

Abstract
Autoimmune B cells play a major role in mediating tissue damage in multiple sclerosis (MS). In MS, B cells are believed to cross the blood-brain barrier and undergo stimulation, antigen-driven affinity maturation and clonal expansion within the supportive CNS environment. These highly restricted populations of clonally expanded B cells and plasma cells can be detected in MS lesions, in cerebrospinal fluid, and also in peripheral blood.
In phase II trials in relapsing MS, monoclonal antibodies that target circulating CD20-positive B lymphocytes dramatically reduced disease activity. These beneficial effects occurred within weeks of treatment, indicating that a direct effect on B cells—and likely not on putative autoantibodies—was responsible. The discovery that depletion of B cells has an impact on MS biology enabled a paradigm shift in understanding how the inflammatory phase of MS develops, and will hopefully lead to development of increasingly selective therapies against culprit B cells and related humoral immune system pathways.
More broadly, these studies illustrate how lessons learned from the bedside have unique power to inform translational research. They highlight the essential role of clinician scientists, currently endangered, who navigate the rocky and often unpredictable terrain between the worlds of clinical medicine and biomedical research.
Keywords: Multiple sclerosis, Charcot Lecture
Introduction
Those of us engaged in biomedical research understand well the slow pace of discovery, the disappointments, and the myriad constraints that complicate our lives. For scientists who are also clinicians, the joys of clinical medicine are an ever-present draw and a pull away from the laboratory; rewards of clinical work are reliable, providing the opportunity to help 10 or maybe 20 patients in a busy day, to experience a series of professional successes before retiring each night, and to be spared the disappointment of rejected manuscripts, unfunded grants, or most crushingly when months or even years of work lead to less than expected progress.
These downsides of a career in research are overwhelmed, however, by the thrill of learning something that has never been known before, and especially by the joy of making a medically useful discovery, something that happens, if one is fortunate, perhaps a few times over the course of a career. For those of us involved in the B cell story in multiple sclerosis (MS), such a happening occurred on a crisp Friday afternoon in northern California; it was September 2006, and the event was the unblinding of the phase II anti-CD20 rituximab (RTX) study (Figure 1).1 Looking back, the joy was twofold. First, we saw evidence of a potentially powerful new approach for therapy of relapsing MS. Second, despite this success, it was also evident that our scientific rationale behind the clinical testing of RTX for MS was almost certainly incorrect. Looking back, this was the best possible result of a translational research experiment. We identified a novel approach that appeared to offer significant benefit for patients, and yet the data also sent us back to the laboratory in new and unexpected research directions. Indeed, the rubber meets the road when ideas born in the laboratory are formally road-tested at the bedside, and when data from real-life patients send us back to the laboratory armed with new hypotheses. Translational medicine is most effective when information flow is bidirectional, linking the laboratory with the clinic.
Figure 1.

The phase IIB trial of RTX in relapsing MS. Patients were treated on day 0 and again on day 15 with either 1000 mg intravenous RTX or placebo. In the figure, the mean number of gadolinium (Gd)-enhancing lesions is shown by week for the RTX (blue) and placebo (black) groups. The primary endpoint was the total composite number of enhancing lesions detected at weeks 12, 16, 20, and 24. There was a 91% relative reduction in new enhancing lesions in the RTX group. Adapted from Hauser et al.1 RTX: rituximab; MS: multiple sclerosis.
The early days
An early experience occurred in the late 1970s during my neurology residency. A number of us were sitting around a conference room on the ninth floor of the old Vincent Burnham building at Massachusetts General Hospital with our chair of neurology, Raymond D. Adams. A postdoctoral fellow from Massachusetts Institute of Technology (MIT) was presenting some work in experimental autoimmune encephalomyelitis (EAE) induced by myelin basic protein (MBP). Adams noted, with a touch of sarcasm, that the paralysis observed in the rodents likely resulted from peripheral nerve, and not central nervous system (CNS), disease. Indeed, he emphasized that the pathology of EAE and MS were quite different (Figure 2). In particular, T cell-mediated acute EAE models in mice were dominated by an inflammatory panencephalitis with relatively sparse demyelination, unlike the primary macrophage-mediated demyelinating pathology typical of human MS. This experience motivated me to dig deeper into the comparative pathology of the two conditions, and eventually to embark on a long-term effort to model MS-like pathology in the laboratory.
Figure 2.

Raymond D. Adams: Conventional wisdom can be wrong.
Developing a better disease model
This effort began in partnership with Norman Letvin (1949–2012), a medical school classmate and postdoctoral lab mate whose interest had gravitated toward the immunology of non-human primates, and who would soon become director of the Harvard-affiliated New England Primate Research Center in Massachusetts, where he would develop new reagents to tackle this problem, and also discover the simian immunodeficiency virus in the process. We began immunizing different species of non-human primates to search for pathologies that closely mimicked MS.2 We were particularly hopeful that a model could be generated in marmosets, and in particular the New World marmoset Callithrix jacchus (C. jacchus), a small primate approximately the size of a guinea pig (400 mg) but with a remarkable and defining characteristic, unique among primates. C. jacchus pregnancies are typically multiple, with gestation of several non-identical embryos at a time. Each fetus shares a common fetal blood supply, leading to establishment of a permanent, stable, lifelong bone marrow chimerism among fraternal twins or triplets. We found that this chimeric state, as predicted, permitted the transfer of T lymphocytes from one sibling to another without eliciting any immune response (alloresponse) in the recipient. These data set the stage, at least in theory, for the adoptive transfer of encephalitogenic T cells in a species phylogenetically close to humans, analogous to earlier experiments in inbred mice that were critical for defining the immunology of murine EAE. If we could produce an MS-like condition in C. jacchus, we'd have the necessary tools to dissect its immune architecture. Indeed, after several years of starts and stops, this was successfully accomplished by Luca Massacesi in 1995.3 The key event in this discovery, which eluded us before Luca's arrival, was the creative use of different immune adjutants.4 Following a single immunization with a myelin extract in incomplete Freund's adjuvant, and later with myelin oligodendrocyte glycoprotein (MOG), animals developed a mild relapsing–remitting disease, and an acute pathology characterized by large concentric areas of macrophage-mediated demyelination with relative axonal sparing and foci of remyelination; the myelin membrane was destroyed and reconstituted into vesicular fragments (Figure 3), a pattern termed vesicular demyelination and previously described in MS lesions by John Prineas.5 The day that we reviewed the pathology slides from C. jacchus EAE we literally gasped—our first eureka moment. We had replicated the MS-like pathology that we had sought for a decade.
Figure 3.
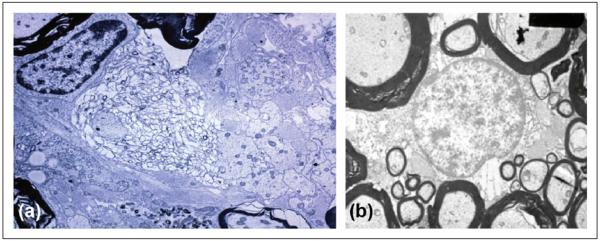
Ultrastructural features of C. Jacchus EAE. In (a), primary demyelination with preservation of axons, macrophage infiltration (macrophage nucleus visible at the top right), and astrogliosis is present. In the center, morphologic changes of myelin dissolution and fasciculation are visible. In (b), findings in chronic C. Jacchus EAE are shown, illustrating areas of thin, compact myelin-encircling axons, indicative of remyelination. C. Jacchus: Callithrix jacchus; EAE: experimental autoimmune encephalomyelitis.
However, when we adoptively transferred MOG-reactive T cell clones from an immunized C. jacchus animal into a chimeric sibling, we replicated the acute murine pathology of panencephalitis but not the distinctive MS-like pathology of vesicular demyelination.3 The explanation for this apparent conundrum was quickly solved by another superb scientist and postdoctoral fellow at the time, Claude Genain. Claude discovered that only by the co-administration of encephalitogenic T cells plus pathogenic Abs could the MS-like demyelinating phenotype be reconstituted. This led us to focus on the concept that an MS-like, demyelinating lesion required both pathogenic T cells plus autoantibodies; the autoantibodies alone were nonpathogenic, presumably because they required encephalitogenic T cells to open the blood-brain barrier (BBB) and permit their passage into the CNS.6,7 Our confidence that these mechanisms were operational in MS was strengthened by older literature in guinea pig optic neuritis first described by Appel and Bornstein in 1964,8 and later by Linington, Olssen, and Wekerle in work with rat EAE models.9,10
In 1999 we completed a deeper dive into the immunohistochemistry of the lesion with the superb experimental neuropathologist Cedric Raine, revealing the presence of bound Abs in the demyelinated lesions of C. jacchus that recognized the immunizing antigen (Ag) MOG. However, when we then turned our collective attention to human MS tissue, we found that deposited Abs were also bound to the myelin membrane but had specificities that were far more diverse than in EAE.11,12 This suggested that the humoral immune response in chronic MS is composed of autoantibodies with multiple specificities, and that in consequence a highly focused immunotherapy is unlikely to be successful.
Back to the bedside
Given the heterogeneous nature of the antibody (Ab) repertoire associated with myelin destruction in MS, it became clear that targeting any specific protein or epitope was a dubious therapeutic strategy. Thus we turned to methods that could deplete or inactivate a broad range of Abs, plasma cells, or perhaps their progenitors, B lymphocytes. The first two options were not feasible with available therapeutics, and we had previously found that indiscriminate Ab removal via plasmapheresis had little meaningful effect on chronic MS,13,14 thus our thoughts turned to B cell-based therapy and specifically the anti-CD20 monoclonal Ab RTX.
RTX was synthesized by Bill Rastetter at Idec Pharmaceuticals in 1986. IDEC entered into a co- development partnership with Genentech in 1995, and two years later RTX, marketed as Rituxan, received Food and Drug Administration (FDA) approval for treatment of B cell lymphoma. In 2001, I began discussions with Genentech around RTX therapeutics for MS after our failed application to the National Institutes of Health (NIH), championed by Claude Genain with Michael Racke and Nancy Monson at University of Texas Southwestern Medical Center (UT Southwestern) left us little hope that public resources could be found to support this trial. The referee comments from the application were dismissive, reflecting profound skepticism in the proposition that humoral immune mechanisms might be central to MS pathogenesis. Across much of academia at the time, MS was dominated by concepts of T cell mediation, analogy to murine EAE models, and a belief that CNS immunoglobulins (Igs), including oligoclonal bands (OCBs), represented meaningless, “nonsense” Abs.15 The field was simply not ready.
Industry proved to be a more flexible, and less risk- averse, partner. Discussions with Genentech progressed well, although it was the company's belief that our chance of success was “less than 15%.” Even if one accepted that autoantibodies were responsible for MS, a B cell-based therapy would not immediately knock down Ab production by long-lived plasma cells. Their experience with RTX indicated that IgG Ab levels were largely unchanged following treatment, although lower-affinity IgM was modestly reduced by approximately 15%. At least in theory, one would need many years of treatment to reduce circulating levels of Ig. Our original plan was to begin with a placebo-controlled phase IIB clinical trial of two courses of RTX spaced six months apart, and a primary endpoint at 12 months, or six months after the final infusion. However, the FDA balked at this design, advising us that it was, in their view, unethical to maintain a group of active relapsing MS patients on placebo therapy for one year. In response to these concerns the trial was scaled back; fewer patients would be enrolled, only a single course of RTX would be administered, and the primary endpoint would be measured at six months. Our prospects for success seemed ever dimmer.
As described in the introduction, when the data were unblinded, we learned that there was a dramatic and almost immediate 91% reduction in gadolinium-enhancing magnetic resonance imaging (MRI) activity (the primary endpoint) plus a significantly reduced rate of new relapses.1 This was our second eureka moment. In addition to the effect size, the rapid-onset of the benefit conferred by RTX was possibly the most stunning aspect of the trial. Because the clinical effects happened so quickly they was almost certainly not due to any reduction of long-lived Abs, but were more likely to be explained by some direct effect on B cells themselves. In many respects, this was the best of all possible results for a clinical experiment. The data were extremely positive, raising hope that an impactful new approach to MS therapy would result, but also perplexing, sending us back to the laboratory with information that the underlying hypothesis behind the clinical trial was almost certainly wrong. We returned to the laboratory with a new focus on B cell biology.
The multifunctional B cell
B cells are extremely diverse members of the universe of adaptive immunity. Although targeting of autoantibodies provided the original conceptual framework for testing RTX in relapsing MS, the resulting data made it likely that the robust efficacy was somehow related to a direct effect on B cells themselves.16 B cells have numerous effector functions independent of their differentiation from Ab-secreting plasma cells (Figure 4). B cells are highly effective Ag-presenting cells (APCs), but unlike other conventional APCs that are promiscuous Ag presenters, B cells are most efficient in presenting Ag that is initially recognized by their surface B cell receptor (BCR), i.e. the clonally specific Ig molecule. Thus they can be viewed as extremely selective APCs. Ag initially bound to surface BCR is internalized, complexed in endosomes with class II major histocompatibility complex (MHC) molecules, and returned to the surface for Ag presentation to T cells. B cells are also highly motile, and in secondary lymphoid structures they play a role in Ag shuttling, a process in which Ag is grabbed from macrophages by B cells via the BCR and transported to follicular dendritic cells (DCs), another class of APCs. Through secretion of cytokines B cells can also regulate, as bystanders, various effector immune functions mediated by both B and T cells. Some B cells support pro-inflammatory function through secretion of tumor necrosis factor alpha (TNFα) and lymphotoxin, whereas a different interleukin (IL)-10-producing B cell population has a regulatory, anti-inflammatory, role. Interestingly, MS B cells may be inherently polarized toward a proinflammatory functional phenotype.17 As noted above, the rapid response of B cell depletion therapy on focal disease activity in relapsing MS indicated that the mechanism of action was likely not, as initially hypothesized, through inhibiting autoantibodies but more likely by blocking B cell APC function and subsequent T cell activation, or perhaps via bystander effects on adaptive immunity. However, the potential inhibition of an as-yet unidentified autoantibody in MS cannot be completely excluded; Abs of the Ig4 isotype class are often synthesized by short-lived plasmablasts, and in IgG4-mediated autoimmune diseases rapid clinical responses to RTX can be associated with a rapid decline in autoantibody titers.18
Figure 4.

An overview of the diverse functional roles of B cells.
IL-interleukin; TNF-α: tumor necrosis factor alpha; LT-α: lymphotoxin-alpha; MHC: major histocompatibility complex; TCR: T cell receptor; DC: dendritic cell.
B cell development begins in the bone marrow and proceeds through stages of pro- and pre-B cells prior to exiting into the circulation as naïve,Ag-inexperienced B cells. The vast majority of B cells are located in follicles in secondary lymphoid tissues, including lymph nodes and spleen, and in mucosal sites. Binding of Ag through the BCR triggers activation, proliferation and somatic hypermutation (SHM) of BCRs (see below), resulting in maturation to memory (Ag-experienced B cells) and differentiation from Ab-secreting plasma cells. Remarkably, B cells are believed to reside in lymphoid follicles for approximately one day only before returning to the circulation, highlighting the dynamic nature of the process of B cell Ag capture, activation, and SHM of the BCR.
It is thought that both memory B cells and Ab-secreting plasmablasts and plasma cells can cross the BBB and enter the CNS in low numbers, and once there can reside in protective niches for long periods of time, a concept that has become increasingly relevant to concepts of progressive MS (see below).
CD20 is in several respects an ideal target for B cell- targeted immunotherapy. The CD20 molecule is expressed on pre-B cells and throughout the life cycle of naïve and memory B cells; CD20 is not expressed on stem cells or pro-B cells at the earliest stages of the B cell differentiation program, nor is it expressed on plasmablasts or terminally differentiated plasma cells (Figure 5 top). Following removal of CD20 B cells with RTX, there is consistent repletion from early B cell progenitors residing in the bone marrow, generally beginning four to six months after treatment (Figure 5 bottom). As long-lived plasma cells are unaltered, at least some Ab responses to infectious agents or to vaccinations are preserved during periods of B cell depletion. This feature may also explain the favorable safety record (after approximately 3 million doses) of RTX. In addition, monoclonal Abs against CD20 do not effectively remove B cells residing in protective niches within secondary lymphoid structures. Circulating B cells, representing only 2% or so of the total B cell pool in humans, are the B cell com partment most efficiently depleted by these agents.
Figure 5.

The effects of RTX on recirculating B cells. The top figure summarizes the life cycle of B cells destined for the CNS. The bottom highlights the effects of depletion of circulating B cells with anti-CD20 therapy; B cells residing in lymphoid tissues and the CNS are likely to be resistant to depletion with RTX. RTX: rituximab; CNS: central nervous system OCB: oligoclonal band; CSF: cerebrospinal fluid.
If B cells residing in pathogenic niches, such as the CNS in MS, or synovium in rheumatoid arthritis (RA), are relatively protected from anti-CD20 therapy, then how does the treatment work? The most likely explanation is that sustained depletion of circulating B cells, which in autoimmune disease likely includes recirculating, restimulated memory B cells destined to return to the target tissue, prevents their re-entry into focal white matter regions in MS or joint tissue in RA.19
Technology moves faster than clinical research
It took 18 months for the RTX data to find their way into final print,1 and by this time, to the surprise and disappointment of many, the prospects for advancing to phase III clinical trials of RTX were dead. The reasons for this were multiple but included complex governance of the RTX franchise between the two participating pharmaceutical companies, Biogen/Idec and Genentech; the development of a new fully humanized anti-CD20 monoclonal Ab, ocrelizumab (OCR) by Genentech; and Roche's acquisition of Genentech in 2009. A plan was put forward to no longer pursue RTX but instead to initiate an OCR phase IIB trial.
Different monoclonal Abs are not necessarily biologically identical even if they target the same molecule; this is certainly the case for Abs that target CD20. RTX and OCR target different epitopes of CD20 and kill B cells through different cytolytic pathways. RTX has stronger complement-dependent cytotoxicity (CDC) and less Ab-dependent cell-mediated cytotoxicity (ADCC), whereas the converse is true for OCR. Greater ADCC activity by OCR results from a higher affinity of Fc binding to the Fc gamma receptor IIIa (FcγRIIIa) on host natural killer (NK) cells. Importantly, FcγRIIIa genotypes also influence ADCC; at position 158, a substitution of valine for phenylalanine increases affinity for the Fc receptor and ADCC activity. The dose of Ab used, and the frequency of administration, may also influence ADCC activity. These differences between RTX and OCR may help to explain a complication observed in a trial of OCR as add-on therapy for RA in which several serious opportunistic infections developed in older Asian RA patients treated with high doses of OCR.20 This complication in the OCR trial was quite unexpected, as no safety signal of this type had been noted in the nearly 200,000 RA patients treated with RTX as add-on therapy.21 Although the RA trial of OCR was halted, the MS phase II trial of low-dose OCR as monotherapy proceeded, and when the results were unblinded, a robust treatment response identical to that found for RTX was observed.22 Furthermore, no adverse safety signal was observed. Also important, our hope that OCR, a fully humanized Ab, would produce a lower incidence of infusion reactions compared with the chimeric (mouse-human) RTX was dramatically confirmed, making OCR a far more attractive agent for chronic use. All involved breathed a deep collective sigh of relief as we advanced to the pivotal phase III clinical trials.
Additional insights from the phase II trials
We had observed in the original RTX study in MS that focal disease activity remained reduced even after B cells had returned to the peripheral blood (PBL). This point was driven home in a preliminary open-label, open extension phase of the OCR phase II study. After four courses of treatment with OCR, MRI and clinical disease activity remained quiescent 18 months after the last dose. Equally important, in the phase II studies no evidence of rebound was present at any time point. These data suggested that anti-CD20 treatment might reset the immune system in some way, and confer protection against the development of new focal MS lesions beyond the period of B cell depletion. Studies of PBL in RTX-treated individuals indicated that, following repletion, there are persistent changes in both B and T cell subpopulations that could, at least in theory, promote immune homeostasis and reduce proinflammatory responses. Repleting B cells express predominantly naïve and immature (CD5, CD38hi) phenotypes;23 proinflammatory Th1 and Th17 T cells are decreased;17 and CD25+FoxP3+ regulatory T cells are increased.24 Reductions in proinflammatory immune cells are also present in cerebrospinal fluid (CSF), with reduced numbers of T and B cells25,26 and a predominance of resting, CD19-bright, B cells.27
Another surprise—CD20+ T cells
For more than 20 years there have been scattered reports that some human T cells bear surface CD20 Ag, but these reports have generally relied on uncertain methods or study of abnormal, e.g. malignant, T cells. Recently, Christian von Büdingen led an effort that produced unequivocal evidence that CD20 T cells indeed exist in the healthy human circulation.28 This heterogeneous population, representing about 7% of total mature circulating T cells, is composed of numerous T cell subsets including both CD4 helper and CD8 cytotoxic T cells as well as well as naïve and various memory T cell populations. Notably, CD20+T cells have a lower surface density of CD20 compared to B cells, hence the designation CD3, CD20dim, but nonetheless the vast majority of these cells are depleted from the peripheral circulation with anti-CD20 therapy. Thus it remains possible, and would certainly be ironic if true, that the effects of anti-CD20 therapy on MS results from elimination of a pathogenic CD20-positive T cell population.
Back to the bench
In a series of remarkable experiments, Scott Zamvil's laboratory has clarified how B cells functioning as APCs, and in the absence of any contribution of Ab, might lead to MS. His laboratory constructed mixed bone marrow (BM) chimeric mice containing B cells that were selectively deficient in expression of class II major histocompatibility complex (MHC II) molecules; other APCs, including DCs and monocytes, expressed MHC II normally. Following immunization with the extracellular region of mouse MOG, or with an immunodominant p35–55 MOG peptide, BM chimera mice lacking MHC II on B cells developed EAE normally. However, in response to immunization with recombinant human MOG, these mice were entirely resistant to EAE induction, and susceptibility could not be restored by administration of MOG Ab.29 How can one interpret the finding that B cells competent to serve as APCs were absolutely required for EAE against human—but not against murine—MOG? It is likely that this B cell dependence can be attributed to a single amino acid change in the immunodominant region of MOG, e.g. a substitution of proline in place of serine at position 42 in human MOG. In a second series of experiments, Zamvil created transgenic mice that express surface MOG-reactive Ig on their B cells but could not secrete Ab. When crossed with a transgenic MOG-reactive T cell line, progeny developed spontaneous opticospinal EAE, associated with Th17 polarization, B cell activation, and formation of ectopic germinal centers in the meninges (see below), all in the absence of secreted Ab. These experiments provide unequivocal data that B cell APC function, in the absence of autoantibodies, are sufficient to promote T cell activation and an MS-like disorder.
In EAE, B cells tend to be involved as APCs when the immunization regimen employs whole myelin proteins such as human MOG (hMOG), but not when myelin peptides (e.g. p35–55 MOG) are used. Peptide immunization models are B cell independent because the BCR that binds mostly conformational rather than short linear epitopes is not involved in Ag capture.30,31 Interestingly, in EAE induced by whole MOG protein, B cell depletion is protective, but in MOG peptide EAE, B cell depletion worsens disease severity, probably by depleting IL-10-secreting regulatory B cells.32 In humans, a recently published trial of atacicept, a decoy receptor for the B cell growth factors B cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL), paradoxically worsened MS, possibly by altering regulatory B cell tone.33 These cautionary data emphasize that B cell depletion can be deleterious in some situations, and they also highlight the potential clinical relevance of information gleaned from EAE even when the models are imperfect representations of human MS.
Thus, B cells can be proinflammatory or regulatory, and the predominance of one or another function is a critical determinant of the outcome of an ongoing immune response. Clearly what is needed is to better understand B cell biology as it relates specifically to MS. One unbiased approach to this problem is through genetics and functional genomics. Gene variants that are expressed by B cells comprise an important component of the 110 variants thus far known to be associated with inherited risk for MS (Figure 6).34,35 Similarly, a number of functional changes in B cells have been described in MS patients, including changes in cytokine profiles indicating an inherent proinflammatory bias, and a defect in inducing B cell tolerance in the PBL.36
Figure 6.

The genetic landscape of multiple sclerosis. More than 110 genetic variants have now been identified as conferring susceptibility to MS. Most are thought to have roles in adaptive immunity. This figure summarizes MS risk genes known to be expressed by human B cells. The distance from the center is inversely proportional to the odds ratio in the GWAS. The color is representative of the level of expression in B cells (red=high, blue=low, gray=intermediate); white means that no expression data are available. Some labels are drawn larger to emphasize the most highly expressed genes. Data courtesy of the International Multiple Sclerosis Genetics Consortium (IMSGC). MS: multiple sclerosis; GWAS: genome-wide association studies; HLA: human leukocyte antigen, equivalent to the MHC. See reference 34 for additional details.
Identifying and tracking culprit B cells
In addition to modeling B cell mediation in EAE models, another approach to understanding the role of B cells in MS is to dissect the molecular properties of activated B cells directly in people with MS. BCRs are heterodimeric proteins with the Ag-binding portion formed by the variable regions of heavy and light chains. With respect to Ag-recognition, the heavy chain variable region (VH) is generally believed to play the primary role; VH results from the splicing of three gene segments into a mature transcript; one copy of a variable (V), diversity (D), and joining (J) gene segment.37 V, D, and J genes exist as multiple copies in each genome, contributing significantly to the diversity of Ab transcripts. Most Ab diversity is generated by variation in how the gene segments splice together, and especially by somatic mutations in the complementarity determining regions (CDR) of V genes that shape the Ab response.38 Following Ag contact in secondary, and possibly also in ectopic, lymphoid tissues, BCRs undergo somatic diversification as their phenotype advances from naïve to memory B cells, and then to Ab-secreting plasmablasts and plasma cells. We first reported in 2010 through genomic analysis of monozygotic twins that the propensity of B cells to select some members of V gene families over others is highly heritable, but that the clonal repertoire of BCR molecules expressed by any individual is stoichastic in origin and not influenced by differences in the architecture of germline genes.39,40
The von Büdingen laboratory has established an effort to probe the clonal nature of B cells expressed in the CNS, and understand how these relate to the PBL compartment. As expected from earlier work, sequencing of IgG-VH repertoires in MS patients revealed that CSF B cells represent a clonally restricted population that had undergone highly selective activation and affinity maturation within the CNS compartment. With parallel sequencing of many thousands of IgG-VH transcripts per sample, it was possible to construct lineage trees representing clonally related CSF B cells defined by their BCRs, and identify clonally related BCR sequences from PBL in the same individual. Results revealed a deep connection of these highly selected, clonally related B cells between the CSF and PBL compartments (Figure 7).41,42 Furthermore, when CSF IgG-VH sequences were matched with mass-spectrometric proteomic analyses of isoelectric focused (IEF) CSF IgG, remarkably the proteomic data and IgG-VH transcripts matched.43 Most peptides sequenced from OCBs could be shown to map to CSF-derived IgG-VH sequences, and furthermore in a given individual different bands comprising the OCBs were shown to be clonally related; i.e. they belong to the same BCR lineage tree.41,43,44 Thus there is evidence for ongoing stimulation and maturation to clonally restricted Ab-expressing B cells occurring primarily inside the CNS compartment. In some individuals, B cells participating in OCB production can also be identified in PBL; these cells appear to migrate across the BBB and may also undergo further Ag stimulation in the periphery.42,44 Thus OCB are not merely the terminal result of a focused immune response in MS but represent a component of active B cell immunity that is dynamically supported on both sides of the BBB. Although it is unclear where in the periphery activation and/or SHM of B cells responding to brain Ags might occur, recent data from Kevin O'Connor's group suggests that draining cervical lymph nodes represent one such potential site.45
Figure 7.

Intimate connections between CNS and peripheral B cells in MS. Representative lineages of clonally related IgG-VH found in CSF (a), or in CSF and PBL ((b)–(d)) of MS patients as calculated by IgTree software and visualized in Cytoscape 3.1 (organic layout). Each round node represents at least one unique IgG-VH sequence ranging from at least the 5' end of H-CDR1 to the 3' end of H-CDR3; larger nodes represent up to hundreds of identical sequences. Blue nodes represent CSF-derived IgG-VH sequences, red nodes are PBL-derived, and green nodes represent identical sequences found in both compartments. Putative germline sequences represent the lineage root and are labeled black, hypothetical intermediates calculated by IgTree are beige. Triangular nodes contain two or more singleton sequences in leaves. In (a), intrathecal affinity maturation is represented; (b) represents an IgG-VH lineage with predominantly PBL-derived IgG-VH suggestive of B cell migration from the CNS to the PBL or seeding from the PBL into the CNS; (c) is suggestive of B cell migration from the PBL into the CNS, with traces of the clusters remaining in the PBL and with extensive intrathecal B cell SHM; D suggests ongoing B cell exchange across the BBB, or affinity-maturation occurring in both compartments in parallel.
CNS: central nervous system; MS: multiple sclerosis; IgG: immunoglobulin G; VH: heavy chain variable region; CSF: cerebrospinal fluid; PBL: peripheral blood; SHM: somatic hypermutation; BBB: blood-brain barrier.
Returning to the inherited nature of gene segment usage by BCRs, our work in CSF,41,42,44 and that of others studying CSF46 and brain tissue47 clearly show that the activated B cell clones in CNS of MS patients display a bias in terms of increased usage of members of the IgG VH4 family. These data raise the possibility that even more selective therapies based on targeting restricted populations of B cells defined by their surface Ab receptors could be effective.
B cells and progressive MS
As discussed earlier, the anti-CD20 therapies eliminate mostly circulating B cells, leaving B cells in secondary lymphoid organs and other sites largely unaffected. This feature could account for their favorable safety profile, but at least in theory could also pose a challenge to effectively treating progressive MS.48 If established B cell nests residing in lymphoid follicle-like structures in the meninges are drivers of a chronic neurodegenerative process that ultimately results in progressive MS,49 anti-CD20 therapy would likely fail to deplete B cells from these sites. This resistance of B cells in protective niches could explain the relatively meager response of RTX in primary progressive MS,50 and our observation that individuals with relapsing MS can evolve to secondary progressive MS despite ongoing RTX treatment. Of course, it is also possible that long-lived, CD20-negative plasma cells and their Ab products play some role in progressive MS. We, and others, have limited evidence that OCBs can persist in the CSF even after chronic treatment with RTX, indicating that aberrant humoral immune responses have not been eliminated from the CNS. Targeting resident CNS B cell populations might require development of modified anti-CD20 monoclonal Abs that can disrupt protective niches,51 or penetrate the BBB more effectively, for example, by modifying the Ab with a ferritin shuttle. Another area of great promise is to develop small molecules that inhibit critical B cell signaling pathways.52 The first generation of B cell inhibitors have now been developed, ibrutinib53 and idealisib;54 both interact with the phosphoinositide (PI)3 kinase pathway downstream from BCR signaling, but whether either can cross the BBB effectively remains to be determined.
Conclusions
Looking back to that distant seminar room in Boston, it would have been impossible to imagine that 35 years later B cells would rest, arguably, at the epicenter of MS immunology. The B cell saga in MS has provided a cornucopia of surprises, thrilling insights, several disappointments, numerous still-to-be-solved conundrums, and also a few generic lessons. Foremost among the latter is the importance of road-testing ideas developed in the laboratory in real-life clinical situations, and vice versa. It argues for the essential role of clinician-scientists who walk the walk between these two worlds.28 Finally, the long path from conception, to an initial clinical suggestion of efficacy, to completing the definitive clinical trials amply demonstrates, as many have previously noted, the bumpy and uncertain road that accompanies forays between academia and industry, a bidirectional process that must be made more efficient if we are to effectively translate new discoveries into treatments and cures for our patients.
There are too many people to thank here, so I will close by thanking the Multiple Sclerosis International Federation and European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) for this singular honor, which I accept on behalf of so many others who have contributed to this story, shared with me the joys of watching it unfold, and await, as I do, its next chapter.
Acknowledgements
I am extremely grateful to Andrew Barnecut, who provided invaluable editorial assistance with preparation of the manuscript and figures.
Funding This research is supported by the National Institutes of Health (R01 NS049477; 1U19A1067152), and the National Multiple Sclerosis Society (RG 2899-D11).
Footnotes
Conflict of interest Dr. Hauser currently serves on the scientific advisory boards of Symbiotix and Bionure.
References
- 1.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 2.Genain CP, Hauser SL. Creation of a model for multiple sclerosis in Callithrix jacchus marmosets. J Mol Med (Berl) 1997;75:187–197. doi: 10.1007/s001090050103. [DOI] [PubMed] [Google Scholar]
- 3.Massacesi L, Genain CP, Lee-Parritz D, et al. Active and passively induced experimental autoimmune encephalomyelitis in common marmosets: A new model for multiple sclerosis. Ann Neurol. 1995;37:519–530. doi: 10.1002/ana.410370415. [DOI] [PubMed] [Google Scholar]
- 4.Genain CP, Hauser SL. Allergic encephalomyelitis in common marmosets: Pathogenesis of a multiple sclerosis-like lesion. Methods. 1996;10:420–434. doi: 10.1006/meth.1996.0120. [DOI] [PubMed] [Google Scholar]
- 5.Prineas JW, Connell F. The fine structure of chronically active multiple sclerosis plaques. Neurology. 1978;28:68–75. doi: 10.1212/wnl.28.9_part_2.68. [DOI] [PubMed] [Google Scholar]
- 6.Genain CP, Nguyen MH, Letvin NL, et al. Antibody facilitation of multiple sclerosis-like lesions in a nonhuman primate. J Clin Invest. 1995;96:2966–2974. doi: 10.1172/JCI118368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Genain CP, Abel K, Belmar N, et al. Late complications of immune deviation therapy in a nonhuman primate. Science. 1996;274:2054–2056. doi: 10.1126/science.274.5295.2054. [DOI] [PubMed] [Google Scholar]
- 8.Appel SH, Bornstein MB. The application of tissue culture to the study of experimental allergic encephalomyelitis. II. Serum factors responsible for demyelination. J Exp Med. 1964;119:303–312. doi: 10.1084/jem.119.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorentzen JC, Issazadeh S, Storch M, et al. Protracted, relapsing and demyelinating experimental autoimmune encephalomyelitis in DA rats immunized with syngeneic spinal cord and incomplete Freund's adjuvant. J Neuroimmunol. 1995;63:193–205. doi: 10.1016/0165-5728(95)00153-0. [DOI] [PubMed] [Google Scholar]
- 10.Linington C, Berger T, Perry L, et al. T cells specific for the myelin oligodendrocyte glycoprotein mediate an unusual autoimmune inflammatory response in the central nervous system. Eur J Immunol. 1993;23:1364–1372. doi: 10.1002/eji.1830230627. [DOI] [PubMed] [Google Scholar]
- 11.Genain CP, Cannella B, Hauser SL, et al. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. 1999;5:170–175. doi: 10.1038/5532. [DOI] [PubMed] [Google Scholar]
- 12.Raine CS, Cannella B, Hauser SL, et al. Demyelination in primate autoimmune encephalomyelitis and acute multiple sclerosis lesions: A case for antigen-specific antibody mediation. Ann Neurol. 1999;46:144–160. doi: 10.1002/1531-8249(199908)46:2<144::aid-ana3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 13.Hauser SL, Fosburg M, Kevy S, et al. Plasmapheresis, lymphocytapheresis, and immunosuppressive drug therapy in multiple sclerosis. Prog Clin Biol Res. 1982;106:239–254. [PubMed] [Google Scholar]
- 14.Hauser SL, Dawson DM, Lehrich JR, et al. Intensive immunosuppression in progressive multiple sclerosis. A randomized, three-arm study of high-dose intravenous cyclophosphamide, plasma exchange, and ACTH. N Engl J Med. 1983;308:173–180. doi: 10.1056/NEJM198301273080401. [DOI] [PubMed] [Google Scholar]
- 15.Mattson DH, Roos RP, Arnason BG. Isoelectric focusing of IgG eluted from multiple sclerosis and subacute sclerosing panencephalitis brains. Nature. 1980;287:335–337. doi: 10.1038/287335a0. [DOI] [PubMed] [Google Scholar]
- 16.von Büdingen HC, Bar-Or A, Zamvil SS. B cells in multiple sclerosis: Connecting the dots. Curr Opin Immunol. 2011;23:713–720. doi: 10.1016/j.coi.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol. 2010;67:452–461. doi: 10.1002/ana.21939. [DOI] [PubMed] [Google Scholar]
- 18.Khosroshahi A, Bloch DB, Deshpande V, et al. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62:1755–1762. doi: 10.1002/art.27435. [DOI] [PubMed] [Google Scholar]
- 19.Silverman GJ, Boyle DL. Understanding the mechanistic basis in rheumatoid arthritis for clinical response to anti-CD20 therapy: The B-cell roadblock hypothesis. Immunol Rev. 2008;223:175–185. doi: 10.1111/j.1600-065X.2008.00627.x. [DOI] [PubMed] [Google Scholar]
- 20.Rigby W, Tony HP, Oelke K, et al. Safety and efficacy of ocrelizumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: Results of a forty-eight-week randomized, double-blind, placebo-controlled, parallel-group phase III trial. Arthritis Rheum. 2012;64:350–359. doi: 10.1002/art.33317. [DOI] [PubMed] [Google Scholar]
- 21.Rubbert-Roth A, Tak PP, Zerbini C, et al. Efficacy and safety of various repeat treatment dosing regimens of rituximab in patients with active rheumatoid arthritis: Results of a Phase III randomized study (MIRROR) Rheumatology (Oxford) 2010;49:1683–1693. doi: 10.1093/rheumatology/keq116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378:1779–1787. doi: 10.1016/S0140-6736(11)61649-8. [DOI] [PubMed] [Google Scholar]
- 23.Duddy M, Niino M, Adatia F, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol. 2007;178:6092–6099. doi: 10.4049/jimmunol.178.10.6092. [DOI] [PubMed] [Google Scholar]
- 24.Vallerskog T, Gunnarsson I, Widhe M, et al. Treatment with rituximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clin Immunol. 2007;122:62–74. doi: 10.1016/j.clim.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 25.Cross AH, Stark JL, Lauber J, et al. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. 2006;180:63–70. doi: 10.1016/j.jneuroim.2006.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piccio L, Naismith RT, Trinkaus K, et al. Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch Neurol. 2010;67:707–714. doi: 10.1001/archneurol.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monson NL, Cravens PD, Frohman EM, et al. Effect of rituximab on the peripheral blood and cerebrospinal fluid B cells in patients with primary progressive multiple sclerosis. Arch Neurol. 2005;62:258–264. doi: 10.1001/archneur.62.2.258. [DOI] [PubMed] [Google Scholar]
- 28.Palanichamy A, Jahn S, Nickles D, et al. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J Immunol. 2014;193:580–586. doi: 10.4049/jimmunol.1400118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molnarfi N, Schulze-Topphoff U, Weber MS, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. 2013;210:2921–2937. doi: 10.1084/jem.20130699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyons JA, San M, Happ MP, et al. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol. 1999;29:3432–3439. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 31.Fillatreau S, Sweenie CH, McGeachy MJ, et al. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3:944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 32.Weber MS, Prod'homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 2010;68:369–383. doi: 10.1002/ana.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kappos L, Hartung HP, Freedman MS, et al. Atacicept in multiple sclerosis (ATAMS): A randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014;13:353–363. doi: 10.1016/S1474-4422(14)70028-6. [DOI] [PubMed] [Google Scholar]
- 34.Beecham AH, Patsopoulos NA, Xifara DK, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45:1353–1360. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farh KK, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. doi: 10.1038/nature13835. Epub ahead of print 29 October 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kinnunen T, Chamberlain N, Morbach H, et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest. 2013;123:2737–2741. doi: 10.1172/JCI68775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol. 2013;131:959–971. doi: 10.1016/j.jaci.2013.01.046. [DOI] [PubMed] [Google Scholar]
- 38.Neuberger MS. Antibody diversification by somatic mutation: From Burnet onwards. Immunol Cell Biol. 2008;86:124–132. doi: 10.1038/sj.icb.7100160. [DOI] [PubMed] [Google Scholar]
- 39.Baranzini SE, Mudge J, van Velkinburgh JC, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351–1356. doi: 10.1038/nature08990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glanville J, Kuo TC, von Büdingen HC, et al. Naive antibody gene-segment frequencies are heritable and unaltered by chronic lymphocyte ablation. Proc Natl Acad Sci U S A. 2011;108:20066–20071. doi: 10.1073/pnas.1107498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.von Büdingen HC, Kuo TC, Sirota M, et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J Clin Invest. 2012;122:4533–4543. doi: 10.1172/JCI63842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palanichamy A, Apeltsin L, Kuo TC, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med. 2014;6:248ra106. doi: 10.1126/scitranslmed.3008930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Obermeier B, Mentele R, Malotka J, et al. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat Med. 2008;14:688–693. doi: 10.1038/nm1714. [DOI] [PubMed] [Google Scholar]
- 44.Bankoti J, Apeltsin L, Hauser SL, et al. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann Neurol. 2014;75:266–276. doi: 10.1002/ana.24088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stern JN, Yaari G, Vander Heiden JA, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med. 2014;6:248ra107. doi: 10.1126/scitranslmed.3008879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Owens GP, Winges KM, Ritchie AM, et al. VH4 gene segments dominate the intrathecal humoral immune response in multiple sclerosis. J Immunol. 2007;179:6343–6351. doi: 10.4049/jimmunol.179.9.6343. [DOI] [PubMed] [Google Scholar]
- 47.Owens GP, Kraus H, Burgoon MP, et al. Restricted use of VH4 germline segments in an acute multiple sclerosis brain. Ann Neurol. 1998;43:236–243. doi: 10.1002/ana.410430214. [DOI] [PubMed] [Google Scholar]
- 48.Hauser SL, Chan JR, Oksenberg JR. Multiple sclerosis: Prospects and promise. Ann Neurol. 2013;74:317–327. doi: 10.1002/ana.24009. [DOI] [PubMed] [Google Scholar]
- 49.Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130:1089–1104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- 50.Hawker K, O'Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66:460–471. doi: 10.1002/ana.21867. [DOI] [PubMed] [Google Scholar]
- 51.Radford J, Davies A, Cartron G, et al. Obinutuzumab (GA101) plus CHOP or FC in relapsed/refractory follicular lymphoma: Results of the GAUDI study (BO21000) Blood. 2013;122:1137–1143. doi: 10.1182/blood-2013-01-481341. [DOI] [PubMed] [Google Scholar]
- 52.Puri KD, Di Paolo JA, Gold MR. B-cell receptor signaling inhibitors for treatment of autoimmune inflammatory diseases and B-cell malignancies. Int Rev Immunol. 2013;32:397–427. doi: 10.3109/08830185.2013.818140. [DOI] [PubMed] [Google Scholar]
- 53.Byrd JC, Brown JR, O'Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371:213–223. doi: 10.1056/NEJMoa1400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997–1007. doi: 10.1056/NEJMoa1315226. [DOI] [PMC free article] [PubMed] [Google Scholar]