

ABSTRACT
Glioblastoma is a terminal disease with no effective treatment currently available. Among the new therapy candidates are oncolytic viruses capable of selectively replicating in cancer cells, causing tumor lysis and inducing adaptive immune responses against the tumor. However, tumor antiviral responses, primarily mediated by type I interferon (IFN-I), remain a key problem that severely restricts viral replication and oncolysis. We show here that the Semliki Forest virus (SFV) strain SFV4, which causes lethal encephalitis in mice, is able to infect and replicate independent of the IFN-I defense in mouse glioblastoma cells and cell lines originating from primary human glioblastoma patient samples. The ability to tolerate IFN-I was retained in SFV4-miRT124 cells, a derivative cell line of strain SFV4 with a restricted capacity to replicate in neurons due to insertion of target sites for neuronal microRNA 124. The IFN-I tolerance was associated with the viral nsp3-nsp4 gene region and distinct from the genetic loci responsible for SFV neurovirulence. In contrast to the naturally attenuated strain SFV A7(74) and its derivatives, SFV4-miRT124 displayed increased oncolytic potency in CT-2A murine astrocytoma cells and in the human glioblastoma cell lines pretreated with IFN-I. Following a single intraperitoneal injection of SFV4-miRT124 into C57BL/6 mice bearing CT-2A orthotopic gliomas, the virus homed to the brain and was amplified in the tumor, resulting in significant tumor growth inhibition and improved survival.
IMPORTANCE Although progress has been made in development of replicative oncolytic viruses, information regarding their overall therapeutic potency in a clinical setting is still lacking. This could be at least partially dependent on the IFN-I sensitivity of the viruses used. Here, we show that the conditionally replicating SFV4-miRT124 virus shares the IFN-I tolerance of the pathogenic wild-type SFV, thereby allowing efficient targeting of a glioma that is refractory to naturally attenuated therapy vector strains sensitive to IFN-I. This is the first evidence of orthotopic syngeneic mouse glioma eradication following peripheral alphavirus administration. Our findings indicate a clear benefit in harnessing the wild-type virus replicative potency in development of next-generation oncolytic alphaviruses.
INTRODUCTION
Glioblastoma (GBM) is the most common primary brain tumor and a devastating disease with a median survival of only 15 months despite best available therapy (1). Oncolytic virotherapy provides a novel option to treat malignant central nervous system (CNS) tumors, as many of the potential oncolytic viruses are tumor homing, self-amplifying, and may elicit antitumor T-cell responses (2). Oncolytic viruses harnessed recently in virotherapy of human glioblastoma include herpes simplex virus (3), reovirus (Reolysin) (4), Newcastle disease virus (NDV-HUJ) (5), and poliovirus (PVS-RIPO) (6). Apart from anecdotal reports of successful cases and despite a relatively good tolerability of the vectors by the patients, the therapeutic efficacy of viral therapies has been disappointing. Recent findings indicate that the poor treatment efficacies may derive from both biological and physical barriers to oncolytic viruses (reviewed in reference 7). GBM extracellular matrix and resident stromal cells may block infection and virus spread within the tumor. In addition, GBM cells, GBM stem cell-like cells, and infiltrating leukocytes may mount a strong innate response against the virus. In particular, viruses whose selectiveness for cancer tissue relies on defective type I interferon (IFN-I) signaling in tumor cells may completely lose efficacy (8).
Semliki Forest virus (SFV) is an enveloped, positive-sense, single-stranded RNA [(+)ssRNA] virus of the Alphavirus genus. Like most alphaviruses, SFV is able to enter the CNS upon systemic delivery, a feature which we have shown can be exploited with a neuroattenuated strain of SFV, VA7, to target brain tumors (9, 10). However, in accordance with results showing that SFV infectivity and amplification in nonneuronal CNS cells are regulated by IFN-I (11), both the viral replication and therapeutic efficacy of neuroattenuated SFV vector VA7 were dismal in IFN-I-responsive syngeneic mouse glioma models (10, 12). Recent attempts to increase VA7 tumor infectivity and its replication rate by administering to tumor-bearing mice either rapamycin or cyclophosphamide, both of which are known to reduce tumor protection against IFN-I-sensitive vesicular stomatitis virus, were unsuccessful (13), as neither of these drugs led to increased tumor permissiveness to VA7. Thus, other means of achieving tumor infection are needed.
For different strains of neurotropic alphaviruses, the degree of pathogenicity is primarily determined by access to the CNS and rate of replication in neurons. Importantly, the increased neurovirulence of some virus strains correlates with their increased resistance to IFN-I-mediated antiviral effects in nonneuronal cells (14–16), implying that such strains might be able to replicate even in IFN-I signaling-proficient tumors. However, the neurotoxicity of virulent alphaviruses precludes use of them as oncolytic agents. In this regard, we previously demonstrated that the replication of virulent SFV4 in neurons can be inhibited by inserting into the viral nonstructural genome multiple target sequences for the microRNA (miRNA) miR-124 (SFV4-miRT124) (17). On the other hand, the miR-124 target sites do not interfere with the expression of viral genes in cells lacking miR-T124 expression (17), notably gliomas (18). Thus, the rationale for using SFV4-miRT124 as a therapeutic virus is that the neurovirulent SFV4-associated resistance to antiviral cytokine signaling is preserved, allowing robust oncolytic replication in glioma cells, while the SFV4-associated neurotoxicity is limited by reduced replication in normal healthy neurons.
In this work, we report that SFV4-miRT124, which is based on the neurovirulent strain SFV4, shows potency to replicate in and lyse IFN-I-competent syngeneic mouse CT-2A glioma cells, regardless of their elicited IFN-I response. In contrast to the attenuated SFV strain VA7 that was used in previous studies, SFV4-miRT124 displayed enhanced oncolytic potency against CT-2A tumors in vivo and was able to more efficiently destroy human primary GBM cell lines pretreated with IFN-I.
MATERIALS AND METHODS
Cell lines.
C57BL/6 mouse glioma CT-2A cells (from Thomas Seyfried, Boston College) and firefly luciferase expressing-CT-2A-Fluc cells (provided by Jan Brun, Children's Hospital of Eastern Ontario) were cultured in RPMI 1640 medium (Sigma-Aldrich) supplemented with 10% fetal calf serum (FCS; Autogen Bioclear), 1% penicillin-streptomycin (Sigma-Aldrich), and 1% l-glutamine (Sigma-Aldrich). African green monkey kidney Vero(B) cells (University of Turku, Finland) were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich) supplemented with 5% FCS, 1% penicillin-streptomycin, 1% l-glutamine, and 10 mM HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; Sigma-Aldrich]. BHK-21 (Syrian hamster kidney cells), GL261 (C57BL/6 mouse glioma cells; provided by G. Safrany), and B16-F10-LacZ (C57BL/6 mouse melanoma) cells were cultured in DMEM with 10% FCS, 1% penicillin-streptomycin, and 1% l-glutamine.
Glioblastoma cell lines were derived from 3 patients (2 males and 1 female, aged 67 to 68 years) who underwent surgery in Kuopio University Hospital between July 2012 and February 2014. Samples were collected and analyzed according to ethical guidelines approved by the Research Ethics Committee of the Hospital District of Northern Savo. After removal, glioblastoma samples were immediately transported to a cell culturing laboratory in cooled Opti-MEM reduced serum medium (Gibco). Blood-containing tissue parts were removed. The remaining sample was cut using a scalpel and a McIlwain tissue chopper (thickness, 500 μm) and then passed through a 21-gauge needle. After trituration and centrifugation, red blood cells were lysed by using ammonium chloride (Stem Cell Technologies). After 2 sequential centrifugation steps and washings, cells were suspended into a small volume of culture medium, and cell counts were calculated based on trypan blue exclusion. Cells were seeded in uncoated flasks (25 cm2; Greiner CellStar) and cultured in Dulbecco's modified Eagle's medium–F-12 medium (Gibco) containing 1% Glutamax-I (Gibco), 1% penicillin-streptomycin, 1.25 μg/ml amphotericin B (Fungizone; Gibco), and 10% heat-inactivated FBS at 37°C in 5% CO2. To promote growth, cells were regularly switched to another flask (coated with ECM gel, 1:20 dilution; Sigma-Aldrich) after 1 to 7 days in vitro. Cells were treated with Accutase (Sigma-Aldrich) during passaging. Cells at passages 9 to 10 were used in this study.
Viruses.
Generation of VA7-EGFP (enhanced green fluorescent protein construct) and SFV4-miRT124 has been previously described (17, 19). Production of infectious virus particles was done by transfecting (using TransIT [Mirus]) BHK-21 cells with in vitro-transcribed (mMessage mMachine; Ambion) full-length virus (+)RNA. Initial virus propagation (primary virus preparation) was done in a 6-well plate followed by larger-scale infection using the previously collected primary preparation. Virus production was performed in HEPES-buffered DMEM [the same medium as that used for culture of Vero(B) cells], and the final virus stock was cleared from cell debris by centrifugation (1,000 relative centrifugal force, 4 min) followed by filtration through a 0.2-μm syringe filter. L10 virus was propagated from original infectious stock (kindly provided by John Fazakerley). Virus titration was done by plaque assay in Vero(B) cells as described elsewhere (8).
Quantification of IFN-β by ELISA.
For quantification of virus-induced IFN-β, CT-2A-Fluc cells were seeded on 12-well plates (4 × 105 cells/well) and infected at a multiplicity of infection (MOI) of 0.01 the next day. The amount of cell-secreted IFN-β was analyzed by using the VeriKine mouse IFN-β enzyme-linked immunosorbent assay (ELISA) kit (PBL Assay Science) for supernatant samples collected 24 h postinfection (p.i.). Pooled medium samples from three infected wells were analyzed as triplicates. The experiment was replicated and the results were combined.
CT-2A-Fluc plaque assay.
The plaque-forming capabilities of VA7-EGFP, SFV4, and SFV4-miRT124 were analyzed in CT-2A-Fluc cells as follows. CT-2A-Fluc cells seeded on 12-well plates were infected with 40 PFU of virus and incubated for 1 h at +37°C, followed by replacement of culture medium with 0.4% agarose-containing culture medium. The cell layer was stained with crystal violet 3 days after infection to visualize the plaques. For testing JAK inhibition of plaque-forming potency of VA7-EGFP, 250,000 CT-2A cells were seeded in 6-well plates. The next day, cell monolayers were overlaid with culture medium containing 1% agarose and 2× penicillin-streptomycin. When the agarose had solidified, an ∼3-mm-diameter cylindrical piece of agarose was carefully removed using a cut pipette tip. A total of 5,000 PFU of VA7-EGFP virus in 10 μl medium with or without JAK inhibitor I (sc-204021; Santa Cruz Biotechnology, USA) or the appropriate amount of dimethyl sulfoxide (DMSO; 0.1% [vol/vol]) was immediately pipetted into the resulting hole. Virus spread was observed for 4 days by using a fluorescence stereomicroscope (Leica M165 C) with the lids off.
Cell viability measurements.
Cell viability measurements were done in a 96-well format by plating 1 × 104 human glioblastoma cells (derived from the patient samples) or 4 × 104 CT-2A-Fluc cells and then infecting cells with various MOIs (as indicated in the figure legends) on the following day. Viability was assessed using Cell Proliferation kit I (Roche) according to the manufacturer's instructions at 48 or 72 h after infection. For measuring the effect of IFN on cell-killing potency, CT-2A-Fluc or human glioblastoma cells (plated as described above) were administered increasing doses of mouse recombinant IFN-β (Sigma-Aldrich) or human recombinant IFN-β (Nordic Biosite) followed by virus infection. Cell viability was measured 48 h postinfection.
Fluorescence microscopy.
CT-2A-Fluc cells seeded on 12-well plates (4 × 105 cells/well) were infected at an MOI of 0.01. Phase-contrast and fluorescence images were captured with an Axio Observer.Z1 (Zeiss) inverted microscope (10× objective) at the indicated time points.
Western blotting.
For detection of activated STAT1, 2 × 105 Vero(B) or 4 × 105 CT-2A-Fluc cells were seeded on 12-well plates and infected with SFV (rA774, VA7-EGFP, L10, SFV4, or SFV4-miRT124) at an MOI of 10. At the indicated time points after infection, culture medium was replaced with fresh medium containing 1,000 units/ml of human [for Vero(B) cells] recombinant IFN-β (Nordic Biosite) or mouse (for CT-2A-Fluc cells) recombinant IFN-β (Sigma-Aldrich) and incubated at +37°C for 20 min to induce phosphorylation of STAT1. After incubation, cells were washed with cold PBS, lysed by adding radioimmunoprecipitation assay buffer (RIPA; containing 50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 1% IGEPAL [MP Biomedicals], 0.5% deoxycholate [Sigma-Aldrich], 0.1% SDS with Complete Mini protease inhibitor cocktail [Roche], and phosphatase inhibitor cocktail [Roche]), scraped, and collected into microcentrifuge tubes. Samples were stored at −20°C until analyzed.
The protein concentrations of centrifuged samples were determined from sample supernatants by using the Bradford Bio-Rad protein assay dye reagent and a standard of bovine serum albumin. Samples were boiled at 95°C with 1× loading buffer (10× stock of 0.45 M Tris-HCl [pH 6.5], 0.5 M dithiothreitol, 10% SDS, bromophenol blue, <50% glycerol). A total of 25 μg of protein was loaded per well and separated by 10% SDS-PAGE. Wet transfer onto Hybond-ECL membranes (GE Healthcare) was performed, followed by detection with antibodies against STAT1 (rabbit polyclonal; catalog number 61011; BD Transduction Laboratories), Tyr701-phosphorylated STAT1 (rabbit monoclonal, D4A7; Cell Signaling Technology), and β-actin (mouse monoclonal, C4; Santa Cruz Biotechnology) coupled to anti-rabbit–Cy5 and anti-mouse–Cy3 secondary antibodies (Amersham ECL Plex Western blotting system; GE Healthcare). Imaging of membranes was performed with a Typhoon scanner (GE Healthcare). Band intensities were quantified with ImageJ and normalized to the intensity of the β-actin band. Results were plotted as the ratio of P-STAT1 in infected versus mock-infected samples. Average results from triplicate samples, ± standard deviations (SD), are presented.
For detection of activated IRF-3, CT-2A-Fluc cells were infected at an MOI of 0.01, followed by sample collection 24 h p.i. and analysis as described above. Antibody against Ser396 -phosphorylated IRF-3 (rabbit monoclonal, 4D4G; Cell Signaling Technology) and β-actin (as described above) were used for detection.
Animal experiments.
All animal experiments were conducted under biosafety level 2 containment, following the guidelines of the National Committee of Animal Welfare. Intracranial CT-2A-Fluc gliomas were induced into adult (>4 weeks) female C57BL/6JOlaHsd mice (bred in the Kuopio animal facility) as described previously (12). Briefly, 5 × 104 CT-2A-Fluc cells were implanted, using a Hamilton syringe, into the caudate putamen of anesthetized mice (75 mg/kg of body weight ketamine [Intervet], 1 mg/kg medetomidine [Orion Pharma], and isoflurane gas [Baxter]). Mice were given the antisedative atipamezole (1 mg/kg; Orion Pharma) after the operation. Analgesia was applied via subcutaneously administered carprofen (5 mg/kg; ScanVet).
For measurement of CT-2A-Fluc tumor growth, mice were injected intraperitoneally with 150 mg/kg d-luciferin (as the potassium salt diluted in PBS; Caliper). Ten minutes after the d-luciferin injection, mice were imaged using an IVIS Lumina II (Caliper) bioluminescence imaging apparatus under isoflurane gas anesthesia. Images were taken using exposure times ranging from 2 to 60 s and with medium or large binning for best sensitivity. Additionally, ketamine/medetomidine anesthesia (as described above) was used to induce more stable sedation when required (e.g., when removing the fur from a mouse before imaging). Imaging was repeated at the indicated time points post-tumor induction. Image analysis and quantitation of the luciferase signal were done using Living Image software (Caliper). Magnetic resonance imaging (MRI) was conducted on mice under isoflurane anesthesia with a 9.4-T vertical magnet (Oxford Instruments) using a T2-weighted imaging sequence.
Mice showing a detectable luminescence signal at day 2 post-tumor induction were divided into groups that received an intraperitoneal injection of 1 × 106 PFU SFV4-miRT124, VA7-EGFP, or PBS. Single-dose virus or PBS was administered at day 3, after which mice were monitored daily for neurological symptoms or distress. Upon appearance of symptoms, such as paralysis, loss of over 20% of body weight, or notable tumor-caused distress, the mice were sacrificed. Additionally, mice were sacrificed for histological samples at days 4, 5, and 6 post-virus injection. Sacrificed mice were perfused with PBS. Collected tissues were immersed and fixed with cold 4% paraformaldehyde (PFA)–PBS overnight at +4°C. Fixed tissues were kept in 70% ethanol at +4°C until they were embedded into paraffin.
Immunohistochemistry.
Paraffin-embedded tissues were sliced into 7-μm sections by using a rotation microtome and stained using the Vectastain ABC kit (rabbit IgG; Vector Laboratories) and polyclonal rabbit antibody reactive against SFV structural proteins (produced in-house). A color reaction was achieved with horseradish peroxidase-conjugated secondary antibody (in the Vectastain kit) and 3,3′-diaminobenzidine (SigmaFast DAB; Sigma-Aldrich).
Immunocytochemistry.
For immunofluorescence staining, cells were grown on glass coverslips (in 24-well plates) coated with 10 μg/ml laminin (Sigma-Aldrich). Cells were fixed for 10 min with 4% PFA in PBS, washed with cold PBS, and kept at +4°C (in PBS) until staining. Free aldehyde groups were blocked by quenching with 50 mM NH4Cl (in PBS) for 10 min, followed by washing with 0.2% BSA–PBS. Permeabilization was done via a 2-min incubation in 0.1% Triton X-100 (in PBS) followed by washing. Serum samples from CT-2A-Fluc tumor-rejecting mice were pooled, diluted 1:100 in 3% BSA–PBS, and incubated for 1 h at room temperature, followed by washing with PBS. Pooled serum from three unimplanted mice was used as control serum. A secondary rabbit antibody conjugated to Alexa 594 (Life Technologies) was diluted 1:250 in PBS and incubated for 1 h at room temperature followed by washing with PBS. Nuclei were stained with 5 μM 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes) in PBS for 15 min followed by washing with PBS. Coverslips were mounted with Fluoroshield (Sigma-Aldrich) and stored at +4°C until analyzed via confocal microscopy (Zeiss LSM 700, 40× oil immersion objective).
Statistical analysis.
Survival was plotted via the Kaplan-Meier estimator of the Prism software (GraphPad Software). Mice that were sacrificed for reasons unrelated to CT-2A-Fluc glioma (i.e., no tumor detected by IVIS, MRI, or histology) were marked as censored. The number of mice that responded to the therapy was analyzed with Fisher's exact test (GraphPad Software). The increases in the bioluminescence signals (with the day 2 signal as the stranding point) for individual mice were quantified and plotted as geometric means ± the SD of the therapy group. In cases of signal disappearance, a value of 0.1 was assigned. Statistical analyses were performed with GraphPad Prism, using a two-tailed, unpaired Student's t test.
RESULTS
Recombinant SFV strains differ in type I interferon induction and sensitivity in glioma cells.
In order to assess the relative capacity of the parental and engineered SFV clones to induce secretion of IFN-I, we infected syngeneic C57/BL6 glioma CT-2A-Fluc cells with the attenuated vector VA7-EGFP, neurovirulent SFV4, or the derived SFV4-miRT124 (17) at an MOI of 0.01 and quantified the IFN-β present in cell culture supernatants 24 h later. As shown in Fig. 1A, SFV4 and SFV4-miRT124 induced greater amounts of IFN-β from CT-2A-Fluc cells than did VA7-EGFP. The increased IFN-β production correlated with IRF3 activation (Fig. 1B), which has been shown to mediate pattern recognition signaling in SFV infection (20).
FIG 1.

SFV4-miRT124 replicates despite the presence of IFN-I. (A) IFN-β measured from cell culture medium of infected CT-2A-Fluc cells. VA7-EGFP-induced IFN-β was significantly reduced compared to that with SFV4. SFV4-miRT124 induced significantly more IFN-β than VA7-EGFP or SFV4 (P < 0.001). Data are means of three replicates ± SD. (B) Ser396-phosphorylated IRF3 detected from infected or uninfected (mock) CT-2A-Fluc cells by Western blotting. (C) Plaque assay of CT-2A-Fluc cells. Both SFV and SFV4-miRT124 formed plaques in CT-2A-Fluc cultures (under agarose cover). (D) Plaque expansion of VA7-EGFP is increased when virus is administered with Jak inhibitor 1 (inh.1). (E) SFV4 and SFV4-miRT124 potently induce a cytopathic effect in mouse IFN-β-treated CT-2A-Fluc cells. Cells were infected with an MOI of 10. Cell viability was measured in an MTT assay. Data are presented as means (from 5 replicates) ± SD.
Because under standard cell culture conditions both the virus and secreted proteins, such as IFN-I, may freely diffuse into the supernatant from infected cells, potential low-level paracrine IFN-I signaling between neighboring cells (operating in solid tumors in vivo) may escape detection (10). To test this, we infected monolayers of CT-2A-Fluc cells in 6-well plates with 40 PFU of VA7-EGFP, SFV4, or miRT-124 and overlaid the cells with agarose to assess plaque formation over 3 days. We found that under agarose, VA7-EGFP infection was indeed self-limiting; producing smaller plaques than SFV4 and the microRNA-regulated SFV4-miRT124, which both produced large plaques with similar efficacies in the indicated time periods (Fig. 1C).
To affirm the role of IFN-I in restricting virus spread in CT-2A-Fluc cells under agarose, we performed a plaque expansion assay with VA7-EGFP in the presence or absence of JAK inhibitor I, a known IFN-I signaling blocker, as previously described (10). We found a marked enhancement of VA7-EGFP spread under agarose by this IFN-I-antagonist, supporting the hypothesis that in CT-2A-Fluc cells, VA7-EGFP spread is self-limiting due to IFN-I-dependent paracrine signaling (Fig. 1D). On the other hand, uninhibited plaque formation by SFV4 and its miRT derivative in CT-2A-Fluc cells (Fig. 1C) further showed that, despite induction of IFN-I (Fig. 1A), these viruses possess a greater capacity to resist its antiviral effects than attenuated VA7-EGFP. Quantification of the relative virus resistance to IFN-I was performed in a cell viability assay on infected CT-2A-Fluc cells treated with increasing doses of external murine IFN-β. This experiment revealed 50% protection with less than 0.5 U/ml of IFN-β for VA7-EGFP, whereas for SFV4 or SFV4-miRT124, protection was not reduced below 50% even at saturating IFN-β doses (Fig. 1E).
Virulent SFV4 and SFV4-miRT124 inhibit STAT1 phosphorylation.
In order to identify the underlying mechanisms that might contribute to the observed differential resistance to IFN-I, we analyzed the initial reactions of IFN-I activation in viral infection in CT-2A-Fluc and Vero(B) cells. Vero(B) cells were selected for the study as they show a defect in their IFN-I production, thus obviating the question about secondary IFN-I signaling triggered by infection or by external recombinant IFN-β (as shown in Fig. 1A). To address potential links between virulence in vivo and the capacity to resist the antiviral effects of IFN-I in Vero(B) cells in vitro, we used nonvirulent strain rA774 (19) and virulent strain SFV4, but we also included the SFV strain L10, which shows more severe neurovirulence than SFV4 (21). Despite our not having observed any phenotypic differences between VA7-EGFP and rA774, unmodified rA774 was selected to allow comparison with strains SFV4 and L10, which do not contain the EGFP expression cassette.
We found a statistically significant reduction in STAT1 Tyr701 phosphorylation (P-STAT1) in response to exogenous IFN-β at 6 h postinfection in cells infected with the SFV4, SFV4-mIRT124, or L10 virus compared to that with the attenuated rA774 virus (Fig. 2A and B). Notably, similar to Vero(B) cells, decreased levels of P-STAT1 were observed also in IFN-β-treated (1,000 U/ml) CT-2A-Fluc cells infected with SFV4 or SFV4-miRT124. Similar to parental rA774 virus, the recombinant expression vector VA7-EGFP was unable to block STAT-1 phosphorylation upon IFN-β treatment (Fig. 2C).
FIG 2.
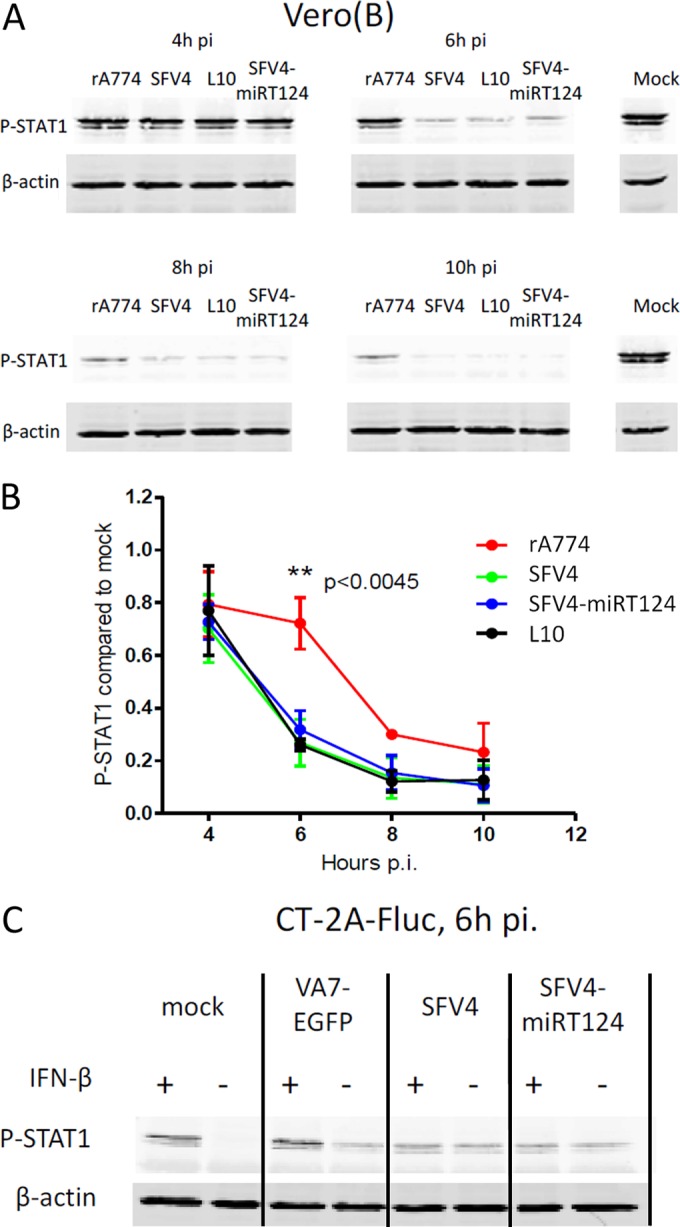
Virulent SFV inhibits STAT1 activation in infected cells. (A) Tyr701-phosphorylated STAT1 quantified in infected Vero(B) cells after stimulation with 1,000 U/ml human IFN-β. (B) rA774 showed significantly reduced potency for inhibiting STAT1 activation. Band intensities were quantified by using ImageJ. Data presented are means of three replicate analyses ± SD. (C) SFV4 and SFV4-miRT124 but not VA7-EGFP show P-STAT1 inhibition in CT-2A-Fluc cells. Results show phosphorylated STAT1 detected from CT-2A-Fluc cells that were either treated or not with 1,000 U/ml mouse IFN-β 6 h postinfection.
Taken together, the data in Fig. 1 and 2 show that virulent SFV strains SFV4, SFV4-mIRT124, and L10 are capable, on one hand, of tolerating the antiviral effectors induced by IFN-I signaling, and on the other hand, of blocking signal transduction from the IFN-I receptor during infection.
SFV4 nonstructural proteins are required for IFN-I-resistant replication.
As our findings highlighted clear differences in IFN-I tolerance between the SFV strains, we sought to find out whether the nonstructural (nsp) genes shown previously to govern SFV neuronal replication (22, 23) also mediate the IFN-I-tolerant phenotype. For this, we assessed the replication in CT-2A-Fluc cells untreated or pretreated with recombinant mouse IFN-β of three different SFV chimeras bearing SFV4 genes for nonstructural proteins nsp1-2, nsp3, or nsp3-nsp4 in the genome backbone of the neuro-attenuated rA774 (Fig. 3A). As shown in Fig. 3B, the increased capacity to resist the antiviral effects of IFN-β, measured by increased virus cytotoxicity, of SFV4 compared to rA774 was recapitulated in the CMW34 chimera, which had the rA774 nsp3 and nsp4 genes replaced with equivalents from the neurovirulent SFV4. In contrast, the chimeras CMW12 and CMW3 were unable to kill the IFN-I-treated cells, similar to rA774 (Fig. 3B). These results showed that the capacity of SFV4 to resist the antiviral effects of IFN-β resides within the nsP3-nsp4 region.
FIG 3.

SFV4 nonstructural genes required for IFN-I-resistant replication in CT-2A cells. (A) Schematic of the chimeric viruses we used. Segments marked in black were derived from neurovirulent SFV4. Numbers indicate nucleotide positions. (B) Cell viability was measured in an MTT assay 48 h after infection, with (black bars) or without (white bars) pretreatment with 1,000 units/ml IFN-β. Means ± SD from 4 parallel wells are presented.
SFV4-miRT124 retains the parental SFV4 oncolytic capacity in murine glioma cells in vitro.
As our aim was to exploit the increased IFN-I tolerance of neurovirulent SFV4 to target IFN-I-responsive tumors, we first confirmed in vitro that the microRNA manipulation did not reduce the oncolytic capacity of SFV4-miRT124 compared to parental SFV4 or VA7-EGFP. We infected CT-2A-Fluc cells under normal culture conditions with an MOI of 0.01 with VA7-EGFP, SFV4, or SFV4-miRT124 and analyzed the cells under the microscope. As shown in Fig. 4A, all three viruses exhibited extensive cytopathic effects (CPE) at 48 h to 72 h postinfection. All three viruses replicated in and killed confluent CT-2A-Fluc cultures with a similar efficacy, even at low MOIs, as quantified in a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay for cell viability (Fig. 4B) and with plaque titration for virus replication (Fig. 4C).
FIG 4.

VA7-EGFP, SFV4, and SFV4-miRT124 showed similar replication potencies in CT-2A-Fluc cells under normal culture conditions. (A) Phase-contrast and fluorescence microscopy images (overlaid) were taken before and 24, 48, and 70 h after infection (MOI, 0.01). (B) Cell viability was measured in an MTT assay 72 h postinfection with an MOI of 0.01. Means of 4 replicates ± SD are presented. (C) Infectious titers were determined 24 h postinfection from culture medium collected from 12-well plates seeded with 4 × 105 CT-2A-Fluc cells. Titers were measured from a pooled sample of three wells infected in parallel for the plaque assay using Vero(B) cells. Means of 3 replicate measurements ± SD are presented.
SFV4-miRT124 displays increased oncolytic potency in murine gliomas in vivo above that with attenuated VA7.
Prompted by the implications of our in vitro findings, we evaluated the infectivity and oncolytic capacities of SFV4-miRT124 virus in orthotopic CT-2A-Fluc tumors in a syngeneic C57BL/6 mouse model. A total of 5 × 104 CT-2A-Fluc cells were implanted intracranially into adult c57BL/6OlaHsd mice, and tumor development was monitored by bioluminescence imaging and small-animal MRI. Mice with a detectable tumor luminescence signal at day 2 post-tumor implantation (pti) were divided into therapy groups and administered intraperitoneally (i.p.) either 1 × 106 PFU of SFV4-miRT124 (n = 16) or VA7-EGFP (n = 15) in PBS 3 days after tumor implantation. A control group (n = 14) received an i.p. injection of PBS. VA7-EGFP was used as a control virus to SFV4-miRT124, instead of the parental strain SFV4, which is neuropathogenic.
A steady increase in luminescent signal was evident in all experimental groups between days 2 and 6 pti (Fig. 5A and C). At day 9 pti (6 days after virus injection), a complete disappearance of the luminescent signal was witnessed in 8/16 SFV4-miRT124-treated mice (Fig. 5A; Table 1), whereas in mice administered VA7-EGFP or PBS (control), the signal decayed in 3/15 and 2/14 mice, respectively (Table 1). MRI of therapy mice was performed to ensure that signal disappearance was not related to loss of Fluc expression in tumors (Fig. 5B). Indeed, the cessation of bioluminescence increase in treated mice revealed simultaneous reduction of tumor growth in mice infected with SFV4-miRT124 compared to those administered PBS or VA7-EGFP (Fig. 5C). In correlation with the reduced Fluc signals, there was an increase in survival for SFV4-miRT124 compared to the PBS group (Fisher's exact test, P = 0.03450 (Fig. 5D). The correlation between luminescence signals and the tumor sizes according to MRI results was good (Fig. 5E). For comparison, toluidine blue staining of a fully developed orthotopic CT-2A-Fluc glioma tumor is shown in Fig. 5F.
FIG 5.

Tumor growth and mouse survival. (A) Representative IVIS images of animals treated with SFV4-miRT124 or VA7-EGFP (1 × 106 PFU i.p. at day 3). Signal disappearance can be seen between day 6 and day 9 following SFV4-miRT124 therapy. (B) Representative MRIs of SFV4-miRT124- and VA7-EGFP-treated mice at day 22. A large tumor mass was clearly detectable following VA7-EGFP therapy (arrow). (C) The tumor-emitted luminescence signal was quantified as the average radiance (photons per second per square centimeter per steradian). The increase was measured as the fold change compared to the first measurement, performed at day 2. Data are plotted as geometric means ± SD. Undetectable signals were given a value of 0.1. Statistical analysis was done using an unpaired, two-tailed t test. Comparison results for VA7-EGFP or PBS treatments versus SFV4-miRT124 treatment are marked with stars or circles, respectively: *** or •••, P < 0.001; ** or ••, P < 0.01. (D) Kaplan-Meier survival plot for the mice. Statistical analysis used Fisher's exact test. *. P < 0.05. (E) Comparison of IVIS and MRI signal development in an untreated mouse. (F) Toluidine blue staining of tumor mass in mouse displaying endpoint symptoms.
TABLE 1.
Number of mice with complete responses (no tumors) or neurological symptoms
| Treatment (PFU) | No. (%) of mice showing: |
|
|---|---|---|
| Complete responsea | Severe neurological symptoms | |
| VA7-EGFP (1 × 106) | 3/15 (20) | None |
| SFV4-miRT124 (1 × 106) | 8/16 (50) | 3/24 (12.5) |
| PBS | 2/14 (13.4) | None |
A complete response was one in which the tumors disappeared.
SFV4-miRT124 infects orthotopic CT-2A-Fluc tumors but also shows sporadic replication in brain and spinal cord neurons.
Immunohistochemical analysis of mouse brains confirmed that the SFV4-miRT124 virus was able to reach and infect the intracranial CT-2A-Fluc tumors following intraperitoneal administration (Fig. 6A and B). This was in contrast to VA7-EGFP or PBS (uninfected control), for which we could not detect virus antigen in tumor cells in the brain (Fig. 6C and D).
FIG 6.

Immunohistochemical analysis of mouse brains; virus antigens in CT-2A-Fluc glioma tissue were measured from brain samples collected 5 days post-i.p. virus injection. (A) Tumor mass of SFV4-miRT124; (B) magnification of the inset marked in panel A. (C and D) Results in VA7-EGFP-treated mice (C) and PBS-treated mice (D). (E and F) Detection of virus replication in the brain and spinal cord following i.p. injection of SFV4 miRT124. Samples were collected from a mouse suffering from neurological symptoms (E) and an asymptomatic mouse (F).
As previously observed in BALB/c mice (17), intraperitoneal injection of SFV4-miRT124 resulted in neurological symptoms, including severe paralysis, in 12.5% of C57BL/6JOlaHsd mice in the present study (Table 1). In correlation with the neurological symptoms elicited by SFV4-miRT124 (Table 1), virus replication was detected in normal cells in the brains and spinal cords of these mice (Fig. 6E), suggesting viral escape from the miRNA control in a fraction of the infected mice. No SFV antigen was detected in normal CNS cells in asymptomatic mice (Fig. 6F).
Mice surviving CT-2A glioma show antitumor immune responses.
Long-term survival in response to oncolytic virotherapy has been shown to be associated with priming of a tumor-reactive adaptive immune response (24–26). To test whether this was also occurring in the present model, we rechallenged mice surviving the initial CT-2A-Fluc implantation with a second intracranial implantation of 5 × 104 CT-2A-Fluc cells. All mice showed a clear initial Fluc signal up to 2 days after tumor induction, indicative of successful tumor implantation, but signals rapidly decayed by day 6 pti (Fig. 7A). In order to confirm induction of a tumor-reactive adaptive immune response, we measured antibodies recognizing CT-2A-Fluc cells in sera collected from rechallenged mice at day 14 pti (Fig. 7B). The strongest reactivity was observed against CT-2A-Fluc cells, but notable antibody reactivity was also detected against CT-2A cells not expressing the firefly luciferase marker gene, and also against GL261 mouse glioma cells (Fig. 7B). A clearly weaker antibody response was obtained against B16-F10-LacZ melanoma cells.
FIG 7.

Mice surviving the initial CT-2A-Fluc inoculation become resistant to tumor rechallenge. (A) Mice imaged with the IVIS system after CT-2A-Fluc rechallenge. (B) C57BL/6 mouse tumor cell-reactive antibodies detected with immunofluorescence from sera of rechallenged mice. Blue, cell nuclei (stained with DAPI); red, tumor cell-reactive antibodies.
SFV4-miRT124 shows enhanced oncolysis in human glioblastoma cell lines despite exogenous IFN-β.
To estimate the possible clinical relevance of our findings with VA7-EGFP and SFV4-miRT124, we infected three malignant cell lines derived from clinical specimens from patients with GBM. Virus-induced CPE was measured in the presence and absence of human IFN-β to evaluate the IFN-I sensitivity of cell killing. Results were similar to those obtained with CT-2A-Fluc cells (Fig. 1E); SFV4-miRT124 displayed markedly increased cytotoxicity compared to VA7-EGFP, independent of IFN-β administration (Fig. 8). This suggested that human gliomas may possess a functional IFN-I defense, warranting engineering and use of oncolytic viruses, which display sufficient tolerance to antiviral tumor response, as effective therapeutics.
FIG 8.

SFV4-miRT124 induces increased CPE in human glioblastoma cell lines. Cell viability was measured in an MTT assay 48 h postinfection (MOI, 10). Human recombinant IFN-β was administered simultaneously with virus. Means of 4 replicates ± SD are presented. The statistical analysis was done with Student's t test: ***, P < 0.001; **, P < 0.01.
DISCUSSION
In order to avoid unwanted replication in healthy cells, virulence factors of oncolytic viruses are commonly deleted or mutated. As these factors typically confer resistance to IFN-I-mediated antiviral responses, such oncolytic viruses are exquisitely dependent on the lack of antiviral IFN-I responses in cancer cells (27). However, the human GBM cell lines analyzed by us (Fig. 8) and others (13) show functional IFN-I signaling. Of note, even a small fraction of cells in the heterogeneous tumor tissue capable of IFN-I signaling can promote resistance to virotherapy (8).
To address these issues, in this study we explored whether we could harness the IFN-I-tolerant phenotype of a wild-type virulent alphavirus to target IFN-I-responsive tumors. Neurovirulent SFV4 has previously shown a mild benefit against subcutaneous K-BALB and CT26 tumors when administered intratumorally (28). However, in order to protect the animals from SFV4-associated neurotoxicity, the animals in that study were preimmunized with replication-deficient SFV, which likely limited virus replication during actual therapy and may also affect the antitumor immune responses in unexpected ways. In the present study, we used a neuronally detargeted derivative of the SFV4 virus, SFV4-miRT124, whose replication in nonneuronal (glioma) cells was expected to be uncompromised in targeting orthotopic glioma in naive animals. The current results indeed confirmed that the SFV4-miRT124 virus retained full replicative capacity and the IFN-I-tolerant phenotype of its parental strain (Fig. 1 to 4), and it was able to infect and destroy IFN-I-responsive gliomas in adult immunocompetent mice (Fig. 5). This was in striking contrast to our previous oncolytic virus candidate VA7-EGFP, which was based on an IFN-I-sensitive strain of SFV.
The molecular mechanisms behind the IFN-I-resistant phenotype of SFV4 remain incompletely understood. Our results indicate that SFV4 and SFV-miRT124, similar to neurovirulent Sindbis virus (14) and Venezuelan equine encephalitis virus (15), are capable of inhibiting cell signaling via the JAK/STAT pathway (Fig. 2). However, this effect was seen at later time points after infection, and thus is unlikely to explain the increased replication seen also in IFN-I-pretreated CT-2A-Fluc and human GBM cells (Fig. 1E and 8). By using previously described SFV chimeras (22), we could identify viral genomic regions responsible for mediating IFN-I insensitivity. We found that a region containing the nsp3 and nsp4 genes was necessary (Fig. 3). These genes encode a multidomain protein with unknown function and the viral RNA polymerase, respectively. As expected due to its lethal phenotype (22), the chimera bearing both nsp3 and nsp4 showed IFN-I tolerance. Interestingly, however, the CMW3 chimera, carrying solely the nsP3 gene from the virulent SFV4, was found to be sensitive to IFN-I (Fig. 3) despite being highly neurovirulent for adult mice (22). Thus, the nsp3-dependent neurovirulence factors are not sufficient and perhaps not at all overlapping with those governing SFV replication in IFN-I-stimulated glioma cells. This notion is in agreement with earlier results showing that the replication of nonvirulent SFV A7(74) is dramatically increased in peripheral tissues and nonneuronal CNS cells, but not in neurons of IFN-I receptor-deficient mice (11, 29). Moreover, we found no difference in STAT1 inhibition between L10 and SFV4 that would explain the increased neurovirulence of L10 (Fig. 2A and B).
Neurovirulence of other alphaviruses may also be independent of IFN-I signaling, as exemplified by Yin et al., who observed robust neuronal replication of Venezuelan equine encephalitis virus despite active IFN-I signaling in these cells (30). Thus, efficient replication of SFV4 and SFV-miRT124 viruses in CT-2A-Fluc glioma cells pretreated with IFN-I could be related to resistance of these viruses to one or more specific antiviral interferon-stimulated genes (ISGs), such as ZAP, Viperin, IRF1, ISG20, and ISG15, previously shown to possess an antialphavirus effector function (31–33). Alternatively, the neurovirulent SFV4 virus may be resistant to IFN-I antiviral effects in glioma cells as a result of adaptation to IFN-I-responsive cells (oligodendrocytes and astrocytes) by extensive passaging in adult mouse brains, as opposed to results with an attenuated VA7 virus when it was serially passaged in suckling mouse brains (34).
In correlation with the stronger IRF-3 phosphorylation, SFV4-miRT124 induced significantly larger amounts of IFN-β in CT-2A cells than did VA7-EGFP or parental SFV4 (Fig. 1A and B). As the replication rate of all viruses under normal culture conditions was found equal by both MTT assay and titration (Fig. 4), the results lead to the conclusion that the inserted miRT124 element promotes cytoplasmic recognition of the virus (likely mediated by RIG-I and MDA5). Of note, IFN-I expression has been shown to strongly enhance tumor antigen cross-presentation to naive CD8+ T cells by dendritic cells, thus promoting cytotoxic T cell priming and tumor rejection (35). In this context, a replicating therapy virus capable of inducing strong IFN-I production in tumor cells, while still being resistant to the antiviral effects imposed by IFN-I, would offer an attractive tool for immunovirotherapy.
When given i.p. to orthotopic CT-2A-Fluc tumor-bearing mice, SFV4-miRT124, but not VA7-EGFP, caused significant retardation of tumor growth (Fig. 5). Additionally, with SFV4-miRT124 treatment, we witnessed a complete disappearance of tumor signal in 50% of the mice (Fig. 5; Table 1). As the reduction of tumor luminescence signal was evident 5 to 6 days post-virus injection, overlapping with the peak CNS viremia, it is likely that the growth inhibition was primarily mediated via direct virus replication in the tumor cells. Supporting this hypothesis, viral antigens could be detected in tumors of SFV4-miRT124-treated mice, whereas VA7-EGFP was unable to infect the tumors (Fig. 6). The tumor clearance was accompanied by a resistance to rechallenge and generation of CT-2A-reactive antibodies in the mice (Fig. 7). The small fraction of animals treated with VA7-EGFP or injected with PBS that showed tumor eradication could possibly be explained by the immunogenicity of the engineered CT-2A-Fluc cells. In fact, resistance to CT-2A-Fluc rechallenge was observed in all mice that survived the initial tumor inoculation, and antitumor antibodies emerged in these animals (Fig. 7B). The presence of multiepitope antitumor antibodies in the sera of virus-treated mice, combined with the larger number of surviving animals in the SFV4-miRT124 group compared to PBS controls, suggests that the SFV4-miRT124 therapy promotes host immune reactivity against the CT-2A-Fluc glioma. However, the contribution of the adaptive immune responses in tumor eradication relative to virus replication-associated oncolysis remains to be elucidated in future studies.
There are only a few reports of oncolytic virotherapy resulting in long-term survival in syngeneic glioma models. Barnard et al. observed long-term survival in 40% of CT-2A-bearing mice following intratumoral injection of oncolytic herpes simplex virus expressing immunostimulatory Flt3-L (36). Similarly, Muik et al. showed long-term survival in 50% of CT-2A-bearing mice treated with intracranial dosing of neuroattenuated vesicular stomatitis virus (37). Before the present study, only parvovirus H-1 virus was shown to be efficient against syngeneic gliomas when administered intravenously. As shown by Geletneky et al., a long-term response was gained in 6/9 RG2 glioma-bearing rats when they were administered the virus on 8 consecutive days (38). These studies corroborated the findings in our present report that indicated robust virus replication in the tumor tissue is a prerequisite for effective therapy.
As noted also in our previous work (17), SFV4-miRT124 infection caused neurological symptoms in a small fraction of the mice (Table 1). Virus antigen was detected in brains and spinal cords of these affected mice, suggesting that either miR124 expression was limited in these CNS regions (39) or that the vector had lost its miR-target sequence or the sequence had mutated. Of note, there was no positive correlation between neurological symptoms and tumor clearance, indicating that miRT124-controlled SFV nonpathogenic replication is sufficient for the observed therapeutic response. In our previous work, we could isolate virus from mouse brain with deleted miR124 target sites (17). Although such events were rare, our future efforts will be directed to the evaluation of the stability of the insertions, and we seek to test whether alternative and multiple/mixed miRNA targets could improve the vector safety. An alternative (nonexclusive) approach would be to create a novel IFN-I-tolerant, neuroattenuated SFV4/VA7 chimera, which is supported by the observation that IFN-I resistance is not prerequisite for neurovirulence.
Taken together, to our knowledge this is the first time that a single dose of oncolytic virus administered peripherally has been shown to reach and replicate in a syngeneic IFN-I-responsive murine glioma.
ACKNOWLEDGMENTS
We thank the A. I. Virtanen Institute NMR group, led by Olli Gröhn, for support with MRIs, Kirsi Rilla (University of Eastern Finland) and Kirsi Hellström and Tero Ahola (University of Helsinki) for help with confocal microscopy techniques, and also research nurses Katariina Helin, Marita Voutilainen, and Minna Rautiainen (Kuopio University Hospital) for coordinating issues related to patient samples.
This research was partially funded by the Academy of Finland, University Strategic Funding for the Cancer Center of Eastern Finland, Oskar Öflund Foundation, Kuopio University Foundation, State Funding for University Hospitals, Medicinska Understödsföreningen Liv och Hälsa, Finnish Cancer Foundations, Foundation for Research on Viral Diseases, Maud Kuistila Memorial Foundation, Emil Aaltonen Foundation, Cancer Society of North Savo, Finnish Cultural Foundation North Savo Regional Fund, and the Doctoral Program of Molecular Medicine (A. I. Virtanen Institute for Molecular Sciences).
REFERENCES
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Chiocca EA, Rabkin SD. 2014. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res 2:295–300. doi: 10.1158/2326-6066.CIR-14-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, Nabors LB, Markiewicz M, Lakeman AD, Palmer CA, Parker JN, Whitley RJ, Gillespie GY. 2009. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre- and post-tumor resection for recurrent GBM. Mol Ther 17:199–207. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kicielinski KP, Chiocca EA, Yu JS, Gill GM, Coffey M, Markert JM. 2014. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol Ther 22:1056–1062. doi: 10.1038/mt.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freeman A, Zakay-Rones Z, GomoriI J, Linetsky E, Rasooly L, Greenbaum E, Rozenman-Yair S, Panet A, Libson E, Irving C, Galun E, Siegal T. 2006. Phase I/II trial of Intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther 13:221–228. doi: 10.1016/j.ymthe.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 6.Goetz C, Gromeier M. 2010. Preparing an oncolytic poliovirus recombinant for clinical application against glioblastoma multiforme. Cytokine Growth Factor Rev 21:197–203. doi: 10.1016/j.cytogfr.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vähä-Koskela M, Hinkkanen A. 2014. Tumor restrictions to oncolytic virus. Biomedicines 2:163–194. doi: 10.3390/biomedicines2020163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruotsalainen JJ, Kaikkonen MU, Niittykoski M, Martikainen MW, Lemay CG, Cox J, De Silva NS, Kus A, Falls TJ, Diallo J-S, Le Boeuf F, Bell JC, Ylä-Herttuala S, Hinkkanen AE, Vähä-Koskela MJ. 2015. Clonal variation in interferon response determines the outcome of oncolytic virotherapy in mouse CT26 colon carcinoma model. Gene Ther 22:65–75. doi: 10.1038/gt.2014.83. [DOI] [PubMed] [Google Scholar]
- 9.Heikkilä JE, Vähä-Koskela MJV, Ruotsalainen JJ, Martikainen MW, Stanford MM, McCart JA, Bell JC, Hinkkanen AE. 2010. Intravenously administered alphavirus vector VA7 eradicates orthotopic human glioma xenografts in nude mice. PLoS One 5:e8603. doi: 10.1371/journal.pone.0008603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vähä-Koskela MJV, Le Boeuf F, Lemay C, De Silva N, Diallo J-S, Cox J, Becker M, Choi Y, Ananth A, Sellers C, Breton S, Roy D, Falls T, Brun J, Hemminki A, Hinkkanen A, Bell JC. 2013. Resistance to two heterologous neurotropic oncolytic viruses, Semliki Forest virus and vaccinia virus, in experimental glioma. J Virol 87:2363–2366. doi: 10.1128/JVI.01609-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fragkoudis R, Breakwell L, McKimmie C, Boyd A, Barry G, Kohl A, Merits A, Fazakerley JK. 2007. The type I interferon system protects mice from Semliki Forest virus by preventing widespread virus dissemination in extraneural tissues, but does not mediate the restricted replication of avirulent virus in central nervous system neurons. J Gen Virol 88:3373–3384. doi: 10.1099/vir.0.83191-0. [DOI] [PubMed] [Google Scholar]
- 12.Ruotsalainen J, Martikainen M, Niittykoski M, Huhtala T, Aaltonen T, Heikkilä J, Bell J, Vähä-Koskela M, Hinkkanen A. 2012. Interferon-β sensitivity of tumor cells correlates with poor response to VA7 virotherapy in mouse glioma models. Mol Ther 20:1529–1539. doi: 10.1038/mt.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alain T, Lun X, Martineau Y, Sean P, Pulendran B, Petroulakis E, Zemp FJ, Lemay CG, Roy D, Bell JC, Thomas G, Kozma SC, Forsyth PA, Costa-Mattioli M, Sonenberg N. 2010. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc Natl Acad Sci U S A 107:1576–1581. doi: 10.1073/pnas.0912344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simmons JD, Wollish AC, Heise MT. 2010. A determinant of Sindbis virus neurovirulence enables efficient disruption of Jak/STAT signaling. J Virol 84:11429–11439. doi: 10.1128/JVI.00577-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simmons JD, White LJ, Morrison TE, Montgomery SA, Whitmore AC, Johnston RE, Heise MT. 2009. Venezuelan equine encephalitis virus disrupts STAT1 signaling by distinct mechanisms independent of host shutoff. J Virol 83:10571–10581. doi: 10.1128/JVI.01041-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deuber SA, Pavlovic J. 2007. Virulence of a mouse-adapted Semliki Forest virus strain is associated with reduced susceptibility to interferon. J Gen Virol 88:1952–1959. doi: 10.1099/vir.0.82264-0. [DOI] [PubMed] [Google Scholar]
- 17.Ylösmäki E, Martikainen M, Hinkkanen A, Saksela K. 2013. Attenuation of Semliki Forest virus neurovirulence by microRNA-mediated detargeting. J Virol 87:335–344. doi: 10.1128/JVI.01940-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karsy M, Arslan E, Moy F. 2012. Current progress on understanding microRNAs in glioblastoma multiforme. Genes Cancer 3:3–15. doi: 10.1177/1947601912448068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vähä-Koskela MJV, Tuittila MT, Nygårdas PT, Nyman JK-E, Ehrengruber MU, Renggli M, Hinkkanen AE. 2003. A novel neurotropic expression vector based on the avirulent A7(74) strain of Semliki Forest virus. J Neurovirol 9:1–15. doi: 10.1080/13550280390173382. [DOI] [PubMed] [Google Scholar]
- 20.Baigent SJ, Zhang G, Fray MD, Flick-Smith H, Goodbourn S, McCauley JW. 2002. Inhibition of beta interferon transcription by noncytopathogenic bovine viral diarrhea virus is through an interferon regulatory factor 3-dependent mechanism. J Virol 76:8979–8988. doi: 10.1128/JVI.76.18.8979-8988.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atkins GJ, Sheahan BJ, Liljeström P. 1999. The molecular pathogenesis of Semliki Forest virus: a model virus made useful? J Gen Virol 80:2287–2297. [DOI] [PubMed] [Google Scholar]
- 22.Tuittila MT, Santagati MG, Röyttä M, Määttä JA, Hinkkanen AE. 2000. Replicase complex genes of Semliki Forest virus confer lethal neurovirulence. J Virol 74:4579–4589. doi: 10.1128/JVI.74.10.4579-4589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuittila M, Hinkkanen AE. 2003. Amino acid mutations in the replicase protein nsP3 of Semliki Forest virus cumulatively affect neurovirulence. J Gen Virol 84:1525–1533. doi: 10.1099/vir.0.18936-0. [DOI] [PubMed] [Google Scholar]
- 24.Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, De Vleeschouwer S, Agostinis P, Graf N, Van Gool SW. 2015. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer 136:E313–E325. doi: 10.1002/ijc.29202. [DOI] [PubMed] [Google Scholar]
- 25.Jiang H, Clise-Dwyer K, Ruisaard KE, Fan X, Tian W, Gumin J, Lamfers ML, Kleijn A, Lang FF, Yung W-KA, Vence LM, Gomez-Manzano C, Fueyo J. 2014. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS One 9:e97407. doi: 10.1371/journal.pone.0097407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grekova SP, Raykov Z, Zawatzky R, Rommelaere J, Koch U. 2012. Activation of a glioma-specific immune response by oncolytic parvovirus minute virus of mice infection. Cancer Gene Ther 19:468–475. doi: 10.1038/cgt.2012.20. [DOI] [PubMed] [Google Scholar]
- 27.Russell SJ, Peng K-W, Bell JC. 2012. Oncolytic virotherapy. Nat Biotechnol 30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smyth JWP, Fleeton MN, Sheahan BJ, Atkins GJ. 2005. Treatment of rapidly growing K-BALB and CT26 mouse tumours using Semliki Forest virus and its derived vector. Gene Ther 12:147–159. doi: 10.1038/sj.gt.3302390. [DOI] [PubMed] [Google Scholar]
- 29.Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 30.Yin J, Gardner CL, Burke CW, Ryman KD, Klimstra WB. 2009. Similarities and differences in antagonism of neuron alpha/beta interferon responses by Venezuelan equine encephalitis and Sindbis alphaviruses. J Virol 83:10036–10047. doi: 10.1128/JVI.01209-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karki S, Li MMH, Schoggins JW, Tian S, Rice CM, MacDonald MR. 2012. Multiple interferon stimulated genes synergize with the zinc finger antiviral protein to mediate anti-alphavirus activity. PLoS One 7:e37398. doi: 10.1371/journal.pone.0037398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Burke CW, Ryman KD, Klimstra WB. 2007. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J Virol 81:11246–11255. doi: 10.1128/JVI.01282-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teng T-S, Foo S-S, Simamarta D, Lum F-M, Teo T-H, Lulla A, Yeo NKW, Koh EGL, Chow A, Leo Y-S, Merits A, Chin K-C, Ng LFP. 2012. Viperin restricts Chikungunya virus replication and pathology. J Clin Invest 122:4447–4460. doi: 10.1172/JCI63120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bradish CJ, Allner K, Maber HB. 1971. The virulence of original and derived strains of Semliki Forest virus for mice, guinea-pigs and rabbits. J Gen Virol 12:141–160. doi: 10.1099/0022-1317-12-2-141. [DOI] [PubMed] [Google Scholar]
- 35.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD. 2011. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barnard Z, Wakimoto H, Zaupa C, Patel AP, Klehm J, Martuza RL, Rabkin SD, Curry WT. 2012. Expression of FMS-like tyrosine kinase 3 ligand by oncolytic herpes simplex virus type I prolongs survival in mice bearing established syngeneic intracranial malignant glioma. Neurosurgery 71:741–748. doi: 10.1227/NEU.0b013e318260fd73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muik A, Stubbert LJ, Jahedi RZ, Geiβ Y, Kimpel J, Dold C, Tober R, Volk A, Klein S, Dietrich U, Yadollahi B, Falls T, Miletic H, Stojdl D, Bell JC, von Laer D. 2014. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res 74:3567–3578. doi: 10.1158/0008-5472.CAN-13-3306. [DOI] [PubMed] [Google Scholar]
- 38.Geletneky K, Kiprianova I, Ayache A, Koch R, Herrero Calle YM, Deleu L, Sommer C, Thomas N, Rommelaere J, Schlehofer JR. 2010. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro Oncol 12:804–814. doi: 10.1093/neuonc/noq023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pena JTG, Sohn-Lee C, Rouhanifard SH, Ludwig J, Hafner M, Mihailovic A, Lim C, Holoch D, Berninger P, Zavolan M, Tuschl T. 2009. miRNA in situ hybridization in formaldehyde and EDC-fixed tissues. Nat Methods 6:139–141. doi: 10.1038/nmeth.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]