Pan KM, Groth D, Mirenda C, Torchia M, Yang SL, Serban D, Carlson GA. 1990. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63:673–686. doi: 10.1016/0092-8674(90)90134-Z. [DOI] [PubMed] [Google Scholar]
- 29.Bessen RA, Marsh RF. 1992. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol 73(Pt 2):329–334. [DOI] [PubMed] [Google Scholar]
- 30.Kimberlin RH, Walker CA, Fraser H. 1989. The genomic identity of different strains of mouse scrapie is expressed in hamsters and preserved on reisolation in mice. J Gen Virol 70(Pt 8):2017–2025. doi: 10.1099/0022-1317-70-8-2017. [DOI] [PubMed] [Google Scholar]
- 31.Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. 1993. Mice devoid of PrP are resistant to scrapie. Cell 73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 32.Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M, Burton D, Yang SL, DeArmond SJ. 1993. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci U S A 90:10608–10612. doi: 10.1073/pnas.90.22.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruce ME, McConnell I, Fraser H, Dickinson AG. 1991. The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J Gen Virol 72(Pt 3):595–603. [DOI] [PubMed] [Google Scholar]
- 34.Stephenson DA, Chiotti K, Ebeling C, Groth D, DeArmond SJ, Prusiner SB, Carlson GA. 2000. Quantitative trait loci affecting prion incubation time in mice. Genomics 69:47–53. doi: 10.1006/geno.2000.6320. [DOI] [PubMed] [Google Scholar]
- 35.Tamguney G, Giles K, Glidden DV, Lessard P, Wille H, Tremblay P, Groth DF, Yehiely F, Korth C, Moore RC, Tatzelt J, Rubinstein E, Boucheix C, Yang X, Stanley P, Lisanti MP, Dwek RA, Rudd PM, Moskovitz J, Epstein CJ, Cruz TD, Kuziel WA, Maeda N, Sap J, Ashe KH, Carlson GA, Tesseur I, Wyss-Coray T, Mucke L, Weisgraber KH, Mahley RW, Cohen FE, Prusiner SB. 2008. Genes contributing to prion pathogenesis. J Gen Virol 89:1777–1788. doi: 10.1099/vir.0.2008/001255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Race RE, Priola SA, Bessen RA, Ernst D, Dockter J, Rall GF, Mucke L, Chesebro B, Oldstone MB. 1995. Neuron-specific expression of a hamster prion protein minigene in transgenic mice induces susceptibility to hamster scrapie agent. Neuron 15:1183–1191. doi: 10.1016/0896-6273(95)90105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Race R, Oldstone M, Chesebro B. 2000. Entry versus blockade of brain infection following oral or intraperitoneal scrapie administration: role of prion protein expression in peripheral nerves and spleen. J Virol 74:828–833. doi: 10.1128/JVI.74.2.828-833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kercher L, Favara C, Chan CC, Race R, Chesebro B. 2004. Differences in scrapie-induced pathology of the retina and brain in transgenic mice that express hamster prion protein in neurons, astrocytes, or multiple cell types. Am J Pathol 165:2055–2067. doi: 10.1016/S0002-9440(10)63256-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartz JC, Kincaid AE, Bessen RA. 2003. Rapid prion neuroinvasion following tongue infection. J Virol 77:583–591. doi: 10.1128/JVI.77.1.583-591.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bessen RA, Robinson CJ, Seelig DM, Watschke CP, Lowe D, Shearin H, Martinka S, Babcock AM. 2011. Transmission of chronic wasting disease identifies a prion strain causing cachexia and heart infection in hamsters. PLoS One 6:e28026. doi: 10.1371/journal.pone.0028026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shearin H, Bessen RA. 2014. Axonal and transynaptic spread of prions. J Virol 88:8640–8655. doi: 10.1128/JVI.00378-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bessen RA, Wilham JM, Lowe D, Watschke CP, Shearin H, Martinka S, Caughey B, Wiley JA. 2012. Accelerated shedding of prions following damage to the olfactory epithelium. J Virol 86:1777–1788. doi: 10.1128/JVI.06626-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bessen RA, Shearin H, Martinka S, Boharski R, Lowe D, Wilham JM, Caughey B, Wiley JA. 2010. Prion shedding from olfactory neurons into nasal secretions. PLoS Pathog 6:e1000837. doi: 10.1371/journal.ppat.1000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bessen RA, Martinka S, Kelly J, Gonzalez D. 2009. Role of the lymphoreticular system in prion neuroinvasion from the oral and nasal mucosa. J Virol 83:6435–6445. doi: 10.1128/JVI.00018-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langeveld JP, Jacobs JG, Erkens JH, Bossers A, van Zijderveld FG, van Keulen LJ. 2006. Rapid and discriminatory diagnosis of scrapie and BSE in retro-pharyngeal lymph nodes of sheep. BMC Vet Res 2:19. doi: 10.1186/1746-6148-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williamson RA, Peretz D, Pinilla C, Ball H, Bastidas RB, Rozenshteyn R, Houghten RA, Prusiner SB, Burton DR. 1998. Mapping the prion protein using recombinant antibodies. J Virol 72:9413–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crowell J, Wiley JA, Bessen RA. 2015. Lesion of the olfactory epithelium accelerates prion neuroinvasion and disease onset when prion replication is restricted to neurons. PLoS One 10:e0119863. doi: 10.1371/journal.pone.0119863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pirisinu L, Di Bari M, Marcon S, Vaccari G, D'Agostino C, Fazzi P, Esposito E, Galeno R, Langeveld J, Agrimi U, Nonno R. 2010. A new method for the characterization of strain-specific conformational stability of protease-sensitive and protease-resistant PrP. PLoS One 5:e12723. doi: 10.1371/journal.pone.0012723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilham JM, Orru CD, Bessen RA, Atarashi R, Sano K, Race B, Meade-White KD, Taubner LM, Timmes A, Caughey B. 2010. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog 6:e1001217. doi: 10.1371/journal.ppat.1001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, Onwubiko HA, Priola SA, Caughey B. 2008. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods 5:211–212. doi: 10.1038/nmeth0308-211. [DOI] [PubMed] [Google Scholar]
- 51.Dougherty R. 1964. Animal virus titration techniques, p 183–186. In Harris R. (ed), Techniques in experimental virology. Academic Press, Inc, New York, NY. [Google Scholar]
- 52.Bartz JC, Bessen RA, McKenzie D, Marsh RF, Aiken JM. 2000. Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J Virol 74:5542–5547. doi: 10.1128/JVI.74.12.5542-5547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marsh RF, Hanson RP. 1978. The Syrian hamster as a model for the study of slow virus diseases caused by unconventional agents. Fed Proc 37:2076–2078. [PubMed] [Google Scholar]
- 54.Browning SR, Mason GL, Seward T, Green M, Eliason GA, Mathiason C, Miller MW, Williams ES, Hoover E, Telling GC. 2004. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J Virol 78:13345–13350. doi: 10.1128/JVI.78.23.13345-13350.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tamguney G, Giles K, Bouzamondo-Bernstein E, Bosque PJ, Miller MW, Safar J, DeArmond SJ, Prusiner SB. 2006. Transmission of elk and deer prions to transgenic mice. J Virol 80:9104–9114. doi: 10.1128/JVI.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bartz JC, Kramer ML, Sheehan MH, Hutter JA, Ayers JI, Bessen RA, Kincaid AE. 2007. Prion interference is due to a reduction in strain-specific PrPSc levels. J Virol 81:689–697. doi: 10.1128/JVI.01751-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bessen RA, Marsh RF. 1992. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol 66:2096–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Telling GC, Scott M, Hsiao KK, Foster D, Yang SL, Torchia M, Sidle KC, Collinge J, DeArmond SJ, Prusiner SB. 1994. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci U S A 91:9936–9940. doi: 10.1073/pnas.91.21.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL, Welch J, Hill AF, Lloyd SE, Wadsworth JD, Collinge J. 2002. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J 21:6358–6366. doi: 10.1093/emboj/cdf653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scott MR, Will R, Ironside J, Nguyen HO, Tremblay P, DeArmond SJ, Prusiner SB. 1999. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci U S A 96:15137–15142. doi: 10.1073/pnas.96.26.15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D, Yuan J, Zheng M, Bai H, Deng H, Chen K, Jenny AL, O'Rourke K, Belay ED, Schonberger LB, Petersen RB, Sy MS, Chen SG, Gambetti P. 2005. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci 25:7944–7949. doi: 10.1523/JNEUROSCI.2467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hecker R, Taraboulos A, Scott M, Pan KM, Yang SL, Torchia M, Jendroska K, DeArmond SJ, Prusiner SB. 1992. Replication of distinct scrapie prion isolates is region specific in brains of transgenic mice and hamsters. Genes Dev 6:1213–1228. doi: 10.1101/gad.6.7.1213. [DOI] [PubMed] [Google Scholar]
- 63.Raymond GJ, Raymond LD, Meade-White KD, Hughson AG, Favara C, Gardner D, Williams ES, Miller MW, Race RE, Caughey B. 2007. Transmission and adaptation of chronic wasting disease to hamsters and transgenic mice: evidence for strains. J Virol 81:4305–4314. doi: 10.1128/JVI.02474-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC. 2011. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog 7:e1001317. doi: 10.1371/journal.ppat.1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karapetyan YE, Saa P, Mahal SP, Sferrazza GF, Sherman A, Sales N, Weissmann C, Lasmezas CI. 2009. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS One 4:e5730. doi: 10.1371/journal.pone.0005730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe JG. 1990. Transgenic mice expressing beta-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron 5:187–197. doi: 10.1016/0896-6273(90)90308-3. [DOI] [PubMed] [Google Scholar]
- 67.Ford MJ, Burton LJ, Li H, Graham CH, Frobert Y, Grassi J, Hall SM, Morris RJ. 2002. A marked disparity between the expression of prion protein and its message by neurones of the CNS. Neuroscience 111:533–551. doi: 10.1016/S0306-4522(01)00603-0. [DOI] [PubMed] [Google Scholar]
- 68.Bailly Y, Haeberle AM, Blanquet-Grossard F, Chasserot-Golaz S, Grant N, Schulze T, Bombarde G, Grassi J, Cesbron JY, Lemaire-Vieille C. 2004. Prion protein (PrPc) immunocytochemistry and expression of the green fluorescent protein reporter gene under control of the bovine PrP gene promoter in the mouse brain. J Comp Neurol 473:244–269. doi: 10.1002/cne.20117. [DOI] [PubMed] [Google Scholar]
- 69.Laine J, Marc ME, Sy MS, Axelrad H. 2001. Cellular and subcellular morphological localization of normal prion protein in rodent cerebellum. Eur J Neurosci 14:47–56. doi: 10.1046/j.0953-816x.2001.01621.x. [DOI] [PubMed] [Google Scholar]
- 70.Haeberle AM, Ribaut-Barassin C, Bombarde G, Mariani J, Hunsmann G, Grassi J, Bailly Y. 2000. Synaptic prion protein immunoreactivity in the rodent cerebellum. Microsc Res Tech 50:66–75. doi:. [DOI] [PubMed] [Google Scholar]
- 71.Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. 2006. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci U S A 103:3416–3421. doi: 10.1073/pnas.0511290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raeber AJ, Race RE, Brandner S, Priola SA, Sailer A, Bessen RA, Mucke L, Manson J, Aguzzi A, Oldstone MB, Weissmann C, Chesebro B. 1997. Astrocyte-specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO J 16:6057–6065. doi: 10.1093/emboj/16.20.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. 2003. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 74.Ayers JI, Kincaid AE, Bartz JC. 2009. Prion strain targeting independent of strain-specific neuronal tropism. J Virol 83:81–87. doi: 10.1128/JVI.01745-08. [DOI] [PMC free article] [PubMed] [Google Scholar]