

ABSTRACT
The vaccinia virus B1R gene encodes a highly conserved protein kinase that is essential for the poxviral life cycle. As demonstrated in many cell types, B1 plays a critical role during viral DNA replication when it inactivates the cellular host defense effector barrier to autointegration factor (BAF or BANF1). To better understand the role of B1 during infection, we have characterized the growth of a B1-deficient temperature-sensitive mutant virus (Cts2 virus) in U2OS osteosarcoma cells. In contrast to all other cell lines tested to date, we found that in U2OS cells, Cts2 viral DNA replication is unimpaired at the nonpermissive temperature. However, the Cts2 viral yield in these cells was reduced more than 10-fold, thus indicating that B1 is required at another stage of the vaccinia virus life cycle. Our results further suggest that the host defense function of endogenous BAF may be absent in U2OS cells but can be recovered through either overexpression of BAF or fusion of U2OS cells with mouse cells in which the antiviral function of BAF is active. Interestingly, examination of late viral proteins during Cts2 virus infection demonstrated that B1 is required for optimal processing of the L4 protein. Finally, execution point analyses as well as electron microscopy studies uncovered a role for B1 during maturation of poxviral virions. Overall, this work demonstrates that U2OS cells are a novel model system for studying the cell type-specific regulation of BAF and reveals a role for B1 beyond DNA replication during the late stages of the viral life cycle.
IMPORTANCE The most well characterized role for the vaccinia virus B1 kinase is to facilitate viral DNA replication by phosphorylating and inactivating BAF, a cellular host defense responsive to foreign DNA. Additional roles for B1 later in the viral life cycle have been postulated for decades but are difficult to examine directly due to the importance of B1 during DNA replication. Here, we demonstrate that in U2OS cells, a B1 mutant virus escapes the block in DNA replication observed in other cell types and, instead, this mutant virus exhibits impaired late protein accumulation and incomplete maturation of new virions. These data provide the clearest evidence to date that B1 is needed for multiple critical junctures in the poxviral life cycle in a manner that is both dependent on and independent of BAF.
INTRODUCTION
Poxviruses are complex viruses containing linear double-stranded DNA genomes with the unique characteristic of undergoing viral replication in the cytoplasm of host cells. Vaccinia virus, the most well studied poxvirus, has a genome that is 192 kb in size and encodes approximately 200 proteins. The vaccinia virus life cycle includes a temporally regulated cascade of early gene expression, DNA replication, and intermediate and late stages of gene expression (1). This cascade culminates in the production of the structural proteins needed for the assembly and maturation of new virions in a process referred to as morphogenesis (2).
Viral DNA replication is orchestrated by a number of early proteins, including the catalytic subunit of the viral DNA polymerase (the product of the viral E9 gene) (3–6), a heterodimeric processivity factor (A20/D4) (7–9), a single-stranded DNA (ssDNA)-binding protein (I3) (10, 11), a DNA-independent nucleotide triphosphatase (D5) (12–14), a putative scaffolding protein (H5) (15), and a serine/threonine protein kinase (B1) (6, 16–18). B1 is highly conserved within the members of the Poxviridae family that infect mammals, with the only exceptions being the Molluscipox and Parapox genera (19). It is well established that the vaccinia virus B1 protein kinase is essential for productive infection. This conclusion is drawn from studies of temperature-sensitive mutant viruses with lesions in the B1 locus (Cts2 and Cts25 viruses), the progeny of which are severely reduced in number during infection at nonpermissive temperatures, due to critical defects in viral DNA replication (16, 20). Interestingly, there is evidence that the severity of the Cts2 virus phenotype is cell type dependent. For example, in L929 murine fibroblasts, Cts2 virus production at the nonpermissive temperature is reduced by 95%, with a correlative decrease in the amount of viral DNA accumulation to <5% of the amount of viral DNA produced during a permissive infection being found (16). In contrast, in BSC40 primate epithelial cells, the Cts2 viral yield is also reduced to ∼15% of wild-type (WT) viral titers, but viral DNA replication is less restricted, with the virus producing 67% of the amount of viral DNA relative to the amount produced during permissive infection (16). Together, these previous studies have led to a speculation that B1 and/or its substrates may be impacted by the host environment and that B1 may be needed during stages of the viral life cycle after DNA replication.
The B1 protein is a viral homolog of a family of mammalian kinases known as the vaccinia virus-related kinases (VRKs) (21, 22). These viral and cellular kinases are also functionally conserved, as demonstrated by (i) evidence that VRK1 can rescue the Cts2 viral DNA replication defect when expressed from the Cts2 virus genome (23) and (ii) the discovery that B1 and VRK1 share the same cellular substrate, the barrier to autointegration factor (BAF/BANF1) (24). BAF is a highly conserved DNA-binding protein with essential functions in eukaryotic cells. BAF is needed for the survival and differentiation of both human and mouse embryonic stem cells (25), and the depletion or knockout of BAF in Caenorhabditis elegans and Drosophila melanogaster is embryonically lethal (26, 27). BAF has also been implicated in human disease, as a point mutation within the BAF-coding region has been identified in two patients harboring a hereditary progeroid syndrome called Nestor-Guillermo progeria syndrome (28). In addition to these cellular functions, BAF is also capable of strongly inhibiting vaccinia virus DNA replication (24). The host defense activity of BAF against vaccinia virus is dependent on its DNA-binding property, which can be blocked through phosphorylation by B1, thus highlighting the importance of this kinase during poxviral DNA replication (29).
Herein, we further explore the cell type specificity of the B1-BAF axis during vaccinia virus infection. In this study, we found that the Cts2 virus is unimpaired during viral DNA replication in U2OS human epithelial osteosarcoma cells. Interestingly, the Cts2 viral yield in these cells was still significantly diminished, indicating that B1 is required at a postreplicative stage(s) of the vaccinia virus life cycle in these cells. Our investigation of B1-BAF signaling further indicated that endogenous BAF in U2OS cells appears to have little impact on viral DNA replication or viral production, even though its protein expression and wild-type mRNA sequence are comparable to that in other cell lines. Further investigation revealed that U2OS cells can be made nonpermissive for Cts2 viral DNA replication by overexpressing BAF or by fusion of U2OS cells with a naturally nonpermissive cell type, suggesting that the permissive phenotype is not dominant in those assays.
We took advantage of this uniquely permissive nature of U2OS cells to inquire whether B1 is needed during later stages of the vaccinia virus life cycle. Interestingly, examination of vaccinia virus late gene products demonstrated that B1 is required for the optimal production of the A11 protein as well as the complete processing of the L4 protein in infected U2OS cells. Furthermore, execution point analyses of the Cts2 virus using rifampin (RIF) treatment coupled to temperature shift procedures were also performed. Together with examination of Cts2 virus-infected cells by electron microscopy, these data reveal for the first time that B1 is likely involved in late steps in infection and that in its absence viral morphogenesis is impaired.
MATERIALS AND METHODS
Cell culture.
African green monkey BSC40, mouse fibroblast L929, and human epithelial osteosarcoma U2OS cells were obtained from ATCC, maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals) and penicillin-streptomycin, and incubated at 37°C in a 5% CO2 atmosphere. Infected cell cultures were incubated at 31.5°C or 39.7°C (for simplicity, written 32°C and 40°C, respectively), as indicated below.
Plasmids.
The transfer vector pLL3.7-GFP was used in the production of lentivirus expressing green fluorescent protein (GFP). The transfer vector pLV-mCherry was obtained from Addgene (catalog number 36084) and was used in the production of lentivirus expressing mCherry.
Viruses and viral infections.
The following viruses were used: wild-type vaccinia virus (WR strain), B1-deficient Cts2 virus (30), recombinant fluorescent protein (RFP)-tagged WT (WT-RFP) and Cts2-RFP viruses (24), D5 mutant and E9 mutant Cts24 viruses (gifts from Rich Condit, University of Florida), and recombinant viruses Cts2/B1 and Cts2/VRK1 (kindly provided by Paula Traktman, Medical College of Wisconsin). The Cts2-revB1 virus was produced by infecting BSC40 cells with Cts2 (multiplicity of infection [MOI] = 0.03) and cotransfecting a linearized DCT.VV.ORF92 plasmid (Addgene), which contains the wild-type WR B1R open reading frame (ORF) (31). These infected/transfected cells were incubated at 40°C for 24 h, harvested, and titrated on BSC40 cells at 40°C for isolation of individual plaques. The Cts2-revB1 virus was plaque purified twice before being expanded on BSC40 cells and purified using a sucrose cushion.
For viral spread analysis, U2OS cells were infected with WT-RFP or Cts2-RFP virus at an MOI of 0.02 at 40°C. Cells were then fixed at 24 or 48 hpi with 4% paraformaldehyde (PFA) prior to fluorescence imaging. For immunofluorescence assays, U2OS cells were infected with WT or Cts2 virus at an MOI of 3 at 40°C for 9 h prior to fixation with 4% PFA. For viral DNA replication assays, U2OS cells were infected with WT or Cts2 virus at an MOI of 3 at 40°C. Cells were harvested at 0, 8, or 24 h postinfection (hpi). For viral DNA replication with Cts virus mutants, U2OS cells were infected with WT, Cts2, Cts24, Cts42, Cts2/B1, or Cts2/VRK1 virus at an MOI of 3 at 40°C for 24 h. Following cell harvest, cells were pelleted and resuspended in phosphate-buffered saline (PBS) prior to DNA purification. For viral yield analysis, U2OS cells were infected with WT or Cts2 virus at an MOI of 3 at 32°C or 40°C for 24 h. For viral yield analysis in BAF-depleted cells, U2OS cells were infected with WT or Cts2 virus at an MOI of 3 at 40°C for 24 h prior to harvest. Following cell harvest, cells were pelleted and resuspended in 10 mM Tris (pH 9). Samples were freeze-thawed three times prior to titration on BSC40 cells at 32°C for plaque assays. For immunoblotting analysis, the cell types indicated below were infected with WT or Cts2 virus at an MOI of 3 at 40°C for 24 h. For transmission electron microscopy (TEM) analysis, U2OS cells were infected with WT or Cts2 virus at an MOI of 5 at 40°C for 19 to 20 h.
Generation of cells overexpressing 1× FLAG-BAF, depleted of BAF, or expressing GFP or mCherry.
Lentiviral expression vectors for 1× FLAG-tagged BAF (FLAG-BAF) (32) were used to generate lentivirus as previously described (29). The stable overexpression of BAF was accomplished by transducing cells with these lentivirus preparations as described previously (32). Transduced cells were selected with 200 μg/ml of hygromycin prior to further analysis.
The stable depletion of BAF was performed using lentivirus expressing BAF-specific short hairpin RNA (shRNA) or control (scrambled) shRNA as previously described (29). Those transduced cells were selected with 10 μg/ml of puromycin prior to further study.
Cells stably expressing GFP or mCherry were generated via transduction with lentiviral vectors carrying those proteins. Stable fluorophore expressions were monitored via fluorescence microscopy.
Transient heterokaryon assay.
L929 cells were seeded overnight on a 12-well dish. On the following day, U2OS cells were trypsinized, quantified, and added to the dish containing L929 cells. After 1 h of coculture, the cells were washed with 1× PBS followed by treatment with 50% (wt/vol) polyethylene glycol (PEG) 1000 (Rigaku) for 1 min to induce cell-cell fusion. The cells were then washed with 1× PBS twice and with medium once. Immediately after fusion, the cells were infected with WT or Cts2 virus at an MOI of 5 at 40°C for 10 h prior to fixation.
Immunofluorescence.
For the assays whose results are shown in Fig. 1, following fixation with 4% PFA, cells were permeabilized with 0.5% saponin in PBS for 5 min at room temperature. Cells were then washed twice with 0.05% saponin in PBS and incubated with I3 antibody (custom) at a 1:300 dilution at room temperature. After incubation with primary antibody, the cells were washed and incubated with Alexa Fluor 488-conjugated goat anti-rabbit immunoglobulin antibody (Invitrogen) at room temperature for 2 h, followed by nuclear staining with DAPI (4′,6-diamidino-2-phenylindole). Fluorescence images were taken by indirect fluorescence on an inverted confocal microscope (Olympus IX 81), and final images were obtained by pseudocoloring using ImageJ software.
FIG 1.
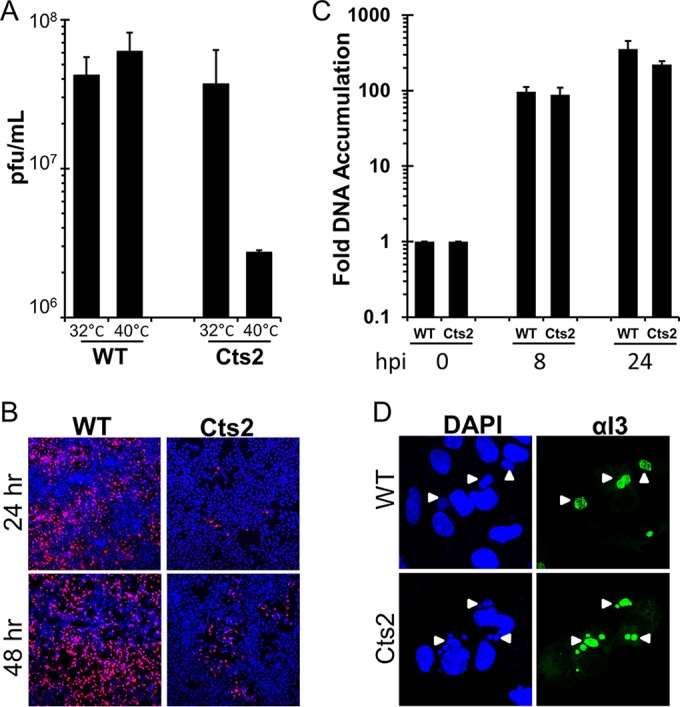
Cts2 virus production and viral spread, but not viral DNA replication, were reduced in U2OS cells at the nonpermissive temperature. (A) Viral yield analysis of WT- and Cts2 virus-infected U2OS cells. Cells were infected at an MOI of 3 at 32°C or 40°C for 24 h. The data from three independent experiments were averaged, and error bars represent standard deviations. (B) Viral spread analysis of U2OS cells infected with WT-RFP and Cts2-RFP virus at an MOI of 0.02 at 40°C. At 24 h or 48 hpi, cells were fixed and stained with DAPI. Representative plaques containing red fluorescent cells are shown. (C) Relative viral DNA accumulation by U2OS cells infected with WT and Cts2 virus at an MOI of 3 at 40°C for 0 h, 8 h, and 24 h. Viral DNA accumulation was analyzed via qPCR by using vaccinia virus DNA-specific primers. The fold DNA accumulation relative to that at 0 hpi was calculated for each virus. The data from three independent experiments were obtained, and error bars represent standard deviations. (D) Immunofluorescence analyses of cells infected with WT and Cts2 virus at an MOI of 3 at 40°C for 9 h. Following fixation, cells were then stained with an anti-I3 antibody (Alexa Fluor 488 conjugated; green) and DAPI. Arrowheads, viral replication factories marked by both DAPI- and I3-positive sites.
In transient heterokaryon assays, cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature. After washing with PBS, the cells were then incubated with I3 antibody in PBS at a 1:500 dilution at room temperature for 1 h. Afterward, the cells were washed with PBS and incubated with Alexa Fluor 647-conjugated goat anti-rabbit immunoglobulin secondary antibody (Invitrogen) in PBS at room temperature for 1 h. Fluorescence images were taken using an EVOS FL auto cell imaging system (Life Technologies).
Immunoblotting analysis.
To check for protein expression, U2OS cells (1 × 106) were harvested and resuspended in 300 μl of SDS sample buffer supplemented with 10 units of Benzonase. A volume of lysate equivalent to that from 105 cells was resolved on an 18% SDS-polyacrylamide gel, followed by protein transfer to a polyvinylidene difluoride membrane overnight. The blots were then incubated with the appropriate antibodies prior to signal development with chemiluminescent reagents. Quantifications of the chemiluminescence signal were performed by using Bio-Rad ImageLab software. The primary antibodies used were as follows: antibody against GAPDH (glyceraldehyde-3-phosphate dehydrogenase; 1:500; catalog number sc-25778; Santa Cruz Biotechnology), antibody against BAF (custom) (32), and custom antibodies against vaccinia virus proteins I3, A11, F17, and L4.
DNA purification and qPCR.
Viral DNA was extracted and purified using a GeneJET whole-blood genomic DNA purification minikit (catalog number K0782; Thermo Scientific). Quantitative PCR (qPCR) was performed using Bio-Rad iTaq universal SYBR Green supermix (catalog number 172-5121). Serial dilutions were included in each qPCR run to develop a standard curve and determine the PCR efficiency of the primer sets in that experiment set. qPCR analyses were performed using 1 μl of purified DNA and 1 μM vaccinia virus DNA-specific primers HA F (5′-CATCATCTGGAATTGTCACTACTAAA-3′) and HA R (5′-ACGGCCGACAATATAATTAATGC-3′).
Rifampin experiment.
U2OS cells (1 × 106) were infected with WT or Cts2 virus at an MOI of 3. After 30 min, the viral inoculum was removed and replaced by medium containing either a 1% dimethyl sulfoxide control or 100 μg/ml rifampin (catalog number BP2679-1; Fisher Scientific). The cells were then incubated at 32°C for 12 h. At 12 hpi, the cells were either maintained at 32°C or shifted to 40°C. For some treatments, the medium containing rifampin was removed and cell cultures were fed with fresh medium or with medium containing 50 μM 1-β-d-arabinofuranosylcytosine (araC). The cells were then harvested at 21 hpi and lysates were titrated on BSC40 cells at 32°C for plaque assays.
Transmission electron microscopy.
Following infection with WT or Cts2 virus as described above, the cells were washed with PBS and fixed in 0.1 Sorenson's phosphate buffer (0.1 M NaH2PO4·2H2O, 0.1 M Na2HPO4, pH 7.4) containing 2.5% electron microscopy-grade glutaraldehyde for 1 h at room temperature. The cells were then harvested by scraping and pelleted by centrifugation at 2,000 rpm for 5 min at 4°C. The cell pellets were dehydrated in a graduated ethanol series and embedded in Epon 812 (Electron Microscopic Sciences, Fort Washington, PA). Next, thin sections (80 nm) were stained with uranyl acetate and lead citrate and observed under a transmission electron microscope (model H7500-I; Hitachi) at the University of Nebraska—Lincoln (UNL) Microscopy Core Facility.
Statistics.
The error bars shown in the figures represent the standard deviations from the mean. The P values indicated were calculated using the Student t test.
RESULTS
In U2OS cells, the B1-defective Cts2 virus displays a temperature-dependent reduction in viral progeny but no detectable block in viral DNA replication.
The studies described herein have grown out of our efforts to characterize the Cts2 virus life cycle in various cell lines to better understand the cell type-specific behavior previously observed with this temperature-sensitive virus. In the course of these studies, we examined whether Cts2 virus production is impaired upon infection of human U2OS cells at the nonpermissive temperature, 40°C. Indeed, following infection at an MOI of 3, we found that Cts2 virus production at 40°C was reduced by 13-fold relative to the level of virus production at 32°C (Fig. 1A), while WT vaccinia virus exhibited no change in yield at the higher temperature. Analysis of infections with either the WT or Cts2 virus expressing the red fluorescent protein at an MOI of 0.02 further confirmed that the production and spread of virus in Cts2-infected cells at 24 or 48 hpi were significantly reduced (Fig. 1B) compared to the production and spread in the WT-infected controls. To determine if the defect in Cts2 virus production correlated with impaired viral DNA replication, we monitored the accumulation of viral DNA at 0, 8, or 24 hpi (MOI = 3) at 40°C. Viral DNA accumulation was quantified via real-time qPCR using vaccinia virus DNA-specific primers, and the fold increases relative to the amount of DNA isolated at 0 hpi were calculated. This analysis revealed that viral DNA accumulates to similar levels in WT- and Cts2 virus-infected cells at 8 or 24 hpi (Fig. 1C). Next, the size of the viral factories at 9 hpi at 40°C was assessed by immunofluorescence analysis using an antibody specific for the viral I3 ssDNA-binding protein. The assembly of I3-positive factories is known to be dependent on the replication of DNA and thus can be used as an assay complementary to our qPCR analysis (10). This study showed the accumulation of viral replication factories of equal size in both WT- and Cts2 virus-infected cells (Fig. 1D). Altogether, these data indicate that U2OS cells provide a unique system wherein B1 is apparently not required during viral DNA replication but is required during the later stage of the vaccinia virus life cycle. The data presented above led us to formulate three possible models to explain the role of the B1-BAF axis during infection of U2OS cells (1). Endogenous BAF is not present in U2OS cells. Therefore, B1 is not needed for DNA replication but is needed at a later stage of the viral life cycle, presumably to target another substrate (2). Endogenous BAF is expressed but is somehow inactive in U2OS cells. Therefore, B1 is not needed for DNA replication but is needed at a later stage of the viral life cycle to target another substrate (3). The host defense activity of BAF is delayed in some way. Therefore, B1 is still needed to inactivate BAF later in the viral life cycle. In an effort to discriminate between these three models, we pursued the experiments described below.
Overexpression of BAF leads to the sharply reduced accumulation of Cts2 viral DNA.
The activity of the B1 kinase is required during DNA replication in many cell lines in order to inactivate the cellular DNA-binding protein BAF (24). As such, the inhibition of Cts2 viral DNA accumulation can be exacerbated in cells that overexpress BAF (24, 29, 32). To test the impact of BAF overexpression in U2OS cells, cell lines that stably express a FLAG epitope-tagged form of BAF were generated using a lentiviral transduction system as previously described (29, 32). BAF expression levels were analyzed by immunoblotting with BAF-specific antibody (Fig. 2A) to verify the expression of FLAG-tagged BAF and the endogenous BAF. Next, to test whether BAF can exert an impact on Cts2 viral DNA replication in U2OS cells if it is overexpressed, we analyzed the accumulation of viral DNA in control cells or in FLAG-BAF-expressing cells. In addition to WT and Cts2 viruses, we utilized Cts24 and Cts42 viruses, as well as the recombinant viruses Cts2/B1 and Cts2/VRK1. The Cts24 and Cts42 viruses have been shown to exhibit a significant reduction of viral DNA accumulation at the nonpermissive temperature, as the proteins mutated in these viruses, D5 and E9, respectively, are essential for viral DNA replication (14, 17). The Cts2/B1 and Cts2/VRK1 viruses express the wild-type B1 kinase and the human VRK1 kinase, respectively, from the J2R locus of the Cts2 virus genome, and both kinases have previously been found to inactivate BAF's host defense activity (23, 24). Following infection with these viruses at an MOI of 3 at 40°C, viral DNA accumulation was analyzed via qPCR, and all values were normalized to the level of DNA in WT-infected control cells. With regard to WT virus infection, we observed similar levels of viral DNA accumulation in both control and FLAG-BAF-expressing cells, thus indicating that DNA replication is independent of the BAF protein expression level in the presence of the B1 kinase (Fig. 2B). Both the Cts24 and Cts42 viruses exhibited 100-fold less accumulation of viral DNA than the WT (Fig. 2B), consistent with their behavior in other cell types (14, 17). This reduction in viral DNA was independent of the BAF protein expression level, as expected if BAF is not involved in D5- or E9-regulated processes. Interestingly, during Cts2 virus infection, viral DNA accumulation was dramatically reduced (>50-fold) in cells expressing additional FLAG-BAF protein but not in control cells (Fig. 2B). The BAF-mediated inhibition of Cts2 virus was not observed during Cts2/B1 or Cts2/VRK1 virus infection of FLAG-BAF cells, indicating that the expression of those kinases can inactivate BAF, as we have previously observed in other studies (24). In sum, the data shown in Fig. 2 demonstrate that the nonpermissive phenotype of Cts2 virus common to other cell types can also be observed in U2OS cells upon BAF overexpression.
FIG 2.
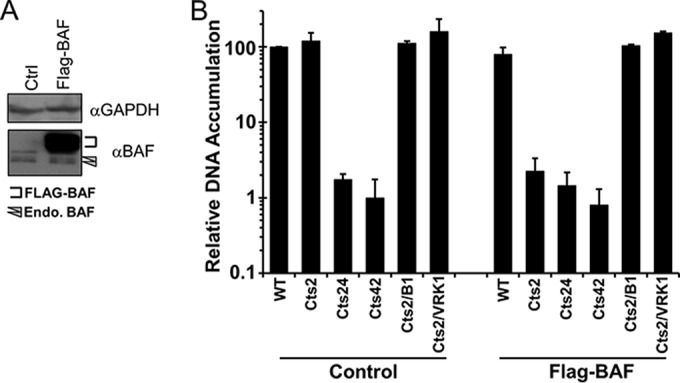
The accumulation of Cts2 viral DNA during infection of U2OS cells can be inhibited by BAF overexpression. (A) Representative Western blot analysis of whole-cell lysates from control U2OS cells or cells stably overexpressing FLAG-tagged WT BAF. Anti-BAF antibody recognizes both endogenous BAF protein and FLAG-tagged proteins. Anti-GAPDH antibody was used as a loading control. (B) Relative viral DNA accumulation by control and FLAG-BAF cells infected with WT, Cts2, Cts24 (D5 mutant), Cts42 (E9 mutant), Cts2/B1, or Cts2/VRK1 virus. Cells were infected at an MOI of 3 at 40°C for 24 h. The fold DNA accumulation relative to that by WT-infected control cells, for which the value was set equal to 100, was calculated. Error bars represent standard deviations.
Homokaryons of U2OS form viral replication factories, whereas homokaryons of L929 cells do not form viral replication factories during Cts2 virus infection.
Next, we inquired whether there may be insufficient levels of endogenous BAF in U2OS cells to combat Cts2 virus or whether the cellular regulation of BAF phosphorylation may be different in U2OS cells. We have previously published our observation that similar BAF protein levels are present in U2OS, L929, and BSC40 cells (20). We confirmed and extended these observations by performing immunoblotting analysis to determine the levels of both total and phosphorylated BAF in these three cell lines. As shown in Fig. 3A, similar total and phosphorylated BAF protein levels were present in U2OS, L929, and BSC40 cells, thus arguing against the possibility that Cts2 viral DNA replication proceeds in infected U2OS cells simply because BAF is absent. In addition, these data also demonstrate that there are similar levels of BAF phosphorylation in these three cell lines (Fig. 3A), therefore suggesting that the accumulation of Cts2 viral DNA in infected U2OS cells is not due to impaired host defense activity caused by enhanced BAF phosphorylation. Finally, sequencing analysis of BAF cDNA made from RNA extracted from U2OS cells showed the presence of transcripts containing the same sequences annotated in PubMed (GenBank accession numbers NM_001143985.1 and NM_003860.3) for human BANF1 transcripts (data not shown). These sequence data indicate that the absence of BAF-mediated inhibition of Cts2 viral DNA replication is not due to any mutation within the BANF1 coding region that may impair its functions.
FIG 3.

Evaluation of BAF protein levels and the formation of viral factories in unfused and homotypic cell fusions of permissive and restrictive cells. (A) Western blot analysis of BAF protein expression in BSC40, L929, and U2OS cells. Equal loading from 100,000 cells is shown. A representative blot from three independent experiments is shown. (C) Experimental design of somatic cell fusion assay. (B, D) Immunofluorescence analyses, as outlined in panel C, of WT- and Cts2 virus-infected cells that remained unfused (B) or fused with the same cell types (D). Cocultured L929-GFP and U2OS-mCherry cells were allowed to fuse with treatment with 50% PEG, followed by infection with virus at an MOI of 5 at 40°C for 10 h. After fixation, the cells were then stained with an anti-I3 antibody (Alexa Fluor 647 conjugated; blue). *, either GFP-expressing L929 cells (green) or mCherry-expressing U2OS cells (red); arrows, viral replication factories that are present in infected cells. Bar = 50 μm. (E) Quantification of the homotypic cell-cell fusion described in the legend to panel D. The presence or absence of viral factories within the L929-L929 or the U2OS-U2OS cell fusions was calculated and displayed as the percent distribution. n, the total number of infected cells (positive I3 signal) in which the viral factories were either present or absent. Five independent experiments which represent three sets of data from coculturing of L929-GFP and U2OS-mCherry cells and three sets of data from coculturing of L929-mCherry and U2OS-GFP were performed.
As the protein expression, phosphorylation, and mRNA sequence of BAF from U2OS cells appear to be no different from those of BAF from other cell types, we posited that permissive Cts2 viral DNA replication in U2OS cells is due to the presence of an additional factor that is differentially expressed in U2OS cells compared to nonpermissive cells, such as L929 cells. Such a regulatory factor could either activate or repress BAF in nonpermissive or permissive cells, respectively, and may function via a variety of potential molecular mechanisms. To better understand the cell type-dependent activity of BAF, we sought to determine which phenotype would be dominant in a cell fusion assay.
With this goal, we established a polyethylene glycol (PEG)-mediated somatic cell fusion assay using U2OS and L929 cells in which we could assay the viral DNA replication occurring in fused cells. L929 cells were selected for our fusion assays because they are known to be nonpermissive for Cts2 viral DNA replication but replicate wild-type vaccinia virus to high titers. To visually distinguish between the two different cell types, we generated cells that stably express either green fluorescent protein (GFP) or mCherry fluorescent protein. L929-GFP and U2OS-mCherry cells were cocultured in the same dish prior to cell-cell fusion by PEG treatment and infection with WT or Cts2 virus at an MOI of 5 at 40°C for 10 h (Fig. 3C). As a control, experiments were performed in which the opposite pairing of cell line and fluorescent protein were used (fusion between L929 cells and mCherry and U2OS cells and GFP or between L929 cells and GFP and U2OS cells and mCherry). However, as the results between the two pairs were indistinguishable, only the L929-GFP and U2OS-mCherry cell fusion data were presented. Following PEG treatment and infection, we first analyzed the formation of viral replication factories in either L929-GFP or U2OS-mCherry cells that remained unfused (Fig. 3B). As a marker for viral DNA replication, we utilized a primary antibody against the vaccinia virus ssDNA-binding protein I3, which is expressed early and localizes to cytoplasmic viral factories (10, 11). Importantly, these factories are dependent on viral DNA replication for their formation (10, 33). Thus, by examining whether factories have formed in fused cells, we can assay whether they are permissive for DNA replication. During WT infection, we observed the formation of viral factories in both the L929-GFP and U2OS-mCherry cells (Fig. 3B, arrows). However, during Cts2 virus infection, viral factories were absent in L929-GFP cells but were still present in U2OS-mCherry cells (Fig. 3B), as described above (Fig. 1D).
Next, to ensure that PEG-mediated fusion does not impair viral factory formation, we analyzed cell fusion events between cells of the same type: L929 homokaryons or U2OS homokaryons. Similar to our observations in unfused cells, we found that in WT infection, viral factories are present in both the L929-GFP homokaryons and the U2OS-mCherry homokaryons (Fig. 3D, arrows). Further quantifications of these fusion events reflect this conclusion, as both of these cell types predominantly contained factories (Fig. 3E). In the case of Cts2 virus infection, we found that viral factories were absent in L929-GFP homokaryons but were still predominant in U2OS-mCherry homokaryons (Fig. 3D and E). Overall, these results confirm that PEG-mediated cell fusions do not impair the formation of WT viral factories and retain the Cts2 virus host range phenotypes of the unfused cells.
Heterokaryons formed between U2OS and L929 cells lack the ability to form viral replication factories during Cts2 virus infection.
To determine whether fusions of permissive U2OS and restrictive L929 cells are restrictive or permissive for Cts2 viral DNA replication, we analyzed the heterokaryons of L929-GFP and U2OS-mCherry cells that were infected with WT or Cts2 virus. We found that following WT infection, 93% of heterokaryons that were infected, as indicated by I3 expression, contained viral factories (Fig. 4A and B). This is in contrast to the findings for Cts2 virus-infected cells, where the vast majority of infected heterokaryons (96% out of 115 total heterokaryons) were devoid of viral replication factories (Fig. 4A and B). In parallel assays, we also performed cell-cell fusions between L929-mCherry and U2OS-GFP cells and obtained results similar to those obtained with the L929-GFP and U2OS-mCherry cell fusions (individual data not shown; cumulative data are shown in Fig. 4B). Altogether, these results demonstrate that the nonpermissive phenotype exhibited by L929 cells is clearly dominant in L929-U2OS heterokaryons, leading to restricted Cts2 viral DNA replication in those cells.
FIG 4.

A heterokaryon assay shows that the restrictive L929 cell phenotype (absence of factories) is dominant in L929-U2OS fused cells. (A) Immunofluorescence analyses of WT- and Cts2 virus-infected L929-GFP (green) and U2OS-mCherry (red) fused cells. Arrows, viral replication factories that are present in infected cells (positive for I3 staining [blue]). Two representative images from each virus infection are shown. Bar = 50 μm. (B) Quantification of L929-U2OS cell-cell fusions described in the legend to panel A. The presence or absence of viral factories within the fused cells was calculated and is displayed as the percent distribution. n, the total number of infected cells (positive I3 signal) in which the viral factories were either present or absent. Five independent experiments which represent three sets of data from coculturing of L929-GFP and U2OS-mCherry cells and three sets of data from coculturing of L929-mCherry and U2OS-GFP cells were performed.
Depletion of endogenous BAF shows no impact on Cts2 virus production.
Although endogenous BAF in U2OS cells does not impact Cts2 viral DNA replication, we asked whether it was responsible for the decreased Cts2 virus production in these cells, perhaps affecting a postreplicative stage of the life cycle. To address this question, we infected control cells or cells that were depleted of native BAF (Fig. 5A) with WT or Cts2 virus at an MOI of 3 at 40°C. As shown in Fig. 5B, BAF depletion led to a very modest (<2-fold) increase in Cts2 virus production compared to that in the control cells. This difference was not statistically significant and was also observed during WT infection (Fig. 5B). In addition, infection of these cells at a lower MOI of 0.01 for 48 h also yielded similar results (data not shown). These data argue against the hypothesis that endogenous BAF is responsible for the decrease in viral progeny observed during Cts2 virus infection by blocking another stage in the viral life cycle.
FIG 5.
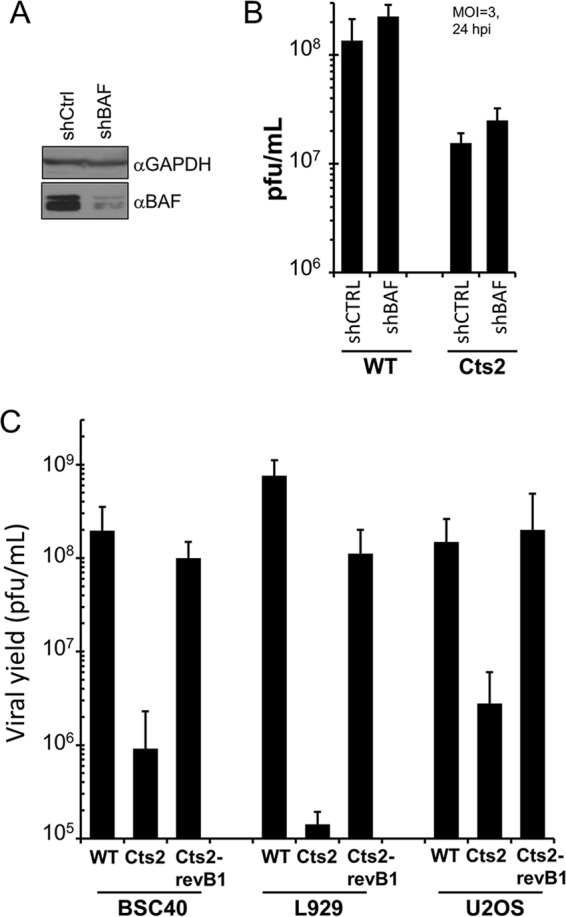
Cts2 virus production in U2OS cells is rescued by repair of the B1 locus but not by the depletion of endogenous BAF. (A) Representative Western blot analysis of whole-cell lysates from control cells or cells depleted of endogenous BAF. Anti-GAPDH antibody was used as a loading control. shCtrl, control shRNA; shBAF, shRNA against BAF. (B) Analysis of WT and Cts2 viral yields following infection of either U2OS control cells or cells that were depleted of endogenous BAF. Cells were infected at an MOI of 3 at 40°C for 24 h. The data from three independent experiments were averaged, and error bars represent standard deviations. (C) Viral yields of BSC40, L929, or U2OS cells infected with WT, Cts2, or Cts2-revB1 virus. Cells were infected at an MOI of 3 at 40°C for 24 h. Error bars represent standard deviations.
Next, we asked whether the mutated B1 locus within Cts2 virus was sufficient to cause the decreased viral yield in U2OS cells. We therefore examined whether the Cts2 virus phenotype can be rescued by replacement of the mutated Cts2 virus B1 locus with a wild-type B1 sequence. For this purpose, the Cts2 B1 revertant virus (Cts2-revB1 virus) was produced by infecting BSC40 cells with the Cts2 virus (MOI = 0.03) and cotransfecting these cells with a plasmid encoding the wild-type WR B1 ORF. Virus harvested from these infections/transfections yielded large plaques, which were then subjected to multiple rounds of plaque purification, before expansion and preparation of large viral stocks for use in the follow-up assays. As shown in Fig. 5C, infection of BSC40, L929, or U2OS cells with Cts2 virus led to a significant reduction in virus production compared to that in WT virus infection. L929 cells displayed the most restrictive phenotype, with an almost 4-log-unit reduction in virus production being detected (Fig. 5C). BSC40 cells and U2OS cells were also restrictive, with 2-log-unit and 1.5-log-unit reductions in virus production, respectively, being detected (Fig. 5C). The viral yield was dramatically recovered in BSC40, L929, or U2OS cells infected with the Cts2-revB1 virus. Both the U2OS and BSC40 cells that were infected with the Cts2-revB1 virus displayed viral yields that were similar to those in WT virus infection, suggesting a complete rescue of the defects in virus production in the Cts2 virus (Fig. 5C). Similarly, Cts2-revB1 virus-infected L929 cells also displayed a rescue of the Cts2 virus defects, although the viral yield was slightly less (∼7-fold) than that from the WT infection (Fig. 5C). Taken together, these results demonstrate that reversion of the Cts2 B1 virus ORF to the wild-type sequence is sufficient to rescue the viral yield in multiple cell lines, including U2OS cells. This suggests that the decreased Cts2 viral yield observed in U2OS cells is directly linked to the expression of a mutant B1 and is not likely due to the presence of another uncharacterized mutation within the Cts2 virus genome.
B1 contributes to the early and late stages of virion morphogenesis in U2OS cells.
As B1 is apparently not required during viral DNA replication but is required during another stage of the vaccinia virus life cycle, we sought to determine the postreplicative stage of the Cts2 viral life cycle that is impaired in U2OS cells. As such, we examined the production or processing of vaccinia virus late gene products A11, F17, and L4. A11 localizes to the viral factories and the endoplasmic reticulum and is essential for the formation of membrane crescents and immature virions (IVs) (34–36), F17 is a core phosphoprotein essential for the maturation of virions (37, 38), and L4 is also a core protein that is proteolytically cleaved from its precursor and is required for the production of infectious particles during viral assembly (39–42). Immunoblot analyses were performed following infection of L929, BSC40, or U2OS cells with WT or Cts2 virus at an MOI of 3 at 40°C. As an indicator of early gene expression, the accumulation of the I3 protein was also monitored. First, we monitored the expression of the three late proteins A11, F17, and L4 in the L929 and BSC40 restrictive cells. Upon WT infection of cells of either cell line, these late proteins accumulated to comparable levels at 16 or 24 hpi (Fig. 6A). This is in contrast to the findings obtained with Cts2 virus infection, where all three late proteins were absent in both L929 and BSC40 cells at 16 and 24 hpi (Fig. 6A). This is expected, as DNA replication arrests during infection at 40°C (16).
FIG 6.
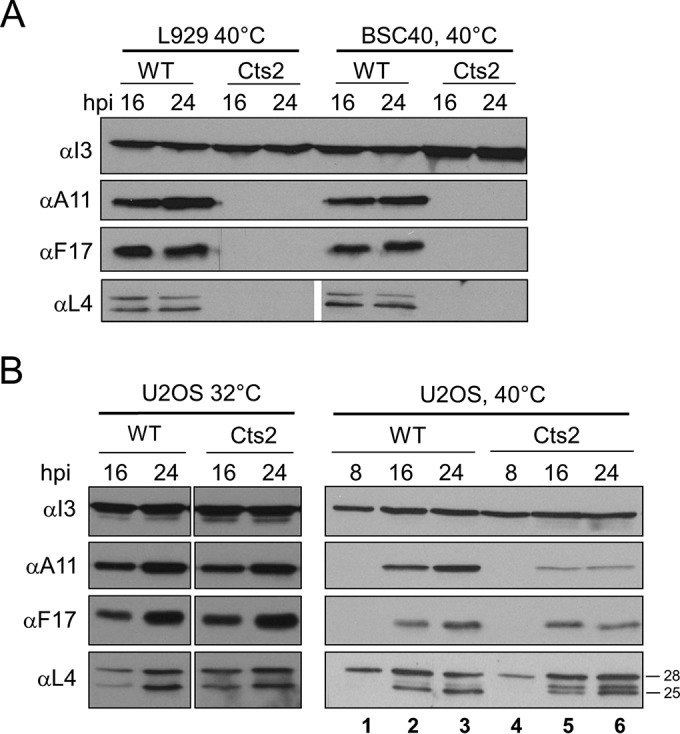
Late genes are expressed during Cts2 virus infection of U2OS cells, but protein accumulation and/or processing is reduced. (A, B) Western blot analyses of late vaccinia virus proteins upon infection of BSC40 and L929 cells (A) or U2OS cells (B). Cells were infected with WT or Cts2 virus at an MOI of 3 for 8, 16, and 24 h. Antibodies specific for I3, A11, F17, and L4 were used as indicated. Data were obtained from three independent experiments, and a representative blot is shown. The numbers to the right of the gel are molecular masses (in kilodaltons).
Next, we monitored the expression of the three late proteins A11, F17, and L4 in U2OS cells. During infection at the permissive temperature of 32°C, all three late proteins accumulated to comparable levels at 16 or 24 hpi in cells infected with either the WT or Cts2 virus (Fig. 6B). Similar observations could also be seen during WT infection performed at the nonpermissive temperature of 40°C (Fig. 6B). In contrast, in Cts2 virus-infected U2OS cells, the level of A11 protein was reduced at 16 and 24 hpi compared to that in cells infected with the WT (Fig. 6B). Not all late proteins exhibited diminished expression, though, as the levels of both F17 and L4 were similar between WT- and Cts2 virus-infected cells. Interestingly, the proteolytic processing of the L4 protein appeared to be incomplete in Cts2 virus-infected cells at both 16 and 24 hpi (Fig. 6B, lanes 5 and 6). Specifically, despite the similar levels of accumulation of the 28-kDa L4 precursor protein in both WT- and Cts2 virus-infected cells, the L4 cleavage product of the 25-kDa species was predominant only in the WT-infected cells (Fig. 6B, lanes 2 and 3). In Cts2 virus-infected cells, an additional cleavage product of 26 kDa could also be observed to accumulate to a level similar to that of the 25-kDa species (Fig. 6B, lanes 5 and 6). Taken together, these results suggest that B1 is required for WT levels of A11 protein accumulation and the complete processing of the L4 protein in U2OS cells.
Cleavage of the L4 precursor protein, as well as other precursor proteins, occurs within the IVs and can be an indicator for the efficiency of assembly of nascent virions (39, 43). As such, the incomplete processing of precursor L4 during Cts2 virus infection suggests that B1 may be involved during the assembly of virus particles. In order to assess the contribution of B1 during viral morphogenesis more directly, we utilized the chemical agent rifampin (RIF) in our infection experiments. RIF targets the vaccinia virus late protein D13, which is a scaffolding protein required for determining the shape of spherical immature virus particles (44–47). RIF treatment leads to a synchronous arrest in morphogenesis as well as an accumulation of irregularly shaped precursor membranes that can acquire their normal morphology and eventually assemble into mature virions (MVs) when cells are released from RIF (48, 49). When combined with a temperature shift-up protocol, a release from RIF-mediated synchronization can reveal whether a temperature-sensitive mutation impacts vaccinia virus morphogenesis during or after immature virion assembly. Thus, if B1 plays a role only before IV formation, then releasing Cts2 virus-infected cells from RIF treatment will result in the recovery of a viral yield that is the same as that during WT infection. However, if a loss of recovery is observed following the RIF release of Cts2 virus-infected cells at the nonpermissive temperature, then B1 is likely needed during the later stages that proceed after the formation of IV particles.
To test these possibilities, cells were infected at 32°C with WT or Cts2 virus (MOI = 3) in the presence or absence of RIF. At 12 hpi, cells either remained untreated, were released from RIF, were released from RIF and shifted to 40°C for an additional 9 h, or released from RIF and treated with 50 μM the nucleoside analog 1-β-d-arabinofuranosylcytosine (araC). As expected, continuous RIF treatment resulted in minimal virus production in both WT and Cts2 virus infections (Fig. 7). When cells were released from RIF at 12 hpi, the viral yield significantly increased by 3 orders of magnitude compared to that for the cells maintained in RIF (Fig. 7). Similarly, WT-infected cells that were released from RIF and further shifted up to 40°C also yielded a significant increase in viral titer, although to a lesser degree (∼3-fold) than the cells released from RIF and maintained at 32°C (Fig. 7). This is in contrast to the findings for the Cts2 virus-infected cells that were released from RIF and further shifted up to 40°C. Under this condition, the titer of Cts2 virus only partially recovered (10-fold less) compared to that of WT virus recovered from the WT-infected cells treated identically (Fig. 7). The partial recovery of Cts2 mutant virus suggests that B1 is likely involved in both earlier and later steps of morphogenesis. Finally, in light of previous data demonstrating the importance of B1 in DNA replication, we examined whether inhibition of DNA replication late in infection would be sufficient to cause only a partial recovery of viral yield, as was found in Cts2 virus infections in the presence of RIF shifted to 40°C. In testing this, we found that WT-infected cells that were released from RIF and further treated with araC also recovered and produced viruses with yields similar to those produced by cells released from RIF (Fig. 7). Similar to these findings for the WT infection, Cts2 virus-infected cells also produced comparable viral titers after those two treatments (Fig. 7). These results further confirm that viral DNA replication in Cts2 virus-infected cells is completed by 12 hpi and that no new DNA synthesis is required for virus production.
FIG 7.
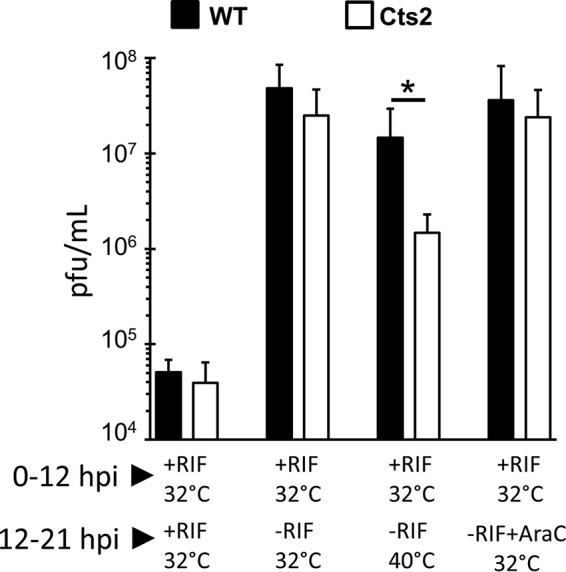
Rifampin release experiments indicate that Cts2 virus is impaired during virion morphogenesis. Cells were infected at an MOI of 3 at 32°C for 12 h in the presence or absence of 100 μg/ml RIF, as indicated. At 12 hpi, one set of cells was released from RIF by supplementing the cells with fresh medium without RIF, a second set of cells was placed at 40°C as well as released from RIF, and a third set was released from RIF but further supplemented with 50 μM araC. After 21 hpi, infected cells were harvested and titrated on BSC40 cells. The data were obtained from three independent experiments, and error bars represent standard deviations, *, P < 0.05 from unpaired t-test analysis.
Immature virions that are devoid of nucleoid structures accumulate in Cts2 virus-infected U2OS cells.
To determine the stage(s) within morphogenesis that is affected by the loss of B1, TEM analysis was performed with WT- or Cts2 virus-infected cells. U2OS cells infected (MOI = 5) at 40°C were fixed at 19 to 20 hpi. In WT-infected cells, all stages of virion morphogenesis—crescents (Cs), IVs, immature virions with nucleoids (IVNs), and MVs—could be observed (Fig. 8A). In Cts2 virus-infected cells, virosomes (Vs), Cs, and IV could readily be observed (Fig. 8B to D). However, only a very few IVNs and MVs were present (Fig. 8B to D). Dense spherical and dense half-filled particles could also be observed (Fig. 8C and D). In addition, some Cts2 virus IV particles appeared to have double membranes (Fig. 8C), a characteristic also observed in other studies in which viral morphogenesis was blocked (50, 51). Together, these results suggest that the initial steps of viral particle assembly are not affected by the lack of the B1 protein; however, they indicate that the B1 protein is needed for the formation of IVNs in U2OS cells.
FIG 8.
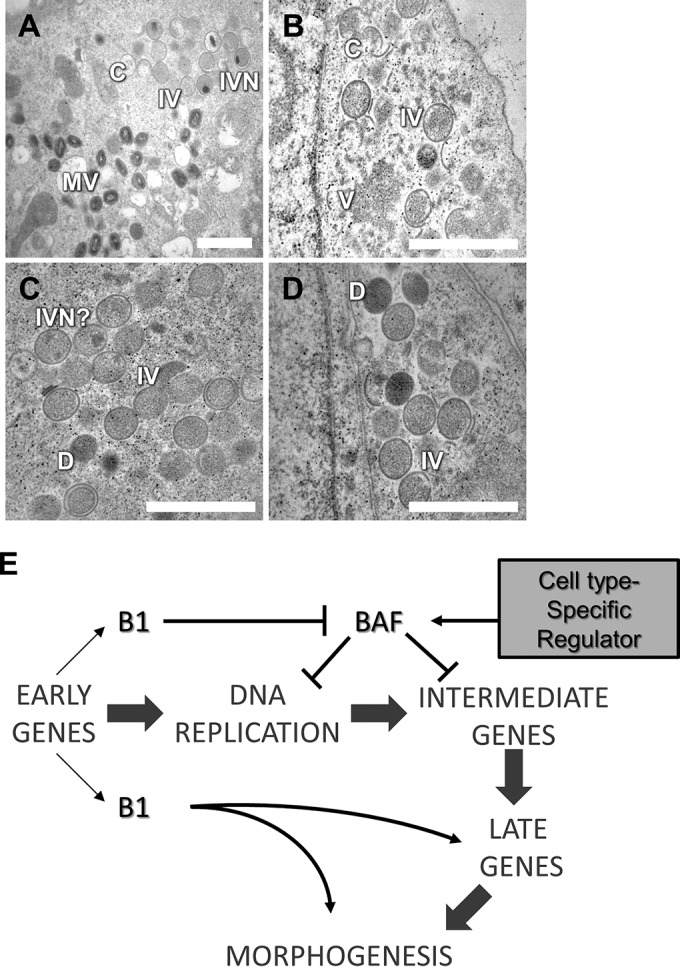
The accumulation of immature virions devoid of nucleoid can be observed in Cts2 virus-infected U2OS cells. (A to D) Representative TEM images from WT virus-infected (A) and Cts2 virus-infected (B to D) U2OS cells. Magnifications, ×8,000 (bar = 2 μm) (A) and ×15,000 (bars = 1 μm) (B to D). C, crescent; IV, immature virion; IVN, immature virion with nucleoid; MV, mature virion; D, dense virions. (E) Working model of the involvement of B1 at various stages in the vaccinia virus life cycle.
DISCUSSION
Conditional lethal mutants are invaluable tools for the dissection of gene function during viral infection. Examination of the life cycle of a mutant under nonpermissive conditions can reveal the initial execution point for a mutant, thus uncovering the first stage at which a gene is needed during infection. In the case of the B1-deficient Cts2 virus, execution point analysis demonstrated a critical role for the B1 kinase during viral DNA replication. The importance of B1 at this stage has been demonstrated in mouse L929 cells, nonhuman primate CV1 and BSC40 cells, and human 293 HEK and THP1 cells (16, 24, 29) (data not shown) and has been directly linked to the ability of B1 to phosphorylate and inactivate the host antiviral effector BAF, which can otherwise bind viral DNA and repress DNA replication (24, 32). While the discovery of the B1-BAF axis provided insight into the role of B1 during DNA replication, herein we address two complementary questions: is this axis critical in every cell type, and, if not, does B1 play other roles during the viral life cycle in those cells.
In this study, we found that in U2OS cells, the Cts2 virus exhibits a temperature-dependent decrease in viral progeny but, interestingly, no detectable impediment during DNA replication. Subsequent experiments demonstrated that while late gene expression is completely absent during Cts2 virus infection of L929 cells or BSC40 cells, expression of late proteins during Cts2 virus infection in U2OS cells was robust in the case of the F17 or L4 protein or only moderately decreased in the case of the A11 protein. Of note, the migration of L4 during Cts2 virus infection was altered compared to that of L4 expressed during WT virus infection. Changes in L4 migration have previously been linked to the cleavage of this protein by the I7 protease, which is a hallmark processing event known to occur during virion assembly and maturation (43, 52). We therefore compared WT- and Cts2 virus-infected U2OS cells by TEM, to determine whether any differences in the intermediate forms that arise during morphogenesis of new viral particles could be found. All stages of virion assembly were observed during WT infection, and in Cts2 virus-infected cells, viral membrane crescents and immature virions were abundant. However, IVN particles were much less common and mature virions were quite rare compared to the amounts in which they were present in WT-infected cells. These data add to a growing body of evidence that after DNA replication is completed, B1 continues to play a vital function later in the vaccinia virus life cycle.
A postreplicative role for B1 has been postulated for more than 20 years but has been difficult to examine because of the impact that B1 has on DNA replication. The suggestion that B1 plays a role later in the vaccinia virus life cycle was first based on the astute observation that the decrease in Cts2 viral progeny in BSC40 cells is more severe than the defect in DNA replication (16). Evidence that B1 has a long half-life also supported the possibility that B1 could act at late times of infection (17). Later, by using plasmid promoter/reporter constructs to study gene expression independently of viral DNA replication, it was demonstrated that B1 is needed for optimal intermediate gene transcription (53). More recent studies revealed that B1 is needed to inactivate the host DNA-binding protein BAF, which can otherwise bind viral or plasmid DNA in the cytoplasm, thus impeding replication and/or transcription from that DNA (20, 29, 32). The fact that U2OS cells are permissive for Cts2 viral DNA replication suggests that the intrinsic immune capability of BAF to block DNA replication is inactive in these cells. Furthermore, our observation that the depletion of BAF has no significant impact on Cts2 viral yield suggests that BAF does not affect a later stage in the viral life cycle either, leading us to conclude that B1 is probably needed to phosphorylate another substrate, as summarized in the model in Fig. 8E.
Why BAF is not acting as an antiviral effector in U2OS cells is an intriguing question. In attempting to answer it, we found that BAF is indeed endogenously expressed in U2OS cells at levels similar if not identical to those found in L929 and BSC40 cells (20), where it can act as a potent repressor of poxviral DNA replication. Subcellular fractionation assays also indicate that endogenous BAF is present in the U2OS cell cytoplasm at levels similar to those in these other cell types (data not shown) and U2OS-BAF cells do not exhibit a basal level of phosphorylation greater than that in the other cell types (Fig. 3). Furthermore, DNA sequencing analysis of BAF cDNA from U2OS cells did not uncover any mutations within the endogenous BAF mRNA that might render the protein nonfunctional (A. Jamin and M. S. Wiebe, unpublished observations). It also remains a formal possibility that the Cts2 virus mutant B1 protein exhibits residual activity at a high temperature in U2OS cells and is able to contribute to BAF inactivation. However, the fact that an increase in BAF phosphorylation is not observed by Western blot analysis of Cts2 virus-infected U2OS cell lysates (24) (data not shown) argues against this possibility. In sum, these data suggest that an upstream regulator of BAF that is unique in comparison with other previously characterized regulatory mechanisms, such as localization and phosphorylation, exists. It is plausible that this regulatory activity could be either a repressor or an activator of BAF present in Cts2 virus-permissive or -nonpermissive cells, respectively.
Our cell fusion experiments provide some important clues regarding the nature of this regulatory activity. Specifically, our observation that 97% of L929-U2OS heterokaryons are devoid of viral DNA replication factories following Cts2 virus infection supports the conclusion that the restrictive phenotype of the L929 cells is strongly dominant. This result likely indicates that L929 cell BAF remains fully active in fused cells and provides an antiviral defense throughout the mixed cytoplasm of the two cells. However, it is also possible that the U2OS cell BAF becomes activated following fusion through either the action of an L929 cell-specific activator or the dilution of a U2OS cell-specific repressor of BAF. Indeed, the possibility that a repressive factor exists in U2OS cells at limiting amounts would be consistent with the fact that overexpression of BAF can make U2OS cells nonpermissive, perhaps because the overexpressed BAF outcompetes the repressor. Future studies will be needed to discriminate between these models and are likely to reveal new insights into the U2OS-specific regulation of BAF's antiviral function.
It is interesting to note that this is not the first evidence that U2OS cells may have repressed intrinsic immune effectors and can thus support the replication of mutant viruses. For example, this cell type is permissive for the replication of herpes simplex virus 1 (HSV-1) lacking the ICP0 protein (HSV-1 ΔICP0) (54). HSV-1 ICP0 is hypothesized to stimulate viral gene expression partly via inactivation of host defense proteins involved in silencing of viral gene expression in the nucleus (55). While it is unknown exactly why these cells support HSV-1 ΔICP0 growth (54), U2OS cells have served as a useful tool for the dissection of host signaling pathways that act during HSV-1 infection, as they do now for our vaccinia virus studies.
In addition to providing new insights regarding the cell type-specific regulation of BAF, the data presented herein also demonstrate that in U2OS cells B1 plays an important role in the later stages of the viral life cycle. For example, the decreased level of the A11 protein that we observed in Cts2 virus-infected cells may be an indicator that B1 is needed for full expression of the gene for this protein and perhaps the genes for other late proteins. Alternatively, it may be that the stability of the A11 protein is diminished by the lack of B1, similar to the loss of A11 stability observed during infection with mutant viruses lacking the L2 protein (56). Whatever the exact reason for the low levels of A11 in Cts2 virus-infected cells is, it is unlikely that decreased A11 levels alone are sufficient for the block that we observed during morphogenesis. We base this conclusion on the findings of previous studies demonstrating that during infection with a virus lacking A11, membrane crescent formation is blocked and large areas of unencapsidated virosomal material can be found (34, 35). That A11-specific phenotype is in contrast to the findings of our TEM examination of Cts2 virus-infected cells, where, despite the presence of crescents, large areas of virosome were not seen and little IVN maturation from the IV could be observed. This same block at the IV-to-IVN transition was apparent in Cts2 virus-infected U2OS cells depleted of BAF (data not shown), further indicating that BAF is not causing this block.
To determine whether the Cts2 virus block in morphogenesis that we observed by TEM was the indirect result of B1's contribution at an earlier stage in the viral life cycle or could be due to a direct involvement of B1 during virion assembly, we employed a temperature shift and rifampin release protocol during viral yield studies. This approach allows premorphogenesis events to proceed at the permissive temperature in the presence of RIF. This was then followed by a synchronous shift to a high temperature and release from the RIF-mediated block of crescent formation. As virion assembly resumed efficiently once RIF was removed, the incomplete rescue of viral yield provides evidence of a later role for a gene, as revealed for F10, H5, A30, and G7 (15, 48, 57). When applying this method to the Cts2 virus, we observed only a partial rescue of viral yield compared to that in WT-infected cells. This reduction in viral yield is not likely due to a block in Cts2 viral DNA replication after the temperature shift because RIF-treated cells that were released from RIF and then treated with araC exhibited no decrease in viral yield. Thus, we conclude that B1 may act at multiple stages in the viral life cycle during both late gene expression and virion assembly. Future studies will be needed to elucidate whether this role for B1 late in infection is critical only in a subset of cell types, such as U2OS cells, or if it is of broad importance.
Although it remains to be directly tested, we speculate that the catalytic activity of B1 is likely required for its postreplicative role during infection. Little is known about whether late proteins can be targeted by B1, although in vitro studies suggest that B1 cannot phosphorylate G7, a viral protein needed for IV formation (57). One intriguing candidate to consider is the viral H5 protein, which has previously been identified to be a B1 substrate both in vitro and in infected cells (58–60). H5 is a constitutively expressed protein with roles in DNA replication, postreplicative gene expression, as well as virion assembly (15, 61, 62). H5 is also known to be dynamically phosphorylated by both viral and cellular kinases, although how this modification regulates H5 function is poorly understood (2, 60). Regarding the role of H5 during morphogenesis, it is interesting that TEM studies of H5 temperature-sensitive mutants revealed a role for this protein during the IV-to-IVN transition (15, 61), just as we now show for Cts2 virus. Future experiments to explore how the phosphorylation of H5 or other B1 substrates contributes to morphogenesis will be of significant interest. Such studies are now possible with this cell system and will provide valuable insights into how posttranslation modification controls poxvirus assembly.
ACKNOWLEDGMENTS
This research was supported through NIH grants to M.S.W. (R56AI099062) and the Nebraska Center for Virology (P30GM103509), which supported certain aspects of these studies, in particular, the Morrison Microscopy Core Facility at UNL.
The contents of this report are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
We thank Paula Traktman (Medical College of Wisconsin) for her gift of the antibodies against vaccinia virus proteins A11, F17, and L4. We also thank Clinton Jones and Jason Mercer for helpful discussions and Han Chen for his help on the TEM images.
REFERENCES
- 1.Bernard Moss. 2001. Poxviridae: the viruses and their replication, p 2849–2883. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res 66:31–124. doi: 10.1016/S0065-3527(06)66002-8. [DOI] [PubMed] [Google Scholar]
- 3.McDonald WF, Klemperer N, Traktman P. 1997. Characterization of a processive form of the vaccinia virus DNA polymerase. Virology 234:168–175. doi: 10.1006/viro.1997.8639. [DOI] [PubMed] [Google Scholar]
- 4.Sridhar P, Condit RC. 1983. Selection for temperature-sensitive mutations in specific vaccinia virus genes: isolation and characterization of a virus mutant which encodes a phosphonoacetic acid-resistant, temperature-sensitive DNA polymerase. Virology 128:444–457. doi: 10.1016/0042-6822(83)90269-6. [DOI] [PubMed] [Google Scholar]
- 5.Taddie JA, Traktman P. 1991. Genetic characterization of the vaccinia virus DNA polymerase: identification of point mutations conferring altered drug sensitivities and reduced fidelity. J Virol 65:869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Traktman P, Anderson MK, Rempel RE. 1989. Vaccinia virus encodes an essential gene with strong homology to protein kinases. J Biol Chem 264:21458–21461. [PubMed] [Google Scholar]
- 7.Punjabi A, Boyle K, DeMasi J, Grubisha O, Unger B, Khanna M, Traktman P. 2001. Clustered charge-to-alanine mutagenesis of the vaccinia virus A20 gene: temperature-sensitive mutants have a DNA-minus phenotype and are defective in the production of processive DNA polymerase activity. J Virol 75:12308–12318. doi: 10.1128/JVI.75.24.12308-12318.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stanitsa ES, Arps L, Traktman P. 2006. Vaccinia virus uracil DNA glycosylase interacts with the A20 protein to form a heterodimeric processivity factor for the viral DNA polymerase. J Biol Chem 281:3439–3451. doi: 10.1074/jbc.M511239200. [DOI] [PubMed] [Google Scholar]
- 9.Ishii K, Moss B. 2001. Role of vaccinia virus A20R protein in DNA replication: construction and characterization of temperature-sensitive mutants. J Virol 75:1656–1663. doi: 10.1128/JVI.75.4.1656-1663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greseth MD, Boyle KA, Bluma MS, Unger B, Wiebe MS, Soares-Martins JA, Wickramasekera NT, Wahlberg J, Traktman P. 2012. Molecular genetic and biochemical characterization of the vaccinia virus I3 protein, the replicative single-stranded DNA binding protein. J Virol 86:6197–6209. doi: 10.1128/JVI.00206-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rochester SC, Traktman P. 1998. Characterization of the single-stranded DNA binding protein encoded by the vaccinia virus I3 gene. J Virol 72:2917–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans E, Traktman P. 1992. Characterization of vaccinia virus DNA replication mutants with lesions in the D5 gene. Chromosoma 102:S72–S82. doi: 10.1007/BF02451789. [DOI] [PubMed] [Google Scholar]
- 13.Evans E, Klemperer N, Ghosh R, Traktman P. 1995. The vaccinia virus D5 protein, which is required for DNA replication, is a nucleic acid-independent nucleoside triphosphatase. J Virol 69:5353–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyle KA, Arps L, Traktman P. 2007. Biochemical and genetic analysis of the vaccinia virus d5 protein: multimerization-dependent ATPase activity is required to support viral DNA replication. J Virol 81:844–859. doi: 10.1128/JVI.02217-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D'Costa SM, Bainbridge TW, Kato SE, Prins C, Kelley K, Condit RC. 2010. Vaccinia H5 is a multifunctional protein involved in viral DNA replication, postreplicative gene transcription, and virion morphogenesis. Virology 401:49–60. doi: 10.1016/j.virol.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rempel RE, Anderson MK, Evans E, Traktman P. 1990. Temperature-sensitive vaccinia virus mutants identify a gene with an essential role in viral replication. J Virol 64:574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rempel RE, Traktman P. 1992. Vaccinia virus B1 kinase: phenotypic analysis of temperature-sensitive mutants and enzymatic characterization of recombinant proteins. J Virol 66:4413–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin S, Chen W, Broyles SS. 1992. The vaccinia virus B1R gene product is a serine/threonine protein kinase. J Virol 66:2717–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senkevich TG, Koonin EV, Bugert JJ, Darai G, Moss B. 1997. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology 233:19–42. doi: 10.1006/viro.1997.8607. [DOI] [PubMed] [Google Scholar]
- 20.Ibrahim N, Wicklund A, Jamin A, Wiebe MS. 2013. Barrier to autointegration factor (BAF) inhibits vaccinia virus intermediate transcription in the absence of the viral B1 kinase. Virology 444:363–373. doi: 10.1016/j.virol.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nezu J, Oku A, Jones MH, Shimane M. 1997. Identification of two novel human putative serine/threonine kinases, VRK1 and VRK2, with structural similarity to vaccinia virus B1R kinase. Genomics 45:327–331. doi: 10.1006/geno.1997.4938. [DOI] [PubMed] [Google Scholar]
- 22.Nichols RJ, Traktman P. 2004. Characterization of three paralogous members of the mammalian vaccinia related kinase family. J Biol Chem 279:7934–7946. doi: 10.1074/jbc.M310813200. [DOI] [PubMed] [Google Scholar]
- 23.Boyle KA, Traktman P. 2004. Members of a novel family of mammalian protein kinases complement the DNA-negative phenotype of a vaccinia virus ts mutant defective in the B1 kinase. J Virol 78:1992–2005. doi: 10.1128/JVI.78.4.1992-2005.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiebe MS, Traktman P. 2007. Poxviral B1 kinase overcomes barrier to autointegration factor, a host defense against virus replication. Cell Host Microbe 1:187–197. doi: 10.1016/j.chom.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox JL, Mallanna SK, Ormsbee BD, Desler M, Wiebe MS, Rizzino A. 2011. Banf1 is required to maintain the self-renewal of both mouse and human embryonic stem cells. J Cell Sci 124:2654–2665. doi: 10.1242/jcs.083238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furukawa K, Sugiyama S, Osouda S, Goto H, Inagaki M, Horigome T, Omata S, McConnell M, Fisher PA, Nishida Y. 2003. Barrier-to-autointegration factor plays crucial roles in cell cycle progression and nuclear organization in Drosophila. J Cell Sci 116:3811–3823. doi: 10.1242/jcs.00682. [DOI] [PubMed] [Google Scholar]
- 27.Margalit A, Neufeld E, Feinstein N, Wilson KL, Podbilewicz B, Gruenbaum Y. 2007. Barrier to autointegration factor blocks premature cell fusion and maintains adult muscle integrity in C. elegans. J Cell Biol 178:661–673. doi: 10.1083/jcb.200704049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puente XS, Quesada V, Osorio FG, Cabanillas R, Cadinanos J, Fraile JM, Ordonez GR, Puente DA, Gutierrez-Fernandez A, Fanjul-Fernandez M, Levy N, Freije JM, Lopez-Otin C. 2011. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet 88:650–656. doi: 10.1016/j.ajhg.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ibrahim N, Wicklund A, Wiebe MS. 2011. Molecular characterization of the host defense activity of the barrier to autointegration factor against vaccinia virus. J Virol 85:11588–11600. doi: 10.1128/JVI.00641-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Condit RC, Motyczka A, Spizz G. 1983. Isolation, characterization, and physical mapping of temperature-sensitive mutants of vaccinia virus. Virology 128:429–443. doi: 10.1016/0042-6822(83)90268-4. [DOI] [PubMed] [Google Scholar]
- 31.Tscharke DC, Karupiah G, Zhou J, Palmore T, Irvine KR, Haeryfar SM, Williams S, Sidney J, Sette A, Bennink JR, Yewdell JW. 2005. Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J Exp Med 201:95–104. doi: 10.1084/jem.20041912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jamin A, Wicklund A, Wiebe MS. 2014. Cell- and virus-mediated regulation of the barrier-to-autointegration factor's phosphorylation state controls its DNA binding, dimerization, subcellular localization, and antipoxviral activity. J Virol 88:5342–5355. doi: 10.1128/JVI.00427-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tolonen N, Doglio L, Schleich S, Krijnse Locker J. 2001. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol Biol Cell 12:2031–2046. doi: 10.1091/mbc.12.7.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Resch W, Weisberg AS, Moss B. 2005. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J Virol 79:6598–6609. doi: 10.1128/JVI.79.11.6598-6609.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maruri-Avidal L, Weisberg AS, Moss B. 2013. Association of the vaccinia virus A11 protein with the endoplasmic reticulum and crescent precursors of immature virions. J Virol 87:10195–10206. doi: 10.1128/JVI.01601-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu X, Meng X, Yan B, Rose L, Deng J, Xiang Y. 2012. Vaccinia virus virion membrane biogenesis protein A11 associates with viral membranes in a manner that requires the expression of another membrane biogenesis protein, A6. J Virol 86:11276–11286. doi: 10.1128/JVI.01502-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang YF, Moss B. 1991. Vaccinia virus morphogenesis is interrupted when expression of the gene encoding an 11-kilodalton phosphorylated protein is prevented by the Escherichia coli lac repressor. J Virol 65:6101–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wickramasekera NT, Traktman P. 2010. Structure/function analysis of the vaccinia virus F18 phosphoprotein, an abundant core component required for virion maturation and infectivity. J Virol 84:6846–6860. doi: 10.1128/JVI.00399-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.VanSlyke JK, Franke CA, Hruby DE. 1991. Proteolytic maturation of vaccinia virus core proteins: identification of a conserved motif at the N termini of the 4b and 25K virion proteins. J Gen Virol 72(Pt 2):411–416. doi: 10.1099/0022-1317-72-2-411. [DOI] [PubMed] [Google Scholar]
- 40.Wilcock D, Smith GL. 1994. Vaccinia virus core protein VP8 is required for virus infectivity, but not for core protein processing or for INV and EEV formation. Virology 202:294–304. doi: 10.1006/viro.1994.1346. [DOI] [PubMed] [Google Scholar]
- 41.Wilcock D, Smith GL. 1996. Vaccinia virions lacking core protein VP8 are deficient in early transcription. J Virol 70:934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jesus DM, Moussatche N, Condit R. 2014. Vaccinia virus mutations in the L4R gene encoding a virion structural protein produce abnormal mature particles lacking a nucleocapsid. J Virol 88:14017–14029. doi: 10.1128/JVI.02126-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ansarah-Sobrinho C, Moss B. 2004. Role of the I7 protein in proteolytic processing of vaccinia virus membrane and core components. J Virol 78:6335–6343. doi: 10.1128/JVI.78.12.6335-6343.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baldick CJ Jr, Moss B. 1987. Resistance of vaccinia virus to rifampicin conferred by a single nucleotide substitution near the predicted NH2 terminus of a gene encoding an Mr 62,000 polypeptide. Virology 156:138–145. doi: 10.1016/0042-6822(87)90444-2. [DOI] [PubMed] [Google Scholar]
- 45.Bahar MW, Graham SC, Stuart DI, Grimes JM. 2011. Insights into the evolution of a complex virus from the crystal structure of vaccinia virus D13. Structure 19:1011–1020. doi: 10.1016/j.str.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McNulty-Kowalczyk A, Paoletti E. 1993. Mutations in ORF D13L and other genetic loci alter the rifampicin phenotype of vaccinia virus. Virology 194:638–646. doi: 10.1006/viro.1993.1303. [DOI] [PubMed] [Google Scholar]
- 47.Tartaglia J, Piccini A, Paoletti E. 1986. Vaccinia virus rifampicin-resistance locus specifies a late 63,000 Da gene product. Virology 150:45–54. doi: 10.1016/0042-6822(86)90264-3. [DOI] [PubMed] [Google Scholar]
- 48.Punjabi A, Traktman P. 2005. Cell biological and functional characterization of the vaccinia virus F10 kinase: implications for the mechanism of virion morphogenesis. J Virol 79:2171–2190. doi: 10.1128/JVI.79.4.2171-2190.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sodeik B, Griffiths G, Ericsson M, Moss B, Doms RW. 1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J Virol 68:1103–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jesus DM, Costa LT, Goncalves DL, Achete CA, Attias M, Moussatche N, Damaso CR. 2009. Cidofovir inhibits genome encapsidation and affects morphogenesis during the replication of vaccinia virus. J Virol 83:11477–11490. doi: 10.1128/JVI.01061-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiu WL, Chang W. 2002. Vaccinia virus J1R protein: a viral membrane protein that is essential for virion morphogenesis. J Virol 76:9575–9587. doi: 10.1128/JVI.76.19.9575-9587.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Byrd CM, Bolken TC, Hruby DE. 2002. The vaccinia virus I7L gene product is the core protein proteinase. J Virol 76:8973–8976. doi: 10.1128/JVI.76.17.8973-8976.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kovacs GR, Vasilakis N, Moss B. 2001. Regulation of viral intermediate gene expression by the vaccinia virus B1 protein kinase. J Virol 75:4048–4055. doi: 10.1128/JVI.75.9.4048-4055.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao F, Schaffer PA. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J Virol 69:6249–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hancock MH, Corcoran JA, Smiley JR. 2006. Herpes simplex virus regulatory proteins VP16 and ICP0 counteract an innate intranuclear barrier to viral gene expression. Virology 352:237–252. doi: 10.1016/j.virol.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 56.Maruri-Avidal L, Weisberg AS, Moss B. 2011. Vaccinia virus L2 protein associates with the endoplasmic reticulum near the growing edge of crescent precursors of immature virions and stabilizes a subset of viral membrane proteins. J Virol 85:12431–12441. doi: 10.1128/JVI.05573-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mercer J, Traktman P. 2005. Genetic and cell biological characterization of the vaccinia virus A30 and G7 phosphoproteins. J Virol 79:7146–7161. doi: 10.1128/JVI.79.11.7146-7161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beaud G, Beaud R. 2000. Temperature-dependent phosphorylation state of the H5R protein synthesised at the early stage of infection in cells infected with vaccinia virus ts mutants of the B1R and F10L protein kinases. Intervirology 43:67–70. doi: 10.1159/000025025. [DOI] [PubMed] [Google Scholar]
- 59.Beaud G, Sharif A, Topa-Massé A, Leader DP. 1994. Ribosomal protein S2/Sa kinase purified from HeLa cells infected with vaccinia virus corresponds to the B1R protein kinase and phosphorylates in vitro the viral ssDNA-binding protein. J Gen Virol 75:283–293. doi: 10.1099/0022-1317-75-2-283. [DOI] [PubMed] [Google Scholar]
- 60.Beaud G, Beaud R, Leader DP. 1995. Vaccinia virus gene H5R encodes a protein that is phosphorylated by the multisubstrate vaccinia virus B1R protein kinase. J Virol 69:1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeMasi J, Traktman P. 2000. Clustered charge-to-alanine mutagenesis of the vaccinia virus H5 gene: isolation of a dominant, temperature-sensitive mutant with a profound defect in morphogenesis. J Virol 74:2393–2405. doi: 10.1128/JVI.74.5.2393-2405.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boyle KA, Greseth MD, Traktman P. 2015. Genetic confirmation that the H5 protein is required for vaccinia virus DNA replication. J Virol 89:6312–6327. doi: 10.1128/JVI.00445-15. [DOI] [PMC free article] [PubMed] [Google Scholar]