

Abstract
Bacterial toxin or viral entry into the cell often requires cell surface binding and endocytosis. The endosomal acidification induces a limited unfolding/refolding and membrane insertion reaction of the soluble toxins or viral proteins into their translocation competent or membrane inserted states. At the molecular level, the specific orientation and immobilization of the pre-transitioned toxin on the cell surface is often an important prerequisite prior to cell entry. We propose that structures of some toxin membrane insertion complexes may be observed through procedures where one rationally immobilizes the soluble toxin so that potential unfolding ↔ refolding transitions that occur prior to membrane insertion orientate away from the immobilization surface in the presence of lipid micelle pre-nanodisc structures. As a specific example, the immobilized prepore form of the anthrax toxin pore translocon or protective antigen can be transitioned, inserted into a model lipid membrane (nanodiscs), and released from the immobilized support in its membrane solubilized form. This particular strategy, although unconventional, is a useful procedure for generating pure membrane-inserted toxins in nanodiscs for electron microscopy structural analysis. In addition, generating a similar immobilized platform on label-free biosensor surfaces allows one to observe the kinetics of these acid-induced membrane insertion transitions. These platforms can facilitate the rational design of inhibitors that specifically target the toxin membrane insertion transitions that occur during endosomal acidification. This approach may lead to a new class of direct anti-toxin inhibitors.
Keywords: Anthrax protective antigen, GroEL, Lipid nanodiscs, Biolayer interferometry, Electron microscopy
Introduction
Many bacterial infections involve numerous pore-forming bacterial toxins that have evolved to traffic different substrates across lipid bilayers, ranging from small inorganic ions to large proteins or polypeptides. These trafficked substrates often disrupt cellular homeostasis and in some instances can lead to lethality. Interestingly, many of these toxin structures initially exist as soluble states that then bind to specific cellular membranes or receptors. Upon endocytosis of the bound toxins, the endosome is acidified and the bound toxins transition from their soluble to membrane inserted forms at the membrane surface. Structures of many of the soluble toxins have been solved using X-ray crystallography yet only a few membrane-inserted structures have been similarly resolved. This gap in structural knowledge of the inserted toxins has hindered our understanding of structure based protein translocation mechanisms. In this short report, an alternative approach based on replicating a specific immobilization and orientation and insertion into smaller bilayer structures such as lipid nanodiscs is highlighted. The validity of this method is demonstrated with the successful construction of the anthrax toxin pore translocon nanodisc structures for EM analysis. In particular, protein toxin assembly and transitions often occur following capture of the protein toxins onto membrane surfaces. In some instances, soluble bacterial toxin binding to cell surface receptor proteins proceeds endocytosis and acidification. For example, prior to entry, the receptor-binding domain of the diphtheria toxin binds to the heparin binding epidermal growth factor precursor (hb-EGF) (Naglich et al. 1992), and the receptor-binding domains of the anthrax toxin protective antigen (PA) primarily binds to the capillary morphogenesis protein (CMG2) (Liu et al. 2009). Likewise, Clostridia tetani neurotoxin, cholera toxin, shiga toxins and a host of viral proteins all bind to cell surface glycolipids prior to cellular entry (Esko and Sharon 2009). Armed with the knowledge that toxin binding is orientation specific with respect to membranes, it is useful to explore the possibility that methods aimed at orientating and recapitulating this toxin transitioning reaction toward membrane surfaces is a worthwhile approach to generate large quantities of transitioned toxins inserted into membranes. Thus far, most successful efforts where structures of transitioned toxins (e.g., alpha-hemolysin, Hemolytic lectin CEL-III, Aeromonas toxin proaerolysin) have been resolved rely on classic detergent solubilization approaches to generate two dimensional arrays for X-ray crystallographic analysis or negative stain electron microscopy (Parker et al. 1994; Song et al. 1996; Unno et al. 2014). In these particular instances, the assembly of the oligomeric states and the transition to the membrane inserted state appears to occur directly on membrane surfaces. With the recent revolution in cryo-electron microscopy, improving one’s ability to prepare large quantities of purified membrane-inserted toxins will be crucial for resolving the structures of toxins inserted into authentic lipid bilayers to generate translocation competent states. The development of this method followed a circuitous path that started with the notion that one could prevent aggregation of the transitioning toxins with chaperone proteins.
Embracing the Unconventional: Using GroEL as an Orientation Platform for Anthrax Prepore to Pore Transitions
The tetradecameric chaperonin GroEL contains a large 45 Å diameter hydrophobic binding site that is wide enough to accommodate de-lipidated membrane proteins (Deaton et al. 2004a, b; Sun et al. 2005). Following capture and ATP addition, GroEL can release these membrane proteins in their membrane insertable states as evidenced by their reinsertion into vesicles. Based on these experimental observations, it was surmised by Collier and Fisher that GroEL may be a useful protein capture system or serve as an alternative membrane protein solubilizer to prevent aggregation of the anthrax toxin prepore during its transition to its pore state because the transitioned membrane hydrophobic tip may insert into the hydrophobic GroEL binding cavity. As predicted, GroEL was able to capture the prepore state of the PA heptamer (Katayama et al. 2008) but surprisingly through an entirely different molecular interaction surface. It turns out that the heptameric PA of anthrax contains a predominant positive electrostatic surface on the PA prepore cap region that than binds through electrostatic interactions onto the top of the negative electrostatic potential that surrounds the GroEL hepatmeric binding cavity (Coyle et al. 1997) with a sevenfold symmetry match. This interaction is easily diminished by increasing the ionic strength of the solution. More convincingly, it determined that a specific arginine mutant (R178A) located on the surface of the anthrax prepore cap region abolishes lethal factor binding to the heptamer pore or prepore and greatly diminished GroEL complex formation (Katayama and Janowiak unpublished results). With this PA R178A mutant, GroEL was no longer able to capture the prepore and avoid aggregation of PA during transition. In the Fig. 1, the approximate orientation of anthrax toxin protective antigen pore (PA pore) binding to the GroEL heptamer surface, based on EM negative stain EM single particle reconstructions, is modeled based on negative stain electron microscopy averages of GroEL PA particles (Fig. 1 far left). It is evident that the interaction of the transitioned PA pore transition with GroEL interacts through the PA cap region.
Fig. 1.
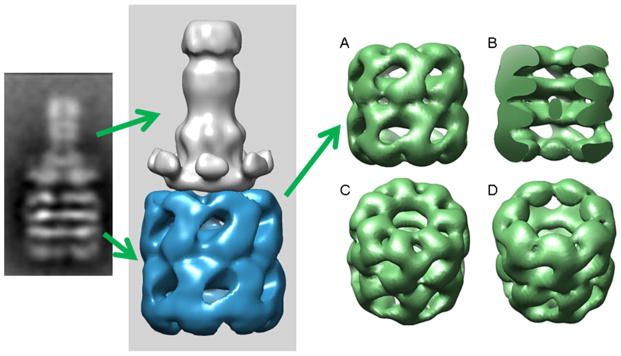
Raw averaged image (left) of GroEL–PA pore electrostatic complexes from negative stain electron microscopy. Middle-EM reconstruction of GroEL PA pore complex (approximate) results in a 1.24 MDa complex. Less than 1 % of all GroEL PA complexes showed two PA pore binding to one GroEL suggesting negative cooperative binding of PA to the GroEL chaperonin. A view of the 25A reconstruction of the GroEL alone (structures in green) that was complexed with the PA pore depicts the PA binding interface in the far right GroEL images a (full) and b (half slice). A 45°tilt of the top c and bottom (face opposite PA binding interface) indicates that the central binding cavity of GroEL on the binding site opposite the PA binding site is drastically expanded (d)
Once bound, the transition of the anthrax toxin to its pore translocon competent state on the GroEL surface was accomplished by destabilizing the prepore state at 37 °C with the addition of 1 M urea. This did not disrupt the GroEL protein substrate interaction and the unfolding refolding transition of part of domain 2 resulted in a clearly EM resolved 100 Å extended beta barrel (Katayama et al. 2008). It was important to point out that this was done without detergents or detergent screens. This observation leads to the realization that orientation specific toxin transitions could be possible. The reason that GroEL was a workable platform is probably due to its large size. This increase in complex size diminishes diffusional collision rates, allowing the pore to transition without interference from other transitioning pores.
It is interesting to note that GroEL only binds one pore although both binding sites are initially available.
The structure of the GroEL that was bound to the pore was reconstructed from 1,379 particles using a simple cylinder with a radius of 80 Å. Reconstructions converged to the GroEL structure presented in Fig. 1 (far right-hand side). In agreement with the two dimensional average of the GroEL–pore complex, no density is observed in the substrate binding cavity (cis GroEL ring), and it appears that the pore is interacting with the exterior surface of GroEL (Fig. 1 right-hand side a–c). No significant changes could be observed at the cis ring, but an enlarged opening and density thinning in the apical domain of the trans ring are apparent (Fig. 1d). Second, substantial movements occur at the heptamer ring apical domains trans to the cis heptamer PA7 binding site, resulting in distinct inter-ring asymmetry (compare Fig. 1c and d). Curiously, this large asymmetry arises from protein substrate interactions with the surface of the GroEL apical domains rather than within the GroEL cavity. Such large structural changes could result from heterogeneous asymmetric movements of the apical domains, resulting in the observed loss of apical density during sevenfold averaging (Fig. 1d). Since the density thinning in the trans ring is substantial in the GroEL-pore structure compared to trans rings from existing GroEL-substrate protein reconstructions (Falke et al. 2004; Elad et al. 2007), the asymmetric movements of the trans ring in the GroEL–pore complexes may be considerable when sevenfold symmetry operations are not imposed. Nonetheless, the GroEL binding interaction with the pore, even at 1:1 GroEL to PA pore oligomer ratios, only populates singly bound GroEL species, suggesting that this specific surface interaction between PA and GroEL electrostatic interfaces dramatically demonstrates significant negative cooperativity between the GroEL rings. Similar differences in ring diameters with GroEL were previously observed in instances where ATP hydrolysis created heterogeneous populations resulting in a similar thinning of apical densities on one GroEL ring (Roseman et al. 2001). In the GroEL PA complex, these differences in the GroEL binding cavity could reflect a significant increase in flexibility in the opposite ring. Large structural changes such as this suggest that the binding interactions of the PA pore cap face to the GroEL surface results in long range changes in the GroEL platform (~140 Å).
As stated above, one reasonable explanation for the diminished aggregation during prepore to pore transitions may be related to the increase in complex mass, increase in complex time, which in turn, substantially reduces the diffusion rates of the complexes. The increased lifetime of the complex and the slower collision rates allowed the PA prepore to transition on the surface of GroEL before unproductive aggregation could occur. Once formed, the exposed hydrophobic tips of the PA pore can still interact with other PA exposed hydrophobic tips as evidenced by the negative stain images where complexes make dimers, trimers, and tetramers via the tip–tip interactions (See Fig. 2 in Katayama et al. 2008; Fig. 3 in Naik et al. 2013).
In the efforts to produce a soluble portion of the PA pore, a loopless version was created where the beta sheet and turn residues that inserted into the membrane bilayer were removed. The resulting truncation mutant (Δ 303–324 PA or loopless PA) is predicted to form a smaller barrel region that may be soluble. However, using acidic or 1 M urea/37 °C transition schemes this truncated pore still aggregates during its transition. Therefore, GroEL was again added to potentially stabilize this form and the complexes were evaluated using negative stain EM (Fig. 2). Interestingly, the EM micrograph shows distinct differences in the loop-less PA GroEL complexes. While the transitioned loopless PA could be readily observed (Fig. 2b, c), the aggregation properties of this GroEL–loopless PA showed drastic differences. The wild type PA pore aggregates via the exposed hydrophobic tip (EM field; Fig. 2a) while the GroEL loopless complexes formed long chains in a manner where the PA loopless is between two GroEL molecules. This interaction may indicate that GroEL can bind both the cap of the truncated PA as well as the now shorter barrel. This is also in keeping with the notion that some of these interactions are electrostatic in nature because the PA loopless is missing the prominent negatively charged (interior lumen) membrane insertion tips and is left with a shorter barrel that contains a prominent positive charge (Wynia-Smith et al. 2012). This barrel is no longer repelled from the GroEL binding site that is surrounded by a negative electrostatic potential (Coyle et al. 1997). Following the selection of 1,400 images of the GroEL PA loopless complexes, the resulting two dimensional averages showed a distinct truncation of the extended PA barrel. In comparison with the wild type PA pore, the PA loopless structure clearly lacks the hydrophobic membrane insertion tip that excludes negative stain (Fig. 2c).
Fig. 2.

Comparison of full length PA pore with the loopless PA pore. Electron micrographs of a GroEL PA complexes and b GroEL-loopless PA indicate that the aggregation complexes between these two pore forms are different. The expanded insets in a and b reveal differences in aggregation complexes. The GroEL PA loopless complexes form long chains while the GroEL PA wild type complexes show aggregation at the tips of the PA pore. c Comparison of the two dimensional averages of GroEL PA wildtype (Katayama et al. 2008) and 1400 GroEL PA loopless particles show distinct length differences between these two forms
Construction of Truncated Lethal Factor-Protective Antigen Pore Inserted into Lipid Nanodiscs
Although GroEL was able to capture and stabilize the PA pore, the release of this pore structure from the GroEL surface still resulted in excessive aggregation even when a compatible detergent was present (e.g., FOS14 see Viener et al. 2009). Likewise, upon release from its GroEL platform, the truncated PA pore also failed to remain monodisperse. At this point, the option of using nanodisc technologies to solubilize the transitioned pore complex was explored.
Nanodiscs are stable lipid bilayer constructions derived from truncated apolipoprotein A-1 (reviewed in Ritchie et al. 2009). The truncated protein product, called the matrix scaffold protein or MSP, is thought to form an antiparallel alpha-helical belt that provides scaffolding for lipid bilayer. The ease of formation of nanodiscs, coupled with very detailed published protocols to insert solubilized membrane proteins into these nanodiscs has led to the solubilization of a large number of membrane proteins inserted or tethered into these unique discoid lipid bilayers. To date, reports of successful insertion of membrane proteins numbers into these nanodiscs are approaching ~500 publications. The particular appeal of this method is that one may be able to easily generate monodisperse membrane proteins that are inserted into authentic bilayers which are soluble.
Published nanodisc construction protocols rely on initial detergent solublization of the protein (Ritchie et al. 2009; Bayburt and Sligar 2010). This solubilization is crucial for membrane protein-nanodisc insertion success since even the formation of soluble aggregates may result in failure. The lipid make up of these bilayers can be highly variable and can be tailored to mimic biologically relevant membranes (e.g., lipid mixes in endosomes). Our first attempts at producing an anthrax toxin pore inserted into a nanodisc were reliant on finding a suitable solubilizing detergent. An extensive detergent screen performed by Vernier et al. (2009) determined that the detergent FOS14 was deemed the best detergent out of 140 detergents to successfully solubilize the PA pore as verified by solubility properties, functional insertion, and negative stain electron microscopy. This detergent was then used in tandem with the nanodisc construction protocol but the solution based method failed to generate detectable concentrations of PA pore nanodisc complexes. Often, failure at this stage can be due to the existence of small soluble aggregates. Indeed, the observation that the transitioned pore still aggregates via its membrane insertion tip may negatively impact nanodisc assembly methods in solution. In addition to this failure, one also has to be aware of the possibility that detergent induced destabilization of the extra-membranous soluble domains of membrane proteins can sometimes compromise the protein structure (Palazzo et al. 2010; Yang et al. 2014).
Given that the transition of soluble PA prepore to its membrane insertion competent PA pore form can be accomplished without a detergent solubilization step, this lead to the simple prediction that one may be able to immobilize the PA prepore in a similar orientation (i.e., immobilizing the PA prepore complex with the cap facing the immobilizing surface) so that the PA barrel pore transition reaction occurs away from an immobilizing surface. This in turn avoids large-scale aggregation that accompanies solution based approaches (including ATP-dependent release from the GroEL platform). The unconventional aspect of this approach relies on unfolding and folding the protein to its membrane insertion state in the absence of detergents. It is worth noting that this particular approach with the anthrax toxin may not work for other membrane insertion requiring toxins, particularly those toxins that require interaction with the membrane interface prior to insertion (e.g. Diphtheria toxin, Clostridia tetani neurotoxin).
A successful platform was engineered by Pentelute and Collier who constructed a specific thiol immobilized lethal factor N-terminal domain that is positioned to specifically interact with the prepore cap region, enabling one to capture and properly orientate the PA prepore heptamer (Akkaladevi et al. 2013). Introduction of conditions that induce the prepore to pore transition (pH or 1 M urea/ 37 °C at pH 7.5) as PA is bound to this capture platform positions the unfolding and refolding reaction of the PA prepore in a direction positioned away from the immobilizing surface. Since these complexes were eventually used for EM analysis, the use of the neutral pH transition conditions was used to avoid any potential unfolding of the truncated lethal factor. Specifically, the formation of the extended 100 Å beta barrel occurs opposite the biospecific affinity surface (See Fig. 1 in Akkaladevi et al. 2013; Fig. 3 below). Thus, once the prepore binds to the lethal factor, the prepore can be transitioned to its pore conformation and a pre-nanodisc micelle consisting of cholate, POPC, and the MSP micelles surround the newly exposed hydrophobic membrane insertion tip. Once the cholate is dialyzed away from the micelle, nanodisc formation occurs directly on the immobilized pore (Fig. 3 below). The reversible thio linkage is cleaved and the released LFN-PA pore nanodisc complex was then applied onto EM grids for negative stain EM or cryo-EM analysis. Figure 3 below shows the illustrated protocol for constructing cryo-EM quality purified PA nanodiscs based on the immobilization of the prepore and transitioning the prepore to its membrane insertion PA pore state. Figure 4 shows the progression of structures that have been obtained using our various immobilization pore transition protocol.
Fig. 3.

Illustrated protocol for constructing purified protective antigen pores inserted into lipid nanodiscs. The red outlined boxes illustrate the steps involved in capturing the prepore, transitioning this prepore to the pore form, adding defined prenanodisc micelles, removing cholate and collapsing the micelle to its nanodisc form. The EM grid on the right was collected at the National center for Imaging Macromolecules (NCMI) at Baylor and clearly shows purified PA inserted into nanodiscs
Fig. 4.

Evolution of the EM analysis of the anthrax protective antigen pore structure inserted into lipid nanodiscs. The top row represents published data where the procedures to generate the initial negative stain EM reconstructions of the pore alone and pore nanodisc complexes culminated a low resolution cryo-EM structure. The latest images (bottom row) of the lipid solubilized PA pore at pH 7.5 was reconstructed from image field collected at the NCMI facility at Baylor under the direction of Steve Ludtke and Wah Chiu. The progression of reconstructed images shows that one is able to easily refine the sample preparation of the nanodisc inserted complexes
Construction of Purified PA Pore Nanodiscs Alone Verified by Negative Stain EM
The ability to capture the PA pore in lipid nanodiscs is convenient for generating structures under various solution conditions. For example, the current lethal factor bound-PA pore EM structures have been generated at pH 7.5 (urea/37 °C induced transition) which allows one to easily change solution conditions to any pH value. Theoretically, it should be feasible to examine structures where translocation is not observed and compare these with pore structures that are capable of translocating the lethal or edema factors (lower pH conditions).
In the next phase of structural analysis, one can also determine if bound lethal or edema factors influence PA pore structure. This also requires that one can construct both LF or EF bound and PA pore free structures. In order to construct PA pore structures that could be inserted into lipid nanodiscs while positioned on an immobilized support, three potential cysteine attachment sites were engineered into the anthrax PA prepore cap alone. Of these mutants, two cysteine PA heptamer variants (S186C and S206C) were successfully immobilized using thio-sepharose beads and these immobilized PA pores were transitioned at pH 7.5, 1 M urea as shown previously (Katayama et al. 2010; Akkaladevi et al. 2013; Gogol et al. 2013). Prior to assembly, these cysteine PA pores, these constructs were shown to be functionally competent to transport K+ ions across a potential gradient. To avoid multiple thiol attachments onto the thio-sepharose bead supports which in turn may inhibit effective release, hetero-heptameric complexes were formed using procedures developed by Janowiak et al. 2009, 2011. Addition of pre-nanodisc cholate–POPC–MSP micelles surrounded the exposed hydrophobic tips and upon dialysis with biobeads (Bio-RAD), the pre-nanodiscs collapsed to nanodisc structures. Each of these released cys mutant PA pore nanodiscs were then visualized using negative stain electron microscopy (Figs. 5, 6). The samples were homogenous, pure, and no aggregates were observed. Once again, using a rationally designed immobilization strategy leads to the easy formation of nanodisc inserted complexes for eventual structural analysis.
Fig. 5.

a Preparations of PA nanodisc with engineered S186C and S204C PA mutants. a Top view surface of serine to cysteine mutants that are used to immobilize the PA prepore in preferred orientation for transition and nanodisc assembly. b Representative field of PA S186C negatively stained EM micrograph showing transitioned PA pores inserted into partial and full lipid nanodiscs. The scale bar is 100 nm
Fig. 6.

a Electron microcopy field of released S204C PA inserted into lipid nanodiscs. The particles are primarily monodisperse and are easily distinguished as single particles. b Initial two dimensional classification and averaging of ~150 particles using EMAN 2.1 revealed virtually identical classified side view populations inserted into distinct lipid complexes
It is apparent that structure information from soluble pre-transitioned toxins is useful in designing rational construction platforms to capture and insert transitioned toxins into lipid nanodiscs. The usefulness of this immobilization approach can also be extended to monitor assembly and transition kinetics of protein–protein interactions and pore transition reaction using label-free platforms to develop inhibitors of these interactions and transitions. In addition, rational immobilization construction can also lead to the development of micro-scale sampling techniques for eventual structural analysis. This is particularly important in monitoring and evaluating membrane protein-nanodisc construction. These developments are discussed in the next two sections.
Recapitulating Endosomal Toxin Assembly and Transitions on Immobilized Label-Free Biosensor Platforms
In the previous sections, it was demonstrated that one can successfully assemble the anthrax toxin components, insert these into lipid nanodiscs and release of these components as purified complexes for EM analysis. Based on this obvious success, it was also surmised that this construction methodology could be used to successfully measure the kinetics of the pore assembly and the pore transition itself using biolayer interferometry (BLI) label-free biosensor platform technologies. The principle of BLI depends on detecting changes in thickness of a biolayer matrix that result from the addition of biomass or in some reactions presented here, changes in conformation. This instrumentation detects changes in the biolayer thickness by comparing the reflective interference pattern from an internal reference layer with the biomatrix sample layer. As the biomatrix layer changes thickness due to interactions or conformational transitions, real time kinetic and quantitative changes can be observed as a phase shift (Δ in nanometers). Surface plasmon resonance (SPR) and BLI technologies appear to be completely complementary. In the following sections, BLI technology is used to demonstrate that one can extend the rational immobilization of toxins to follow the assembly, transition, and membrane insertion of anthrax toxin complexes.
The most important aspect of this immobilization technology lies in the fact that one can mimic certain toxin assembly and transition reactions that occur within the endosome. This immobilization once again avoids solution limited aggregation reactions. Our laboratories developed a number of novel and entirely unique BLI based platform technologies that will eventually lead to the direct assessment of novel antimicrobial compounds targeting specific molecular transition reactions. The advantages and comparisons of these direct measurement approaches for novel drug discovery are extensively discussed in Naik et al. (2013). More importantly, however, useful test compounds that specifically inhibit pH induced transitions will be unique, novel anti-microbial agents because they can potentially be designed to directly target the initial pH dependent molecular transitions that lead to protein toxin entry and in some cases, cell lethality.
The preceding toxin assembly schemes were similarly applied toward immobilizing specifically orientated anthrax toxin pre-transition complexes that normally form during anthrax infections (Target #3 Fig. 7 below) on BLI biosensor tips (Naik et al. 2013). This approach enables one to directly visualize and target the actual molecular transitions that facilitate toxin entry through the PA pore into the cell (Fig. 7 far right). In vivo, Bacillus anthracis infection involves the production of three different toxins. The assembly of this defined set protein toxins occurs in a concerted manner (Fig. 7 Targets 1–3 below), culminating in the eventual translocation of the entire lethal and/or edema factor proteins (~80 kDa) from the endosome into the cell, leading to increased cell stress, a compromised immune system, or cell lethality. It has been suggested that as little as one protein toxin molecule (e.g., lethal factor) is sufficient to kill an entire cell. Thus, the lethal doses of the combined anthrax toxin are small. Specifically, we successfully constructed the assembled pre-endosomal complex (green arrows) using the cysteine mutant N-terminal portion of the anthrax lethal factor, previously developed to obtain structures for EM structure analysis (Akkaladevi et al. 2013; Gogol et al. 2013). Using a similar covalent attachment protocol, the N-terminal lethal factor (LFN) E126C was linked to the biosensor tip through a mixed disulfide to orient the LF binding site on the BLI biosensor surface so that the next components of the anthrax toxin can be assembled in a stepwise manner (Fig. 8 top left corner; see also Fig. 1 in Naik et al. 2013).
Fig. 7.
Sequence of anthrax toxin translocation into the cell. The immobilization of the anthrax toxin complex on the cell surface involves anthrax toxin assembly, receptor binding, pore transition following endocytosis, and subsequent translocation of the enzymic toxins (in this case, lethal factor). This shows that there are a number of potential interaction and transition reactions that are potential targets to develop novel anti-toxin small molecules therapeutics. (Figure was fashioned from Feld et al. 2012)
Fig. 8.

The complete BLI biosensogram following toxin assembly, receptor binding, pore transition, and the decrease in receptor binding immobilized on the biosensor surface. Assembly of the toxin complexes, receptor binding and endosomal transition outlined in Fig. 6 provide kinetic readouts where the effectiveness of rational in silico drug design (Docking approaches using Ray Casting—DARC) can be evaluated (Khar et al. 2013). Each targeted reaction is outlined below the representative kinetic traces
The stepwise assembly and transition scheme allows one to visualize the BLI binding signals so specific reaction inhibitors can be developed against various toxin assembly steps (Figs. 7, 8; Target #1 PA-LF/EF interaction, Target #2 PA-receptor assembly). The uniquely novel feature of this platform is that one can follow the direct pH induced (pH 5.0) molecular transition of toxin transition reaction (Target #3) thus allowing us to directly test small molecule inhibitors of this transition at endosomal pH 5.0. The novel aspect of this platform is that we can actually test and validate authentic inhibitors for two of the anthrax toxin assembly reactions, including the pH induced transition all using one platform. The acid-induced transition is responsible for triggering intracellular transport of the enzymic anthrax components (LF or EF) into the cell cytoplasm (Fig. 1-Targeted Step #3). The multi-target nature of this anthrax toxin platform offers a unique approach aimed at rapidly developing novel direct antitoxin inhibitors. Although there are many instances where molecules have been developed to inhibit the anthrax enzymic targets (for EF—Laine et al. 2012; for LF—Liao et al. 2014; to name just few recent examples), drug entry into the cell is always a crucial roadblock in drug development. Leppela and colleagues developed a novel; more directed approach that targeted the translocon function, specifically the prepore to pore transition directly, and involved the use of a novel HTS type cell based reporter assays screening approach. Unfortunately in this latter instance, the lead compounds that were identified targeted the endosome acidification processes rather than the pre-pore to pore transition directly. Since endosomal acidification is a necessary cellular process, this fact alone rules out its use as a general anthrax inhibitor (Zhu et al. 2009). What appears to be missing in most compound evaluations or assay developments targeting anthrax toxin entry is the ability to follow the protein structural changes that occur during the simulated endosome acidification step. If one can target this specific acid-induced transition, one may be able to develop specific easily administered prophylactic drug(s) compound that bind(s) tightly (~sub-nanomolar Kds) to the anthrax component of interest under both physiological and endosomal pH conditions. Following small molecule binding, one could subsequently determine if this (these) compound(s) can inhibit toxin translocation into the cell (inhibits pore formation, blocks LF or EF unfolding, or inhibits receptor binding). The immobilization of the toxin components on these biosensor surfaces avoids the complications of large-scale aggregation of soluble components that occurs as solutions are acidified. In general, this platform approach may enable researchers to validate drugs that directly target and stabilize toxins prior to or during cell entry and may provide easier routes toward developing novel anti-toxin compounds.
The entire transition can be monitored using a single channel fortebio BLI system (Sample output; Fig. 8). This single channel unit allows one to follow the kinetics of the anthrax toxin assembly and pore transition reactions (Figs. 7, 8; also see Naik et al. 2013). It is worth noting that the acid-induced structural changes accompanying the PA prepore to pore transition alone yield substantial and reproducible signals, allowing one to follow changes in these large observable changes as potential read-outs for inhibitor development (Fig. 9). This transition signal is amplified when pre-nanodisc micelles are added and bind to the transitioned pore directly (Fig. 8). As an added bonus, the next section outlines the procedures where validation of BLI detected transition signals can be coupled with direct structural screens using EM analysis to confirm complex formation and pore transitions.
Fig. 9.

Scheme for evaluating the assembly of toxin complexes using biolayer interferometry, releasing the complexes from the BLI biosensor into small volume, depositing the complexes onto EM grids, and validating complex formation and transitions using negative stain electron microscopy (scheme replicated from data presented in Naik et al. 2013)
Accelerating and Validating EM Sample Preparation of Transitioned Toxins Inserted into Lipid Nanodiscs Using BLI Technologies
One of the laborious aspects of assembling membrane proteins into nanodiscs for structural analysis involves extensive searches for detergent solubilization, optimizing detergent exchange with pre-nanodisc micelles and removal of cholate or other detergents by dialysis (Ritchie et al. 2009) to facilitate nanodisc formation. In this section, we introduce an accelerated BLI biosensor method to evaluate lipid inserted toxin complexes in μl-sized volumes as a rapid screen to identify superior sample preparation for EM analysis.
As mentioned in the previous section, one can easily follow the kinetics of the assembly, toxin transition and micelle insertion on one platform using BLI (Fig. 7) provided that these transitions are orientated away from the biosensor surface. This method also works for the assembly of other large complexes. Most importantly, the success of this micro-assembly approach depends on immobilizing complexes using attachment chemistries can be reversed and released into micro-volumes. Once assembled complexes are released from the small BLI biosensor tips into small volumes (e.g., 2–3 μl), the formation of large complexes (>100 kDa) can be easily verified using electron microscopy (Fig. 9; Naik et al. 2013, 2014).
The BLI-based assembly process is also useful in rapidly evaluating membrane proteins inserted into nanodiscs. For instance, one required step in nanodisc-membrane protein construction involves estimating how many phospholipid molecules are needed to insert the protein into the nanodisc. For example, membrane proteins containing seven transmembrane alpha helices will displace a certain amount of lipid in the nanodiscs and one can approximate the correct lipid ratio to MSP for adequate membrane protein-nanodisc construction. In other instances, it is necessary for one to test multiple ratios of the lipid:MSP:membrane protein to assemble target protein–nanodisc complexes. The protocols outlined by Ritchie et al. (2009) recommend that one should test at least 3–5 different lipid:MSP ratios. If the protein of interest is large enough to visualize, then one can use negative stain EM to evaluate the protein–nanodisc complex (Akkaladevi et al. 2013). The advantage of using the BLI to track micelle assembly and nanodisc formation is that this screening process can be done on a micro-volume scale. Furthermore, it is entirely possible that one can use EM validation of membrane protein insertion into lipid nanodiscs with small membrane proteins because insertion can be confirmed kinetically and by EM analysis. In the latter case, one could construct lipid nanodisc–small membrane protein complexes and validate the insertion of the protein into the nanodisc using specific monoclonal antibodies against the small membrane protein since antibody molecules are easily distinguished in an EM image field (Hernández-Rocamora et al. 2012; Naik et al. 2014). It is important to note that some commercially available MSPS that are used to form nanodiscs contain poly-histidine tags, enabling one with the ability to capture newly formed nanodiscs that can bind to Ni NTA BLI biosensor surfaces. In this instance, one can release membrane protein inserted into nanodiscs from Ni BLI tips into micro-volumes for EM or mass spectroscopy analysis using standard metal chelation procedures.
Once the complex formation has been replicated and scaled up using larger bead support systems, adequate numbers of particles can be used to obtain a single particle reconstruction of the solubilized complex (Naik et al. 2013). One can easily dialyze away the detergents, collapse the micelle into a nanodisc and evaluate this complex directly using electron microscopy. The advantage of using this micro-scale volume release approach is obvious since it can decrease testing times for optimal complex formation and can accelerate visualization of complex formation. This particular strategy of validating complexes using micro-scale samples for EM analysis from BLI platforms has also been used to evaluate and visualize protein–protein interactions between GroEL and pre-aggregates of therapeutic proteins (Naik et al. 2014).
Future Prospects
The ability to both observe and capture bacterial toxin transition states in lipid bilayer nanodiscs offers a unique platform to visualize and reconstruct higher resolution membrane protein structures at 3.3 Å (see Perspective Kühlbrandt 2014). A cryo-EM reconstruction of the programmed translating 70S ribosome structure at 7.1-Å resolution inserted into a nanodisc was recently observed by Beckmann and colleagues (Frauenfeld et al. 2011). In this structure, the SEC–YEG complex inserted into clearly defined nanodiscs positioned below the translocating ribosome 70S ribosome structure. Smaller membrane protein insertions such as Bax appear to form distinct channels within nanodiscs as visualized by negative stain electron microscopy (Xu et al. 2013). Improving on previous successful pore insertion (Akkaladevi et al. 2013), our own laboratory has recently acquired the entire anthrax toxin pore translocon structure at pH 7.5 solubilized with lipid nanodiscs at 22.5 Å resolution. Nanodisc technologies can also be used as foundation membrane structures for larger complex formation (Hernández-Rocamora et al. 2012) and to monitor conformational changes that occur during membrane protein–soluble protein interactions (Ye et al. 2010).
In the course of developing this method, experimental experience with multiple preparations indicate that obtaining adequate populations of protein–nanodisc complexes is routinely robust using negative stain EM (Fig. 6). Curiously, earlier cryo-EM imaging of the anthrax pore complex on quantifoil grids containing a thin carbon layer over the holes showed a preferential end-on view of the nanodiscs (Gogol et al. 2013). Many anthrax pore nanodisc complexes were observed but the contrast of the protein was diminished due to the background carbon layer. The end-on view of nanodiscs ranging from 100 to 400 Å were readily observed. Without the carbon layer, fewer anthrax pore–nanodisc complexes were routinely resolved. For reasons that are not clear at the moment, it appears as if the presence of a carbon layer, either with carbon coated negative stain or cryo-EM grids with thin carbon layers, partitions side-views nanodiscs onto these layers or coatings from solution. Recent discussions among those seeking to produce adequate numbers of protein complexes inserted into nanodiscs to solve structures using cryo-EM also noted the same decrease in resolved nanodisc protein complexes. We speculate that one potential reason for this disparity may result from a time-dependent alignment (partitioning) of nanodisc at the air–water interface (Wadsäter et al. 2011). To increase nanodisc populations with more random orientations within the water layer may require the addition of fluorinated surfactants to prevent partitioning at the air–water interface. Fluorinated surfactants can prevent aggregation of membrane proteins in solution and chaperone their insertion into the membranes without partitioning themselves into lipid bilayers (Rodnin et al. 2008; Kyrychenko et al. 2012). Interestingly, Holzenburg and colleagues used ammonium perfluoroonate to solubilize the V-type H+-ATPase for 2D crystallization (Reviakine et al. 1996). In a most recent paper, Raunser and colleagues (Efremov et al. 2014) obtained a 6.1 Å resolution of the ryanodine receptor that is clearly inserted into nanodiscs. The simple addition of a 0.2 % of fluorinated octyl-maltoside prior to vitrification on carbon grids resulted in a much improved random distribution of complex orientation allowing these investigators to obtain more conformational classes which lead to better 3D reconstructions.
The recent revolution in cryo-EM imaging using direct electron detector systems, along with new Zernike phase plate construction for better contrast (Nagayama and Danev 2010) will potentially allow structural biologists to rapidly resolve high resolution structures of membrane proteins inserted into lipid nanodiscs once these samples are constructed at high enough purity. The ability to observe complex formation using label-free methods, removing the complex from these platforms, and depositing these directly onto electron microscopy grids will allow easy validation of assembly and insertion methods for scale up, eventually enabling one to rapidly generate pure samples to obtain high resolution structures (Naik et al. 2013, 2014). By immobilizing and orientating soluble toxins prior to their insertion into nanodisc micelles, the crucial transition steps can be captured, allowing one to acquire high atomic resolution of toxin translocation competent states. Furthermore, the ease at which one can change solution conditions between neutral and endosomal pH environments will be useful in deciphering the necessary structural adjustments that accompany many directional translocation events of toxin proteins. As a more general method, it also may be feasible that this immobilization, orientation, and release approach may be successful with other membrane proteins preparations provided that one can acquire the correct fold on the immobilized surface prior to forming membrane protein–nanodisc complexes.
Acknowledgments
This work was supported grant funds from National Institutes of Health (NIH) R56 R56AI090085 (MTF), NIH R01AI090085 (MTF), and NIH SR37AI022021 (RJC). Some initial cryo-EM results presented in Figs. 3 and 4 were collected at the National Center for Imaging Macromolecules, supported by NIH Grant NIGMS P41 GM103832 (Wah Chiu). Additionally, some of the recombinant proteins used in the study were prepared in the Biomolecule Production Core of the New England Regional Center of Excellence (NERCE), supported by NIH Grant Number AI057159.
Contributor Information
Narahari Akkaladevi, Department of Biochemistry and Molecular Biology, University of Kansas Medical Center, Kansas City, KS, USA.
Srayanta Mukherjee, Department of Biochemistry and Molecular Biology, University of Kansas Medical Center, Kansas City, KS, USA.
Hiroo Katayama, Department of Biochemistry and Molecular Biology, University of Kansas Medical Center, Kansas City, KS, USA.
Blythe Janowiak, Department of Biology, Saint Louis University, St. Louis, MO 63101, USA.
Deepa Patel, Department of Microbiology and Molecular Genetics, Harvard University, Boston, MA, USA.
Edward P. Gogol, School of Biological Sciences, University of Missouri-Kansas City, Kansas City, MO, USA
Bradley L. Pentelute, Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02193, USA
R. John Collier, Department of Microbiology and Molecular Genetics, Harvard University, Boston, MA, USA.
Mark T. Fisher, Email: mfisher1@kumc.edu, Department of Biochemistry and Molecular Biology, University of Kansas Medical Center, Kansas City, KS, USA
References
- Akkaladevi N, Hinton-Chollet L, Katayama H, Mitchell J, Szerszen L, Mukherjee S, Gogol EP, Pentelute BL, Collier RJ, Fisher MT. Assembly of anthrax toxin pore: lethal-factor complexes into lipid nanodiscs. Protein Sci. 2013;22:492–501. doi: 10.1002/pro.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayburt TH, Sligar SG. Membrane protein assembly into nanodiscs. FEBS Lett. 2010;584(9):1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JE, Jaeger J, Gross M, Robinson CV, Radford SE. Structural and mechanistic consequences of polypeptide binding by GroEL. Fold Des. 1997;2(6):R93–R104. doi: 10.1016/S1359-0278(97)00046-1. [DOI] [PubMed] [Google Scholar]
- Deaton J, Sun J, Holzenburg A, Struck DK, Berry J, Young R. Functional bacteriorhodopsin is efficiently solubilized and delivered to membranes by the chaperonin GroEL. Proc Natl Acad Sci USA. 2004a;101(8):2281–2286. doi: 10.1073/pnas.0307759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton J, Savva CG, Sun J, Holzenburg A, Berry J, Young R. Solubilization and delivery by GroEL of megadalton complexes of the lambda holing. Protein Sci. 2004b;13(7):1778–1786. doi: 10.1110/ps.04735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov RG, Leitner A, Aebersold R, Raunser S. Architecture and conformational switch mechanism of the ryanodine receptor. Nature. 2014 doi: 10.1038/nature13916. [DOI] [PubMed] [Google Scholar]
- Elad N, Farr GW, Clare DK, Orlova EV, Horwich AL, Saibil HR. Topologies of a substrate protein bound to the chaperonin GroEL. Mol Cell. 2007;26(3):415–426. doi: 10.1016/j.molcel.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko JD, Sharon N. Microbial lectins: hemagglutinins, adhesins and toxins. In: Varki A, Cummings RD, Esko JD, editors. Essentials of glycobiology. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2009. [PubMed] [Google Scholar]
- Falke S, Tama F, Brooks CL, 3rd, Gogol EP, Fisher MT. The 13 angstroms structure of a chaperonin GroEL–protein substrate complex by cryo-electron microscopy. J Mol Biol. 2004;348(1):219–230. doi: 10.1016/j.jmb.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Feld GK, Brown MJ, Krantz BA. Ratcheting up protein translocation with anthrax toxin. Protein Sci. 2012;21(5):606–624. doi: 10.1002/pro.2052. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfeld J, Gumbart J, Sluis EO, Funes S, Gartmann M, Beatrix B, Mielke T, Berninghausen O, Becker T, Schulten K, Beckmann R. Cryo-EM structure of the ribosome–SecYE complex in the membrane environment. Nat Struct Mol Biol. 2011;18:614–621. doi: 10.1038/nsmb.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogol EP, Akkaladevi N, Szerszen L, Mukherjee S, Chollet-Hinton L, Katayama H, Pentelute BL, Collier RJ, Fisher MT. Three dimensional structure of the anthrax toxin transloconlethal factor complex by cryo-electron microscopy. Protein Sci. 2013;22:586–594. doi: 10.1002/pro.2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Rocamora VM, García-Montañés C, Rivas G, Llorca O. Reconstitution of the Escherichia coli cell division ZipA–FtsZ complexes in nanodiscs as revealed by electron microscopy. J Struct Biol. 2012;180(3):531–538. doi: 10.1016/j.jsb.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Janowiak BE, Finkelstein A, Collier RJ. An approach to characterizing single-subunit mutations in multimeric prepores and pores of anthrax protective antigen. Protein Sci. 2009;18:348–358. doi: 10.1002/pro.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowiak BE, Jennings-Antipov LD, Collier RJ. Cys–Cys cross-linking shows contact between the N-terminus of lethal factor and Phe427 of the anthrax toxin pore. Biochemistry. 2011;50(17):3512–3516. doi: 10.1021/bi1017446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Janowiak BE, Brzozowski M, Jurcyk J, Falke S, Gogol EP, Collier RJ, Fisher MT. GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nat Struct Mol Biol. 2008;15:754–760. doi: 10.1038/nsmb.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Wang J, Tama F, Chollet L, Gogol EP, Collier RJ, Fisher MT. Three-dimensional structure of the anthrax toxin pore inserted into lipid nanodiscs and lipid vesicles. Proc Natl Acad Sci USA. 2010;107:3453–3457. doi: 10.1073/pnas.1000100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühlbrandt W. The resolution revolution. Science. 2014;343:1443–1444. doi: 10.1126/science.1251652. [DOI] [PubMed] [Google Scholar]
- Kyrychenko A, Rodnin MV, Posokhov YO, Holt A, Pucci B, Killian JA, Ladokhin AS. Thermodynamic measurements of bilayer insertion of a single transmembrane helix chaperoned by fluorinated surfactants. J Mol Biol. 2012;416(3):328–334. doi: 10.1016/j.jmb.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine E, Martínez L, Ladant D, Malliavin T, Blondel A. Molecular motions as a drug target: mechanistic simulations of anthrax toxin edema factor function led to the discovery of novel allosteric inhibitors. Toxins (Basel) 2012;4(8):580–604. doi: 10.3390/toxins4080580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H-S, Liu H-L, Chen W-H, Ho Y. Structure-based pharmacophore modeling and virtual screening to identify novel inhibitors for anthrax lethal factor. Med Chem Res. 2014;23:3725–3732. [Google Scholar]
- Liu S, Crown D, Miller-Randolph S, Moayeri M, Wang H, et al. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc Natl Acad Sci USA. 2009;106:12424–12429. doi: 10.1073/pnas.0905409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama K, Danev R. Phase contrast enhancement with phase plates in biological electron microscopy. Microsc Today. 2010;18(4):10–13. [Google Scholar]
- Naglich JG, Metherall JE, Russell DW, Eidels L. Expression cloning of a diphtheria toxin receptor: identity with a heparin-binding EGF-like growth factor precursor. Cell. 1992;69:1051–1061. doi: 10.1016/0092-8674(92)90623-k. [DOI] [PubMed] [Google Scholar]
- Naik S, Brock S, Akkaladevi N, Tally J, Gao P, Zhang N, Pentelute BL, Collier RJ, Fisher MT. Monitoring the kinetics of the pH driven transition of the anthrax toxin prepore to the pore by biolayer inferferometry and surface plasmon resonance. Biochemistry. 2013;52:6335–6347. doi: 10.1021/bi400705n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Kumru OS, Cullom M, Telikepalli SN, Lindboe E, Roop TL, Joshi SB, Amin D, Gao P, Middaugh CR, Volkin DB, Fisher MT. Probing structurally altered and aggregated states of therapeutically relevant proteins using GroEL coupled to bio-layer interferometry. Protein Sci. 2014;23(10):1461–1478. doi: 10.1002/pro.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo G, Lopez F, Mallardi A. Effect of detergent concentration on the thermal stability of a membrane protein: the case study of bacterial reaction center solubilized by N,N-dimethyldodecylamine-N-oxide. Biochim Biophys Acta. 2010;804(1):137–146. doi: 10.1016/j.bbapap.2009.09.021. [DOI] [PubMed] [Google Scholar]
- Parker MW, Buckley JT, Postma JP, Tucker AD, Leonard K, Pattus F, Tsernoglou D. Structure of the Aeromonas toxin proaerolysin in its water-soluble and membrane-channel states. Nature. 1994;367:292–295. doi: 10.1038/367292a0. [DOI] [PubMed] [Google Scholar]
- Reviakine I, Stoylova S, Holzenburg A. Surfactosomes: a novel approach to the reconstitution and 2-D crystallisation of membrane proteins. FEBS Lett. 1996;380(3):296–300. doi: 10.1016/0014-5793(96)00063-4. [DOI] [PubMed] [Google Scholar]
- Ritchie TK, Grinkova YV, Bayburt TH, Denisov IG, Zolnerciks JK, Atkins WM, Sligar SG. Chapter 11—reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 2009;464:211–231. doi: 10.1016/S0076-6879(09)64011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnin MV, Posokhov YO, Contino-Pépin C, Brettmann J, Kyrychenko A, Palchevskyy SS, Pucci B, Ladokhin AS. Interactions of fluorinated surfactants with diphtheria toxin T-domain: testing new media for studies of membrane proteins. Biophys J. 2008;94(11):4348–4357. doi: 10.1529/biophysj.107.126235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseman AM, Ranson NA, Gowen B, Fuller SD, Saibil HR. Structures of unliganded and ATP-bound states of the Escherichia coli chaperonin GroEL by cryoelectron microscopy. J Struct Biol. 2001;135(2):115–125. doi: 10.1006/jsbi.2001.4374. [DOI] [PubMed] [Google Scholar]
- Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- Sun J, Savva CG, Deaton J, Kaback HR, Svrakic M, Young R, Holzenburg A. Asymmetric binding of membrane proteins to GroEL. Arch Biochem Biophys. 2005;434(2):352–357. doi: 10.1016/j.abb.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Unno H, Goda S, Hatakeyama T. Hemolytic lectin CEL-III heptamerizes via a large structural transition from α-helices to a β-barrel during the transmembrane pore formation process. J Biol Chem. 2014;289(18):12805–12812. doi: 10.1074/jbc.M113.541896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernier G, Wang J, Jennings LD, Sun J, Fischer A, Song L, Collier RJ. Solubilization and characterization of the anthrax toxin pore in detergent micelles. Protein Sci. 2009;18(9):1882–1895. doi: 10.1002/pro.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsäter M, Simonsen JB, Lauridsen T, Tveten EG, Naur P, Bjørnholm T, Wacklin H, Mortensen K, Arleth L, Feidenhans’l R, Cárdenas M. Aligning nanodiscs at the air–water interface, a neutron reflectivity study. Langmuir. 2011;27(24):15065–15073. doi: 10.1021/la203100n. [DOI] [PubMed] [Google Scholar]
- Wynia-Smith SL, Brown MJ, Chirichella G, Kemalyan G, Krantz BA. Electrostatic ratchet in the protective antigen channel promotes anthrax toxin translocation. J Biol Chem. 2012;287(52):43753–43764. doi: 10.1074/jbc.M112.419598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XP, Zhai D, Kim E, Swift M, Reed JC, Volkmann N, Hanein D. Three-dimensional structure of Bax-mediated pores in membrane bilayers. Cell Death Dis. 2013;4:e683. doi: 10.1038/cddis.2013.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wang C, Zhou Q, An J, Hildebrandt E, Aleksandrov LA, Kappes JC, DeLucas LJ, Riordan JR, Urbatsch IL, Hunt JF, Brouillette CG. Membrane protein stability can be compromised by detergent interactions with the extramembranous soluble domains. Protein Sci. 2014;23(6):769–789. doi: 10.1002/pro.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Hu G, Taylor D, Ratnikov B, Bobkov AA, McLean MA, Sligar SG, Taylor KA, Ginsberg MH. Recreation of the terminal events in physiological integrin activation. J Cell Biol. 2010;188(1):157–173. doi: 10.1083/jcb.200908045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Hobson JP, Southall N, Qiu C, Thomas CJ, Lu J, Inglese J, Zheng W, Leppla SH, Bugge TH, Austin CP, Liu S. Quantitative high-throughput screening identifies inhibitors of anthrax-induced cell death. Bioorg Med Chem. 2009;17(14):5139–5145. doi: 10.1016/j.bmc.2009.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]