

ABSTRACT
Simian immunodeficiency virus (SIV)-specific CD8+ T cells kill SIV-infected CD4+ T cells in an major histocompatibility complex class I (MHC-I)-dependent manner. However, they are reportedly less efficient at killing SIV-infected macrophages. Since the viral accessory protein Nef has been shown to downregulate MHC-I molecules and enhance cytotoxic T lymphocyte (CTL) evasion in human immunodeficiency virus type 1 (HIV-1)-infected CD4+ T cells, we examined whether Nef played a role in protecting SIV-infected macrophages from killing by SIV-specific CD8+ T cells. To explore the role of Nef in CD8+ T cell evasion, we compared the ability of freshly sorted SIV-specific CD8+ T cells to readily suppress viral replication or eliminate CD4+ T cells or monocyte-derived macrophages infected with SIV variants containing wild-type (WT) or mutated nef genes. As expected, SIV-specific CD8+ T cells suppressed viral replication and eliminated the majority of SIV-infected CD4+ T cells, and this killing was enhanced in CD4+ T cells infected with the nef variants. However, macrophages infected with nef variants that disrupt MHC-I downregulation did not promote rapid killing by freshly isolated CD8+ T cells. These results suggest that mechanisms other than Nef-mediated MHC-I downregulation govern the resistance of SIV-infected macrophages to CD8+ T cell-mediated killing. This study has implications for viral persistence and suggests that macrophages may afford primate lentiviruses some degree of protection from immune surveillance.
IMPORTANCE Myeloid cells are permissive for HIV/SIV replication in vitro and may contribute to viral persistence in vivo. While many studies have been geared to understanding how CD8+ T cells control viral replication in CD4+ T cells, the role of these cells in controlling viral replication in macrophages is less clear. Primary, unstimulated CD8+ T cells insignificantly suppress viral replication or eliminate SIV-infected macrophages. Since the viral Nef protein downregulates MHC-I and provides infected cells some degree of protection from CD8+ T cell-mediated effector functions, we evaluated whether Nef may be contributing to the resistance of macrophages to CD8+ T cell suppression. Our results suggest that Nef is not involved in protecting infected macrophages from CD8+ T cell killing and suggest that other mechanisms are involved in macrophage evasion from CD8 surveillance.
INTRODUCTION
Macrophages may play an important role in human immunodeficiency virus type 1/simian immunodeficiency virus (HIV-1/SIV) pathogenesis. Infection of microglia and perivascular macrophages actively mediates entry of HIV-1 into the central nervous system (CNS), resulting in HIV-associated dementia, encephalitis, cognitive disorder, and peripheral neuropathy (1). Accumulating data suggest that tissue macrophages are an important reservoir of virus. Infection of rhesus macaques with a highly pathogenic hybrid simian-human immunodeficiency virus (SHIV) resulted in the rapid depletion of CD4+ T cells such that macrophages were the principal reservoir that sustained high viral loads in the SHIV-infected animals (2). In another study, rhesus macaques depleted of CD4+ T cells exhibited higher viral loads than nondepleted animals, and in situ staining performed on gut tissue revealed that macrophages were the primary source of viral replication (3). It has also been suggested that the level of monocyte turnover is responsible for disease progression in the macaque model of AIDS (4).
Macrophages present unique obstacles to infection by primate lentiviruses. The nondividing status of terminally differentiated macrophages and low deoxynucleoside triphosphate (dNTP) levels have to be accommodated in order for primate lentiviruses to establish a productive infection (5, 6). Additionally, macrophages are resistant to the cytopathic effects of viral replication in comparison to the sensitivity of activated CD4+ T cells (2, 7–9), and HIV-1 has evolved mechanisms to prolong the life span of infected macrophages (9, 10). HIV-1 can also be detected in individuals on suppressive antiretroviral therapy (ART), raising the possibility that these cells serve as a viral reservoir (11). As such, macrophages may be relevant to strategies aimed at elimination of HIV-1 from infected individuals. In this regard, researchers have begun to explore approaches to eradicate persistent viral reservoirs. One method is to stimulate viral activity in the reservoir with the hope that the HIV-infected cells are eliminated by viral cytopathic effects and/or cell-mediated immune clearance (12).
Understanding how CD8+ T cells control viral replication in CD4+ T cells has been the focus of many studies, but little attention has been paid to understanding CD8+ T cell-mediated control of viral replication in infected macrophages. A recent report demonstrated that freshly sorted, SIV-specific CD8+ T cells had the ability to suppress viral replication and eliminate SIV-infected CD4+ T cells after 2 days of infection but were not able to eliminate the majority of SIV-infected macrophages (13). Another study demonstrated that viral replication in HIV-1-infected macrophages could be considerably suppressed 5 to 7 days after infection by ex vivo freshly sorted bulk CD8+ T cells (14). These studies conflict with previous reports that have shown that CD8+ T cell lines or clones can significantly suppress viral replication in HIV/SIV-infected macrophages in a short time frame (15, 16). In order to generate CD8+ T cell lines, B-lymphoblastoid cell lines (BLCLs) are repeatedly pulsed with peptide, and CD8+ T cell lines are repeatedly stimulated in vitro and maintained in interleukin-2 (IL-2)-containing tissue culture medium. These cell lines may have less physiological relevance in the study of CD8+ T cell killing than freshly sorted CD8+ T cells and may not accurately reflect what happens in vivo.
To determine the mechanism responsible for the reported protection of infected macrophages from rapid killing by freshly sorted unstimulated CD8+ T cells, we chose to focus on the effect of the viral accessory Nef protein. Nef can downregulate major histocompatibility complex class I (MHC-I) molecules, thereby facilitating evasion from CD8+ T cell recognition and increased viral replication in infected CD4+ T cells (17, 18). We examined if Nef-mediated MHC-I downregulation underscored the ability of infected macrophages to resist CD8+ T cell surveillance. We examined the capacity of freshly isolated SIV-specific CD8+ T cells to control viral replication in primary target cells (CD4+ T cells and CD14+ monocyte-derived macrophages) infected with SIV harboring a nef variant encoding a point mutation (Y223F) that has been shown to impair MHC-I downregulation and a nef deletion mutant (Δnef strain, with a 181-bp deletion at nucleotide 175 in the beginning of the nef coding sequence) that would be expected to abrogate all activities of Nef, including MHC-I downregulation (19–21). As effector cells, we used freshly sorted SIV-specific CD8+ T cells from elite controller animals. We show that although macrophages infected with SIV nef mutants increase MHC-I expression, this is not sufficient to impact their sensitivity to CD8+ T cell killing.
MATERIALS AND METHODS
Animals.
Indian rhesus macaques (Macaca mulatta) used in this study were housed at the Wisconsin National Primate Research Center. The animals were typed for various MHC-I alleles, including Mamu-B*08, Mamu-A*01, Mamu-A*02, Mamu-B*17, and Mamu-B*03 (Table 1). The animals were cared for according to the regulations and guidelines of the University of Wisconsin Institutional Animal Care and Use Committee.
TABLE 1.
Rhesus macaques used in this study
| Animal no. | Vaccine regimena | SIV insert(s) | Strain used for infection | Viral load at the time of sampling (vRNA copies/ml)e | Relevant MHC-I genotype | Sex |
|---|---|---|---|---|---|---|
| r04106 | EP rDNA + pIL-12/rAd5/rVSV/rRRV | Env, Vif, Nef, Tat | SIV negative | NA | Mamu-A*01 | Female |
| r08061 | EP rDNA + pIL-12/rAd5/rVSV/rRRV | Gag, Vif, Nef, Tat | SIV negative | NA | Mamu-A*01 | Male |
| r04134 | EP rDNA + pIL-12/rAd5/rVSV/rRRV | Env, Vif, Nef, Tat | SIV negative | NA | Mamu-A*02 | Female |
| r00068 | EP rDNA + pIL-12/rYF17D/rAd5 | Nef | SIV negative | NA | Mamu-B*08 | Female |
| r02019a | None | NAd | SIVmac239 | <50 | Mamu-B*08 | Female |
| r96067b | None | NA | SIVmac239 | 4.8 × 102 | Mamu-A*01 Mamu-B*08 | Male |
| r09089 | EP rDNA + pIL-12/rYF17D/rAd5 | Nef | SIVmac239 | 300 | Mamu-B*08 | Male |
| rh2361c | rYF17D/rAd5 | Vif, Nef | SIVmac239 | 428 | Mamu-B*08 | Male |
| rh2352c | rYF17D/rAd5 | Vif, Nef | SIVmac239 | 6.5 × 102 | Mamu-B*08 MamuB*17 | Male |
| r01068 | EP rDNA + pIL-12/rYF17D/rAd5 | Nef | SIVmac239 | 6.2 × 103 | Mamu-B*08 | Male |
| r01049 | EP rDNA + pIL-12/rYF17D/rRRV | Gag | SIVmac239 | 5.5 × 105 | Mamu-A*01 | Male |
| r11039 | rRRV/rAd5/rVSV | Env, Gag, Vif, Nef, Tat | SIVmac239 | ND | Mamu-A*01 | Male |
| r11016 | rRRV/rAd5/rVSV | Empty vectors or irrelevant inserts | SIVmac239 | ND | Mamu-A*01 Mamu-A*02 | Male |
| r98038 | rYF17D/EP rDNA + pIL-12/rAd5/rVSV/rRRV | Empty vectors or irrelevant inserts | SIVmac239 | ND | Mamu-B*17 | Male |
See reference 40. Vaccine abbreviations: EP rDNA, recombinant DNA-expressing plasmid; rAd5, recombinant adenovirus type 5; rVSV, recombinant vesicular stomatitis virus; rRRV, rhesus macaque rhadinovirus; YF17D, yellow fever vaccine virus 17D.
See reference 41.
See reference 42.
NA, not applicable.
vRNA, viral RNA; ND, not determined.
Cell isolation and culture.
To generate target cells, peripheral blood mononuclear cells (PBMC) were freshly isolated from SIV-naive Indian rhesus macaque whole blood by Ficoll-Plaque Plus density centrifugation (GE Healthcare Life Sciences). CD14+ target cells were isolated by positive selection using CD14 microbeads (Miltenyi Biotec) and LS columns (Miltenyi Biotec) and used according to the manufacturer's instructions. CD14+ cells were resuspended in macrophage medium comprising Dulbecco's modified Eagle's medium (DMEM) (Gibco) containing 10% heat-inactivated human serum (Sera Care Life Sciences), 2 mM l-glutamine (Gibco), 10 μg/ml gentamicin (Sigma-Aldrich), and 10 ng/ml rhesus monocyte colony-stimulating factor (rhMCSF; R&D System); cells were seeded at 1 million cells per well in a 24-well plate (Corning) and cultured for 6 days at 37°C with 5% CO2. CD4+ T target cells were isolated from the CD14-negative PBMC fraction using positive selection with CD4 microbeads (Miltenyi Biotec) and LS columns. Target CD4+ T cells were activated for 3 days with 5 mg/ml phytohemagglutinin (PHA) (Roche Diagnostics) and 10 U of interleukin-2 (IL-2)/ml and further cultured in R10-100 medium (RPMI 1640 medium [Gibco] containing 10% fetal bovine serum [FBS; HyClone], supplemented with 2 mM l-glutamine [Gibco], 100 IU/ml penicillin [Gibco], 100 μg/ml streptomycin [Gibco], and 100 U/ml IL-2). IL-2 was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
SIV-specific CD8+ T cells were purified from PBMC isolated from SIV-infected elite controller, progressor, or vaccinated macaques (Table 1) using Ficoll-Plaque Plus density centrifugation. T cells were isolated by negative selection using a Pan-T cell isolation kit (Miltenyi Biotec) and LS columns and used according to the manufacturer's instructions. The T cell fraction was resuspended at 40 million cells/ml in R10-100 medium and incubated with one of the following tetramers for 1 h at 37°C: Gag181–189 CM9-APC (where APC is allophycocyanin) (MBL), Nef137–146RL10-APC, Vif97–104WY8-PE (where PE is phycoerythrin), Vif66–73HW8-APC, or Vif100–109VL10-APC from the NIH Tetramer Core Facility (Table 2). Tetramer-bound CD8+ T cells were enriched using APC or PE microbeads (Miltenyi Biotec), depending on the fluorophore used, and incubated overnight in R15-100 medium (prepared similarly to R10-100 but containing 15% FBS) at 2 million cells/ml in a 24-well plate. Surface staining was performed postsorting to ensure that epitope-specific CD8+ T cells were at least 50% of the purified cells.
TABLE 2.
Freshly sorted SIV-specific CD8+ T cells used in this study
| Protein | Amino acid position | Core epitope | MHC-I restriction | Epitope sequence |
|---|---|---|---|---|
| Gag p27 capsid | 181–189 | CM9 | Mamu-A*01 | CTPYDINQM |
| Nef | 137–146 | RL10 | Mamu-B*08 | RRHRILDIYL |
| Vif | 100–109 | VL10 | Mamu-A*01 | VTPNYADILL |
| Vif | 97–104 | WY8 | Mamu-A*02 | WTDVTPNY |
| Vif | 66–73 | HW8 | Mamu-B*17 | HLEVQGYW |
Generation of SIV and spinoculation infection.
SIVmac239wt (wild type [WT]), SIVmac239Δnef (Δnef), and SIVmac239-Y223F (Y223F) viruses pseudotyped with vesicular stomatitis virus glycoprotein (VSV-G) were generated by cotransfection of 293T cells using Lipofectamine 2000 (Invitrogen) containing 12 μg of SIV proviral plasmids and 1 μg of the pCMV-VSV-G (where CMV is cytomegalovirus) plasmid. The SIVmac239wt and SIVmac239Δnef plasmids were kindly provided by Ronald Desrosiers, and the SIVmac239-Y223F plasmid was obtained from Frank Kirchhoff. Virus-containing supernatants were harvested at 48 h and 72 h posttransfection. Monocyte-derived macrophages (macrophages) were infected with 50 ng of Gag p27 of WT, Y223F, or Δnef (VSV-G-pseudotyped) virus per million cells for 6 h at 48 h prior to the cocultures. Subsequent experiments were completed with macrophages infected with 2 ng of p27gag to reduce infection levels to approximately 35%, referred to here as a low multiplicity of infection (MOI). Viral input was washed, and macrophages were cultured in 1 ml of macrophage medium. CD4+ T cells were infected with 400 ng of p27gag WT, Y223F, or Δnef (VSV-G-pseudotyped) virus per 1 million cells in a 12-well plate (Corning) via spinoculation at 1,200 × g for 2 h at 24 h prior to coculture (22). Cells were allowed to sit for an additional 2 h at 37°C; they were then washed twice and cultured in R15-100 medium. Since, the Nef variants (Y223F or Δnef virus) were available in the SIVmac239 backbone, a lymphocyte-tropic virus, we chose to use pseudotyped virus to facilitate viral infectivity in macrophages. As a control, macrophages were infected in the presence of tenofovir at 400 μM for 72 h to ensure that productive infection was being quantified by flow cytometry in Gag p27+ target cells. Tenofovir was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
Ex vivo viral suppression assay.
The viral suppression assay (VSA) involved the coculture of primary WT-, Y223F-, or Δnef-infected CD4+ T lymphocytes or monocyte-derived macrophages (target cells) with enriched, primary unstimulated SIV-specific CD8+ T cells (effector cells) from several elite controller, infected, or vaccinated animals (Table 2) for 24 h (Fig. 1). In the majority of the cocultures containing WT- and Y223F-infected target cells, freshly sorted Gag181–189 CM9- and Nef137–146RL10-CD8+ T cells (Table 2) were used from elite controller animals (Table 1). The cocultures with Δnef virus-infected target cells or target cells infected at a low MOI were performed with freshly sorted effector cells from infected or vaccinated animals (Table 1) that were elicited from SIV-specific alleles that mounted alternate target peptides (A*01 Gag181–189 CM9, A*02 Vif97–104WY8, A*02 Vif100–109VL10, and B*17 Vif66–73HW8) (Table 2). Suppression of viral replication was determined by an antigen capture assay for Gag p27 (ZeptoMetrix) in culture supernatants. Elimination of infected target cells was assessed by flow cytometric quantification of Gag p27+ target cells in the presence and absence of effectors. SIV-infected target cells cultured in the absence of effectors (target cells only) and SIV-infected target cells isolated from animals with mismatched MHC-I alleles were included as biological controls. Target and effector cells were not autologous but matched in at least one MHC-I allele. Therefore, MHC-I-specific elimination was determined by subtraction of the nonspecific killing observed in mismatched target cell cocultures.
FIG 1.

Ex vivo viral suppression assay scheme. (a) Target cells (macrophages and CD4+ T cells) were isolated from uninfected MHC-I-matched and -mismatched rhesus macaque PBMCs and infected in vitro. (b) Effector SIV-specific CD8+ T cells were enriched from SIV-infected elite controller, progressor, or vaccinated animals using MHC-I tetramers specific for the epitope of interest (Gag181–189 CM9-APC, Nef137–146RL10-APC, Vif97–104WY8-PE, or Vif100–109VL10-APC). (c) Freshly sorted SIV-specific CD8+ T cells were cocultured with SIV-infected macrophages or activated SIV-infected CD4+ T cells at a 1:1 ratio for 24 h. (d) Supernatant was collected from each coculture condition, and p27 content was determined using a Gag p27 ELISA to calculate the percentage of viral suppression. (e) Flow cytometry was used to analyze intracellular staining of Gag p27 and to calculate the percent elimination of target cells (40). mAb, monoclonal antibody.
Infected CD4+ T lymphocytes (2.5 × 104) or infected macrophages (2.5 × 104) were cocultured at 37°C with SIV-specific CD8+ T cells at an effector/target (E/T) ratio of 1:1 in 200 μl of R15-100 or macrophage medium, respectively. The following day, cocultures were centrifuged, and supernatants were collected and frozen for p27 enzyme-linked immunosorbent assays (ELISAs). The cells of the cocultures were harvested and stained with antibodies against CD14 (BV421, clone M5E2; Biolegend) or CD4 (BV421, clone L200; BD), CD3 (PerCPCy5.5, where PerCP is peridinin chlorophyll protein) (clone SP43-2; BD), CD8a (BV605, clone RPA-T8), and HLA-A, B, C (APC, clone W6/32; Biolegend), followed by intracellular Gag p27 staining using BD's Cytofix/Cytoperm with a primary 55-2F12 Gag p27 antibody at 20 μg/ml and 5 ng/ml goat anti-mouse IgG2b R-PE-conjugated secondary antibody (Invitrogen) for 20 min at room temperature. A Live/Dead Fixable Near-IR (infrared) kit (Invitrogen) was used to exclude dead cells from the analysis. Data were collected using an LSR II flow cytometer (BD Biosciences) and analyzed using FlowJo software (version 10.0.6.3). MHC-I-specific elimination of target cells was calculated as the percentage of p27-positive cells eliminated after 24 h of coculture with effector cells, using the following equation: [(percentage of p27-positive cells in target cells only − percentage of p27-positive cells in target cells with fresh CD8+ T cells)/percentage of p27-positive cells in target cells only] ×100 − percent elimination of mismatch controls. Percent maximum suppression of viral replication in target cells was calculated from the concentration of p27gag present in the supernatant collected at 24 h of coculture with effector cells, using the following equation: [(amount of p27 in target cells only − amount of p27 in target cells with fresh CD8+ T cells)/amount of p27 in target cells only] × 100, where the amount of p27 is in nanograms/milliliter. Using tetramer-bound CD8+ T cells in the coculture assays did not prevent their effector function as they were able to kill infected CD4+ T cells. The tetramer-bound CD8+ T cells were not readily phagocytized by infected macrophages as their presence and viability were confirmed using a live/dead gating by flow cytometry, and they were approximately 80% viable (Fig. 2a and b).
FIG 2.

Freshly sorted SIV-specific CD8+ T cells are more effective at eliminating WT-infected CD4+ T cells and suppressing their viral replication than WT-infected macrophages. Dead cells were excluded from the analysis by amine reactive dye staining in cocultures. Staining is displayed for macrophages infected at low MOI (a) and CD4+ T cells cocultured with Nef137–146RL10-CD8+ T cells (b). (c to h) Contour plots representing intracellular p27 staining of infected target cells as indicated above the plots. Contour plots of the target cells were generated by gating on live CD8− T cells or live CD14+ macrophages from one representative experiment. (i and j) Comparison of the MHC-I specific elimination of WT-infected targets and MHC-I specific suppression of viral replication in target cells by tetramer-sorted SIV-specific CD8+ T cells after subtraction of MHC-I-mismatched nonspecific killing. The SIV-specific CD8+ T cells listed in Table 2 were used in this data set. The black line represents the mean percent elimination or suppression of virus, and each data point represents cocultures from different animals. P values: ****, <0.0001; **, 0.001 to 0.01; *, 0.01 to 0.05. FSC, forward scatter; SSC, side scatter.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism, version 6.00, for Windows (GraphPad Software). Unpaired, two-tailed Student's t tests were used for comparing the difference in the suppression levels of viral replication in WT, Y223F, and Δnef virus-infected CD4+ T cells to WT-, Y223F- and Δnef-infected macrophages. Paired, two-tailed Student's t tests were used to compare the difference in results for WT-infected and Y223F-infected CD4+ T cells or for WT-infected and Δnef-infected CD4+ T cells. Paired, two-tailed Student's t tests were used to compare the difference in results for WT-infected and Y223F-infected macrophages or for WT-infected and Δnef-infected macrophages.
RESULTS
SIV-specific CD8+ T cells are less effective at killing WT-infected macrophages.
HIV/SIV-specific CD8+ T cells control viral replication in HIV/SIV-infected CD4+ T cells in an MHC class I (MHC-I)-dependent manner (13, 23–25). However, they are less efficient at controlling viral replication in SIV-infected macrophages (13). To confirm and possibly extend these results, monocyte-derived macrophages and CD4+ T cells were infected in vitro with VSV-G-pseudotyped SIVmac239 (WT), and their elimination and the suppression of viral replication were evaluated by freshly sorted SIV-specific CD8+ T cells ex vivo (Fig. 1). Single-cycle infection of macrophages is very inefficient, even with macrophage-tropic viruses. Therefore, the viruses were pseudotyped with VSV-G, which has the ability to transduce a wide range of mammalian cells, to ensure synchronous and efficient infection of macrophages. Tetramer-sorted SIV-specific CD8+ T cells were effective at eliminating WT-infected MHC-I-matched CD4+ T cells (Fig. 2c and i) but less efficient at killing MHC-I-matched macrophages infected at a high (Fig. 2e and i) or low MOI (Fig. 2g and i) with wild-type virus. The nonspecific killing observed in cocultures with nonautologous MHC-I-mismatched target cells may be a result of an allogeneic reaction by SIV-specific CD8+ T cells, which are mismatched by the MHC-I allele of interest (Fig. 2d, f, and h). Therefore, to evaluate MHC-I-specific elimination, the percentage of elimination was calculated by subtracting the MHC-I-mismatched nonspecific killing (Fig. 2i). It was shown that SIV-specific CD8+ T cells were able to eliminate a mean of 29% of WT-infected CD4+ T cells but only a mean of 3% of WT-infected macrophages, and this difference was statistically significant (P value, <0.0001). Additionally, an increase was observed in MHC-I-specific elimination in WT-infected macrophages with reduced infection (mean of 8%) compared to macrophages with a higher infection (mean of 3%), and this difference was statistically significant (P value, 0.0168). Furthermore, suppression of viral replication was significantly higher (P value, 0.0028) in WT-infected CD4+ T cells than in WT-infected macrophages (Fig. 2j). A mean of 28% maximum suppression was observed in WT-infected CD4+ T cells in contrast to 5% in WT-infected macrophages. Indeed, the majority of WT-infected CD4+ T cells exhibited evidence of suppression in viral replication by CD8+ T cells.
To ensure that the higher levels of infection in macrophages were not saturating the capacity of freshly sorted SIV-specific CD8+ T cells to kill them, cocultures with macrophages infected at a lower MOI were also completed. The resulting lower MOI decreased the infection levels to approximately 35% of the target cells. At the time of these experiments, our access to SIV-specific CD8+ T cells from elite controllers was limited. Therefore, cocultures were completed with macrophages infected at a low MOI using SIV-specific CD8+ T cells isolated from an infected elite controller, progressor, or vaccinated animal as effectors. Lowering the MOI did significantly increase the elimination of macrophages from a mean of 3% to a mean of 8% (P value, 0.0168) (Fig. 2i). However, significant differences (P value, <0.0001) were still observed in the ability of SIV-specific CD8+ T cells to eliminate CD4+ T cells compared to that of macrophages infected at a low MOI (Fig. 2i). Likewise, suppression of viral replication was significantly higher (P value, 0.0092) in WT-infected CD4+ T cells than in macrophages infected at a low MOI (Fig. 2j). No significant differences were detected in the ability of different SIV-specific CD8+ T cells to eliminate or suppress viral replication in WT-infected target cells. As a positive control, SIV-specific CD8+ T cell lines were generated and used to coculture macrophages infected at a high MOI, resulting in a complete elimination of the target cells, as other investigators have previously shown (data not shown) (14, 15).
Impairing MHC-I downregulation does not sensitize infected macrophages to elimination by SIV-specific CD8+ T cells.
HIV-1-infected CD4+ T cells have been shown to be eliminated by HIV-specific CD8+ T cells that were freshly sorted (13) or generated into cell lines (13, 23, 24) primarily in an MHC-I-dependent manner. Therefore, we investigated whether the disruption of the ability of Nef to downregulate MHC-I molecules would sensitize the infected macrophages to elimination and suppression of viral replication by freshly sorted SIV-specific CD8+ T cells early in coculture (23). To validate the ability of Nef variants to disrupt MHC-I downregulation, CD4+ T cells and macrophages were infected with VSV-G-pseudotyped SIVmac239wt (WT), SIVmac239Δnef (Δnef), and SIVmac239-Y223F (Y223F). Y223F virus contains a replacement of a tyrosine with a phenylalanine at position 223 that impairs the downregulation of cell surface MHC-I molecules by nef (19), and Δnef virus contains a 181-bp deletion at nucleotide 175 in the beginning of the nef coding sequence that would be expected to inactivate all activities of nef (20, 21). The Nef variants (Y223F or Δnef) were available in the SIVmac239 backbone, a lymphocyte-tropic virus that replicates efficiently in lymphocytes but poorly in macrophages (26). Infection with VSV-G-pseudotyped viruses facilitates synchronous presentation of peptides on MHC-I molecules. Because of the efficiency of pseudotyped infections, virus spread was not an issue. To confirm productive infection (and not transfer of virus particles) in macrophages infected with pseudotyped Nef variants, macrophages were infected for 72 h in the presence or absence of tenofovir, a protease inhibitor (data not shown). Flow cytometry analysis of MHC-I surface expression on target cells infected with the different Nef variants showed that infection with pseudotyped viruses did not affect the Nef-mediated modulation of MHC-I (Fig. 3). Compared to wild-type SIV, the point mutant and deletion mutant were impaired in their ability to downregulate MHC-I, and this disruption was more pronounced for the deletion mutant (Fig. 3a and b). MHC-I expression in uninfected cells was used for comparison but may differ from that in infected cells due to the activation state of the cell.
FIG 3.

MHC-I expression profile of target cells infected with SIV Nef variants. The MHC-I histograms were generated from gating cells on live infected CD4+ T cells and macrophages infected with WT and Nef variants (Y223F; ΔNef) for 72 h. Data are from a representative experiment. Uninfected cells were used for comparison. The geometric means of the MHC-I profile are included below the histograms.
To validate the ability of freshly isolated tetramer-sorted SIV-specific CD8+ T cells to kill target cells infected with Nef variants capable of disrupting MHC-I downregulation, ex vivo viral suppression assays were completed with infected CD4+ T cells and macrophages isolated from the same animal in parallel. Cocultures of infected CD4+ T cells with effector cells showed a nonsignificant modest increase in the elimination of Y223F-infected cells (36% elimination) compared to that of WT-infected cells (29% elimination) (Fig. 4e). However, in the cocultures with macrophages infected at a high MOI, only a mean of 2% of the WT-infected cells were eliminated, and no increase in the MHC-I-specific elimination of Y223F-infected macrophages was observed (1%). Similar results were seen in WT-infected macrophages (mean of 8%) compared to those in Y223F-infected macrophages (mean of 6%) with reduced infection. Ex vivo-sorted SIV-specific CD8+ T cells that eliminated WT-infected CD4+ T cells (Fig. 4e) eliminated fewer WT-infected macrophages at either high MOI (P value, 0.0025) or low MOI (P value, 0.0093). Moreover, the same effectors significantly eliminated Y223F-infected CD4+ T cells but were inefficient at eliminating Y223F-infected macrophages at either high MOI (P value, 0.0001) or low MOI (P value, 0.0001). A slight increase was observed in MHC-I-specific elimination in WT-infected and Y223F-infected macrophages at low MOI compared to that in macrophages infected at high MOI (P values, 0.0415 and 0.0276, respectively).
FIG 4.

Freshly sorted SIV-specific CD8+ T cells are more effective at eliminating CD4+ T cells than macrophages when infected with Y223F. (a to d) Contour plots representing intracellular p27 staining of MHC-I-matched target cells infected with WT- and Y223F virus as indicated. Contour plots were generated by gating on live CD8− T cells or live CD14+ macrophages from one representative experiment. (e) MHC-I-specific elimination in WT- and Y223F-infected target cells, including macrophages infected at a low MOI, after subtraction of MHC-I-mismatched nonspecific killing. The mean percent elimination of target cells is represented as a black line, and each data point represents an independent experiment. Cocultures of macrophages infected at a high MOI were performed with Gag181–189 CM9- and Nef137–146RL10-CD8+ T cells isolated from multiple elite controller animals. Cocultures with low MOI-infected macrophages included Gag181–189 CM9- and Vif66–73HW8-CD8+ T cells isolated from infected or vaccinated animals. Two experiments included Nef137–146RL10-CD8+ T cells from an elite controller as effector cells. P values: ****, <0.0001; ***, 0.0001 to 0.001; **, 0.001 to 0.01; *, 0.01 to 0.05.
An increase was observed in the mean of MHC-I-specific suppression of viral replication in Y223F-infected CD4+ T cells (mean of 25%) compared to that in WT-infected CD4+ T cells (mean of 9%) (P value, 0.0113) (Fig. 5). However, we did not see a significant increase in the ability of SIV-specific CD8+ T cells to eliminate Y223F-infected macrophages compared to elimination of WT-infected macrophages irrespective of the MOI used in macrophages. Freshly sorted SIV-specific CD8+ T cells that suppressed viral replication in Y223F-infected CD4+ T cells suppressed viral replication to a lesser extent in Y223F-infected macrophages at a high MOI (P value, 0.0003) or low MOI (P value, 0.0023). Due to limited availability of elite controllers for our studies, the majority of coculture experiments completed with macrophages infected at a low MOI were completed with effectors from infected or vaccinated animals, which could explain the reduced elimination and suppression of viral replication observed in WT-infected CD4+ T cells controls (Fig. 5). Taking these results together, it appears that disrupting Nef's ability to downregulate MHC-I does not further sensitize infected macrophages to killing.
FIG 5.
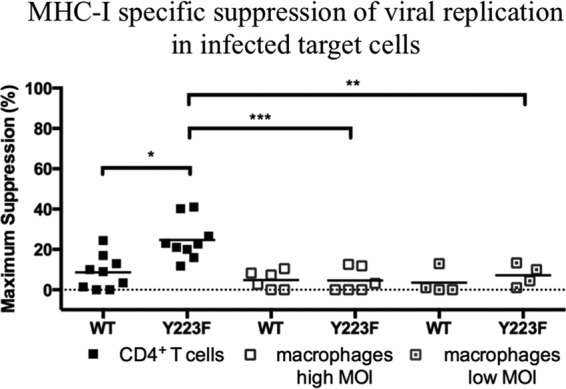
Freshly sorted SIV-specific CD8+ T cells are more effective at suppressing viral replication in CD4+ T cells than macrophages infected with Y223F. A comparison of the MHC-I-specific suppression of viral replication in WT- and Y223F-infected target cells after subtraction of MHC-I-mismatched nonspecific killing is shown. The mean percent suppression of viral replication is represented as a black line, and each data point represents an independent experiment. Cocultures of macrophages infected at a high MOI were performed with Gag181–189 CM9- and Nef137–146RL10-CD8+ T cells isolated from multiple elite controller animals. Cocultures of low-MOI-infected macrophages included Gag181–189 CM9- and Vif66–73HW8-CD8+ T cells isolated from infected progressor or vaccinated animals. Two experiments included Nef137–146RL10-CD8+ T cells from an elite controller as effector cells. P values: ***, 0.0001 to 0.001; **, 0.001 to 0.01; *, 0.01 to 0.05.
Disrupting Nef function is not required for the elimination of infected macrophages by SIV-specific CD8+ T cells.
Infecting target cells with a point mutant that is unable to completely downregulate MHC-I expression did not significantly sensitize infected macrophages to MHC-I-specific elimination or suppression of viral replication by freshly sorted CD8+ T cells (Fig. 5e). Accordingly, it was reasoned that the interference of MHC-I downregulation may not be fully recapitulated in the point mutant (Fig. 3a and b). Flow cytometry analysis on the ability of Δnef and Y223F variants to disrupt MHC-I downregulation proved that the Δnef variant was indeed more effective (Fig. 3a and b). As Nef is also able to downregulate CD4 surface expression (27), in order to accurately calculate the elimination of Δnef-infected CD4+ T cells, both CD4+ p27+ and CD4− p27+ T cell gates were used in the analysis. Since the deletion used to construct the Δnef variant resulted in a frameshift that disrupts the Nef137–146RL10 epitope, cocultures with Δnef-infected target cells were performed with freshly sorted effector cells from infected or vaccinated animals. Cocultures of infected CD4+ T cells with effector cells showed a significant increase in the mean of MHC-I-specific elimination of Δnef-infected CD4+ T cells (29%) compared to that of WT-infected CD4+ T cells (16%) (P value, 0.0064) (Fig. 6e). However, no increase was observed in Δnef-infected macrophages (3%) at a high MOI compared to the level in WT-infected macrophages (2%). Similar results were also seen in macrophages infected at a low MOI when Δnef and WT infections were compared (5% and 8%, respectively). As stated previously, reducing the infection levels slightly increased the MHC-I-specific elimination in WT-infected macrophages (mean difference of 6%), but the difference was not significant. The same was true for Δnef-infected macrophages (mean difference of 2%). As observed in previous experiments (Fig. 2i and 4e), effector cells were more efficient in eliminating WT-infected CD4+ T cells than macrophages infected at high (P value, 0.0064) (Fig. 6e) or low MOI. Similar results were obtained when the percentage of elimination of Δnef-infected CD4+ T cells was compared to that of macrophages infected at a high (P value, 0.0005) or low (P value, 0.0025) MOI. Cocultures with Δnef-infected target cells were completed with SIV-specific CD8+ T cells from infected or vaccinated animals due to the limited availability of elite controller animals with the desired SIV-specific CD8+ T cell responses, which may explain the lower values in viral suppression in CD4+ T cell cocultures.
FIG 6.

Freshly sorted SIV-specific CD8+ T cells are more effective at eliminating Δnef-infected CD4+ T cells than Δnef-infected macrophages. (a to d) Contour plots representing intracellular p27 staining of MHC-I-matched target cells infected with WT- and Δnef virus as indicated. Contour plots were generated by gating on live CD8− T cells or live CD14+ macrophages, and data are from one representative experiment. (e) MHC-I-specific elimination in WT- and Δnef-infected target cells, including macrophages infected at a low MOI, after subtraction of MHC-I-mismatched nonspecific killing. The mean percent elimination of target cells is represented as a black line, and each data point represents an independent experiment. Macrophage cocultures were performed with Gag181–189 CM9-, Vif97–104WY8-, Vif66–73HW8-, or Vif100–109VL10-CD8+ T cells isolated from infected or vaccinated animals and used as effector cells. Cocultures used for the experiments shown in panels a and b or in panels c and d were completed with target cells from the same animal using the same effector cells. P values: ***, 0.0001 to 0.001; **, 0.001 to 0.01; *, 0.01 to 0.05.
Consistently, there was a significant increase in the suppression of viral replication in Δnef-infected CD4+ T cells (mean of 32%; P value, 0.0075) (Fig. 7) compared to that in the WT-infected cells (mean of 6%). Interestingly, ex vivo-sorted SIV-specific CD8+ T cells, which suppressed viral replication in Δnef-infected CD4+ T cells, were not able to suppress viral replication in Δnef-infected macrophages at a high (mean of 0%) or low (mean of 6%) MOI, and these differences were statistically significant (P values of 0.0062 and 0.0098, respectively). Furthermore, reducing the infection levels of macrophages did not reveal significant differences in elimination by SIV-specific CD8+ T cells used in this data set. Therefore, despite the fact that Δnef virus restored MHC-I expression, it did not sensitize infected macrophages to CD8+ T cell elimination or suppression of viral replication after 24 h of coculture (Fig. 7). Therefore, Nef is neither required nor sufficient to control viral replication in infected macrophages by freshly isolated SIV-specific CD8+ T cells.
FIG 7.
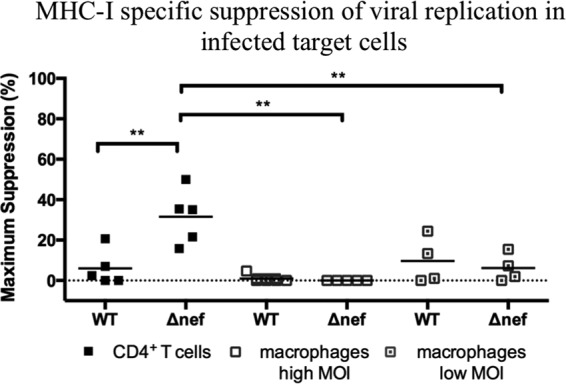
Freshly sorted SIV-specific CD8+ T cells are more effective at suppressing viral replication in Δnef-infected CD4+ T cells than in Δnef-infected macrophages. A comparison of the levels of MHC-I-specific suppression of viral replication in WT- and Δnef-infected target cells after subtraction of MHC-I-mismatched nonspecific killing is shown. The mean percent elimination of target cells is represented as a black line, and each data point represents an independent experiment. Macrophage cocultures were performed with Gag181–189 CM9-, Vif97–104WY8-, Vif66–73HW8-, or Vif100–109VL10-CD8+ T cells isolated from infected or vaccinated animals and used as effector cells. **, P = 0.001 to 0.01.
DISCUSSION
Macrophages potentially constitute a viral reservoir in infected individuals on suppressive ART. Therefore, understanding whether these cells succumb to CD8+ T cell killing is important in the design of strategies to cure HIV-1 infection (1–3, 9, 28). Previously, it has been shown that primary CD8+ T cell clones and CD8+ T cell lines suppress HIV-1 and SIV replication in macrophages (14–16). Interestingly, autologous bulk CD8+ T cells and CD4+ T cells from human elite controllers suppressed viral replication in HIV-1-infected macrophages within 5 to 7 days (14). However, within this window of time macrophages were able to spread infection to CD4+ T cell effectors (14). Therefore, slow killing of macrophages is likely to be of little consequence in cell-mediated control of viral replication in infected individuals. Elimination of macrophages early in the infection cycle is necessary in order to prevent further virus spread. Our data show that freshly sorted SIV-specific CD8+ T cells failed to significantly eliminate infected macrophages.
Based on our results, freshly sorted CD8+ T cells eliminated and suppressed viral replication in WT-infected CD4+ T cells, and most of the killing was MHC-I dependent. We reasoned that the ability of Nef to downregulate MHC-I was protecting macrophages from SIV-specific CD8+ T cell recognition. However, altering the ability of Nef to downregulate MHC-I in infected macrophages, with a point mutant or by entirely disrupting the Nef function, did not further sensitize macrophages to killing by primary effector cells. Therefore, Nef appears to be neither necessary nor sufficient for the resistance of infected macrophages to elimination or suppression of viral replication by unstimulated, freshly sorted SIV-specific CD8+ T cells. HIV may have acquired Nef to protect infected CD4+ T cells from CD8-mediated killing, but macrophages seem to have developed alternate mechanisms for preserving viability in the face of the antiviral CD8+ T cell responses.
We (data not shown) along with others observed the elimination of infected macrophages by HIV/SIV-specific CD8+ T cells lines (14–16) but only marginally by freshly sorted HIV/SIV-specific CD8+ T cells (13). HIV/SIV-infected macrophages may be more resilient to the effector molecules secreted by unstimulated freshly sorted SIV-specific CD8+ T cells. In order to generate peptide-specific CD8+ T cell lines, we used previously described methods (29, 30). Briefly, the Nef137–146RL10-specific CD8+ T cell lines were started using freshly isolated PBMC from an elite controller animal containing Nef137–146RL10-specific CD8+ T cells. The Nef137–146RL10-specific CD8+ T cell lines were expanded for 1 month by repeated stimulation with peptide-pulsed irradiated autologous B-lymphoblastoid cell lines (BLCLs). The freshly sorted Nef137–146RL10-specific CD8+ T cells were not stimulated before coculture assays and were only exposed overnight to IL-2. Cocultures with prestimulated CD8+ T cell lines and freshly isolated unstimulated CD8+ T cells were completed in parallel. The elimination of infected macrophages by the CD8+ T cell lines, which were repeatedly stimulated with the Nef137–146RL10 peptide, may, upon recognition of their specific epitope, elicit abnormal antiviral responses that may overcome defense mechanisms by macrophages. A study completed by Shan et al. revealed the upregulation of granzyme B (GrB), gamma interferon (IFN-γ), CD107α, and perforin production in primary CD8+ T cells stimulated with Gag-specific peptides compared to that in nonstimulated cells (31). It is possible that infected macrophages are targeted more efficiently by the excessively primed Nef137–146RL10-specific CD8+ T cell lines and are able to deliver more robustly effector molecules that may overcome defense mechanisms by infected macrophages, whereas freshly sorted SIV-specific CD8+ T cells may be a more physiological representation of in vivo killing of infected macrophages.
Are macrophages intrinsically resistant to CD8-mediated T cell killing, or does infection of macrophages by HIV-1/SIV induce a protective mechanism? CD8+ T cells are able to lyse peritoneal macrophages infected with lymphocytic choriomeningitis virus (LCMV), suggesting that macrophages can be killed by virus-specific CD8+ T cells (32, 33). If LCMV-infected macrophages are susceptible to killing by primary CD8+ T cells, this would suggest that HIV-1 may employ a mechanism to circumvent recognition or killing by CD8+ T cells. Many viruses have adopted various strategies to preserve host cell viability in the face of cytopathicity and cell-mediated clearance forces. In particular, Kaposi sarcoma-associated human herpesvirus 8 and related gammaherpesviruses prevent the triggering of TRAIL-induced apoptosis by encoding viral FLICE-inhibitory proteins (vFLIPs) that interact with the Fas-associated death domain (FADD) to inhibit active caspase 8 generation (34). Additionally, T cells infected with human-T cell leukemia virus type 1 are resistant to TRAIL-mediated apoptosis by the viral transactivator Tax (35). Furthermore, three proteins (E3 RID) encoded by the human adenovirus type 5 induce the internalization of TRAIL receptors from the cell surface targeting lysosomal degradation (36). Moreover, the human herpesvirus 7 is resistant to TRAIL-mediated cytotoxicity, and this is associated with downregulation of the TRAIL-R1 receptor from the surface of cells (37). Also in B cells, the BHRF1 protein is encoded by the Epstein-Barr virus and is responsible for the inhibition of apoptosis by TRAIL (38, 39).
We have previously demonstrated that HIV-1 induces prosurvival factors that preserve host cell viability in the face of cytopathicity. In HIV-1-infected macrophages, the envelope glycoprotein induces the prosurvival cytokine monocyte colony-stimulating factor (MCSF). This circumvents TRAIL-mediated apoptosis to maintain cell survival in the face of apoptotic stimuli, thereby affording macrophages protection from the cytopathic effects of the virus (9). Apoptotic sensitivity could be restored when these prosurvival effectors were inhibited by treating infected macrophages with imatinib, an anticancer drug that has the ability to block signaling by the MCSF receptor (9). Induction of prosurvival cytokines, such as MCSF, appears to be envelope dependent and independent of Nef and indicates that Nef is dispensable for the resistance of infected macrophages to CD8+ T cell killing. The fact that inactivating mutations in Nef do not restore susceptibility to CD8+ T cell surveillance implies that the induction of prosurvival pathways in infected macrophages is Nef independent. This further suggests that the induction of prosurvival pathways creates a phenotype that overrides the impact of cytotoxic CD8+ T cells on infected macrophages. Therefore, the results in the current study, that Nef does not govern resistance to CD8+ T cell killing, can be reconciled by our previous observations that the viral envelope may harness activities that maintain the viability of the infected macrophage in the face of immune (CD8+ T cell killing) or viral (cytopathicity) assault. Studies are under way to determine whether induction of prosurvival cytokines in infected macrophages underscores the resistance of infected macrophages to CD8+ T cell killing, and we are now exploring whether pharmacologic inhibition of these prosurvival pathways can restore viral cytopathicity and the killing of infected macrophages by CD8+ T cells. Determining the intrinsic factors in HIV/SIV-infected macrophages that give protection from elimination by CD8+ T cells could help in developing drug treatments to eradicate the macrophage reservoir.
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health (NIH) grants R01MH093306-01, 5U19A1096109-04, and 1P01MH100942-02 awarded to M.S. and grants 1P01AI094420-03, 5R37AI052056-13, and 1R01AI108421-02 awarded to D.I.W.
We thank Ronald Desrosiers for providing SIVmac239 and SIVmac239Δnef, Frank Kirchhoff for providing SIVmac239-Y223F, and John Altman for providing the tetramers. We also thank Eva Rakasz for providing the 55-2F12 SIVmac Gag p27 antibody.
REFERENCES
- 1.Tambuyzer BR, Ponsaerts P, Nouwen EJ. 2009. Microglia: gatekeepers of central nervous system immunology. J Leukoc Biol 85:352–370. doi: 10.1189/jlb.0608385. [DOI] [PubMed] [Google Scholar]
- 2.Igarashi T, Brown CR, Endo Y, Buckler-White A, Plishka R, Bischofberger N, Hirsch V, Martin MA. 2001. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4+ T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): implications for HIV-1 infections of humans. Proc Natl Acad Sci U S A 98:658–663. doi: 10.1073/pnas.98.2.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ortiz AM, Klatt NR, Li B, Yi Y, Tabb B, Hao XP, Sternberg L, Lawson B, Carnathan PM, Cramer EM, Engram JC, Little DM, Ryzhova E, Gonzalez-Scarano F, Paiardini M, Ansari AA, Ratcliffe S, Else JG, Brenchley JM, Collman RG, Estes JD, Derdeyn CA, Silvestri G. 2011. Depletion of CD4+ T cells abrogates post-peak decline of viremia in SIV-infected rhesus macaques. J Clin Invest 121:4433–4445. doi: 10.1172/JCI46023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hasegawa A, Liu H, Ling B, Borda JT, Alvarez X, Sugimoto C, Vinet-Oliphant H, Kim WK, Williams KC, Ribeiro RM, Lackner AA, Veazey RS, Kuroda MJ. 2009. The level of monocyte turnover predicts disease progression in the macaque model of AIDS. Blood 114:2917–2925. doi: 10.1182/blood-2009-02-204263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiss KK, Chen R, Skasko M, Reynolds HM, Lee K, Bambara RA, Mansky LM, Kim B. 2004. A role for dNTP binding of human immunodeficiency virus type 1 reverse transcriptase in viral mutagenesis. Biochemistry 43:4490–4500. doi: 10.1021/bi035258r. [DOI] [PubMed] [Google Scholar]
- 6.Gavegnano C, Kennedy EM, Kim B, Schinazi RF. 2012. The impact of macrophage nucleotide pools on HIV-1 reverse transcription, viral replication, and the development of novel antiviral agents. Mol Biol Int 2012:625983. doi: 10.1155/2012/625983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coleman CM, Wu L. 2009. HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology 6:51. doi: 10.1186/1742-4690-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 9.Swingler S, Mann AM, Zhou J, Swingler C, Stevenson M. 2007. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog 3:1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynoso R, Wieser M, Ojeda D, Bonisch M, Kuhnel H, Bolcic F, Quendler H, Grillari J, Grillari-Voglauer R, Quarleri J. 2012. HIV-1 induces telomerase activity in monocyte-derived macrophages, possibly safeguarding one of its reservoirs. J Virol 86:10327–10337. doi: 10.1128/JVI.01495-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doyle T, Geretti AM. 2012. Low-level viraemia on HAART: significance and management. Curr Opin Infect Dis 25:17–25. doi: 10.1097/QCO.0b013e32834ef5d9. [DOI] [PubMed] [Google Scholar]
- 12.Archin NM, Margolis DM. 2014. Emerging strategies to deplete the HIV reservoir. Curr Opin Infect Dis 27:29–35. doi: 10.1097/QCO.0000000000000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vojnov L, Martins MA, Bean AT, Veloso de Santana MG, Sacha JB, Wilson NA, Bonaldo MC, Galler R, Stevenson M, Watkins DI. 2012. The majority of freshly sorted simian immunodeficiency virus (SIV)-specific CD8+ T cells cannot suppress viral replication in SIV-infected macrophages. J Virol 86:4682–4687. doi: 10.1128/JVI.06324-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker-Sperling VE, Buckheit RW III, Blankson JN. 2014. Comparative analysis of the capacity of elite suppressor CD4+ and CD8+ T cells to inhibit HIV-1 replication in monocyte-derived macrophages. J Virol 88:9789–9798. doi: 10.1128/JVI.00860-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujiwara M, Takiguchi M. 2007. HIV-1-specific CTLs effectively suppress replication of HIV-1 in HIV-1-infected macrophages. Blood 109:4832–4838. doi: 10.1182/blood-2006-07-037481. [DOI] [PubMed] [Google Scholar]
- 16.Sacha JB, Giraldo-Vela JP, Buechler MB, Martins MA, Maness NJ, Chung C, Wallace LT, Leon EJ, Friedrich TC, Wilson NA, Hiraoka A, Watkins DI. 2009. Gag- and Nef-specific CD4+ T cells recognize and inhibit SIV replication in infected macrophages early after infection. Proc Natl Acad Sci U S A 106:9791–9796. doi: 10.1073/pnas.0813106106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foster JL, Garcia JV. 2008. HIV-1 Nef: at the crossroads. Retrovirology 5:84. doi: 10.1186/1742-4690-5-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. 1996. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med 2:338–342. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- 19.Munch J, Stolte N, Fuchs D, Stahl-Hennig C, Kirchhoff F. 2001. Efficient class I major histocompatibility complex down-regulation by simian immunodeficiency virus Nef is associated with a strong selective advantage in infected rhesus macaques. J Virol 75:10532–10536. doi: 10.1128/JVI.75.21.10532-10536.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibbs JS, Regier DA, Desrosiers RC. 1994. Construction and in vitro properties of SIVmac mutants with deletions in “nonessential” genes. AIDS Res Hum Retroviruses 10:607–616. doi: 10.1089/aid.1994.10.607. [DOI] [PubMed] [Google Scholar]
- 21.Swigut T, Iafrate AJ, Muench J, Kirchhoff F, Skowronski J. 2000. Simian and human immunodeficiency virus Nef proteins use different surfaces to downregulate class I major histocompatibility complex antigen expression. J Virol 74:5691–5701. doi: 10.1128/JVI.74.12.5691-5701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Doherty U, Swiggard WJ, Malim MH. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol 74:10074–10080. doi: 10.1128/JVI.74.21.10074-10080.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. 1998. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 24.Yang OO, Kalams SA, Trocha A, Cao H, Luster A, Johnson RP, Walker BD. 1997. Suppression of human immunodeficiency virus type 1 replication by CD8+ cells: evidence for HLA class I-restricted triggering of cytolytic and noncytolytic mechanisms. J Virol 71:3120–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swigut T, Alexander L, Morgan J, Lifson J, Mansfield KG, Lang S, Johnson RP, Skowronski J, Desrosiers R. 2004. Impact of Nef-mediated downregulation of major histocompatibility complex class I on immune response to simian immunodeficiency virus. J Virol 78:13335–13344. doi: 10.1128/JVI.78.23.13335-13344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mori K, Ringler DJ, Desrosiers RC. 1993. Restricted replication of simian immunodeficiency virus strain 239 in macrophages is determined by env but is not due to restricted entry. J Virol 67:2807–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia JV, Alfano J, Miller AD. 1993. The negative effect of human immunodeficiency virus type 1 Nef on cell surface CD4 expression is not species specific and requires the cytoplasmic domain of CD4. J Virol 67:1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharova N, Swingler C, Sharkey M, Stevenson M. 2005. Macrophages archive HIV-1 virions for dissemination in trans. EMBO J 24:2481–2489. doi: 10.1038/sj.emboj.7600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loffredo JT, Burwitz BJ, Rakasz EG, Spencer SP, Stephany JJ, Vela JP, Martin SR, Reed J, Piaskowski SM, Furlott J, Weisgrau KL, Rodrigues DS, Soma T, Napoe G, Friedrich TC, Wilson NA, Kallas EG, Watkins DI. 2007. The antiviral efficacy of simian immunodeficiency virus-specific CD8+ T cells is unrelated to epitope specificity and is abrogated by viral escape. J Virol 81:2624–2634. doi: 10.1128/JVI.01912-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogel TU, Friedrich TC, O'Connor DH, Rehrauer W, Dodds EJ, Hickman H, Hildebrand W, Sidney J, Sette A, Hughes A, Horton H, Vielhuber K, Rudersdorf R, De Souza IP, Reynolds MR, Allen TM, Wilson N, Watkins DI. 2002. Escape in one of two cytotoxic T-lymphocyte epitopes bound by a high-frequency major histocompatibility complex class I molecule, Mamu-A*02: a paradigm for virus evolution and persistence? J Virol 76:11623–11636. doi: 10.1128/JVI.76.22.11623-11636.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, Zhang H, Margolick JB, Blankson JN, Siliciano RF. 2012. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zinkernagel RM, Doherty PC. 1975. Peritoneal macrophages as target cells for measuring virus-specific T cell mediated cytotoxicity in vitro. J Immunol Methods 8:263–266. doi: 10.1016/0022-1759(75)90120-9. [DOI] [PubMed] [Google Scholar]
- 33.Zinkernagel RM, Doherty PC. 1975. H-2 compatability requirement for T-cell-mediated lysis of target cells infected with lymphocytic choriomeningitis virus. Different cytotoxic T-cell specificities are associated with structures coded for in H-2K or H-2D. J Exp Med 141:1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. 1997. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 35.Matsuda T, Almasan A, Tomita M, Uchihara JN, Masuda M, Ohshiro K, Takasu N, Yagita H, Ohta T, Mori N. 2005. Resistance to Apo2 ligand (Apo2L)/tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis and constitutive expression of Apo2L/TRAIL in human T-cell leukemia virus type 1-infected T-cell lines. J Virol 79:1367–1378. doi: 10.1128/JVI.79.3.1367-1378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Tollefson AE, Toth K, Doronin K, Kuppuswamy M, Doronina OA, Lichtenstein DL, Hermiston TW, Smith CA, Wold WS. 2001. Inhibition of TRAIL-induced apoptosis and forced internalization of TRAIL receptor 1 by adenovirus proteins. J Virol 75:8875–8887. doi: 10.1128/JVI.75.19.8875-8887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Secchiero P, Mirandola P, Zella D, Celeghini C, Gonelli A, Vitale M, Capitani S, Zauli G. 2001. Human herpesvirus 7 induces the functional up-regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) coupled to TRAIL-R1 down-modulation in CD4+ T cells. Blood 98:2474–2481. doi: 10.1182/blood.V98.8.2474. [DOI] [PubMed] [Google Scholar]
- 38.Kawanishi M, Tada-Oikawa S, Kawanishi S. 2002. Epstein-Barr virus BHRF1 functions downstream of Bid cleavage and upstream of mitochondrial dysfunction to inhibit TRAIL-induced apoptosis in BJAB cells. Biochem Biophys Res Commun 297:682–687. doi: 10.1016/S0006-291X(02)02261-1. [DOI] [PubMed] [Google Scholar]
- 39.Werner AB, de Vries E, Tait SW, Bontjer I, Borst J. 2002. Bcl-2 family member Bfl-1/A1 sequesters truncated bid to inhibit is collaboration with pro-apoptotic Bak or Bax. J Biol Chem 277:22781–22788. doi: 10.1074/jbc.M201469200. [DOI] [PubMed] [Google Scholar]
- 40.Saez-Cirion A, Shin SY, Versmisse P, Barre-Sinoussi F, Pancino G. 2010. Ex vivo T cell-based HIV suppression assay to evaluate HIV-specific CD8+ T-cell responses. Nat Protoc 5:1033–1041. doi: 10.1038/nprot.2010.73. [DOI] [PubMed] [Google Scholar]