

ABSTRACT
The NS1 protein of influenza virus has multiple functions and is a determinant of virulence. Influenza viruses with NS1 deletions (DelNS1 influenza viruses) are a useful tool for studying virus replication and can serve as effective live attenuated vaccines, but deletion of NS1 severely diminishes virus replication, hampering functional studies and vaccine production. We found that WSN-DelNS1 viruses passaged in cells consistently adapted to gain an A14U substitution in the 3′ noncoding region of the M segment of viral RNA (vRNA) which restored replicative ability. DelNS1-M-A14U viruses cannot inhibit interferon expression in virus infected-cells, providing an essential model for studying virus replication in the absence of the NS1 protein. Characterization of DelNS1-M-A14U virus showed that the lack of NS1 has no apparent effect on expression of other viral proteins, with the exception of M mRNAs. Expression of the M transcripts, M1, M2, mRNA3, and mRNA4, is regulated by alternative splicing. The A14U substitution changes the splicing donor site consensus sequence of mRNA3, altering expression of M transcripts, with M2 expression significantly increased and mRNA3 markedly suppressed in DelNS1-M-A14U, but not DelNS1-M-WT, virus-infected cells. Further analysis revealed that the A14U substitution also affects promoter function during replication of the viral genome. The M-A14U mutation increases M vRNA synthesis in DelNS1 virus infection and enhances alternative splicing of M2 mRNA in the absence of other viral proteins. The findings demonstrate that NS1 is directly involved in influenza virus replication through modulation of alternative splicing of M transcripts and provide strategic information important to construction of vaccine strains with NS1 deletions.
IMPORTANCE Nonstructural protein (NS1) of influenza virus has multiple functions. Besides its role in antagonizing host antiviral activity, NS1 is also believed to be involved in regulating virus replication, but mechanistic details are not clear. The NS1 protein is a virulence determinant which inhibits both innate and adaptive immunity and live attenuated viruses with NS1 deletions show promise as effective vaccines. However, deletion of NS1 causes severe attenuation of virus replication during infection, impeding functional studies and vaccine development. We characterized a replication-competent DelNS1 virus which carries an A14U substitution in the 3′ noncoding region of the vRNA M segment. We found that M-A14U mutation supports virus replication through modulation of alternative splicing of mRNAs transcribed from the M segment. Our findings give insight into the role of NS1 in influenza virus replication and provide an approach for constructing replication-competent strains with NS1 deletions for use in functional and vaccine studies.
INTRODUCTION
Influenza A virus is an important human respiratory tract pathogen which causes annual epidemics and occasional pandemics (1, 2). The influenza A virus genome contains eight negative-sense single-stranded RNA segments (3). The segmented genome allows frequent reassortment events to occur between different influenza viruses, which can lead to altered pathogenic and transmission properties in reassortant viruses (1, 4). The influenza A virus replication process is regulated by host and viral factors during the infection cycle (5, 6). Entry of influenza virus into cells is initiated by attachment of viral hemagglutinin (HA) to cellular sialic acid receptors, which mediates an endocytotic process to release the viral genome into the cytoplasm. The viral genome is subsequently imported into the nucleus via an importin-α/importin-β1-dependent nuclear import pathway, where the influenza virus utilizes genome-bound viral RNP polymerase complex and host transcription machinery to replicate the viral genome and express viral mRNA for protein synthesis (6). At least 14 viral proteins have been identified from virus-infected cells to date (3, 7–11). Among these viral proteins, PB1, PB2, PA, NP, HA, NA, M1, M2, NS1, and NS2 (NEP) are regularly expressed in infected cells. Expression of PB1-F2, PB1-N40, and PA-X is less consistently observed and may be associated with viral strain and infection conditions (9–12). Expression of an M2-related protein, M42, was also reported during infection with virus mutated at the mRNA4 splice donor site in the M segment transcript (7). However, one study reported that many influenza A virions may fail to express at least one essential viral protein and become replication-incompetent infectious particles (13). Expression of viral proteins is subject to both viral and host controls. It is believed that differential regulation of viral protein expression results in differences in the replication efficiency of virus, leading to variable pathogenic outcomes of infection.
Among the eight genome segments of influenza A virus, both the NS and M segments express differentially spliced transcripts. Two matured mRNAs, NS1 and NS2 (NEP), and four mRNAs, M1, M2, M3, and M4, are expressed from the NS and M segments, respectively (3). While there are four differentially spliced transcripts from the M segment, only the matrix (M1) and ion channel (M2) proteins have known roles in influenza A virus infection. M1 and M2 have essential functions in viral nuclear export, virion packaging, and budding during virus replication (14–16). No known function has been found for the other two mRNAs derived from the M segment, mRNA3 and mRNA4. A study of viral mRNA kinetics found that accumulation of M1 and M2 mRNA is regulated during virus infection of cells (17). A similar phenomenon is also observed in NS1 and NS2 (NEP) splicing regulation, which is coordinated with the progress of virus infection (18). It is important to understand how these differentially spliced forms of mRNAs are regulated in influenza virus infection. A study by Shih et al. reported that the viral polymerase complex regulates the utilization of alternative 5′ splice sites in influenza virus M1 mRNA, in coordination with the cellular splicing factor SF2/ASF, to control the expression of M2 mRNA during virus replication (19, 20). However, another study showed that the NS1 protein regulates splicing of M segment mRNAs and that this activity requires NS1 to possess RNA binding function (21). Adaptive substitutions were reported to be gained in the M segment through passage of a reassortant virus containing H5 and N1 from A/turkey/Turkey/1/05 and the remaining segments from the A/PR/8/34 strain, but with NS1 deleted (22). While the specific functions of these M segment substitutions were not characterized, the studies seem to suggest that there is an interaction between NS1 and M functions in virus replication (21, 22).
Although the NS1 protein is not essential for viral replication, it has multiple functions, and deletion of the NS1 gene leads to severe attenuation of influenza virus replication (23). Numerous studies on the biology of NS1 have focused on its functions in antagonism of host antiviral activity (24). It is believed that the attenuation of DelNS1 virus may be due to a loss of ability to inhibit host expression of interferon (IFN), since virus without NS1 is able to replicate normally in interferon-deficient cells (25, 26). However, several studies have shown that NS1 may also be involved in regulation of influenza virus transcription and replication through other mechanisms (27, 28). To delineate the mechanism of NS1 protein function in regulation of virus replication, we employed a system where we created DelNS1 influenza viruses derived from the A/WSN/33 and A/PR/8/34 strains. While deletion of the NS1 gene usually led to severe attenuation of viruses in interferon-competent systems, we discovered that an adaptive substitution, A14U, in the 3′ noncoding region (NCR) of the M segment of viral RNA (vRNA) significantly enhances the replication of DelNS1 viruses. Further characterization revealed that DelNS1 viruses are unable to express sufficient amounts of M2 mRNA, probably due to the absence of NS1 function, and that the M-A14U substitution restores M2 expression.
MATERIALS AND METHODS
Cells and viruses.
Human embryonic kidney (HEK) 293T (ATCC) and Vero cells (ATCC) were cultured at 37°C in Dulbecco's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate (Life Technologies), while MDCK (ATCC) cells were grown in Eagle's minimal essential medium (MEM) supplemented with the same amounts of serum and antibiotics. All influenza A viruses used in this study were rescued by reverse genetics and amplified in MDCK cells, as described previously (28, 29). Viruses were purified using Amicon Ultra-15 centrifugal filter units (100KD) (Millipore) to remove the cytokines in the medium. Sendai virus (SeV) was propagated in embryonated chicken eggs.
Plasmid construction.
The NS1 deletion plasmid was constructed according to the protocol described in a previous report (25). In brief, inverse PCR was carried out to delete the intron of the NS gene inserted into the pHW2000 vector (a kind gift provided by Robert Webster of St. Jude Children's Research Hospital, USA), and the plasmid was phosphorylated and self-ligated (29, 30). Primers for inverse PCR were 5′-GACATACTGATGAGGATGTCAAAAATG-3′ (NS-529F) and 5′-CTGAAAGCTTGACACAGTGTTTGG-3′ (NS-56R). For point mutations, the QuikChange II site-directed mutagenesis kit (Stratagene) was used.
Passage of the DelNS1 virus.
Blind serial passage of the DelNS1 virus was performed in this study. The DelNS1 virus was first rescued by cotransfecting eight pHW2000 plasmids containing the eight segments of the influenza virus genome into a HEK293T/MDCK mixed cell culture; the supernatant was subsequently collected at 72 h posttransfection and designated passage 0 (P0) virus. The P0 virus obtained from the rescue procedure was used to infect MDCK cells in a T25 flask at 37°C. Two or three days later, the supernatant was transferred to infect fresh MDCK cells. Virus titers were measured at each passage. After 5 passages, when the virus titer in the subculture had stabilized, a full genome sequence of each of the DelNS1 virus passages was obtained and analyzed.
Reporter assay.
Dual luciferase reporter assays were carried out as previously described (29). HEK293T cells were seeded onto 48-well plates and cotransfected with 50 ng each of the tested plasmids and firefly luciferase reporter containing noncoding regions from the M segment (29), together with 10 ng Renilla luciferase reporter as a control. After 24 h of culture, luciferase activity was measured according to the instructions of the manufacturer (Promega). For the IFN-β promoter activity reporter assay, cells were infected with the indicated viruses at a multiplicity of infection (MOI) of 1 (influenza A virus) or 50 HA units (Sendai virus) at 24 h posttransfection. All firefly luciferase values were normalized using the Renilla luciferase values.
Growth kinetics.
MDCK or Vero cells at 80 to 100% confluence seeded in 24-well plates were infected with the indicated viruses at an MOI of 0.1. After absorption for 1 h, the supernatant was removed and cells washed twice with 500 μl phosphate-buffered saline (PBS). Infected cells were overlaid with MEM containing 1 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Sigma) and incubated at 37°C. Supernatants were collected at the indicated time points and the virus titer determined by plaque assay in MDCK cells.
Plaque assay.
Tenfold serial dilutions of each virus to be tested were made in MEM. Confluent MDCK cells seeded onto 6-well plates were inoculated with virus for adsorption at 37°C for 1 h before supernatant was removed. Cells were washed twice with PBS and then overlaid with 1% MEM agarose containing 1 μg/ml TPCK-treated trypsin. Plates were placed upside down in a 37°C incubator for 48 h. Plates were then fixed with 25% formalin for at least 2 h at room temperature. After staining with 1% crystal violet in 20% ethanol, plates were washed with tap water to remove excess dye. Plaques were visualized with the naked eye and counted. In our hands, the plaque assay detects influenza virus at concentrations of ≥1 PFU/ml.
Western blotting.
Western blotting of influenza virus proteins was performed as previously described (27). Briefly, MDCK cells were infected with the indicated viruses at an MOI of 5 and lysed at different time points with cell lysis buffer (50 mM Tris-HCl, 150 mM NaCl, and 1% Triton X-100, pH 7.4). Cell debris was discarded after centrifugation at a speed of 12,000 × g for 15 min. Native polyacrylamide gel electrophoresis (N-PAGE) was performed as follows. Briefly, a 7 to 8% polyacrylamide gel was made without the addition of SDS and with the stacking gel omitted. Samples for N-PAGE were lysed with the passive lysis buffer used in the luciferase assay, as previously described (29). After mixing with 5× loading buffer (1 M Tris-HCl [pH 6.8], 50% glycerol, 1% bromophenol blue), samples were either stored at −80°C or analyzed immediately, with the gel being prerun for 30 min at a constant current of 40 mA at 4°C before loading samples. Gels were then processed in a manner similar to that for SDS-PAGE. Mouse monoclonal anti-M1 (sc-57881) and rabbit polyclonal anti-IRF3 (sc-9082) antibodies were purchased from Santa Cruz Biotechnology. Mouse monoclonal anti-M2 (ab5416) was purchased from Abcam. Mouse monoclonal anti-β-tubulin was purchased from Sigma. NP, HA, and NS1 were detected using laboratory-made antibodies at dilutions of 1:5,000, 1:3,000, and 1:5,000, respectively.
qRT-PCR.
Quantitative real-time PCR (qRT-PCR) was performed as previously described (29). At the indicated time points after infection or transfection, total RNA was isolated using RNAiso (TaKaRa). DNA contamination was removed by DNase (Ambion) treatment. Approximately 200 ng total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Life Technologies). The Uni12 and oligo(dT) primers were used for preparing mRNA and vRNA, respectively, in reverse transcription reactions. The SYBR Premix Ex Taq kit (TaKaRa) was used for real-time PCR. Primers for WSN-M1 were 5′-CGGTCTCATAGGCAAATGGT-3′ (M-478F) and 5′-CAATATCCATGGCCTCTGCT-3′ (M-616R). Primers for WSN-M2 were 5′-CCGAGGTCGAAACGCCTATC-3′ (WSN-M2-F) and 5′-CTCTGGCACTCCTTCGGTAG-3′ (WSN-M2-R). The forward primer for WSN-mRNA3 was 5′-AGCAAAAGCAGGCCTATC-3′ (WSN-mRNA3-F), and the forward primer for WSN-M4 was 5′-ACCGATCTTGAGGCCTATC-3′. The reverse primer used for WSN-mRNA3 and WSN-M4 was WSN-M2-R. The forward primer for PR8-M1 was the same as the WSN-M1 forward primer, M-478F, and the reverse primer was 5′-CAACCTCCATGGCCTCTGCT-3′. The reverse primer for PR8-M2 and PR8-mRNA3 was 5′-CTTTGGCACTCCTTCCGTAG-3′, and forward primers were WSN-M2-F and WSN-mRNA3-F, respectively. Primers for WSN-NP were 5′-GGTGAGAATGGACGGAGAAC-3′ (NP-625F) and 5′-CCGGCTCTCTCTCACTTGAT-3′ (NP-738R). Primers for canine IFN-β were 5′-CCAGTTCCAGAAGGAGGACA-3′ (forward) and 5′-CCTGTTGTCCCAGGTGAAGT-3′ (reverse). Primers for canine β-actin were 5′-CCCAAGGCCAACCGCGAGAAGAT-3′ (forward) and 5′-GTCCCGGCCAGCCAGGTCCAG-3′ (reverse). The relative mRNA levels were analyzed using an established protocol (31). The amplification specificity of qRT-PCR was confirmed by melting curve analysis at the end of each program.
Mouse infection.
Mouse experiments were performed using 6- to 8 week old female BALB/c mice as described previously (29). To determine viral replication in lung tissues, groups of 3 mice were infected intranasally with 1 × 104 PFU of WSN-WT, WSN-DelNS1-M-WT, or WSN-DelNS1-M-A14U virus, diluted in 25 μl of PBS. Three days later, mice were euthanized and the lungs removed for homogenization in 1 ml PBS. Viral titers were then determined by plaque assay in MDCK cells. To determine the pathogenicity of the viruses, groups of 6 mice were inoculated intranasally with 5 × 104 PFU of wild-type (WT) or DelNS1-M-A14U virus in 25 μl of PBS or with PBS alone, and the body weights of infected and control group mice were recorded daily for 14 days. Mice with body weight losses of greater than 25% of the initial body weight were euthanized, in accordance with animal ethics guidelines. The protocols used in this experiment were approved by the Committee on the Use of Live Animals in Teaching & Research (CULATR-3064-13), University of Hong Kong.
RESULTS
The A14U mutation in the M segment is sufficient to restore the growth of DelNS1 WSN virus.
Although the NS1 protein of influenza virus is not essential for virus replication, viruses that do not express functional NS1 are severely attenuated and can replicate only in IFN-deficient systems (25, 32). Helper viruses which express NS1 in cells have been used to support production of DelNS1 virus for vaccine studies (33). Substitutions in the M and NS segments have been shown to restore the growth of DelNS1 virus replication in a study using a reassortant virus containing HA and NA from an H5N1 virus and internal segments from A/PR/8/34 (22). However, the mechanism for the restoration of growth was not defined. To better understand the role of NS1 in virus replication and to develop a method for making DelNS1 viruses, we made a DelNS1 version of the A/WSN/33 strain (Fig. 1A). Deletion of NS1 was confirmed by examination of the viral genome, and plaque analysis showed DelNS1 viruses to form significantly smaller plaques than wild-type virus (Fig. 1B and C). We found that the replicative ability of the WSN-DelNS1 virus increased within three passages in MDCK cells, with the virus titer rising almost 2 logs compared to that of the original DelNS1 virus (P1) (Fig. 1D). Sequence analysis confirmed that a variant virus had been generated but found only one substitution, A14U, in the 3′ noncoding region of the vRNA M segment (M-A14U); no other mutations were found elsewhere in the genome (Fig. 1E). To confirm that introduction of M-A14U in the M segment is not a random event, the experiment was repeated and the same A14U variant obtained. To further test if the M-A14U mutation is sufficient to increase the growth of DelNS1 virus, we compared efficiencies of WSN-DelNS1 viruses with M-WT or the M-A14U, M-A14U-CM15, M-A14G, or M-A14C mutation rescued using reverse genetics and then plaque titrated the rescued viruses in MDCK cells. A14G is biochemically similar to A14U, and the corresponding virus was also rescued, but not those with the A14C mutation. To test potential disruption of base pairing by A14U, as it has been observed that mutations at positions 12 and 13 can affect virus growth (34), an M-A14U-CM15 mutant which contains an additional complementary mutation at position 15 at the 3′ end of M cRNA was included. It appears that the A14U substitution is unique and requires no complementary mutation. The titer of rescued DelNS1-M-A14U mutant viruses was as high as 1.75 × 105 PFU/ml, about 750-fold higher than that of the M-WT DelNS1 virus (Fig. 1F). To test if the A14U substitution may also arise in other cells, we passaged WSN-DelNS1 virus in Vero cells. However, no mutation was observed after more than eight passages. To further verify that M-A14U also supports replication of other influenza virus strains, we introduced this substitution into the M segment of a DelNS1 version of the A/PR/8/34 strain and, consistent with the observation with WSN, found that M-A14U significantly enhances the titer of virus rescued (Fig. 1G), while PR8-DelNS1 virus without the M-A14U substitution could not be rescued. These results indicate that the M-14U substitution can restore virus replication in the absence of NS1 protein expression.
FIG 1.

Construction and establishment of stabilized WSN-DelNS1 virus. (A) Schematic illustration of the NS segment transcripts and the NS mutant with an NS1 deletion (DelNS1). The DelNS1 plasmid was constructed by deleting the intron region ranging from nt 57 to 528 in the NS segment. NCR, noncoding region. (B) Confirmation of NS1 deletion in rescued DelNS1 viruses. Viral RNA was extracted from P1 virus, and the NS segment was amplified by RT-PCR and analyzed using agarose gel electrophoresis. (C) Plaque sizes of DelNS1 and wild-type A/WSN/33 viruses. (D) Titer (PFU/ml) of DelNS1 virus after each passage. (E) Sequence analysis of the DelNS1 virus genome revealed an A-to-U substitution at nucleotide position 14 in the M segment noncoding region. The noncoding region is marked by a black line, and the A14U mutation is indicated with an arrowhead. (F) Comparison of rescue efficiency for WSN-DelNS1 viruses containing M-WT and M-A14U, M-A14U-CM15, M-A14C, and M-A14G substitutions. NR, not rescued. (G) Rescue efficiency of PR8-DelNS1 viruses containing M-WT or M-A14U. The DelNS1 viruses were rescued with the indicated M-WT or M mutant plasmids in mixed HEK293T and MDCK cell cultures and then titrated by plaque assay. Values plotted are means (± standard deviations [SD]) (n = 3) and are representative of data from at least 5 independent experiments.
The M-A14U substitution supports virus replication but does not suppress IFN expression.
Previous studies showed that DelNS1 influenza virus is unable to replicate in MDCK cells and can be grown only in Vero cells (26, 35). Analysis of growth kinetics showed that M-A14U DelNS1 virus is able to replicate to a titer comparable to that of the wild-type virus (less than one log lower) in both MDCK and Vero cells (Fig. 2A and B). Similarly, the M-A14U substitution supports PR8 DelNS1 virus replication in Vero and MDCK cells, while PR8 DelNS1 virus without this substitution cannot be propagated (Fig. 2C and data not shown). The function of the NS1 protein as a viral antagonist of host antiviral activity is well defined (24). As expected, deletion of NS1 attenuates virus ability to suppress interferon expression, as measured in a reporter assay and by qRT-PCR of IFN-β activation (Fig. 3A and B). The DelNS1-M-A14U virus is unable to suppress activation of IRF3 in infected cells (Fig. 3C). While M-A14U enables DelNS1 virus to grow more efficiently than WSN-DelNS1-M-WT virus, this substitution has no effect on suppression of interferon (Fig. 3D). To further determine if the A14U substitution in the M segment may alter the pathogenic properties of virus in vivo, we compared the virulence and replication in lung tissues of WSN-WT and WSN-DelNS1-M-A14U mutant viruses in mice. Our results show that while wild-type WSN strains cause rapid body weight loss and death in infected mice, no apparent pathogenicity is observed with WSN-DelNS1-M-A14U mutant virus. Estimation of virus titers in the lung tissues of infected mice shows that both WSN-DelNS1-M-A14U and WSN-DelNS1-M-WT mutants replicate poorly in lung tissues, to levels approximately 3 logs lower than are observed for WSN-WT virus (Fig. 3E and F). Taken together, these results suggest that introduction of the M-A14U substitution would not change the avirulent properties possessed by DelNS1 virus that make it suitable for use in vaccine strain development.
FIG 2.

Growth kinetics of the M-A14U-DelNS1 virus in MDCK and Vero cells. (A and B) Column-purified reverse genetic WSN-WT, WSN-DelNS1-M-A14U, and WSN-DelNS1-M-WT viruses were used to infect MDCK cells (A) or Vero cells (B) at a multiplicity of infection (MOI) of 0.1. Supernatants were collected at the indicated time points and virus titrated by plaque assay. (C) Column-purified reverse genetic PR8-WT and PR8-DelNS1-M-A14U viruses were used to infect MDCK cells (left panel) and Vero cells (right panel) at an MOI of 0.1. Supernatants were collected at 24 h postinfection and titrated by plaque assay. The values (mean ± SD; n = 3) plotted are representative data from at least 3 independent experiments.
FIG 3.

Loss of IFN-β suppression activity in WSN-DelNS1 viruses. (A) HEK293T cells were transfected with an IFN-β reporter plasmid 24 h prior to infection with either WSN-WT or WSN-DelNS1-M-A14U at an MOI of 1 or with the positive control, Sendai virus (SeV), at 50 HA units. After 24 h, cells were harvested and cell lysates prepared for estimation of luciferase activity. The luciferase assays were performed in triplicate, and values were normalized to the Renilla luciferase control. (B) IFN-β and viral NP mRNA levels were quantified by qRT-PCR after MDCK cells were infected with WSN-WT or WSN-DelNS1-M-A14U at an MOI of 0.1 and cultured for 16 h. Values were normalized against canine actin. (C) Suppression of IRF3 dimerization in HEK293T cells infected with WSN-WT, WSN-DelNS1-M-A14U, or Sendai virus was analyzed by native gel electrophoresis. (D) Activity in suppressing IFN-β expression was compared in MDCK cells infected with WSN-DelNS1-M-WT or WSN-DelNS1-M-A14U virus. Similar to the experiment described for panel B, MDCK cells were infected with the indicated viruses at an MOI of 0.1, and at 16 h postinfection, IFN-β and NP mRNA levels were quantified by qPCR. Values were normalized to canine actin. (E) Groups of six BALB/c mice, aged 6 to 8 weeks, were intranasally inoculated with 5 × 104 PFU of WSN-WT or WSN-DelNS1-M-A14U mutant virus, and body weight was monitored daily for 14 days postinfection. (F) Replication efficiency of viruses in lung tissues of infected mice. Groups of three mice were infected with 104 PFU of WSN-WT, WSN-DelNS1-M-WT, or WSN-DelNS1-M-A14U mutant viruses and then euthanized at 72 h postinfection, with lung tissues from each mouse being collected and homogenized for virus titration by plaque assay using MDCK cells. Statistical significance was analyzed by one-way analysis of variance (ANOVA) or Student's t test (**, P < 0.01). The bars plotted show means ± SD (n = 3), and the results represent at least three independent experiments.
The M-A14U substitution affects splicing of M transcripts in virus replication.
The NS1 protein is not a component of the viral polymerase complex. The observation that a sole A14U substitution in the M segment restores replication in DelNS1 virus suggests that NS1 may be involved in regulating replication or transcription from the vRNA M segment. To understand the molecular basis for the role of the M-A14U substitution in supporting replication of virus with an NS1 deletion, we examined the expression of virus proteins during infection of MDCK cells with DelNS1-M-A14U virus. While the expression pattern of viral HA and NP proteins is largely similar in WSN-WT and DelNS1-M-A14U virus infections (Fig. 4A), we found that M2 expression was unchanged but that M1 levels were significantly decreased in M-A14U DelNS1 compared to WSN-WT virus infections. Comparison of viral protein expression between DelNS1-M-A14U and DelNS1-M-WT viruses also showed that the A14U substitution had an effect on the M1 and M2 proteins but not on HA and NP (Fig. 4B). However, the enhancement effect of the M-A14U substitution on M2 protein expression is not apparent in WSN virus with intact NS1 function (Fig. 4C), suggesting that M-A14U may be essential for compensation of the loss of NS1 functions in regulation of expression and splicing of M mRNAs.
FIG 4.
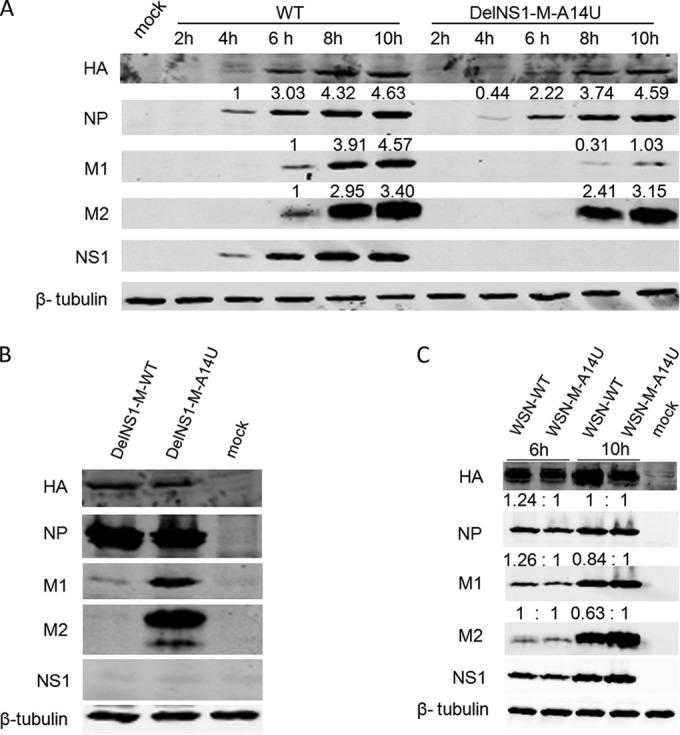
Effect of M-A14U mutation on M1 and M2 protein expression. (A) MDCK cells were infected with WSN-WT or WSN-DelNS1-M-A14U virus at an MOI of 2. Cells were harvested at the indicated time points and cell lysates analyzed by Western blotting with specific antibodies, as described in Materials and Methods. (B) MDCK cells were infected with WSN-DelNS1-M-WT or WSN-DelNS1-M-A14U virus at an MOI of 0.1. At 16 h postinfection, cell lysates were collected for Western blotting with specific antibodies. (C) MDCK cells were infected with WSN-WT or WSN-M-A14U virus at an MOI of 5. Cell lysates were prepared at the indicated time points for Western blotting with specific antibodies. β-Tubulin was included as a loading control. All of the results are representative of three independent experiments.
The M-A14U mutation elevates expression of M2 mRNA through downregulation of mRNA3 during virus replication.
During M segment transcription, differential splicing occurs to generate four transcripts, with M1 and M2 mRNAs expressing matrix (M1) and viral ion channel (M2) proteins, respectively, during influenza virus infection (Fig. 5A). The function of mRNA3 and mRNA4 is unknown. Notably, the A14U mutation is located in the mRNA3 splicing consensus sequence, which covers nucleotides (nt) 9 to 17 at the 5′ end of the noncoding region of M mRNAs. We predict that the A14U substitution alters the splicing donor (SD) site consensus sequence of mRNA3 from CAG/GUA to CAG/GUU and affects production of mRNA3. It seems likely that the A14U substitution in the vRNA M segment supports DelNS1 virus growth by downregulating mRNA3 expression to increase the ratio of M2 to M1 mRNA expression. To verify this hypothesis, we used qRT-PCR analysis to examine mRNA3 levels in virus-infected cells. As expected, among the four M mRNA transcripts, M1, M2, and M4 were significantly increased but mRNA3 was almost completely abolished in WSN-DelNS1-M-A14U virus-infected MDCK cells (Fig. 5B). In contrast, expression of NP mRNA was not affected by M-A14U substitution in DelNS1 viruses (Fig. 5B), suggesting that deletion of NS1 is specifically associated with regulation of expression from the M segment. To further prove that M-A14U affects differential splicing from M1 mRNA, HEK293T cells were transfected with either M-A14U or M-WT segment plasmids which express full-length M mRNA. While levels of M1 mRNA are similar for M-WT and M-A14U plasmids, expression of mRNA3 is markedly downregulated and that of M2 mRNA is significantly upregulated from the M-A14U plasmid (Fig. 5C). Coexpression of NS1 further demonstrated the positive effect of NS1 on transcription of M mRNAs; the A14U substitution may have arisen in the DelNS1 virus to compensate for the NS1-associated enhancement of M mRNA expression. We showed above that the M-A14U substitution has a similar effect in supporting replication of DelNS1 virus derived from the A/PR/8/34 strain (Fig. 1G and 2C). Examination of differentially spliced M transcripts from PR8-DelNS1-M-A14U virus-infected cells found patterns similar to those observed with WSN-DelNS1-M-A14U (Fig. 5D), supporting conservation of function for this nucleotide in both WSN and PR8 strains. Collectively, these results clearly show that M-A14U substitution in the vRNA M segment alters the splicing pattern of M1 mRNA during virus infection, even in the absence of other viral proteins.
FIG 5.

Effect of M-A14U substitution on alternative splicing of M transcripts. (A) Schematic illustration of the M segment transcripts. The sequence of the 3′ noncoding region of the vRNA M segment is shown in red, with the mRNA3 splicing donor (SD) site highlighted in yellow. Splicing consensus sequences for the donor site are indicated in green above the noncoding region sequence (M, A or C; R, A or G). (B) Analysis of levels of different M mRNAs in virus-infected cells. MDCK cells were infected with the indicated viruses at an MOI of 0.1. Total RNA was isolated at 16 h postinfection. The mRNA levels for M1, M2, mRNA3, and M4 were determined by quantitative RT-PCR, as described in Materials and Methods. (C) HEK293T cells were transfected with pHW2000-WSN-M-WT or pHW2000-WSN-M-A14U plasmid with or without cotransfection of pCX-WSN-NS1 for coexpression of NS1. At 48 h posttransfection, total RNAs were isolated. After DNase treatment, the mRNA levels were determined by quantitative RT-PCR. (D) MDCK cells were infected with PR8-WT, PR8-M-A14U, or PR8-DelNS1-M-A14U virus at an MOI of 0.1. At 16 h postinfection, total RNAs were isolated and mRNA levels measured by qPCR. M2/M1 mRNA and mRNA3/M1 mRNA ratios are shown. All the results plotted indicate means ± SD (n = 3) and are representative of three independent experiments.
The M-A14U mutation results in an increased ratio of M2 mRNA to M1 mRNA.
To further explore the molecular mechanism underlying the effect of M-A14U on virus replication in the absence of NS1 expression, we made mutants which downregulate expression of mRNA3. The M-G12C-CM13 mutant virus was reported to markedly downregulate the expression of mRNA3 (34). We confirmed that expression of mRNA3 is reduced in WSN-M-A14U, WSN-M-A14G, and WSN-MG12C-CM13 virus infection (Fig. 6A). However, growth kinetics analysis showed that replication of this virus is attenuated, with the WSN-DelNS1-M-G12C-CM13 virus unable to be efficiently rescued (Fig. 6B), suggesting that downregulation of mRNA3 may not be directly associated with virus growth. The M1 matrix protein is involved in vRNA nuclear export, while M2 has an ion channel function and is involved in the virus uncoating and budding process during influenza virus replication. It is possible that there is coordination of expression of M1 and M2 over the course of the viral replication process through regulation of alternative splicing of M1 mRNA and that NS1 may play a role in this. It seems that the ratio of M1 to M2 is altered in DelNS1 virus infection, as seen in the above results (Fig. 4 and 5). M2 is reported to be selectively expressed during the early hours of virus infection (21), which may suggest a critical role for M2 in the early phase of virus replication. Besides its role in the virus uncoating and budding process, M2 is reported to interact with autophagosomes and inflammasomes (36, 37), suggesting that it may play other roles during virus replication. It is possible that in the absence of NS1, which has multiple functions to antagonize host antiviral activities, DelNS1 viruses may adapt to preferentially express M2 in an attempt to maintain an optimal balance for virus replication. To understand the effect of M-A14U on regulation of differential splicing of M transcripts, the ratio of M2 mRNA to M1 mRNA in virus-infected cells was determined. For this purpose, we compared the M2/M1 ratios in cells infected with WSN-M-A14U, WSN-DelNS1-M-WT, WSN-DelNS1-M-A14U, and WT WSN virus. The M2/M1 splicing efficiency in WSN-DelNS1-M-A14U-infected cells was about 6-fold higher than that in WSN-DelNS1-M-WT-infected cells (Fig. 6C). However, comparison between WSN-WT and WSN-M-A14U M2/M1 ratios revealed that while M2 is upregulated with the A14U substitution, the effect is not as significant (less than a 2-fold difference) as that seen between the WSN-DelNS1 viruses (Fig. 6C). As expected, expression of mRNA3 is diminished and M4 mRNA is downregulated in both WSN-WT-M-A14U and WSN-DelNS1-M-A14U virus infections (Fig. 6D and E). To further test the effect of mRNA3 on virus growth, we expressed mRNA3 from a plasmid, but we found no negative effect on virus titers (Fig. 6F), which suggests that alternative splicing for expression of mRNA3 may be solely for modulating the levels of M1 and M2 mRNAs. These results support the hypothesis that M-A14U mutation leads to increased alternative splicing for production of M2, and perhaps also M1, mRNAs to enhance virus replication in the absence of NS1 expression.
FIG 6.

Effect of M-A14U substitution on M2/M1 mRNA ratio. (A) MDCK cells were infected with WSN-WT, WSN-M-A14U, WSN-M-A14G, or WSN-M-G12C-CM13 virus at an MOI of 5. Total RNAs were extracted at the indicated time points and levels of mRNA3 determined by quantitative RT-PCR. (B) Analysis of growth kinetics of WSN-WT, WSN-M-A14U, WSN-M-A14G, and WSN-M-G12C-CM13 viruses. MDCK cells were infected with these viruses at an MOI of 0.001. Supernatants were collected at the indicated time points and titrated by plaque assay. (C to E) MDCK cells were infected with WSN-WT, WSN-M-A14U, WSN-DelNS1-M-WT, or WSN-DelNS1-M-A14U virus at an MOI of 0.1. At 16 h after infection, total RNAs were isolated, and the M2/M1, mRNA3/M1, and M4/M1 mRNA ratios were determined by quantitative RT-PCR. (F) Effect of mRNA3 on virus replication. HEK293T cells were transfected with pCX-WSN-mRNA3 or a control vector 24 h prior to infection with WSN-DelNS1-M-A14U virus at an MOI of 0.1. Supernatants were collected at the indicated time points and virus titrated by plaque assay. All of the bars and points plotted indicate means ± SD (n = 3) from three independent experiments. **, P = 0.0013 by Student's t test.
The M-A14U mutation enhances alternative splicing of M2 mRNAs and synthesis of M vRNA.
Previous studies have suggested that splicing of M segment mRNAs can be regulated by the viral RNP complex, in conjunction with the host factor SF2 (20), or NS1 (21). Are these mechanisms associated with the M-A14U substitution? We have presented evidence showing that the M-A14U substitution affects splicing efficiency of M1 mRNA and that this property is required to compensate for lack of NS1 expression. In a mechanism where NS1 is involved in the regulation of splicing of M transcripts into M2 mRNA during virus replication, it seems possible that DelNS1 virus may be forced to obtain an adaptive substitution in the regulatory element at the promoter region of the vRNA M segment which normally associates with NS1 in order to compensate for a lack of NS1 function. To test this hypothesis, we examined the effect of restoring NS1 expression on the M2/M1 ratio in DelNS1-M-WT virus-infected cells. To this end, increasing amounts of NS1 expression vector were transfected into HEK293T cells 24 h prior to infection with virus, and expression of M1 and M2 viral mRNAs was estimated by quantitative RT-PCR. The amount of the M2 spliced form rises as levels of NS1 expression increase in DelNS1-M-WT virus-infected cells (Fig. 7A). Using plasmids which express both the vRNA and mRNA of the M segment, we found that even in the absence of other viral proteins, all plasmids with substitutions at position 14 expressed higher levels of M2 protein, but not M1, than M-WT, which supports the hypothesis that M-A14U is associated with upregulation of the M2 spliced form (Fig. 7B). Cotransfection of NS1 plasmid M significantly enhances expression of M1 from all of these plasmids, and levels of M2 are upregulated more significantly for plasmids which have a substitution at the 14th position or at the 12th position to downregulate mRNA3 (G12C-CM13) (34) (Fig. 6A and 7B). This result strongly suggests that NS1 can function to allow preferential expression of M1 and M2 through downregulation of the alterative splicing site for mRNA3, while substitutions at this splicing site allow efficient processing of M1 into the M2 spliced form.
FIG 7.

Regulation of M mRNA splicing host machinery by viral NS1 and polymerase proteins. (A) HEK293T cells were transfected with increasing amounts of pCX-WSN-NS1. At 24 h after transfection, cells were infected with WSN-DelNS1-M-WT, WSN-DelNS1-M-A14U, or WSN-WT virus at an MOI of 0.5. Total RNAs were isolated at 8 h postinfection and M1 mRNA, M2 mRNA, and mRNA3 levels analyzed by quantitative RT-PCR. (B) HEK293T cells were transfected with WT and various mutants of the M segment cloned into pHW2000 plasmids, which express M vRNA, with or without the pCX-WSN-NS1 plasmid. Cell lysates were prepared at 48 h posttransfection for Western blotting with specific antibodies. β-Tubulin was included as a loading control. (C) Effect of A14U substitution on M vRNA replication. MDCK cells were infected with WSN-WT, WSN-M-A14U, or WSN-M-A14G virus at an MOI of 5. At 4 and 10 h postinfection, total RNAs were isolated for qRT-PCR analysis, and relative amounts of vRNA were determined as described above. (D) The A14U substitution enhances M vRNA replication. MDCK cells were infected with WSN-DelNS1-M-WT or WSN-DelNS1-M-A14U virus at an MOI of 0.1. Total RNAs were isolated at 16 h after infection. M and NP vRNA levels were determined by quantitative RT-PCR, as described above. All bars plotted show means ± SD (n = 3). The results represent three independent experiments. **, P < 0.01; ***, P = 0.0002 (by Student's t test).
The A14U substitution occurs in the 3′ noncoding region of the M vRNA segment (5′ of cRNA or 5′ mRNA), which is also the promoter region for replication of the viral genome. It is possible that this mutation affects binding of the viral polymerase complex to enhance vRNA synthesis from the M segment, producing more M1 mRNA for splicing into M2 mRNA during virus replication. We tested the M vRNA synthesis levels in cells infected with mutant viruses containing variations at position 14 but with an intact NS1 gene. We found that the level of M vRNA produced from the WSN-M-A14U mutant was approximately 15-fold higher than that for WSN-WT virus (Fig. 7C). In contrast, relatively lower levels of M vRNA were observed for the WSN-M-A14G mutant than for WSN-WT virus-infected cells (Fig. 7C). We further tested if elevated production of M vRNA is associated with DelNS1-M-A14U virus infection. It is notable that levels of M vRNA are significantly higher in WSN-DelNS1-M-A14U than in WSN-DelNS1-M-WT virus infections, while no similar trend is observed for NP vRNA (Fig. 7D). Collectively, these results strongly suggest that the M-A14U mutation positively affects M vRNA synthesis and that this effect may be required for replication of NS1-deficient viruses.
DISCUSSION
Influenza virus utilizes the viral polymerase complex and host machinery to transcribe and replicate the viral genome in the nucleus. Coordination of expression of viral products for switching from transcription to replication and nuclear export of vRNAs to the cytoplasm is critical for optimal replication efficiency. The viral proteins M1 and NEP (NS2) are involved in nuclear export of vRNPs, but details of the mechanism remain unclear (38). M2 is a structural protein and is involved in the virus uncoating process during the early phase of entry and virion budding in the late stage of virus infection (14, 15). M1/M2 and NS1/NS2 (NEP) viral proteins are expressed through alternatively spliced mRNAs from the M and NS segments, respectively. While there is no direct involvement of proteins from the M or NS segment in the viral polymerase complex, it is suggested that virus replication can be regulated through modulation of alternative splicing of M1/M2 and NS1/NEP (NS2) mRNAs (39, 40). While influenza virus utilizes viral polymerases to replicate and a cap-snatching mechanism to transcribe the viral genome, it is believed that the virus is dependent on host machinery for mRNA splicing (39). Expression of spliced NS and M mRNAs is highly regulated. NS1 was found to inhibit host pre-mRNA splicing through interaction with CPSF and may also have the same effect on viral mRNA (41, 42), but exactly how the NS and M transcripts are regulated is not completely understood. Previous studies showed that both viral and host mechanisms are involved in the regulation of differential splicing of M mRNAs (19–21). Because M1 and M2 proteins have essential functions required for different stages of viral infection, such as RNP nuclear export, virus assembly, and budding processes, control of the timing of M1 and M2 expression to optimize efficiency of viral genome replication is critical for virus infection. The 3′ noncoding region (NCR) of the vRNA M segment contains 25 nucleotides which comprise a promoter for transcription initiation and alternative splicing sites for posttranscriptional processing of mRNA (Fig. 5A). It has been suggested that the viral polymerase complex binds onto the NCR promoter region to block the splicing site for expression of mRNA3, leading to the alternative utilization of another splicing site for M2 mRNA (19, 20). Another study found that it is the NS1 protein which regulates the accumulation of M2 in virus replication (21). However, the regulatory effect of NS1 on accumulation of M2 was not observed in Vero cells infected with DelNS1 virus (43). The question remains as to whether the viral polymerase proteins, NS1, or both regulate the alternative splicing of M transcripts.
In this study, we explored the role of the NS1 protein in virus replication by constructing an NS1 deletion virus derived from the A/WSN/33 influenza virus strain. Interestingly, it was found that a sole A14U substitution in the noncoding region of the M segment arose in DelNS1 virus after a few passages. The DelNS1-M-A14U virus was able to replicate to a level approximately similar to that for the wild-type virus in cells, indicating a functional linkage between the NS1 protein and the transcription or replication of M vRNA. Four mRNAs, M1, M2, mRNA3, and M4, are expressed from M vRNA. We demonstrated that the DelNS1-M-A14U virus expresses elevated levels of M2 spliced from M1 mRNA while suppressing expression of mRNA3, linking the role of NS1 with regulation of the alternative splicing of M transcripts. More direct evidence for the effect of A14U on alternative splicing of M mRNAs came from an analysis of expression of M segments containing A14U and other substitutions, performed in the absence of other viral proteins. The M-A14U segment expresses higher levels of M2 protein, while the level of M1 is unchanged (Fig. 7B), suggesting the A14U substitution favors M2 production by the host splicing machinery. How does the A14U substitution cause expression of M2 to be upregulated? A14U is situated right within the splice donor site for mRNA3 expression (Fig. 5A), and it seems likely that this substitution abolishes or affects the binding of splicing factors to this motif, leading to the selection of the adjacent splice donor site which produces M2 mRNA instead. This hypothesis is supported by the evidence that other substitutions at this site also enhance M2 expression in the absence of other viral proteins (Fig. 7B). Restoration of NS1 expression by transfecting cells with an NS1-expressing plasmid prior to virus infection significantly increased the M2/M1 mRNA ratio in DelNS1-M-WT virus-infected cells, confirming that NS1 is directly involved in the regulation of M2 mRNA splicing.
The A14U substitution may have dual roles, modulating vRNA synthesis and mRNA splicing, as levels of M vRNA were significantly enhanced in DelNS1-M-A14U virus infections compared to DelNS1-M-WT infections, while no similar effect was observed for NP vRNA. Therefore, a compound effect of the A14U substitution in the M vRNA 3′ promoter region would result in higher efficiency of the viral polymerase complex, generating more vRNA for transcription into mRNA, combined with blockage of the mRNA3 splicing site to allow expression of M2 mRNA. While the NS1 protein is not recognized as an essential element for virus replication, it has multiple functions as an antagonist of host antiviral activity. It seems reasonable to suggest that for a virus lacking NS1 to replicate in cells, alternative viral elements to counter host antiviral activity would be required. The M2 protein has been found to interact with inflammasomes and autophagosomes during influenza virus infection. It may be speculated that M2 is required to maintain optimal replication for virus under conditions where NS1 is absent and that the A14U adaptive mutant is thus selected to drive expression of higher levels of M2 for such purposes.
Because the NS1 protein interferes with both innate and adaptive immune responses during virus infection and is an influenza virulence determinant (44–46), DelNS1 virus is regarded as a promising live attenuated vaccine candidate (47–50). Animal experiments show that live attenuated vaccines lacking NS1 may induce better immune responses (45, 46). However, deletion of NS1 severely affects virus replication, and it is difficult to produce high titers of attenuated virus for vaccine applications. Attempts have been made to propagate NS1-deficient viruses to high titers using various strategies (22, 25, 33). The DelNS1 virus was found to be restricted to being amplified in limited systems, either in an IFN-deficient system (25, 47) or in an NS1-expressing system (33, 51). We demonstrate here that a single A14U substitution in the noncoding region of the M segment was sufficient to support replication of DelNS1 viruses to a level close to that of wild-type virus in both MDCK and Vero cells. While the effect of the A14U substitution on DelNS1 virus replication has been confirmed in A/WSN/33 and A/PR/8/34 strains in this study, verification of the influence of this substitution in other influenza virus strains and subtypes is necessary. The mechanism revealed in this study for supporting the replication of DelNS1 virus provides important strategic information relevant to future projects aimed at constructing DelNS1 versions of other influenza virus strains.
ACKNOWLEDGMENTS
This study was supported in part by the Research Grants Council of the Hong Kong SAR (7629/13M and 17103214), the Areas of Excellence Scheme of the University Grants Committee (grant AoE/M-12/06), and a Hong Kong Postgraduate Fellowship (M.Z).
We are grateful to Jane Rayner for editing the manuscript.
REFERENCES
- 1.Taubenberger JK, Kash JC. 2010. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 7:440–451. doi: 10.1016/j.chom.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992. Evolution and ecology of influenza A viruses. Microbiol Rev 56:152–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palese P, Shaw ML. 2006. Orthmyxoviridae: the viruses and their replication, p 1647–1689. In Knipe DM, Howley P (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Nelson MI, Viboud C, Simonsen L, Bennett RT, Griesemer SB, St George K, Taylor J, Spiro DJ, Sengamalay NA, Ghedin E, Taubenberger JK, Holmes EC. 2008. Multiple reassortment events in the evolutionary history of H1N1 influenza A virus since 1918. PLoS Pathog 4:e1000012. doi: 10.1371/journal.ppat.1000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naffakh N, Tomoiu A, Rameix-Welti MA, van der Werf S. 2008. Host restriction of avian influenza viruses at the level of the ribonucleoproteins. Annu Rev Microbiol 62:403–424. doi: 10.1146/annurev.micro.62.081307.162746. [DOI] [PubMed] [Google Scholar]
- 6.Eisfeld AJ, Neumann G, Kawaoka Y. 2015. At the centre: influenza A virus ribonucleoproteins. Nat Rev Microbiol 13:28–41. doi: 10.1038/nrmicro3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wise HM, Hutchinson EC, Jagger BW, Stuart AD, Kang ZH, Robb N, Schwartzman LM, Kash JC, Fodor E, Firth AE, Gog JR, Taubenberger JK, Digard P. 2012. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog 8:e1002998. doi: 10.1371/journal.ppat.1002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wise HM, Foeglein A, Sun J, Dalton RM, Patel S, Howard W, Anderson EC, Barclay WS, Digard P. 2009. A complicated message: identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J Virol 83:8021–8031. doi: 10.1128/JVI.00826-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, Dalton RM, Lo A, Efstathiou S, Atkins JF, Firth AE, Taubenberger JK, Digard P. 2012. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O'Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW. 2001. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med 7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- 11.Wise HM, Barbezange C, Jagger BW, Dalton RM, Gog JR, Curran MD, Taubenberger JK, Anderson EC, Digard P. 2011. Overlapping signals for translational regulation and packaging of influenza A virus segment 2. Nucleic Acids Res 39:7775–7790. doi: 10.1093/nar/gkr487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi M, Jagger BW, Wise HM, Digard P, Holmes EC, Taubenberger JK. 2012. Evolutionary conservation of the PA-X open reading frame in segment 3 of influenza A virus. J Virol 86:12411–12413. doi: 10.1128/JVI.01677-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brooke CB, Ince WL, Wrammert J, Ahmed R, Wilson PC, Bennink JR, Yewdell JW. 2013. Most influenza a virions fail to express at least one essential viral protein. J Virol 87:3155–3162. doi: 10.1128/JVI.02284-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossman JS, Jing X, Leser GP, Lamb RA. 2010. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. 1997. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc Natl Acad Sci U S A 94:11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao S, Liu X, Yu M, Li J, Jia X, Bi Y, Sun L, Gao GF, Liu W. 2012. A nuclear export signal in the matrix protein of influenza A virus is required for efficient virus replication. J Virol 86:4883–4891. doi: 10.1128/JVI.06586-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valcarcel J, Portela A, Ortin J. 1991. Regulated M1 mRNA splicing in influenza virus-infected cells. J Gen Virol 72:1301–1308. doi: 10.1099/0022-1317-72-6-1301. [DOI] [PubMed] [Google Scholar]
- 18.Chua MA, Schmid S, Perez JT, Langlois RA, Tenoever BR. 2013. Influenza A virus utilizes suboptimal splicing to coordinate the timing of infection. Cell Rep 3:23–29. doi: 10.1016/j.celrep.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shih SR, Krug RM. 1996. Novel exploitation of a nuclear function by influenza virus: the cellular SF2/ASF splicing factor controls the amount of the essential viral M2 ion channel protein in infected cells. EMBO J 15:5415–5427. [PMC free article] [PubMed] [Google Scholar]
- 20.Shih SR, Nemeroff ME, Krug RM. 1995. The choice of alternative 5′ splice sites in influenza virus M1 mRNA is regulated by the viral polymerase complex. Proc Natl Acad Sci U S A 92:6324–6328. doi: 10.1073/pnas.92.14.6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robb NC, Fodor E. 2012. The accumulation of influenza A virus segment 7 spliced mRNAs is regulated by the NS1 protein. J Gen Virol 93:113–118. doi: 10.1099/vir.0.035485-0. [DOI] [PubMed] [Google Scholar]
- 22.van Wielink R, Harmsen MM, Martens DE, Peeters BP, Wijffels RH, Moormann RJ. 2012. Mutations in the M-gene segment can substantially increase replication efficiency of NS1 deletion influenza A virus in MDCK cells. J Virol 86:12341–12350. doi: 10.1128/JVI.01725-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hale BG, Randall RE, Ortin J, Jackson D. 2008. The multifunctional NS1 protein of influenza A viruses. J Gen Virol 89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- 24.Krug RM. 2015. Functions of the influenza A virus NS1 protein in antiviral defense. Curr Opin Virol 12C:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, Palese P, Muster T. 1998. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- 26.Egorov A, Brandt S, Sereinig S, Romanova J, Ferko B, Katinger D, Grassauer A, Alexandrova G, Katinger H, Muster T. 1998. Transfectant influenza A viruses with long deletions in the NS1 protein grow efficiently in Vero cells. J Virol 72:6437–6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mok BW, Song W, Wang P, Tai H, Chen Y, Zheng M, Wen X, Lau SY, Wu WL, Matsumoto K, Yuen KY, Chen H. 2012. The NS1 protein of influenza A virus interacts with cellular processing bodies and stress granules through RNA-associated protein 55 (RAP55) during virus infection. J Virol 86:12695–12707. doi: 10.1128/JVI.00647-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen X, Huang X, Mok BW, Chen Y, Zheng M, Lau SY, Wang P, Song W, Jin DY, Yuen KY, Chen H. 2014. NF90 exerts antiviral activity through regulation of PKR phosphorylation and stress granules in infected cells. J Immunol 192:3753–3764. doi: 10.4049/jimmunol.1302813. [DOI] [PubMed] [Google Scholar]
- 29.Song W, Wang P, Mok BW, Lau SY, Huang X, Wu WL, Zheng M, Wen X, Yang S, Chen Y, Li L, Yuen KY, Chen H. 2014. The K526R substitution in viral protein PB2 enhances the effects of E627K on influenza virus replication. Nat Commun 5:5509. doi: 10.1038/ncomms6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A 97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Kistner O, Barrett PN, Mundt W, Reiter M, Schober-Bendixen S, Dorner F. 1998. Development of a mammalian cell (Vero) derived candidate influenza virus vaccine. Vaccine 16:960–968. doi: 10.1016/S0264-410X(97)00301-0. [DOI] [PubMed] [Google Scholar]
- 33.van Wielink R, Harmsen MM, Martens DE, Peeters BP, Wijffels RH, Moormann RJ. 2011. MDCK cell line with inducible allele B NS1 expression propagates delNS1 influenza virus to high titres. Vaccine 29:6976–6985. doi: 10.1016/j.vaccine.2011.07.037. [DOI] [PubMed] [Google Scholar]
- 34.Chiang C, Chen GW, Shih SR. 2008. Mutations at alternative 5′ splice sites of M1 mRNA negatively affect influenza A virus viability and growth rate. J Virol 82:10873–10886. doi: 10.1128/JVI.00506-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Enami M, Enami K. 2000. Characterization of influenza virus NS1 protein by using a novel helper-virus-free reverse genetic system. J Virol 74:5556–5561. doi: 10.1128/JVI.74.12.5556-5561.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ichinohe T, Pang IK, Iwasaki A. 2010. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 11:404–410. doi: 10.1038/ni.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gannage M, Dormann D, Albrecht R, Dengjel J, Torossi T, Ramer PC, Lee M, Strowig T, Arrey F, Conenello G, Pypaert M, Andersen J, Garcia-Sastre A, Munz C. 2009. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6:367–380. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paterson D, Fodor E. 2012. Emerging roles for the influenza A virus nuclear export protein (NEP). PLoS Pathog 8:e1003019. doi: 10.1371/journal.ppat.1003019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dubois J, Terrier O, Rosa-Calatrava M. 2014. Influenza viruses and mRNA splicing: doing more with less. mBio 5(3):e00070-00014. doi: 10.1128/mBio.00070-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manz B, Brunotte L, Reuther P, Schwemmle M. 2012. Adaptive mutations in NEP compensate for defective H5N1 RNA replication in cultured human cells. Nat Commun 3:802. doi: 10.1038/ncomms1804. [DOI] [PubMed] [Google Scholar]
- 41.Nemeroff ME, Barabino SM, Li Y, Keller W, Krug RM. 1998. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol Cell 1:991–1000. doi: 10.1016/S1097-2765(00)80099-4. [DOI] [PubMed] [Google Scholar]
- 42.Garaigorta U, Ortin J. 2007. Mutation analysis of a recombinant NS replicon shows that influenza virus NS1 protein blocks the splicing and nucleo-cytoplasmic transport of its own viral mRNA. Nucleic Acids Res 35:4573–4582. doi: 10.1093/nar/gkm230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salvatore M, Basler CF, Parisien JP, Horvath CM, Bourmakina S, Zheng H, Muster T, Palese P, Garcia-Sastre A. 2002. Effects of influenza A virus NS1 protein on protein expression: the NS1 protein enhances translation and is not required for shutoff of host protein synthesis. J Virol 76:1206–1212. doi: 10.1128/JVI.76.3.1206-1212.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernandez-Sesma A, Marukian S, Ebersole BJ, Kaminski D, Park MS, Yuen T, Sealfon SC, Garcia-Sastre A, Moran TM. 2006. Influenza virus evades innate and adaptive immunity via the NS1 protein. J Virol 80:6295–6304. doi: 10.1128/JVI.02381-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pica N, Langlois RA, Krammer F, Margine I, Palese P. 2012. NS1-truncated live attenuated virus vaccine provides robust protection to aged mice from viral challenge. J Virol 86:10293–10301. doi: 10.1128/JVI.01131-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mueller SN, Langley WA, Carnero E, Garcia-Sastre A, Ahmed R. 2010. Immunization with live attenuated influenza viruses that express altered NS1 proteins results in potent and protective memory CD8+ T-cell responses. J Virol 84:1847–1855. doi: 10.1128/JVI.01317-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Talon J, Salvatore M, O'Neill RE, Nakaya Y, Zheng H, Muster T, Garcia-Sastre A, Palese P. 2000. Influenza A and B viruses expressing altered NS1 proteins: a vaccine approach. Proc Natl Acad Sci U S A 97:4309–4314. doi: 10.1073/pnas.070525997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richt JA, Garcia-Sastre A. 2009. Attenuated influenza virus vaccines with modified NS1 proteins. Curr Top Microbiol Immunol 333:177–195. doi: 10.1007/978-3-540-92165-3_9. [DOI] [PubMed] [Google Scholar]
- 49.Wacheck V, Egorov A, Groiss F, Pfeiffer A, Fuereder T, Hoeflmayer D, Kundi M, Popow-Kraupp T, Redlberger-Fritz M, Mueller CA, Cinatl J, Michaelis M, Geiler J, Bergmann M, Romanova J, Roethl E, Morokutti A, Wolschek M, Ferko B, Seipelt J, Dick-Gudenus R, Muster T. 2010. A novel type of influenza vaccine: safety and immunogenicity of replication-deficient influenza virus created by deletion of the interferon antagonist NS1. J Infect Dis 201:354–362. doi: 10.1086/649428. [DOI] [PubMed] [Google Scholar]
- 50.Wressnigg N, Voss D, Wolff T, Romanova J, Ruthsatz T, Mayerhofer I, Reiter M, Nakowitsch S, Humer J, Morokutti A, Muster T, Egorov A, Kittel C. 2009. Development of a live-attenuated influenza B DeltaNS1 intranasal vaccine candidate. Vaccine 27:2851–2857. doi: 10.1016/j.vaccine.2009.02.087. [DOI] [PubMed] [Google Scholar]
- 51.Kochs G, Martinez-Sobrido L, Lienenklaus S, Weiss S, Garcia-Sastre A, Staeheli P. 2009. Strong interferon-inducing capacity of a highly virulent variant of influenza A virus strain PR8 with deletions in the NS1 gene. J Gen Virol 90:2990–2994. doi: 10.1099/vir.0.015727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]