

ABSTRACT
Cell-to-cell transmission is an efficient mechanism to disseminate human immunodeficiency virus type 1 (HIV-1) and human T cell lymphotropic virus type 1 (HTLV-1). However, it has been challenging to quantify the level of cell-to-cell transmission because the virus-producing cells cannot be easily distinguished from infected target cells. We have previously described replication-dependent vectors that can quantify infection events in cocultured cells. These vectors contain an antisense-oriented promoter and reporter gene interrupted by a sense-oriented intron from the human gamma-globin gene. This strategy prevents expression of the reporter gene in the transfected cells but permits its expression in target cells after infection. However, the gamma-globin intron is not efficiently removed by splicing in the aforementioned vectors, thereby reducing the level of reporter gene expression after transduction into target cells. Here, we used two approaches to improve the replication-dependent vectors. First, we improved the splicing events that remove the gamma-globin intron by optimizing the intron insertion site within the reporter gene. Second, we improved the packaging of the spliced RNA without the gamma-globin intron by targeting the intron-containing RNA via microRNA 30 (miR30)-based short hairpin RNAs. Using two optimized fluorescent reporter vectors and flow cytometry, we determined that multiply HIV-1-infected cells were generated at a higher frequency in coculture than in cell-free infection; furthermore, this increase was dependent upon viruses bearing HIV-1 Env. Compared with previously described vectors, these improved vectors can quantify the infection in lymphocytes and in primary cells with a higher sensitivity and allow the detection and quantitation of multiply infected cells, providing better tools to study retroviral cell-mediated infection.
IMPORTANCE The human-pathogenic retroviruses HTLV-1 and HIV-1 can be transmitted more efficiently in vivo via direct contact of infected cells with healthy target cells than through cell-free virion-mediated infection. Despite its importance, cell-to-cell transmission has been difficult to quantify because the previously infected cells and the newly infected cells are mixed together in the same culture. In the current study, we generated vectors that are significantly improved over the previously described replication-dependent vectors. As a result, these improved vectors can efficiently detect and quantify cell-to-cell transmission or new infection events in cells in mixed culture. These luciferase- or fluorescence protein-based reporter vectors can be used to quantify and study HIV-1 or HTLV-1 cell-mediated infection in a simple one-step transfection/infection assay.
INTRODUCTION
The human-pathogenic retroviruses human T cell lymphotropic virus type 1 (HTLV-1) and human immunodeficiency virus type 1 (HIV-1) can infect target cells either by cell-free virions or by cell-to-cell transmission. In the latter case, viruses are transferred via direct contact of an infected cell with a permissive target cell. Microscopy studies revealed that several types of cell contacts that lead to the transfer of virus particles from infected cells to target cells can be formed. Contacts such as virological synapses (VSs) (1, 2), filopodia (3, 4), biofilm-like structures (5), and polysynapses (6) have been described for different retroviruses. Although cell-to-cell infection is a necessary step for the spread of some pathogens, not all virus transfer events result in the infection of a target cell. For example, nascent virus-like particles (VLPs) without Env can enter target cells via a VS (7; our unpublished data), or exosomes containing viral RNA can be released from HIV-infected cells and transferred to nonpermissive cells (8). These observations raise the question of how often viral synapse formation leads to productive cell-to-cell infection.
Cell-to-cell infection is inherently difficult to quantify. The major challenge is that the virus producer cells and the newly infected target cells cannot be easily distinguished in mixed cell cultures. To solve this issue, we have previously developed reporter vectors based on firefly luciferase (Luc) and yellow fluorescent protein (YFP) to quantify the levels of HIV-1 and HTLV-1 infections in cell cocultures (9). The concept for the design of these vectors was adopted from a retrotransposon detection assay used to study mobile elements (10–12). Specifically, the entire reporter expression cassette, including a promoter and reporter gene, is placed in the antisense orientation relative to the orientation of the viral transcripts so that the reporter gene cannot be expressed from the U3 promoter (Fig. 1). Furthermore, the coding sequence of the reporter gene is disrupted by an intron from the human gamma-globin gene (referred to as the intron in the remaining text) that is placed in the sense orientation. This intron prevents the expression of the functional reporter protein from internal promoter-driven mRNA. However, during the expression of vector RNA from the U3 promoter in virus producer cells, the intron can be removed by splicing; when virus containing such a genome infects a new target cell, a provirus with an intact reporter gene can be generated, allowing the reporter protein to be expressed from the internal promoter. Thus, these vectors are replication dependent because viral replication is required to eliminate the inactivating intron and reconstitute the reporter gene, thereby allowing the reporter to be expressed.
FIG 1.
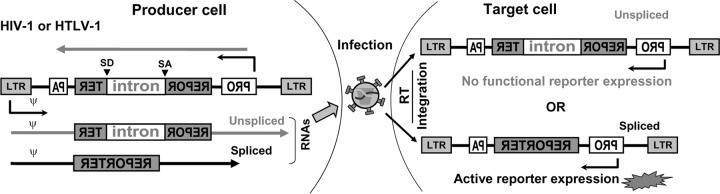
Strategy and design of the replication-dependent retroviral vectors. The reporter gene (REPOR and TER) is inserted in the antisense orientation relative to the orientation of the viral genome, and the coding sequence is disrupted by an intron. During the expression of the viral genome in the producer cell, the intron can be removed by splicing. Both unspliced RNA with the intron and spliced RNA without the intron can be packaged. When infecting the target cells, viral RNA can be reverse transcribed and integrated into the host chromosome to form proviruses. The CMV promoter (PRO) can express the transcript in the antisense orientation; PA, poly(A) sequences. If the provirus has an intron in the middle of the reporter gene, a functional reporter protein cannot be translated. In contrast, if the provirus is derived from the spliced RNA without the intron, the reporter gene can be expressed and detected.
One substantial disadvantage of such a design is that splicing of the gamma-globin intron is not required for viral RNA packaging or the infectivity of the virus. Hence, long terminal repeat (LTR)-driven reporter RNA containing the intron can be efficiently packaged into viral particles and transmitted during infection; however, cells infected by these viruses cannot express the reporter gene, thereby reducing the readout values and sensitivity of the assay. For example, in the previously described vectors, only a small portion of the transduced vectors expressed the reporter gene (9). Thus, inefficient removal of the inactivating intron is a major hindrance for these types of replication-dependent reporter vectors.
Here, we used two approaches to improve the efficiency of the replication-dependent reporter vectors. First, we improved the frequency of reporter gene reconstitution by optimizing the removal of the intron within the reporter gene. Second, we also reduced the packaging of the competing RNAs that retained the intron by enhancing their degradation. We analyzed the secondary RNA structure of different reporter genes and found a positive correlation between the numbers of unpaired nucleotides at the potential intron insertion sites and the levels of splicing that remove the intron in reporter genes. Guided by this observation, we constructed a green fluorescent protein (GFP) turbo-based intron-containing (inGFPt) vector and an mCherry-based intron-containing (inmCherry) vector with a high efficiency of reporter reconstitution. Additionally, using microRNA 30 (miR30)-based short hairpin RNAs (shRNAs) that target the intron, we specifically degraded the unspliced reporter RNA in the cytoplasm and significantly elevated the level of the spliced reporter RNA packaged into viral particles. Thus, these strategies, optimizing intron removal and degrading intron-retaining reporter RNA, can be applied to improve other types of intron-containing vectors. Using the improved inGFPt and inmCherry vectors, we quantified the levels of HIV-1 singly and doubly infected target cells in a suspension lymphoid cell system and demonstrate that multiply infected cells occur at a higher frequency in coculture than in cell-free virion-mediated infection in an HIV-1 Env-dependent manner.
MATERIALS AND METHODS
Cell cultures and reagents.
Human embryonic kidney 293T cells were obtained through the NIH AIDS Research and Reference Reagent Program. The human CD4 T cell line CCRF-CEM and human B cell line Raji were purchased from ATCC. Cells of the Raji/CD4 cell line were kindly provided by Vineet N. KewalRamani (NCI at Frederick). Peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated on a density gradient of Ficoll-Paque (Paneco, Russia), activated with 5 μg/ml of phytohemagglutinin (PHA; Sigma) for 3 days, and grown in the presence of 50 U/ml of recombinant human interleukin-2 (Ronkoleikin, Biotech, Russia). All experiments with the human blood samples were approved by the Human Ethics Committee of the Institute of Immunology (Moscow). All blood donors gave informed consent for the use of their samples in the described experiments. All cells in suspension were maintained in RPMI 1640 medium containing 10% fetal calf serum, 2 mM glutamine, penicillin (100 U/ml), and streptomycin (0.1 mg/ml). In addition, 400 μg/ml of Geneticin (Gibco) was added to the medium used for Raji/CD4 cells to select for CD4 expression. Human 293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum, 50 μM β-mercaptoethanol, 2 mM glutamine, penicillin (100 U/ml), and streptomycin (0.1 mg/ml). 3′-Azido-3′-deoxythymidine (AZT) and phorbol 12-myristate 13-acetate (PMA) were purchased from Sigma.
Plasmid construction.
All modifications of reporter vectors were derived from previously described HIV-1- and HTLV-1-based YFP intron-containing (inYFP) and Luc intron-containing (inLuc) vectors (9). To construct replication-dependent vectors that can express fluorescent proteins, we used GFP turbo (GFPt) and red fluorescent protein (RFP) turbo (RFPt) reporter genes (Eurogen, Russia) and an mCherry reporter gene (Clontech). All sites in the reporter genes containing the A(C)AG/N sequence, where N is any nucleotide, were analyzed using the Mfold RNA-folding web tool. The putative insertion sites with a minimal number of paired nucleotides were identified for further analyses in the context of the intron-containing RNA. The secondary structures of the intron-containing reporter RNAs were analyzed, and the site in the reporter gene where the splice donor (SD) and splice acceptor (SA) had a minimal amount of paired nucleotides was selected for intron cloning. The intron from the human gamma-globin gene and the 5′ and the 3′ portions of reporter genes flanking the intron were amplified and joined together using PCR. These PCR fragments were cloned into the pJet1.2 plasmid (Fermentas), and verified by DNA sequencing. Reporter genes containing the intron were first cloned into the pCRU5-inYFP plasmid via the MluI and BglII restriction sites and then transferred into the pUCHR-inYFP plasmid using the MluI and NheI sites.
miR30-based shRNAs targeting the gamma-globin intron at position 79 (GCAGGGTGTGAGCTGTTTGAA) or 330 (AGGACAAGTATGGTCATTAAA) were cloned into the pGIPZ vector (Open Biosystems); pGIPZ expresses GFP and the puromycin resistance gene (puro), the translation of which is facilitated by an internal ribosomal entry site (IRES) from encephalomyocarditis virus. Vector pGIPZ-P1 containing shRNA targeting phosphoprotein-1 (P1) of respiratory syncytial virus (RSV) has been described previously (13) and was used as a control. The DNA sequences of the miR30-based shRNAs were amplified from the pGIPZ plasmids by PCR with primers 5′-CGTCTAGATGTTTGAATGAGGCTTCAGTAC-3′ and 5′-CGTCTAGAAGTGATTTAATTTATACCATTTTAATTC-3′ and inserted into the intron at the XbaI site. Short hairpin RNAs without an miR30 sequence targeting the intron at position 30 (GCCTTTAGTCTCGAGGCAACT) or 326 (GCAGGACAAGTATGGTCATTA) and with a short loop (TTCAAGAGA) were inserted into the intron at the XbaI site as annealed oligonucleotides.
Transfections and infections.
Human 293T cells were transfected with the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. CEM cells and PHA-activated PBMCs were transfected using a Neon electroporation system (Invitrogen) by pulsing once at 1,230 V for 40 ms, whereas Raji cells were pulsed once at 1,350 V for 30 ms. A one-step transfection/infection assay was conducted in 293T cells using a 12-well plate. To initiate the HIV-1-based assay, cells were cotransfected with 0.6 μg of helper plasmid pCMVΔ8.2R (Addgene accession number 12263), which expresses all HIV-1 proteins except Env; 0.15 μg of the pCMV-VSV-G plasmid (Addgene accession number 8454), which expresses protein G from vesicular stomatitis virus (VSV-G); and 0.9 μg of reporter plasmid. To initiate the HTLV-1-based assay, 293T cells were cotransfected with 0.6 μg of the pCMV HT1-M plasmid, which expresses the full-length HTLV-1 genome (14, 15), and 0.9 μg of a reporter plasmid. The culture medium was replaced at 16 h posttransfection. Samples were harvested and analyzed by flow cytometry or by luminescence at 3 days posttransfection or at the times indicated below. For the luciferase assay, cells were lysed using GLO lysis buffer (Promega), and the luciferase activity was quantified using the Promega luciferase reagent and a GloMax-Multi Jr single-tube multimode reader (Promega).
The cell coculture infection assay was performed in lymphoid cells as described previously (9, 13). Briefly, 106 CEM cells were electroporated with 3 μg of pUCHR-inGFPt and/or 3 μg of pUCHR-inmCherry vector DNA and 2 μg of pCMVΔ8.2R plasmid DNA in the presence or absence of 0.8 μg of the pIIINL-4env plasmid, which expresses Env from HIV-1 strain NL4-3 (16). In some experiments, pCMV-VSV-G was used instead of pIIINL-4env. The cells were washed once with phosphate-buffered saline (PBS) at 5 to 6 h after electroporation and mixed with 5 × 105 Raji/CD4 target cells in 5 ml of culture medium; in this step, AZT was added at a final concentration of 20 μM to samples indicated to be treated with AZT. For cell-free infection, viruses generated from transfected 293T cells were clarified through a filter with a 0.45-μm pore size, concentrated by centrifugation at 100,000 × g for 1 h, and added to a mixture of mock-transfected 106 CEM cells and 5 × 105 Raji/CD4 cells at different dilutions.
To test HIV-1 infection with primary cells, 106 activated PBMCs were transfected with a reporter vector (pUCHR-inLuc or pUCHR-inLuc-mR-int330) along with helper plasmids and cocultured with 5 × 105 Raji/CD4 target cells. Alternatively, 106 Raji cells were transfected with the aforementioned plasmids and mixed with 106 activated PBMCs. Cells were stimulated with 20 nM PMA at 16 h prior to analysis to enhance reporter expression in the infected cells. Samples were analyzed by flow cytometry or by luminescence 72 h after electroporation or exposure to cell-free viruses.
RT-qPCR quantification of RNA splicing efficiency.
To determine the splicing efficiency, 293T cells (1.5 × 106 cells in a 6-cm-diameter dish) were cotransfected overnight with 2 μg of HIV-1 helper plasmid pCMVΔ8.2R or HTLV-1 helper plasmid pCMV HT1-M-ΔXho, each of which contains an inactivating deletion in the env gene (14), and 3 μg of a reporter vector plasmid; the medium was replaced on the following morning. Supernatants were harvested at 48 h posttransfection, centrifuged at 3,000 × g for 15 min to remove the cell debris, and clarified through 0.45-μm-pore-size filters. Viral particles were pelleted by centrifugation of the supernatants at 100,000 × g for 1 h, and virion RNA was isolated using a GeneJET RNA purification kit (Fermentas), followed by DNase I (New England Biolab) treatment. Reverse transcription (RT) with 6-nucleotide random primers and reagents obtained from Sibenzyme (Russia) was performed for 5 min at 25°C and then for 2 h at 37°C. Quantitative PCR (qPCR) was set up in triplicate with ready-for-use qPCR master mixes containing SYBR green I (Syntol, Russia), 10 μM each primer, and 50 to 100 ng of cDNA template. The primers used for amplification of unspliced RNA (intron) were 5′-TGGTGGCCAAACATACATTGC-3′ and 5′-TGTTCCTCACCCCTGGACAT-3′. The splice-specific primers were 5′-TGAAGCTGCCATCCAGATCGT-3′ and 5′-CGACTTCAAGGTGATGGGCA-3′ for inGFPt, 5′-ATTAACGCCCAGCGTTTTCC-3′ and 5′-ACACCCGAGGGGGATGATAA-3′ for inLuc, 5′-CTCGGGGAAGGACAGTTTCAA-3′ and 5′-AAGCTGAAGGTGACCAAGGG-3′ for inmCherry, and 5′-AGTTCACCCCGTTGATCTTG-3′ and 5′-ACACCCGAGGGGGATGATAA-3′ for inRFPt-2. PCR was performed using an iCycler iQ real-time PCR detection system (Bio-Rad) at the following settings: 1 cycle of denaturation at 95°C for 2 min and 45 cycles of amplification (95°C 15 s, 58°C 15 s, and 72°C 15 s). At the end of the reactions, the melting curve for each PCR product was obtained; data were collected and analyzed by using iCycler software. The levels of spliced and unspliced reporter RNAs in the viral particles were quantified for each transfer vector. The efficiency of RNA splicing was expressed as a ratio of the amount of spliced RNA relative to the total amount of RNA (spliced and unspliced) and was calculated by the formula [1/(1 + 2A − B)] · 100, where A is the threshold cycle (CT) for spliced RNA, and B is the CT for unspliced RNA.
Flow cytometry analysis and fluorescence microscopy.
The levels of cell-to-cell infection were determined from the level of expression of the fluorescent reporter gene using flow cytometry or fluorescence microscopy. For flow cytometry, 293T cells were trypsinized, resuspended in medium containing 10% fetal calf serum, washed once with PBS, and resuspended in PBS. Cells were analyzed using a FACSAria SORP instrument (Becton Dickinson). The following laser excitation, emission filter, and bypass filter parameters were used: for GFP turbo and enhanced YFP (eYFP), 488 nm, 515 nm, and 20 nm, respectively; for RFP turbo, 561 nm, 585 nm, and 15 nm, respectively; and for mCherry, 592 nm, 615 nm, and 24 nm, respectively. Flow cytometry results were collected and analyzed using BD FACSDiva and FlowJo software. The fluorescence images of cells were captured with an Olympus IX-71 inverted fluorescence microscope equipped with the following excitation and emission filter units: for GFP turbo and eYFP, 470 to 495 nm and 510 to 550 nm, respectively; for RFP turbo, 540 to 550 nm and 575 to 625 nm, respectively. Microscopy data were analyzed and presented using Olympus cellSens software.
Nucleotide sequence accession numbers.
The HIV-1- and HTLV-1-based inGFPt and inmCherry vectors that generated the best results have been deposited in Addgene under accession numbers 60236 to 60239.
RESULTS
Engineering replication-dependent vectors with improved reporter splicing.
We aimed to develop reporter vectors that can be used to quantify cell-mediated infection by flow cytometry. The previously described eYFP-expressing vector performed poorly; only a small portion of the cells containing this vector can express YFP (9). To improve the replication-dependent vectors, we generated vectors that contain either a GFP turbo gene or a red fluorescent protein gene. For this purpose, we scanned each of the reporter genes to identify potential splice sites containing the A(C)AG/N sequence, where N is any nucleotide, and randomly selected one site in the gfp gene and one site in the rfp gene to insert the intron from the human gamma-globin gene. We first generated two sets of constructs, inGFPt and RFP turbo intron-containing vector 1 (inRFPt-1), in HTLV-1- and HIV-1-based vectors and tested these constructs using a one-step transfection/infection assay. In this assay, 293T cells were cotransfected with one of the reporter vectors along with helper constructs, which supplied viral proteins, including the Env proteins, in trans. The RNA from the reporter vector can be packaged into viral particles that can infect new target cells. If the gamma-globin intron is removed by splicing in the packaged viral genome, the resulting provirus has an intact open reading frame in the reporter gene and can express fluorescent protein, which can be detected by fluorescence microscopy. Our preliminary results indicated drastic differences among the reporter vectors; the new inGFPt vector generated far more fluorescent protein-expressing cells than the inRFPt-1 vector or the previously published inYFP vector. Furthermore, the superior performance of the inGFPt vector was observed for both the HIV-1-based vector (Fig. 2A) and the HTLV-1-based vector (data not shown). These results indicate that the fluorescent protein genes in these vectors were not reconstituted at the same rate; one likely explanation is that the position in which the inactivating intron is inserted affects the efficiency of intron removal by splicing.
FIG 2.

Optimization of intron insertion within the reporter gene in replication-dependent vectors. (A) Transduction efficiencies of the previously described inYFP vector and the new reporter vectors. 293T cells were cotransfected with the HIV-1 reporter vector, packaging plasmid, and a plasmid carrying VSV-G. Images were captured on a fluorescence microscope 3 days later; representative images with equal densities of cells are shown. (B) Schematic representation of intron sequences embedded within various reporter genes. Previously published vectors are noted by asterisks after their names. Nucleotides predicted to be unpaired by the Mfold program are shown in red. (C and D) Kinetics of HIV-1 (C) and HTLV-1 (D) infection measured in 293T cells. Cells were cotransfected as indicated in the legend to panel A and analyzed by flow cytometry on the indicated day posttransfection. Results from at least three independent experiments are shown as averages ± standard deviations. (E and F) Proportion of spliced RNA in total vector RNA in viral particles derived from HIV-1 (E) and HTLV-1 (F). Human 293T cells were cotransfected with one of the reporter vectors and the corresponding env-negative packaging plasmid. Viral particles were harvested at 48 h posttransfection, and RNA samples were isolated. The levels of reporter RNAs with and without introns were quantified by RT-qPCR with intron-specific and spliced RNA-specific primers, respectively. The proportion of spliced RNA transcripts in total vector RNA was calculated; results summarized from at least three independent experiments are shown as averages ± standard deviations.
We then hypothesized that the RNA structures near the splice site can affect the accessibility of RNA for the spliceosome at the splice donor (SD) and splice acceptor (SA) sites, thereby influencing the splicing efficiency and the removal of the intron. Using the Mfold RNA-folding web tool, we analyzed the secondary structures of the RNAs for the tested reporter genes (Luc, YFP, GFPt, RFPt-1) at the sites where the introns were embedded first with reporter RNAs alone and then with reporter RNAs containing the intron sequence. When the sequence of inGFPt was compared with the sequences of inYFP and inRFPt-1, we found that there were more unpaired nucleotides around the SD and SA sites in the inGFPt vector (Fig. 2B) and this vector was more efficient in generating reporter-expressing cells (Fig. 2A). To generate an improved vector expressing red fluorescent protein, we took two approaches. First, we generated a second RFP construct (inRFPt-2) by inserting the intron into the best region of the gene, although this region was far from ideal, where the SD site of the intron had five unpaired nucleotides but the SA site did not contain any unpaired nucleotides (Fig. 2B). The resulting inRFPt-2 vector displayed a slight improvement over the inRFPt-1 vector (Fig. 2A). Second, we analyzed mCherry, a different gene that encodes a red fluorescent protein, by scanning for potential insertion sites using the RNA-folding approach described above. These analyses revealed a site that appeared to be ideal for intron insertion because it contained more than one unpaired nucleotide on both the SD and the SA sites (Fig. 2B). Additionally, placement of the gamma-globin intron at this position did not alter the RNA structure around the SD and SA sites, and both remained unfolded. Based on the findings of these analyses, we generated an inmCherry vector containing the gamma-globin intron. Preliminary studies showed that the mCherry generated from this vector was efficiently reconstituted (data not shown).
To better quantify fluorescent protein expression from the replication-dependent vectors, we performed a one-step transfection/infection assay using 293T cells and quantified the proportion of cells expressing fluorescent protein by flow cytometry. The results from three sets of experiments using the HIV-1- and HTLV-1-based vectors are summarized in Fig. 2C and D, respectively. As shown in Fig. 2C, the HIV-1-based inGFPt and inmCherry vectors expressed fluorescent proteins well, and the infection detected with these reporters spread through the cell culture rapidly. Indeed, by day 3 or day 5, there were at least 10-fold more cells expressing fluorescent proteins in inGFPt- or inmCherry-transduced cultures than in inYFP- or inRFPt-2-transduced cultures. A similar trend was observed with the HTLV-1-based vectors (Fig. 2D); the kinetics of fluorescent protein expression in cells was observed to be much faster in the inGFPt- and inmCherry-transduced cultures than in the inYFP- or inRFPt-2-transduced cultures.
To determine whether the improved reporter gene expression was indeed caused by the increased splicing that removes the inactivating intron, we quantified the levels of spliced and unspliced reporter RNAs in HIV-1 and HTLV-1 particles using RT-qPCR. Each cDNA sample was amplified with a pair of primers, one of which was specific for unspliced RNA and the other of which was specific for spliced RNA. The ratios of spliced RNA to unspliced RNA from the HIV-1-based and HTLV-1-based reporter vectors are summarized in Fig. 2E and F, respectively. The splicing efficiencies varied drastically among the different reporter genes. However, the trends of the splicing efficiencies were similar between the HIV-1- and HTLV-1-based vectors. For example, splicing occurred efficiently in both HIV-1- and HTLV-1-based vectors containing GFP and mCherry; the levels of splicing achieved with the inGFP and inmCherry vectors were 39% and 54%, respectively, for HIV-1 and 53% and 23%, respectively, for HTLV-1. In contrast, the splicing efficiencies for the previously published inLuc vector were 5% for HIV-1 and 3% for HTLV-1. The lowest levels of intron removal were detected for the inRFPt-2 vector (0.02% and 0.03% for HIV-1 and HTLV-1, respectively). These RNA splicing data are consistent with the results obtained from the quantitation of fluorescent protein-expressing cells (Fig. 2C and D). Together, these results indicate that the efficiency with which the intron is removed by splicing dictates the ability of the reporter vector to express the marker gene in the transduced cells. In summary, by optimizing the sites of intron insertion, we generated two improved replication-dependent reporter vectors, inGFPt and inmCherry, with significantly higher levels of splicing and reporter expression after transduction than those for the previously published vectors.
trans-Acting, intron-targeting miR30-based shRNAs enhance the transduction of GFP-expressing vectors.
In an attempt to further improve the replication-dependent vectors using a different approach, we hypothesized that the level of intron-containing RNA can be reduced by using shRNA targeting sequences within the gamma-globin intron and accelerating its degradation. To test this hypothesis, we designed several intron-specific target sequences (see the Materials and Methods section) and cloned them into miR30-based shRNA vector pGIPZ, which expresses a GFP and IRES-puro cassette. As outlined in Fig. 3A, the intron-specific shRNAs or a control shRNA (against the P1 protein of RSV) was stably transduced into 293T cells, followed by puromycin selection, and transduction was confirmed by the detection of GFP expression. These shRNA-transduced cells were then transiently transfected with inRFPt-2 or inLuc, a gag-pol-expressing helper construct, and a plasmid expressing VSV-G that allowed the spread of the replication-dependent vectors. Three days later, RFP or luciferase expression was determined by flow cytometry or luciferase assay, respectively. As shown in Fig. 3B, compared with the expression of control shRNA, miR30-based shRNA targeting the intron starting from position 330 (mR-int330) significantly increased the level of detection of the reporter proteins encoded by the inRFPt-2 (6-fold) and inLuc (4-fold) vectors. miR30-based shRNA targeting the intron starting from position 79 (mR-int79) had a statistically significant effect on the detection of the reporter encoded by the inRFPt-2 vector (4-fold) but not on the detection of the reporter encoded by the inLuc vector compared with the effect of control shRNA. These data demonstrate that when it is expressed in trans, intron-targeting shRNA can efficiently enhance the spread of the reporter-expressing vectors, suggesting that they are targeting and degrading intron-containing RNA.
FIG 3.

Improving replication-dependent vectors via expression of miR30-based shRNAs specific for the intron. (A) Experimental protocol used to evaluate trans-acting intron-specific shRNA. Human 293T cells were stably transduced with various pGIPZ-based shRNA-expressing vectors and selected by puromycin, and GFP expression was measured. Puromycin-resistant cells were transiently cotransfected with the HIV-1-based inLuc or in-RFPt-2 vector along with helper plasmids, and reporter gene expression was determined with a luminometer or by flow cytometry. FACS, fluorescence-activated cell sorter analysis; RLU, relative light units. (B) Effect of trans-expressed, intron-specific shRNAs on levels of reporter expression after transduction with the HIV-1-based inRFPt-2 and inLuc vectors. The expression of the reporter was normalized to that of the P1-specific shRNA controls, which was defined as 1. (C) General structures of the inGFPt vector with inserted sequences: miR30-based shRNA (top) or shRNA (bottom). ψ, packaging signal; GF, 5′ portion of gfp gene; P, 3′ part of gfp gene. (D and E) Relative proportions of GFP+ cells from transduction with inGFPt vectors modified with miR30-based shRNAs (D) and shRNAs without the miR30 sequence (E) normalized to those of GFP+ cells from transduction with inGFPt without modification (control). (F) Effects of miR30-based shRNA modification on the packaging of spliced RNA in viral particles. Results from at least three independent experiments are shown as averages ± standard deviations. *, unless indicated by connector lines, the value is significantly different from that for the control (P < 0.05, Student's t test).
cis-Acting intronic miR30-based shRNAs improves vector quality.
Although it was effective, the trans-acting shRNA strategy described above requires a two-step process, in which the shRNA needs to be introduced into the cells first before the replication-dependent vectors can be tested. To simplify this process, we tested whether intron-targeting shRNA can be effective when expressed in a cis-acting context. For this purpose, we inserted the miR30-based shRNAs into the XbaI site of the intron in the HIV-1-based inGFPt vector. Because the intron-containing region can be expressed from the LTR promoter (sense) and the cytomegalovirus (CMV) promoter (antisense), we generated two sets of vectors expressing shRNA in the sense and the antisense orientations (Fig. 3C). The shRNA-containing vectors were evaluated in 293T cells using the one-step transfection/infection assay, reporter expression was determined after 2 days, and the results were compared with those obtained with the unmodified inGFPt vector. As shown in Fig. 3D, compared to the levels of GFP transduction from the unmodified inGFPt vector, insertion of the sense-oriented sequences of miR30 targeting P1 (mR-P1) or intron (mR-int79 and mR-int330) into the intron resulted in 2-, 2.2-, and 3-fold increases in the levels of GFP transduction, respectively. Insertion of mR-int330 into the intron in the sense orientation resulted in a level of GFP transduction significantly higher than that achieved by insertion of mR-int330 in the antisense direction or mR-int79 in the sense orientation. These results suggest that the orientation of the insert and the target sequences of the shRNA are important for the effect of miR30-based shRNA.
Interestingly, insertion of shRNA that does not specifically target the intron (mR-P1) into inGFPt in both orientations also increased the number of cells that expressed GFP. This observation suggests that miR30 sequences flanking the shRNA may be responsible for this effect. To test this possibility, we inserted only the stem-loop portion of the shRNAs, without the miR30 sequences, in the sense orientation into the XbaI site of the HIV-1-based inGFPt vector (Fig. 3C, bottom). Vectors modified with shRNAs without the miR30 backbone did not have an effect on the transduction of the GFP-expressing vectors (Fig. 3E).
We hypothesized that miR30-based shRNA targets the intron-containing vector RNA, thereby enriching the spliced RNA devoid of the intron and improving the transduction of the vectors that have an intact gfp gene. To verify this hypothesis, we quantified the levels of spliced and unspliced vector RNAs in viral particles by RT-qPCR. As shown in Fig. 3F, the virion RNAs from the HIV-1-based inGFPt vectors with mR-P1, mR-int79, and mR-int330 inserted in the sense orientation contained 56%, 60%, and 75% of the spliced RNA without the gamma-globin intron, respectively. All three values were significantly higher than the value for the unmodified inGFPt reporter (31%). Additionally, virion RNAs from mR-int330-bearing inGFPt contained more spliced RNA than those from mR-P1- or mR-int79-containing inGFPt. These results of virion RNA analysis are consistent with the number of GFP-expressing (GFP+) cells generated by replication-dependent vector transduction (Fig. 3D, solid bars). Taken together, cis-acting miR30-based shRNA targeting the gamma-globin intron at position 330 is effective in reducing the packaging of intron-containing vector RNA, thereby enhancing the number of GFP+ cells in HIV-1-based inGFPt vector transduction.
The mR-int330 modification of the intron improves the packaging of RNAs with intact reporter genes and the readout of transduction events.
To examine whether mR-int330 can improve replication-dependent vectors expressing reporters other than GFPt, we inserted the mR-int330 sequence used in the HIV-1-based inGFPt into the other vectors and examined its effects. The levels of spliced RNA packaging and reporter expression obtained after transduction of these modified vectors were compared with those obtained after transduction of the corresponding vectors without shRNA modification. As shown in Fig. 4A and C, insertion of the mR-int330 sequences increased the packaging of spliced RNAs with intact reporter genes. Significant increases in the packaging of spliced RNAs were obtained with all of the HIV-1- and HTLV-1-based vectors containing mR-int330 compared with the amounts seen with their unmodified control counterparts; however, the levels of the increases varied. In general, the increase in the packaging of the spliced RNA with intact reporter genes correlated with the improvement in the readout of the transduction events. For example, the HIV-1-based inmCherry vector packaged a high level of spliced RNA (54%) (Fig. 4A); the presence of mR-int330 raised the level of packaging of spliced RNA to 80%, resulting in a 1.5-fold increase. The mR-int330 insertion also resulted in a 1.3-fold improvement in the number of cells expressing mCherry after transduction (Fig. 4B), which is similar to the increase in spliced RNA packaging. HIV-1-based inLuc packaged spliced RNA with an intact reporter gene at a low level (∼5%, Fig. 4A); the insertion of mR-int330 resulted in a 3.2-fold increase (∼15.4%) in spliced RNA encapsidation, which is similar to the improvement of the luciferase activity readout (3.4-fold; Fig. 4B) from the transduction. Similar correlations between the packaging of the spliced RNA and the reporter readout after transduction were also observed in HTLV-1-based vectors (Fig. 4C and D). In summary, we generated several substantially improved vectors by inserting mR-int330 into the inactivating intron in both the HIV-1 and the HTLV-1 backbones. These vectors encapsidate high levels (from 47% to 81%) of spliced RNA with intact reporter genes and improve the expression of reporter proteins upon infection of target cells.
FIG 4.

Effects of the mR-int330 shRNA insertion on the packaging of spliced RNA in viral particles and reporter expression after transduction. The proportion of spliced RNA packaged into HIV-1 (A) and HTLV-1 (C) particles and the levels of reporter gene expression after transduction of the HIV-1-based (B) and HTLV-1-based (D) vectors containing mR-int330 compared with those obtained after transduction of the corresponding vectors without modification, for which the values were defined as 1, are shown. Results from at least three independent experiments are shown as averages ± standard deviations. *, the value is significantly different from that for the control (P < 0.05, Student's t test).
Using improved replication-dependent vectors to quantify HIV-1 infection of lymphocytes in coculture experiments.
The purpose for generating improved replication-dependent vectors is to increase the sensitivity of detection of retroviral infection events during cell coculture experiments where both cell-free and cell-to-cell transmission can occur. To examine the ability of the improved vectors to detect infected cells in a mixed cell culture, human CD4+ T cells (CEM cells) were transfected with the HIV-1-based inGFPt and/or inmCherry vector along with helper constructs. As a comparison, we also transfected the previously described inYFP construct with helper plasmids in a separate sample. Transfected cells were mixed with Raji/CD4 target cells, cocultured for 3 days, and analyzed by flow cytometry. Two sets of control experiments were performed in parallel; the Env-expressing construct was omitted during the transfection, or AZT was added to the culture to inhibit virus spread. The results from one set of representative experiments are shown in Fig. 5A. When cells were transfected with inGFPt and/or inmCherry, along with plasmids expressing the gag-pol helper and Env, a significant number of cells (4 to 7%) in the coculture expressed GFP and/or mCherry. In contrast, the level of reporter-positive cells generated from the previously described inYFP vector was about 4-fold lower than that generated from the inGFPt vector (Fig. 5A and B). If cells were transfected with inGFPt, inmCherry, and a packaging plasmid without the Env-expressing construct, only the background level of fluorescent protein expression was found for the cells in the coculture (Fig. 5A, No env). When the nucleoside reverse transcriptase inhibitor AZT was added to the coculture system (Fig. 5A, inGFPt + inmCherry + AZT 20 μM), we observed far fewer GFP- and/or mCherry-positive cells compared with the numbers found in experiments without AZT treatment (Fig. 5A, inGFPt + inmCherry). Thus, these negative controls confirmed that the inGFPt and inmCherry vectors require viral replication for reporter gene expression and thus can be used to detect infection events. These results also demonstrate the advantages of using the improved vectors. The previously described inYFP vector could barely detect the infection event and could not identify cells infected by more than one virus. In contrast, using the two vectors described in this report, not only the signals of the infection were improved but also multiply infected cells could be detected and quantified.
FIG 5.
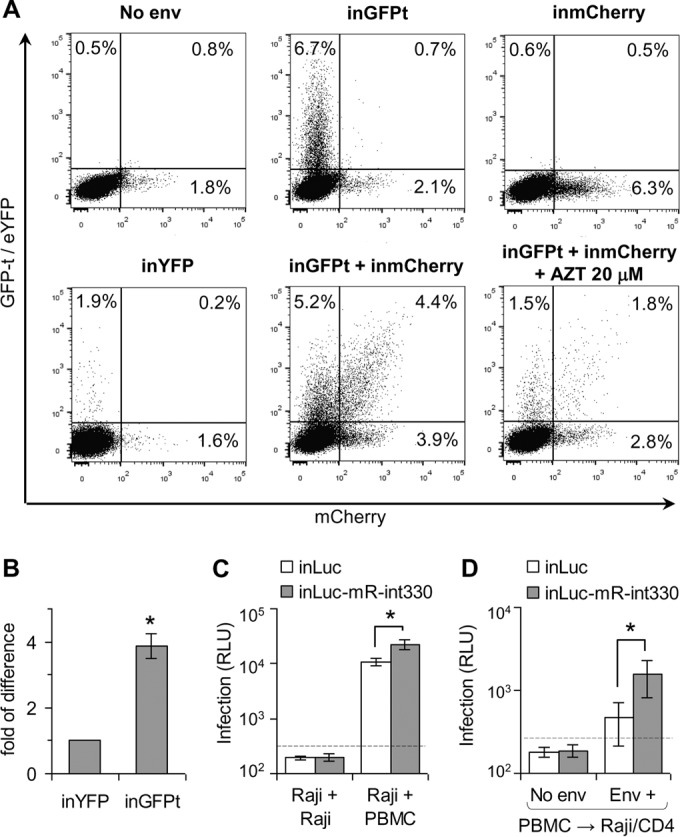
Quantifying HIV-1 infection in cocultures of primary and immortalized lymphocytes. (A) Evaluation of CEM-Raji/CD4 cell cocultures infected with reporter vectors with single or double fluorescence. Representative flow cytometry results from several experiments for cells infected with one (GFP+, mCherry+, or YFP+) or two (GFP+ and mCherry+) reporter vectors are shown. CEM cells were cotransfected with helper plasmids and HIV-1-based reporter vectors, mixed with Raji/CD4 cells, and incubated for 3 days prior to analysis. (B) Comparison of transduction efficiencies of inYFP and inGFPt in CEM-Raji/CD4 cell cocultures. The number of YFP-positive cells from inYFP vector transduction was set equal to 1, and the number of GFP+ cells from inGFPt transduction relative to that from inYFP transduction is shown. (C and D) Measurement of HIV-1 infection with luciferase-based vectors in cocultures of transfected Raji cells with target PBMCs (C) or electroporated PBMCs with Raji/CD4 target cells (D). Background levels of luciferase activity are shown as dashed lines. Results from three independent experiments (B) or for the three healthy donors (C and D) are shown as averages ± standard deviations. *, if not indicated by connector lines, values are significantly different from those for the control (P < 0.05, Student's t test). RLU, relative light units.
To test whether we could detect infection events in primary cells, we used the inLuc vector and the improved inLuc-mR-int330 vector. Raji cells were transfected with a reporter vector along with helper plasmids and cocultured with either activated PBMCs or fresh Raji cells as a negative control because Raji cells do not express CD4 and cannot be infected by viruses pseudotyped with HIV-1 Env. When transfected Raji cells were cultured with fresh Raji cells (Fig. 5C, two left bars), very little luciferase activity which was below the background level was generated (dashed line). In contrast, when transfected Raji cells were cocultured with activated PBMCs, the luciferase activities detected in the samples transfected with the inLuc-mR-int330 vector were approximately 2 orders of magnitude higher than those detected in the negative controls. The previously described inLuc vectors also produced detectable levels of luciferase activity, and the signals were 2-fold lower than those from the improved inLuc-mR-int330 vector.
We also examined whether our reporter vectors could be used to detect viruses produced from the primary cells. For this purpose, we electroporated PBMCs with a reporter vector along with helper plasmids and then mixed them with Raji/CD4 target cells. As shown in Fig. 5D, significant levels of luciferase activity were detected only when the Env expression plasmid was included in the electroporation. In these experiments, the luciferase signal from the improved inLuc-mR-int330 vector was 3.3-fold higher than the readout from the previously described inLuc vector (Fig. 5D).
Taken together, these results show that the improved replication-dependent vectors can be used to quantify HIV-1 infection events in a coculture system using primary cells or immortalized human blood cells. Furthermore, compared with the results obtained with their corresponding previously described vectors, these improved vectors provided higher sensitivities for the detection of infection events and allowed the quantitation of multiply infected cells.
Quantitative comparison of multiply HIV-1-infected cells in cell coculture and cell-free infections.
During cell coculture, HIV-1 infection can occur by cell-free infection and by cell-to-cell infection. To determine the contribution of cell-to-cell infection during cell coculture, we compared HIV-1 infection in cell coculture and cell-free infections using the inGFPt and inmCherry vectors. To generate cell-free viruses, 293T cells were transfected with inGFPt, inmCherry, and HIV-1 helper plasmids, including one that expressed HIV-1 Env. Viruses were harvested from transfected 293T cells, concentrated, serially diluted, and added to a mixture of mock-electroporated CEM cells and Raji/CD4 cells. Cell coculture infection experiments were performed as described in Materials and Methods with electroporated CEM cells and Raji/CD4 target cells. To obtain the different levels of HIV-1 infection in coculture infection experiments, CEM cells were transfected with different amounts of total plasmid DNA, while the same ratio between the reporter plasmids and the helper constructs was maintained. Cells were analyzed by flow cytometry at 3 days after coculture or cell-free infection. As shown in Fig. 6A, both cell coculture and cell-free infections generated similar fractions of cells (7.3%) that expressed fluorescent proteins. However, cell coculture infection experiments generated 2.5-fold more double-positive (GFP+ and mCherry-positive [mCherry+]) cells than cell-free infection. We further studied the relationship between the multiplicity of infection (MOI) and increased double infection in the cell coculture infection experiments. For this purpose, we generated coculture and cell-free infections with different infection rates and compared the fraction of double-positive cells and the total number of positive cells. As shown in Fig. 6B, when similar levels of infection were compared, cell coculture infection experiments (black dots) always generated far more double-positive cells than cell-free infection (open dots). In addition, the mean fluorescence intensity (MFI) of the cell population positive both in the GFP channel and in the mCherry channel was also higher in coculture infection than in infection with cell-free viruses (Fig. 6A). To quantify this difference, we calculated the total MFI value for the double-positive cell population (the MFI value for the cell population positive both in the GFP channel and in the mCherry channel) and divided this value by the total MFI value obtained for the cell population positive in a single channel. As shown in Fig. 6C, the ratio of the MFI for the double-positive cell population to the MFI for the single-positive cell population under coculture conditions was significantly (1.8-fold) higher than that under conditions with cell-free infection. Therefore, both the fraction and the MFI of double-positive cells were significantly different between the coculture and the cell-free infection conditions, indicating that cell-to-cell infection occurs at a significant level in cell coculture infection experiments.
FIG 6.
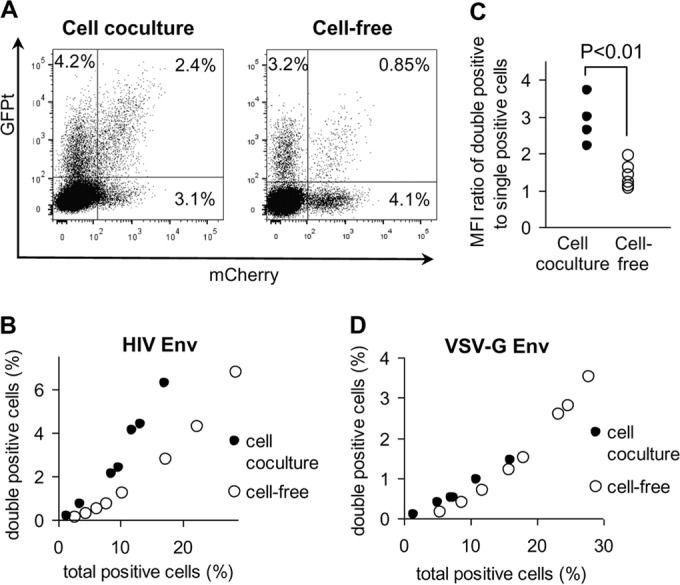
Comparison of HIV-1 cell coculture and cell-free infections with inGFPt and inmCherry vectors. (A) Representative flow cytometry graphs showing the distribution of double- and single-positive cells after coculture of transfected CEM cells with Raji/CD4 targets (left) or after cell-free virus infection of a CEM-Raji/CD4 cell mixture (right). (B and D) Effects of viral titer on the proportion of double-positive cells when HIV-1 Env (B) or VSV-G (D) was used. To obtain different levels of infection, cells were infected with different doses of virus or cocultured with effector cells transfected with various amounts of plasmid DNAs. (C) Increased MFI of the GFP+ mCherry+ cell population detected in cell coculture and cell-free virus infections. Relative values of MFI were calculated for each sample as described in Results.
Interactions between HIV-1 Env and CD4 initiate virological synapse formation, which then triggers cell-to-cell transmission of multiple HIV-1 particles. To determine the role of HIV-1 Env in mediating this process, we compared cell coculture and cell-free infections using HIV-1 pseudotyped with the VSV G protein; the infection tests are described in Fig. 6B. As shown in Fig. 6D, we observed similar results in cell coculture and cell-free infections when VSV-G-pseudotyped HIV-1 was used; i.e., VSV-G-pseudotyped HIV-1 did not generate more multiply infected cells in cell coculture infection experiments. Similarly, the MFI ratios were comparable between cell coculture and cell-free infections (data not shown).
In summary, using two replication-dependent vectors, inGFPt and inmCherry, we have demonstrated that multiply infected cells are generated at a higher frequency in cell coculture infection experiments than in cell-free infection experiments; furthermore, this enhancement is mediated by HIV Env.
DISCUSSION
The mechanisms of transmission from infected cells to target cells can vary among different retroviruses and can be complex. Regardless of the precise mechanism, virus transmission and consequent infection are more efficient when the infected cells are in contact with the target cells than when virus is released into body fluids (9, 17, 18). Furthermore, cell-to-cell infection also alters certain properties of viral infection; for example, it increases the multiplicity of infection (19–21), enhances the resistance to some antivirals (22, 23), allows the virus to bypass potent neutralizing antibodies (24), and contributes to the genetic diversity of HIV (reviewed in reference 25).
We previously developed replication-dependent vectors based on luciferase and YFP reporters to quantify cell-to-cell infection with HIV-1 and HTLV-1 (9). The design of these vectors (detailed in Fig. 1 and the introduction) eliminates the background of reporter gene expression in the producer cells yet allows reporter detection in the target cells. However, the inactivating intron was not efficiently removed in the previously reported inLuc and inYFP vectors, thereby reducing the sensitivity of the assay such that inYFP cannot be used to monitor virus spread by flow cytometry. Additionally, Derse's group also developed inGluc vectors based on the Gaussia luciferase reporter gene (26), which is secreted by cells and can be measured in cell supernatants. However, none of these vectors could be used to determine the proportion of cells infected in coculture and whether double infection occurred because fluorescent protein-based replication-dependent vectors that could be used to follow virus spread by flow cytometry were not available.
In the current study, we aimed to improve the replication-dependent vectors by first optimizing intron removal using selected insertion sites within the reporter genes. We hypothesize that the spliceosome can have better access to unpaired SD and SA sites than to sites that are hidden by secondary RNA structures, thereby increasing the splicing efficiency to remove the inactivating intron. We selected the intron insertion site by analyzing the structures of reporter RNAs and choosing A(C)AG/N motifs with minimal nucleotide pairing. Using this strategy, we generated improved replication-dependent vectors with efficient splicing-mediated intron removal; compared with the splicing of the previously reported inLuc vector, the splicing of the inGFPt and inmCherry vectors was improved by 8- and 11-fold, respectively, in the HIV-1-based vectors and 20- and 9-fold, respectively, in the HTLV-based vectors (Fig. 2).
In addition to increasing the splicing efficiency, we tested several different strategies that would either reduce nuclear export by including the human papillomavirus type 1 AU-rich element (27) or antisense RNA against the HIV-1 Rev-response element targeting 83 nucleotides of stem IA and stem IIA-C, decrease the packaging of the unspliced reporter RNA using antisense RNA against HIV-1 stem-loop 3 of the 5′ untranslated region (28), or enhance intron removal using an exonic splice enhancer from avian sarcoma leukemia virus (29). When these aforementioned cis-acting elements were inserted within the intron of the replication-dependent vector and tested, none of them improved reporter expression in the target cells (data not shown).
We did, however, observe an improvement in reporter expression when we used short RNA interference to reduce the level of unspliced reporter RNAs in the cytoplasm. Rather than using the RNA polymerase III-driven first-generation shRNAs, we employed second-generation shRNAs, which mimic the endogenous microRNA and are processed from an RNA polymerase II-driven long transcript. For gene-knockdown experiments, shRNAs generally target coding sequences or the 5′ and 3′ untranslated regions of mRNAs. An intron is not an ideal target for shRNA in gene-knockdown experiments because the effector siRNA complex interferes with the target RNA in the cytoplasm, and intron-containing, unspliced mRNA is generally located in the nucleus. In the elegant work from Samakoglu et al. (30), promoterless shRNA sequences were placed within the intron to downregulate targeted gene expression in a tissue- and differentiation stage-specific manner. In HIV-1 and HTLV-1, viral proteins Rev and Rex, respectively, mediate the nuclear export of the unspliced or partially spliced viral RNAs. Therefore, the full-length vector genome contains introns, which allows us to target the inactivating intron in the replication-dependent vectors. We observed that the trans-expressed shRNAs targeting the gamma-globin intron reduced the level of unspliced reporter RNAs in the viral particles. Interestingly, when miR30-based shRNAs were placed in cis within the inactivating intron, we observed a reduced level of unspliced RNA packaging compared with that obtained with vectors that did not contain shRNA, even when the miR30-based shRNA did not target virus-specific sequences (Fig. 3D). One possible explanation is that the unspliced reporter RNA containing the miR30 sequence may undergo additional processing, resulting in its degradation in the nucleus.
Using the new, improved HIV-1-based inGFPt and inmCherry constructs, we have demonstrated that these vectors are suitable for evaluating multiply HIV-1-infected cells by quantifying the numbers of double-positive cells in cocultures and determining the levels of fluorescence intensity of the reporters. By comparing cell coculture infection with cell-free infection and pseudotyping virus with VSV-G Env, we demonstrate that multiply HIV-1-infected cells occur at a higher frequency in cell coculture experiments and this enhancement requires expression of HIV Env.
Most of the current studies on HIV cell-to-cell transmission utilize infectious molecular clones that generate a fluorescent or luminescent signal in both producer and target cells. Several different techniques have been used to distinguish target cells from effector cells, such as labeling of cells with fluorescent dyes (21) or mechanical separation of cells by the use of transwells or by continuous shaking of mixed cells (31). However, each of these approaches has its issues. In contrast, the replication-dependent vectors do not express the reporter in producer cells and can be used to precisely quantify infectious events in the target cells. Therefore, the higher levels of performance of the new vectors provide a better tool to study both cell-to-cell and cell-free infection in lymphoid cell lines, as well as in primary human cells.
ACKNOWLEDGMENTS
This work has been supported by grants from the Russian Foundation for Basic Research (NIH-RFBR 13-04-91449, RFBR 12-04-00415-a) and the Russian Science Foundation (RSF 15-15-00135), by the Lomonosov Moscow State University Program of Development, by Intramural Research, Center for Cancer Research, National Cancer Institute, and by funding from the U.S.-Russia Joint Working Group on Biomedical Research Cooperation.
We thank Steven Santos for critical reading of the manuscript.
REFERENCES
- 1.Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CR. 2003. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716. doi: 10.1126/science.1080115. [DOI] [PubMed] [Google Scholar]
- 2.Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J Exp Med 199:283–293. doi: 10.1084/jem.20030648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Horensavitz C, Pypaert M, Mothes W. 2007. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat Cell Biol 9:310–315. doi: 10.1038/ncb1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Prooyen N, Gold H, Andresen V, Schwartz O, Jones K, Ruscetti F, Lockett S, Gudla P, Venzon D, Franchini G. 2010. Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc Natl Acad Sci U S A 107:20738–20743. doi: 10.1073/pnas.1009635107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pais-Correia AM, Sachse M, Guadagnini S, Robbiati V, Lasserre R, Gessain A, Gout O, Alcover A, Thoulouze MI. 2010. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat Med 16:83–89. doi: 10.1038/nm.2065. [DOI] [PubMed] [Google Scholar]
- 6.Rudnicka D, Feldmann J, Porrot F, Wietgrefe S, Guadagnini S, Prevost MC, Estaquier J, Haase AT, Sol-Foulon N, Schwartz O. 2009. Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J Virol 83:6234–6246. doi: 10.1128/JVI.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen P, Chen BK, Mosoian A, Hays T, Ross MJ, Klotman PE, Klotman ME. 2011. Virological synapses allow HIV-1 uptake and gene expression in renal tubular epithelial cells. J Am Soc Nephrol 22:496–507. doi: 10.1681/ASN.2010040379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Columba Cabezas S, Federico M. 2013. Sequences within RNA coding for HIV-1 Gag p17 are efficiently targeted to exosomes. Cell Microbiol 15:412–429. doi: 10.1111/cmi.12046. [DOI] [PubMed] [Google Scholar]
- 9.Mazurov D, Ilinskaya A, Heidecker G, Lloyd P, Derse D. 2010. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog 6:e1000788. doi: 10.1371/journal.ppat.1000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heidmann O, Heidmann T. 1991. Retrotransposition of a mouse IAP sequence tagged with an indicator gene. Cell 64:159–170. doi: 10.1016/0092-8674(91)90217-M. [DOI] [PubMed] [Google Scholar]
- 11.Heidmann T, Heidmann O, Nicolas JF. 1988. An indicator gene to demonstrate intracellular transposition of defective retroviruses. Proc Natl Acad Sci U S A 85:2219–2223. doi: 10.1073/pnas.85.7.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH Jr. 1996. High frequency retrotransposition in cultured mammalian cells. Cell 87:917–927. doi: 10.1016/S0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- 13.Mazurov D, Ilinskaya A, Heidecker G, Filatov A. 2012. Role of O-glycosylation and expression of CD43 and CD45 on the surfaces of effector T cells in human T cell leukemia virus type 1 cell-to-cell infection. J Virol 86:2447–2458. doi: 10.1128/JVI.06993-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derse D, Hill SA, Lloyd PA, Chung H, Morse BA. 2001. Examining human T-lymphotropic virus type 1 infection and replication by cell-free infection with recombinant virus vectors. J Virol 75:8461–8468. doi: 10.1128/JVI.75.18.8461-8468.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell MS, Bodine ET, Hill S, Princler G, Lloyd P, Mitsuya H, Matsuoka M, Derse D. 2007. Phenotypic and genotypic comparisons of human T-cell leukemia virus type 1 reverse transcriptases from infected T-cell lines and patient samples. J Virol 81:4422–4428. doi: 10.1128/JVI.02660-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murakami T, Freed EO. 2000. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc Natl Acad Sci U S A 97:343–348. doi: 10.1073/pnas.97.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carr JM, Hocking H, Li P, Burrell CJ. 1999. Rapid and efficient cell-to-cell transmission of human immunodeficiency virus infection from monocyte-derived macrophages to peripheral blood lymphocytes. Virology 265:319–329. doi: 10.1006/viro.1999.0047. [DOI] [PubMed] [Google Scholar]
- 18.Dimitrov DS, Willey RL, Sato H, Chang LJ, Blumenthal R, Martin MA. 1993. Quantitation of human immunodeficiency virus type 1 infection kinetics. J Virol 67:2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J, Dang Q, Unutmaz D, Pathak VK, Maldarelli F, Powell D, Hu WS. 2005. Mechanisms of nonrandom human immunodeficiency virus type 1 infection and double infection: preference in virus entry is important but is not the sole factor. J Virol 79:4140–4149. doi: 10.1128/JVI.79.7.4140-4149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dang Q, Chen J, Unutmaz D, Coffin JM, Pathak VK, Powell D, KewalRamani VN, Maldarelli F, Hu WS. 2004. Nonrandom HIV-1 infection and double infection via direct and cell-mediated pathways. Proc Natl Acad Sci U S A 101:632–637. doi: 10.1073/pnas.0307636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Del Portillo A, Tripodi J, Najfeld V, Wodarz D, Levy DN, Chen BK. 2011. Multiploid inheritance of HIV-1 during cell-to-cell infection. J Virol 85:7169–7176. doi: 10.1128/JVI.00231-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agosto LM, Zhong P, Munro J, Mothes W. 2014. Highly active antiretroviral therapies are effective against HIV-1 cell-to-cell transmission. PLoS Pathog 10:e1003982. doi: 10.1371/journal.ppat.1003982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, Baltimore D. 2011. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 477:95–98. doi: 10.1038/nature10347. [DOI] [PubMed] [Google Scholar]
- 24.Abela IA, Berlinger L, Schanz M, Reynell L, Gunthard HF, Rusert P, Trkola A. 2012. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog 8:e1002634. doi: 10.1371/journal.ppat.1002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dale BM, Alvarez RA, Chen BK. 2013. Mechanisms of enhanced HIV spread through T-cell virological synapses. Immunol Rev 251:113–124. doi: 10.1111/imr.12022. [DOI] [PubMed] [Google Scholar]
- 26.Aloia AL, Duffy L, Pak V, Lee KE, Sanchez-Martinez S, Derse D, Heidecker G, Cornetta K, Rein A. 2013. A reporter system for replication-competent gammaretroviruses: the inGluc-MLV-DERSE assay. Gene Ther 20:169–176. doi: 10.1038/gt.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan W, Schwartz S. 1995. The Rev protein of human immunodeficiency virus type 1 counteracts the effect of an AU-rich negative element in the human papillomavirus type 1 late 3′ untranslated region. J Virol 69:2932–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chadwick DR, Lever AM. 2000. Antisense RNA sequences targeting the 5′ leader packaging signal region of human immunodeficiency virus type-1 inhibits viral replication at post-transcriptional stages of the life cycle. Gene Ther 7:1362–1368. doi: 10.1038/sj.gt.3301254. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka K, Watakabe A, Shimura Y. 1994. Polypurine sequences within a downstream exon function as a splicing enhancer. Mol Cell Biol 14:1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samakoglu S, Lisowski L, Budak-Alpdogan T, Usachenko Y, Acuto S, Di Marzo R, Maggio A, Zhu P, Tisdale JF, Riviere I, Sadelain M. 2006. A genetic strategy to treat sickle cell anemia by coregulating globin transgene expression and RNA interference. Nat Biotechnol 24:89–94. doi: 10.1038/nbt1176. [DOI] [PubMed] [Google Scholar]
- 31.Duncan CJ, Williams JP, Schiffner T, Gartner K, Ochsenbauer C, Kappes J, Russell RA, Frater J, Sattentau QJ. 2014. High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage-T cell virological synapse. J Virol 88:2025–2034. doi: 10.1128/JVI.03245-13. [DOI] [PMC free article] [PubMed] [Google Scholar]