

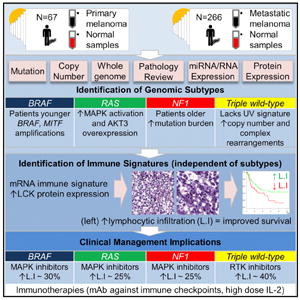
SUMMARY
We describe the landscape of genomic alterations in cutaneous melanomas through DNA, RNA, and protein-based analysis of 333 primary and/or metastatic melanomas from 331 patients. We establish a framework for genomic classification into one of four subtypes based on the pattern of the most prevalent significantly mutated genes: mutant BRAF, mutant RAS, mutant NF1, and Triple-WT (wild-type). Integrative analysis reveals enrichment of KIT mutations and focal amplifications and complex structural rearrangements as a feature of the Triple-WT subtype. We found no significant outcome correlation with genomic classification, but samples assigned a transcriptomic subclass enriched for immune gene expression associated with lymphocyte infiltrate on pathology review and high LCK protein expression, a T cell marker, were associated with improved patient survival. This clinicopathological and multidimensional analysis suggests that the prognosis of melanoma patients with regional metastases is influenced by tumor stroma immunobiology, offering insights to further personalize therapeutic decision-making.
Graphical Abstract
INTRODUCTION
Diagnosis and surgical resection of early-stage primary cutaneous melanoma is often curative for patients with localized disease, but the prognosis is less favorable for patients with regional metastases. Using the technique of lymphatic mapping and sentinel lymph node (SLN) biopsy (Gershenwald and Ross, 2011), early surgical intervention for patients with microscopic regional lymph node metastases (i.e., positive SLNs) has recently been found useful for prognosis, may improve survival in a subgroup of such patients, and serves to guide the use of adjuvant therapy (Morton et al., 2014). Overall, survival has historically been poor for patients with distant metastatic disease, and response to conventional chemotherapy has been infrequent (Balch et al., 2009).
Hot-spot mutations in the V600 codon of BRAF (35%–50% of melanomas) and Q61 codons (less frequently, the G12 or G13 codon) of NRAS (10%–25%) led to the development of highly selective kinase inhibitors that target the MAPK pathway (Tsao et al., 2012). Recent clinical trials have provided proof of principle that therapeutic agents targeting activating mutations for patients with unresectable disease and/or distant melanoma metastases can be identified through genetic analyses. The Food and Drug Administration (FDA) has approved three such inhibitors: vemurafenib, dabrafenib, and trametinib (McArthur and Ribas, 2013). Although antitumor responses have been dramatic, they have rarely been durable. Additional targets and combinatorial treatment strategies are clearly needed.
Recent studies using next-generation sequencing (NGS) have identified additional genetic aberrations (Berger et al., 2012; Hodis et al., 2012; Krauthammer et al., 2012) that provide insights into the biological heterogeneity of melanoma and also have potentially important implications for prognosis and therapy. However, previous biomarker studies in melanoma have either focused on single high-throughput platforms of large sample sets (Hodis et al., 2012; Krauthammer et al., 2012; Winnepenninckx et al., 2006) or multi-platform analyses of fewer samples (Mann et al., 2013; Rakosy et al., 2013). No prior study has integrated multi-platform data from such a large cohort of clinico-pathologically well-annotated samples.
To address this gap, The Cancer Genome Atlas (TCGA) program performed a systematic multi-platform characterization of 333 cutaneous melanomas at the DNA, RNA, and protein levels to create a catalog of somatic alterations and describe their potential biological and clinical significance. We established a genomic/transcriptomic framework of classification that has potential implications for prognosis and therapy and that may relate to recent advances in immunotherapy.
RESULTS
Multi-dimensional Genomic Characterization of Cutaneous Melanoma
Compared to most solid tumors, primary melanomas are generally small at diagnosis; and in routine clinical practice, most or all of primary tumor tissue is used for diagnostic evaluation and is not available for molecular analyses. Accordingly, our study included samples from thick primaries, regional, and distant metastatic sites.
We collected frozen tumor samples from 333 cutaneous primary and/or metastatic melanomas with matched peripheral blood from 331 adult patients from 14 tissue source sites under protocols approved by the relevant Institutional Review Boards. Clinicopathological characteristics are summarized in Table S1A. The samples consisted of 67 (20%) primary cutaneous melanomas (all originating from non-glabrous skin) and 266 (80%) metastases. Of the metastases, 212 were from regional sites (160 from regional lymph nodes and 52 from regional skin/soft tissue), and 35 were from distant sites (Table S1A–S1C). At initial diagnosis, patients had primary tumors (whether or not the primary tumors were included in the TCGA molecular analyses) that were thicker (median and mean, 2.7 mm and 4.9 mm, respectively) than in population-based registry data (Baade et al., 2012; Criscione and Weinstock, 2010). Matched primary and metastatic samples were available for complete molecular analyses from only two patients.
We performed six types of global molecular analysis: solution-based hybrid-capture whole-exome sequencing (WES, n = 320 samples), DNA copy-number profiling by Affymetrix SNP 6.0 arrays (n = 333), mRNA sequencing (n = 331), microRNA sequencing (n = 323), DNA methylation profiling (n = 333), and reverse-phase protein array (RPPA) expression profiling (n = 202). Complete data for all six platforms were available for a core set of 199 samples. TERT promoter mutations at C228T and C250T were assessed by PCR-Sanger sequencing in a subset of 115 samples. Deep-coverage whole-genome sequencing and low-pass whole-genome sequencing were performed on subsets of 38 samples and 119 samples, respectively. Clinico-pathological and molecular data associated with each patient are presented in a patient-centric table (Table S1D); complete methods and results of the analyses are described in the Supplemental Experimental Procedures. The standard data package associated with this report (frozen on November 14, 2013) is available at the GDAC Firehose (http://gdac.broadinstitute.org/runs/stddata__2013_11_14/data/SKCM/20131114). and at Data Portal (https://tcga-data.nci.nih.gov/docs/publications/skcm_2015/).
Identification of Significantly Mutated Genes
WES was performed on paired tumor and germline normal genomic DNA from 318 patients, including primary (n = 58) and metastatic (n = 262) melanomas with a mean exon coverage of 87×, adequate for detecting a single-nucleotide variant (SNV) at an allelic fraction of 0.3 with a power of 80% (Carter et al., 2012) (see Supplemental Experimental Procedures). In total, we identified 228,987 mutations, including both SNVs and indels. Targeted validation of 455 SNVs observed in the significantly mutated genes (see below) in a subset of tumor DNAs (n = 277) revealed an overall validation rate of 96% (see Supplemental Experimental Procedures). The mean mutation rate was 16.8 mutations/Mb, the highest reported for any cancer type thus far analyzed by TCGA (Lawrence et al., 2013) (Figure S1A) and corroborates findings from other NGS melanoma studies (e.g., Hodis et al., 2012) and other ultraviolet (UV)-driven skin cancers such as basal and squamous cell carcinomas (e.g., Jayaraman et al., 2014). Consistent with UV radiation’s mutagenic role in melanoma, most samples showed a high fraction of C>T transitions at dipyrimidines (median 77.7%; interquartile range 69.4%–82.6%) and CC>TT mutations (median 3.9%; interquartile range 2.0%–5.7%) (Figure S1A). We classified samples in which C>T transitions at dipyrimidine sites accounted for more than 60% or CC>TT mutations more than 5% of the total mutation burden as possessing a UV signature (Brash, 2015): 44 (76%) of the 58 primary and 221 (84%) of the 262 metastatic samples had such a signature.
Given the statistical challenge of defining significance against a high background mutation rate, we used two algorithms to define significantly mutated genes (SMGs): MutSig and InVEx (Hodis et al., 2012; Lawrence et al., 2014; Lawrence et al., 2013). MutSig takes into consideration patient-specific mutation frequencies and spectra, mRNA expression levels, and gene-specific DNA replication times; InVEx controls for patient-specific, gene-specific, and nucleotide-context-specific mutation probabilities (see Supplemental Experimental Procedures). WES analysis by InVEx identified 13 SMGs (Bonferroni p < 0.05, or 20 SMGs at Q < 0.1) by either functional mutation burden or loss-of-function tests, all of them among the 42 SMGs identified by MutSig (Q < 0.1) (Tables S2A–S2D and Figure S1B). The 13 SMGs included previously described melanoma oncogenes and tumor suppressors (BRAF, NRAS, CDKN2A, TP53, and PTEN), as well as recently identified mutated genes (RAC1, MAP2K1, PPP6C, and ARID2) (Hodis et al., 2012; Krauthammer et al., 2012; Nikolaev et al., 2012). Our cohort also had sufficient statistical power to annotate several previously implicated melanoma genes as SMGs (NF1, IDH1, and RB1) (Andersen et al., 1993; Draper et al., 1986; Lopez et al., 2010). We also identified DDX3X, a putative RNA helicase, as a novel candidate melanoma SMG (Figures 1A and S1C). SMGs with UV-induced hot-spot mutations included RAC1 (6.9%) and IDH1 (6.2%) (Figure S1C). The RAC1 hot-spot mutation has been linked to resistance to BRAF inhibitors (Van Allen et al., 2014; Watson et al., 2014). Similar to findings in other tumor types, IDH1-mutated samples were enriched in the high CpG island methylator phenotype (CIMP) subgroup (Figures S1D–S1G) (Noushmehr et al., 2010).
Figure 1. Landscape of Driver Mutations in Melanoma.
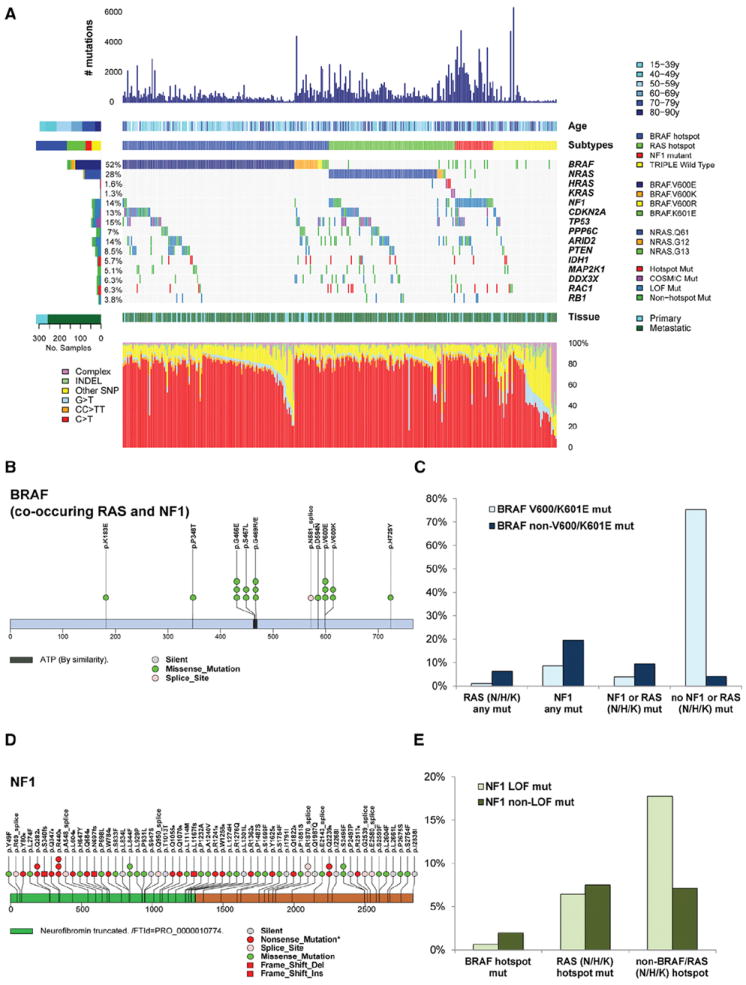
(A) Total number of mutations, age at melanoma accession, and mutation subtype (BRAF, RAS [N/H/K], NF1, and Triple-WT) are indicated for each sample (top). (Not shown are one hyper-mutated and one co-occurring NRAS BRAF hot-spot mutant). Color-coded matrix of individual mutations (specific BRAF and NRAS mutations indicated) (middle), type of melanoma specimen (primary or metastasis), and mutation spectra for all samples (bottom) are indicated. For the two samples with both a matched primary and metastatic sample, only the mutation information from the metastasis was included.
(B) BRAF mutations that co-occur with RAS family member and NF1 mutations are illustrated across the BRAF protein.
(C) Fraction of BRAF V600/K601E and non-V600/K601E co-occurring with the RAS (N/H/K), NF1, NF1/RAS (N/H/K) combined cohort and no NF1/RAS (N/H/K) mutations.
(D) NF1 mutations found in melanoma whole-exome sequencing data across the NF1 protein.
(E) Fraction of NF1 missense and truncating mutations co-occurring with RAS hot-spot or non-BRAF/RAS hot-spot mutations. (Mut, mutation).
See also Figure S1.
Additionally, two genes (MRPS31 and RPS27) that encode ribosomal proteins were identified by MutSig as SMGs. Both possess presumptive UV-induced hot-spot mutations in their 5′ UTR (in ~5% and ~9% of samples, respectively) (Figure S1H). MRPS31 encodes a mitochondrial ribosomal protein not previously associated with cancer; RPS27 is a component of the 40S ribosomal subunit whose overexpression has been reported in melanoma (Santa Cruz et al., 1997). The recurrent mutation in RPS27 was recently shown to expand the 5′ TOP element, a motif known to control mRNA translation regulated through the PI(3)K/AKT and mTOR pathways (Dutton-Regester et al., 2014).
Genomic Classification of Melanoma
One of the most significant successes in clinical practice has been the development of targeted therapies for patients with activating driver mutations (McArthur and Ribas, 2013; Tsao et al., 2012). We therefore classified melanomas based on identified SMGs and their distribution in our cohort (n = 318 cases with WES data; described below, Figure 1A, and Table 1) to create a framework that could be used for personalized therapeutic decisions.
Table 1.
Implications for Clinical Management Based on Features Identified by Comprehensive Molecular TCGA Analysis
| Mutation Subtypes | BRAF | RAS | NF1 | Triple Wild-Type |
|---|---|---|---|---|
| 1MAPK pathway | 1BRAF V600, K601 | 1(N/H/K) RAS G12, G13, Q61 | 1NF1 LoF mut; (BRAF non-hot-spot mut) | 1KIT COSMIC mut/amp, PDGFRa amp, KDR (VEGFR2) amp; rare COSMIC GNA11 mut, GNAQ mut |
| 2Cell-cycle pathway | CDKN2A mut/del/h-meth (~60%); 2(CDK4 COSMIC mut) | CDKN2A mut/del/h-meth (~70%); CCND1 amp (~10%), 2(CDK4 COSMIC mut) | CDKN2A mut/del/h-meth (~70%); RB1 mut (~10%) | CDKN2A mut/del/h-meth (~40%); CCND1 amp (~10%), 2CDK4 amp (15%) |
| 3DNA damage response and cell death pathways | TP53 mut (~10%); 3(note: TP53 wild-type in ~90% of BRAF subtype) | TP53 mut (20%) | TP53 mut (~30%) | 3MDM2 amp (~15%); 3BCL2 upregulation |
| 4PI3K/Akt pathway | 4PTEN mut/del (~20%); 4(rare AKT1/3 and PIK3CA COSMIC mut) | 4AKT3 overexpression (~40%); 4(rare AKT1/3 and PIK3CA COSMIC mut) | 4AKT3 overexpression (~30%) | 4AKT3 overexpression (~20%) |
| 5Epigenetics | 5IDH1 mut, 5(rare EZH2 COSMIC mut); 5ARID2 mut (~15%) | 5IDH1 mut, 5(rare EZH2 COSMIC mut); 5ARID2 mut (~15%) | 5IDH1 mut, 5(EZH2 mut); 5ARID2 mut (~30%) | 5IDH1 mut, 5(rare EZH2 COSMIC mut) |
| Telomerase pathway | Promoter mut (~75%) | Promoter mut (~70%) | Promoter mut (~85%) | Promoter mut (< 10%); TERT amp (~15%) |
| Other pathways | PD-L1 amp, MITF amp, PPP6C mut (~10%) | PPP6C mut (~15%) | ||
| 6High immune infiltration (pathology) | ~30% | ~25% | ~25% | ~40% |
| Class 1: Clinically actionable | 1BRAF inhibitors; 1MEK inhibitors | 1MEK inhibitors | 1C-KIT inhibitors (imatinib, dasatinib, nilotinib, sunitinib); PKC inhibitors (AEB071) | |
| 2CDK inhibitors | 1,2CDK inhibitors | 2CDK inhibitors | ||
| 3MDM2/p53 interaction inhibitors | 3MDM2/p53 interaction inhibitors | |||
| 4PI3K/Akt/mTOR inhibitors | 4PI3K/Akt/mTOR inhibitors | 4PI3K/Akt/mTOR inhibitors | 4PI3K/Akt/mTOR inhibitors | |
| 6immunotherapies (mAb against immune checkpoint proteins, high dose bolus IL-2, interferon-α2b) | ||||
| Class 2: Translationally actionable | 1ERK inhibitors | 1ERK inhibitors | 1MEK inhibitors; 1ERK inhibitors | |
| 5IDH1 inhibitors | 5IDH1 inhibitors | 5IDH1 inhibitors | 5IDH1 inhibitors | |
| (PPP6C) Aurora kinase inhibitors | (PPP6C) Aurora kinase inhibitors | |||
| Class 3: Pre-clinical | 5ARID2 chromatin remodelers (synthetic lethality) | 5ARID2 chromatin remodelers (synthetic lethality) | 5ARID2 chromatin remodelers (synthetic lethality) | 3(BCL2) BH3 mimemitcs |
Prominent mechanisms of pathway alterations in BRAF, RAS, NF1 and Triple Wild-Type (WT) subtypes with potential predictive genetic alterations indicated (1, 2, 3, 4, 5, 6) for Class 1 (clinically actionable alterations), Class 2 (translationally actionable that still require additional data [evidence] to support use in point-of-care decision making), and Class 3 (pre-clinical evidence has demonstrated biological importance but has not yet demonstrated clinical relevance) biomarkers. High immune infiltration (pathology) is percentage of samples in respective mutation subtype with LScores of 5–6. Amp, amplification; del, deletion; mut, mutation, h-meth, hypermethylation.
BRAF Subtype
The largest genomic subtype is defined by the presence of BRAF hot-spot mutations. Of the 318, 52% (n = 166) harbored BRAF somatic mutations. Of those, 145 targeted the well-documented V600 amino acid residue: V600E (n = 124), V600K (n = 18), and V600R (n = 3). The second most frequent BRAF mutation targeted the K601 residue (n = 5). As in previous reports (Pollock et al., 2003), both BRAF V600 and K601 hot-spot mutations were anti-correlated with hot-spot NRAS mutations (Fisher’s exact p < 1e–15). In contrast, BRAF non-hot-spot mutations (including eight exon 11 mutations) co-occurred with RAS (N/H/K) hot-spot and NF1 mutations (Figures 1B and 1C).
RAS Subtype
The second major subtype is defined by the presence of RAS hot-spot mutations, including known amino acid changes with functional consequences, in all three RAS family members (N-, K- and H-RAS). Overall, 28% (n = 88) had NRAS somatic mutations. Of those, 86 had hot-spot mutations, including Q61R (n = 35), Q61K (n = 28), Q61L (n = 11), Q61H (n = 4), 61_62QE > HK (n = 1), G12R/D/A (n = 4), and G13R/D (n = 3). We also identified less-frequent mutations in other RAS family members, including four hot-spot HRAS (G13D, G13S, and Q61K [n = 2]) and three KRAS (G12D, G12R, and Q61R) mutations; all were mutually exclusive with NRAS and BRAF V600 and K601 mutations.
NF1 Subtype
The third most frequently observed SMG in the MAPK pathway was NF1, which was mutated in 14% of samples. More than half of its mutations were predicted to be loss-of-function (LoF) events, including 27 nonsense, 9 splice-site, and 4 frame-shift indels out of 65 mutations (InVEx LoF analysis: p = 1.8e– 11, Q = 9.1e–12) (Figures 1D and 1E). NF1 subtype (n = 28) had the highest mutation prevalence (39 mutations/Mb, more than double that of the other three subtypes). Since NF1 is a GTPase-activating protein known to downregulate RAS activity through its intrinsic GTPase activity, LoF mutation of NF1 can be viewed as an alternative way to activate the canonical MAPK signaling pathway. Indeed, in this cohort, NF1 was mutated in 38.7% of non-hot-spot BRAF/NRAS melanomas (29/75) and in ~70% of non-hot-spot BRAF/NRAS samples with a UV-signature (26/38) (Figure 1A). Furthermore, NF1 mutations were anti-correlated with hot-spot BRAF mutations (p = 1.93–9), but not hot-spot RAS mutations (Figure 1A).
Triple Wild-Type Subtype
We defined the Triple-WT subtype (n = 46) as a heterogeneous subgroup characterized by a lack of hot-spot BRAF, N/H/K-RAS, or NF1 mutations. This lack of hot-spot mutations was not due to lower tumor purity or ploidy, since power calculation taking into account sample-specific purity and ploidy (Carter et al., 2012) showed that our sequencing coverage is powered to detect sub-clonal mutations at a 6% allelic fraction on average in Triple-WT subtype (see Supplemental Experimental Procedures). To identify rare low-frequency driver mutations in this subtype, we cross-referenced all observed SNVs to recurrently mutated base pairs (n > 20) in the COSMIC database v60 and identified 11 additional genes with recurrent COSMIC mutations (Table S2E). Several COSMIC mutations, including known drivers of uveal melanoma—GNAQ (n = 1) and GNA11 (n = 2), KIT (n = 6), as well as CTNNB1 (n = 3) and EZH2 (n = 1)—were found in the Triple-WT subtype.
Molecular Characteristics of the Four Genomic Subtypes
Clinically, patients in the BRAF subtype were younger than patients in the other subtypes, while those in the NF1 subtype were significantly older (rank sum p = 0.008). Regardless of subtype, patients with TP53 mutant melanomas had significantly higher mutation counts and number of C>T transitions (rank sum p = 1.35 e–05 and p = 1.1 e–05, respectively). However, no significant difference was observed in post-accession survival (i.e., survival calculated from date of biospecimen collection/accession to date of last follow-up or death, see Supplemental Experimental Procedures). Therefore, we next explored the molecular heterogeneity among these genomic subtypes by integrative analyses.
UV Signature
We noted that only 30% (14/46) of samples in the Triple-WT subtype harbored a UV signature, compared to 90.7% of samples with a BRAF hot-spot mutation (136/150), 93.5% with a RAS (N-H-K) hot-spot mutation (86/92), and 92.9% of the NF1 subtype (26/28) (Figure S1I) (Fisher’s exact test p = 1e–15). In contrast, Triple-WT samples had more copy-number changes and complex structural arrangements compared to the other groups.
Somatic Copy-Number Alterations
We assessed the patterns of somatic copy-number alteration (CNA) across subtypes. Although global patterns of arm-level alterations were similar, the Triple-WT had significantly more copy-number segments (Figures S2A and S2B) and was enriched for focal amplifications targeting known oncogenes. For example, we found significant 4q12 focal amplification containing the oncogene KIT only in the Triple-WT cohort (Figure 2A). Two other adjacent oncogenes, PDGFRA and KDR (also known as VEGFR2), were frequently co-amplified with KIT (Figure 2B). We also observed high-level focal CNAs containing the oncogenes CDK4 and CCND1 (p < 0.01, FDR < 0.05), consistent with previous studies (Curtin et al., 2005), as well as MDM2 and TERT (p < 0.05, FDR < 0.05) to be significantly enriched in Triple-WT melanomas (Figures 2B and 2C). In contrast, focal amplifications of BRAF, the melanocyte lineage-specific oncogene MITF (p < 0.01, FDR < 0.05), and the ligand for the co-inhibitory immune checkpoint protein PD-1, PD-L1 gene (CD274), were observed at significant frequencies in the BRAF mutant subtype (Figures 2, S2C, and S2D), whereas NRAS amplifications co-occurred in tumors with NRAS mutations (Figure S2C). CD274 amplifications (which encodes PD-L1) are particularly noteworthy given the potential clinical value of PD-L1 expression in predicting response to PD-1 pathway inhibitors (Tumeh et al., 2014).
Figure 2. Landscape of Copy-Number Alterations in Melanoma.
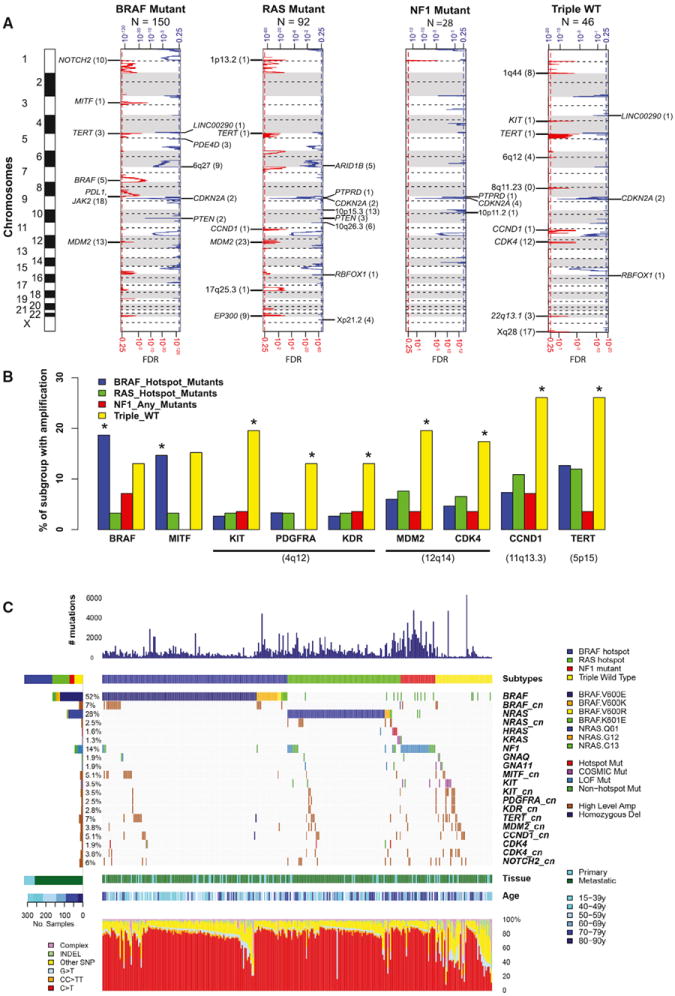
(A) GISTIC 2 analysis across four subtypes with selected highlighted genes from significant minimal common regions.
(B) Fraction of BRAF, RAS (N/H/K), NF1, and Triple-WT subtypes with focal amplifications determined by GISTIC 2 for BRAF and MITF (left) and KIT, PDGFRA, KDR, MDM2, CDK4, CCND1, and TERT (right). Asterisk indicates significant increase in amplification in the indicated mutation subtype compared to the rest by Fisher’s exact test (p < 0.01, FDR < 0.05).
(C) Landscape of mutation subtypes, selected cosmic mutations, and subtype-specific enriched copy-number amplifications. Per sample mutation rate, age, and mutation subtype (BRAF, RAS, NF1, and Triple-WT) (top), color-coded matrix of individual mutations and amplifications (specific BRAF and NRAS mutations indicated) (middle), and type of melanoma specimen (primary or metastasis) and mutation spectra for all samples (bottom) are shown.
See also Figure S2.
Structural Rearrangements
To define fusion events, we performed an integrative analysis using copy-number (n = 333), RNA-seq (n = 331), and whole-genome sequencing (WGS) data complemented by low-pass (n = 119) and deep (n = 38) sequencing. In total, 224 candidate fusion drivers were identified (Table S3A). Although there was only one recurrent fusion (GRM8-CNTNAP2, n = 2), we discovered a number of melanoma-associated genes recurrently fused to various gene partners (Figure S2E), including BRAF (ATG7-BRAF and TAX1BP1-BRAF), RAF1 (TRAK1-RAF1, RAF1-AGGF1, and CLCN6-RAF1), and AKT3 (CEP170-AKT3, AKT3-PLD5, ZEB2-AKT3, and ARHGAP30-AKT3). We also identified three MITF fusions (MITF-FOXP1, CADM2-MITF, and FRMD4B-MITF) and three HMGA2 fusions (PCBP2-HMGA2, TSFM-HMGA2, and SENP1-HMGA2). Eight of the 224 candidate driver fusions (ATG7-BRAF, TAX1BP1-BRAF, LBH-FLT4, LCLAT1-EPHA3, TRAK1-RAF1, CLCN6-RAF1, CPSF4L-ERBB4, and MOBKL1B-EPHB1) possessed a predicted intact kinase domain. Although additional functional studies are required to determine the role of these fusions in melanoma, unbiased pathway analyses of candidate fusions suggest biological functions relevant to melanoma (Tables S3B and S3C).
We saw significant enrichment for the 224 predicted fusion drivers in the Triple-WT subtype (p = 2e–04) (Figure S2F). Using ShatterSeek followed by manual review (see Supplemental Experimental Procedures), we identified complex rearrangement events in 38% of samples (45/117) (Table S1D). Like fusion events, complex structural rearrangements were enriched in the Triple-WT subtype (11/16, Fisher’s exact test p = 0.00098), particularly in those lacking a UV signature (7/7). Taken together with the pattern of somatic CNAs and the lower frequency of samples possessing a UV signature (~30%), these results suggest that, unlike other subtypes, other mutational processes that involve structural rearrangement of the genome drive the malignant phenotype of Triple-WT melanomas.
TERT Promoter Mutations
We confirmed mutually exclusive TERT promoter mutations C228T and C250T (Horn et al., 2013; Huang et al., 2013) in 23.5% and 40.9% of the 115 samples analyzed, respectively. Interestingly, only the C228T mutation was associated with elevated TERT mRNA expression (rank-sum test, p = 0.001) (Figure S2G) and contrasts with glioblastoma (GBM), in which both mutations were linked to increased expression (Brennan et al., 2013). TERT promoter mutations were observed in 75.0% (39/52) of BRAF, 71.9% (23/32) of RAS, and 83.3% (10/12) of NF1 subtypes but in only 6.7% (1/15) of Triple-WT (p = 8e–5, Figure S2H), suggesting an alternative mechanism of TERT activation (e.g., TERT amplification or rearrangement; see above) in the Triple-WT melanomas.
CIMP Phenotype
While a higher frequency of NRAS hot-spot mutations (OR = 2.3, p = 0.003) and a lower frequency of BRAF hot-spot mutations (OR = 0.4, p = 0.0008) were found in the CIMP cluster defined by DNA methylation profiles (EEP), the strongest associations of CIMP were with IDH1 (OR = 4.05, p = 0.005) and ARID2 (OR = 3.5, p = 0.0003) mutations (Figure S1F), both of which are chromatin-remodeling genes. Those observations suggest that, despite the intriguing correlations, the CIMP phenotype is not driven by the events responsible for genotypic subtypes of melanoma.
Signaling Pathways
Classical signaling pathway diagrams suggest that BRAF, RAS (N/H/K), and NF1 subtypes share common downstream signaling. We analyzed RPPA profiles of 181 cancer-related total proteins and phosphoproteins in 200 melanoma samples to further assess downstream signaling among subtypes. Not surprisingly, components of the MAPK, PI(3)K, and apoptotic signaling pathways were differentially activated by BRAF/RAS(N/H/K)/NF1 driver mutations (Figures 3 and S3). Although, for example, the upstream phospho-MAP2K1/MAP2K2 (MEK1/2) S217/S221 was elevated in both BRAF and RAS (N/H/K) hot-spot mutation subtypes (Figure 3A), the highest relative median activation of phospho-T202/Y204 MAPK1/MAPK3 (ERK1/2) was observed in the RAS (N/H/K) mutant subgroup (Figure 3B). As predicted by copy-number analysis, Triple-WT tumors showed the highest median KIT protein abundance (Figure 3C). In contrast, NF1 mutant melanomas had the highest median level of CRAF expression, highlighting differential MAPK activation in this subtype (Figure 3D). Other examples of differential subtype-specific signaling included higher median levels of the anti-apoptotic protein BCL-2 in the Triple-WT subtype (Figure 3E) and regulators of insulin signaling (IGFBP2) in BRAF hot-spot mutants (Figure 3F). Additional proteins involved in the PI(3)K/mTOR and epithelial-mesenchymal transition pathways were also significantly associated with particular mutation subtypes (Figure S3).
Figure 3. Analysis of Protein Expression Levels in Melanoma Samples.
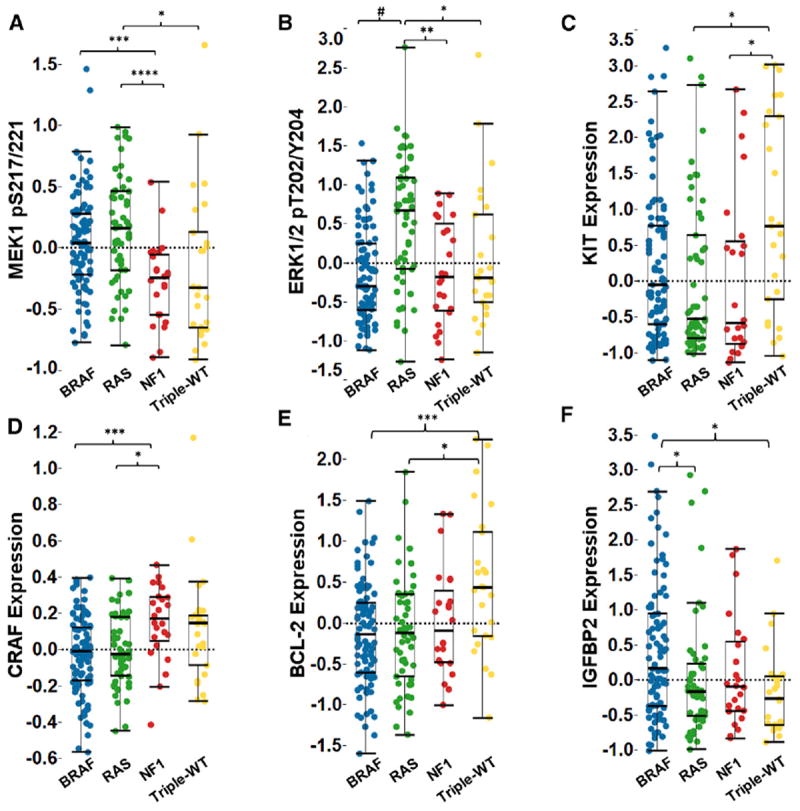
Individual protein levels were determined by RPPA across mutation subtypes.
(A) Phospho-MAP2K1/MAP2K2 (MEK1/2) S217/S221 was elevated in both the BRAF and RAS hot-spot mutation subtypes compared to NF1 and Triple-WT.
(B) Only RAS hot-spot mutant samples showed higher median levels of phospho-T202 Y204 MAPK1/MAPK3 (ERK1/2).
(C) Triple-WT melanomas had the highest median KIT protein expression.
(D and E) (D) NF1 mutant melanomas had a higher median level of CRAF expression, and Triple-WT had higher BCL-2 levels (E) compared to BRAF and RAS subtypes.
(F) Median IGFBP2 levels were highest in BRAF hot-spot mutant samples. Kruskal-Wallis test, and the post hoc Kruskal Nemenyi test for pairwise comparisons.
*p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001, #p = 5.4e–36. See also Figure S3.
Molecular Pathways
To broaden our view of the common molecular processes dysregulated in melanoma, we integrated mutation, copy-number, and methylation data to identify recurrently targeted pathways and signaling interactions involving significantly altered genes in all samples (n = 318) (Figures S4A–S4D). We manually curated the genetic alterations by BRAF, RAS (N/H/K), NF1, and Triple-WT subtypes (Figure 4A) and found that RAS (N/H/K)-MAPK-AKT, RB1/CDKN2A cell-cycle pathways, and MDM2/TP53 apoptosis pathways were altered in 91%, 69%, and 19% of cases, respectively. TP53 mutations were found more frequently in BRAF, RAS, and NF1 tumors, compared to Triple-WT, in which MDM2 amplifications were more frequent. Interestingly, of the 49 TP53 mutations identified, 46 (93.9%) were found in UV signature samples. Although CDKN2A/B alterations were nearly evenly distributed across subtypes, CDK4 and CCND1 amplifications were more frequent in Triple-WTs, and RB1 mutations were detected in a higher fraction of NF1 subtype tumors. Of the 12 RB1 mutations identified in this study, all were in UV signature samples. Finally, as previously reported (Pollock et al., 2003), PTEN mutations and deletions were more frequent in BRAF-mutant melanomas (Figures 4A and 4B), whereas amplification and mRNA overexpression of AKT3 were significantly enriched in RAS (N/H/K), NF1, and Triple-WT compared to the BRAF subtype (p < 0.05) (Figure 4B).
Figure 4. Pathways Altered in Melanoma.
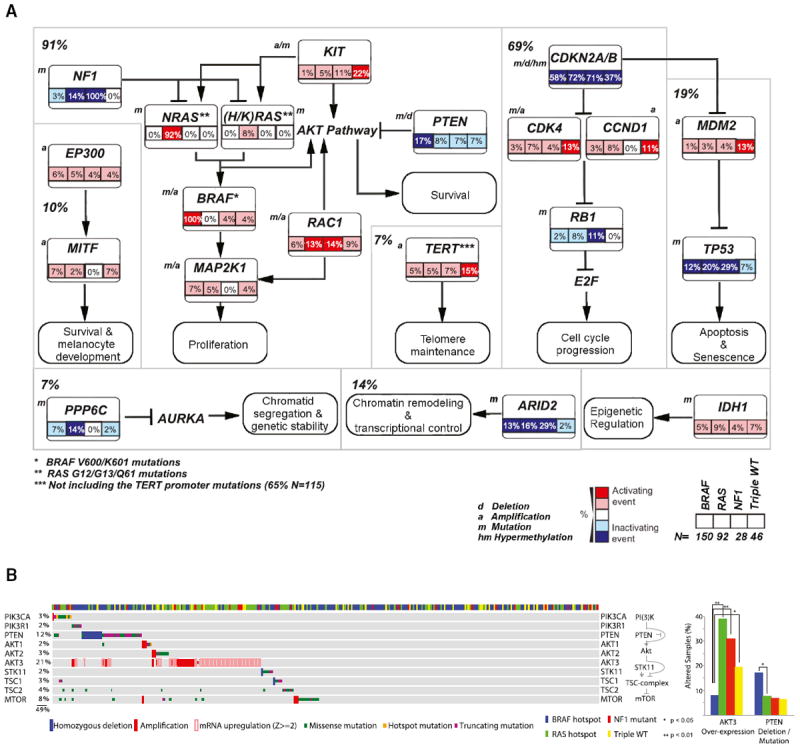
(A) Percentage of recurrently altered pathways in the four melanoma subtypes (BRAF = V600/K601 mutants, RAS [N/H/K] = G12, G13, and Q61 mutants) through integration of mutation, copy-number variation, and hypermethylation data are indicated (n = 316; not shown are one hyper-mutated and one co-occurring BRAF/NRAS hot-spot mutant sample). Manual curated pathway shows percentage of TP53, CDKN2A/RB1, and MAPK/AKT pathway across all samples (note: percentages of alterations of MAPK and AKT pathway are combined, given their high level of interconnectivity). a, amplification; d, deletion, m, mutation.
(B) Co-occurring somatic CNAs, mutations, and mRNA expression (color code indicated on graph) for the PI(3)K/mTOR pathway across the four mutation subtypes (left). Bar graph indicating percentage of fraction of subtypes with AKT3 activation or PTEN inactivation (right). Enrichment of a given alteration in a subgroup is estimated by Fisher’s exact test.
See also Figure S4.
Transcriptomic Classification of Melanoma
We performed consensus hierarchical clustering analysis (TCGA, 2014a) of the 1,500 genes with the most variant expression levels in 329 samples and identified three robust stable clusters. Based on the gene function(s) of discriminatory mRNA transcripts, we named the clusters “immune” (n = 168; 51%), “keratin” (n = 102; 31%), and “MITF-low” (n = 59; 18%) (Figure 5A and Table S4A). Interestingly, post-accession survival of patients with regionally metastatic tumors was significantly different among the three clusters (p = 0.001, Figure 5B), suggesting that these transcriptomically defined subclasses may be biologically relevant and distinct.
Figure 5. Integrative Analysis across Multiple Molecular Data Platforms Provides Insights into the Biology and Prognostic Significance of Immune Infiltrates in Cutaneous Melanoma.
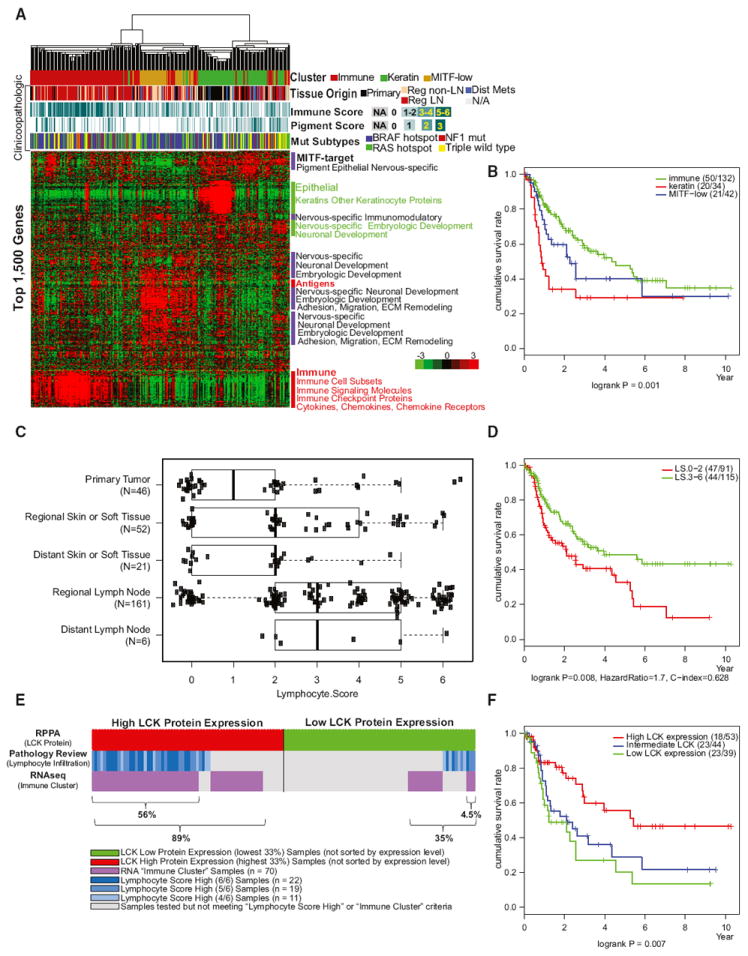
(A and B) (A) Unsupervised clustering of 329 melanoma samples using the top 1,500 genes showing the maximum absolute deviation identify three clusters defined as ‘‘immune-high,” ‘‘keratin-high,” and ‘‘microphthalmia-associated transcription factor (MITF)-low” based on gene function of discriminatory mRNAs and (B) post-accession survival curves for RNA subgroups.
(C) Distribution of lymphocytic scores determined by histopathology analysis according to sample type (described in detail in the Supplemental Experimental Procedures).
(D) Post-accession survival curves for high and low lymphocytic infiltration scores.
(E) Overlap of LCK high and low protein expression obtained from RPPA data with lymphocytic infiltration scores determined by pathology and RNA immune subgroups determined by mRNA clustering analysis.
(F) Association of LCK protein with post-accession survival. Three curves describe cumulative survival rates of three tertile patient subsets (p = 0.007 with log-rank test).
See also Figures S5, S6, and S7 and Data S1.
“Immune” Subclass
A significant number of genes overexpressed in this subclass were associated with immune cell subsets (T cells, B cells, and NK cells), immune signaling molecules, co-stimulatory and co-inhibitory immune checkpoint proteins, cytokines, chemokines, and corresponding receptors (Tables S4A–S4B). As 74% (113/152) of samples in the subclass were procured from regional lymph nodes (Pearson’s chi-square test, p < 0.001), we first assessed whether high expression of immune-related genes reflected the biology of melanoma-infiltrating immune cells or a non-specific admixture of “contaminating” adjacent lymphoid tissue in the samples (Erdag et al., 2012). Specifically, we compared the expression of nine curated immune gene signatures (comprising 793 genes and detailed in Table S4B) in 172 samples from lymph nodes and 157 tumors from other tissues (Figures S5A and S5B). Reassuringly, there was no significant difference in expression of tested immune signatures between the samples from lymph nodes and non-lymph node tissues (Figure S5A), suggesting that the transcriptomic features of the immune subclass were not due to contaminating adjacent lymph node tissue. Patients with regionally metastatic tumors in this subclass showed more favorable post-accession survival than did those in the other two subclasses (log-rank test, p = 0.003), in accordance with previous reports of the host immune response in melanoma (Azimi et al., 2012).
“Keratin” Subclass
This cluster was characterized by high expression of genes associated with keratins, pigmentation, and epithelium, as well as genes associated with neuronal development or other organ-specific embryologic development (Table S4A). Approximately 74% of primary melanomas clustered within this group (Pearson’s chi-square test, p < 0.001) and showed high expression of genes previously reported to be elevated in primary melanomas. Included were several keratins, kallikreins, and other epidermal genes. However, 25 keratin cluster samples were derived from regional lymph nodes, suggesting that expression of the epithelial transcripts was not due solely to admixture of epithelial tissue (such as skin epidermis) with melanoma tumor tissue, at least for this organ site of procurement; indeed, keratins and other epithelial markers have been found in some melanoma cell lines (Shields et al., 2007). Of note, regional metastatic melanomas exhibited worse outcome when compared with stage-matched samples assigned to the immune or MITF-low cluster (log-rank, p = 0.0007) (Figure 5B), supporting the view that the keratin cluster represents, at least in part, a previously unappreciated but biologically distinct melanoma subtype with adverse prognosis.
“MITF-Low” Subclass
The “MITF-low” cluster was characterized by low expression of genes associated with pigmentation and epithelial expression (Table S4A), including several MITF target genes and genes involved in immunomodulation, adhesion, migration, and extra-cellular matrix. This cluster was significantly enriched with genes preferentially expressed within the nervous system and/or associated with neuronal development or other organ-specific embryologic development.
Integrative Molecular Subtypes
Using the iCluster algorithm (see Supplemental Experimental Procedures), we next integrated multiple genomic dimensions (mutation, somatic CNAs, DNA methylation, and expression) to define molecular subtypes and to unravel hidden associations of the various subtypes identified in each genomic dimension (Figures 5A, S1D, S5C, and S7 and Data S1 and Table S4). We observed clear associations between the keratin expression subtype, the CIMP subtype, and a miRNA subgroup (cluster 3), which had a relatively lower frequency of hot-spot BRAF mutations (Figure S5D, iClust 1). Conversely, “MITF-low” cluster samples had a higher percentage of BRAF-hot-spot mutations (compared with “keratin” and “immune” clusters: 66% versus 33% and 45%, respectively; Fisher’s exact test, p = 0.0003 (visualized in Figure S5E). In addition, a lower percentage of tumor samples that were classified as “MITF-low” had no mutations in either BRAF, NRAS, and NF1 compared with “keratin” and “immune” clusters (3% versus 21% and 14%, respectively; Fisher’s exact test, p = 0.006) (Figure S5E). We also discerned associations with the hypomethylation subgroup and the MITF expression class (Figure S5D, iClust 2). Finally, we observed a low copy-number subgroup, a normal-like methylation profile, and enrichment for tumors possessing the immune mRNA expression signature, consistent with the presence of lymphocytic infiltration (Figure S5D, iClust 3).
Clinical Significance of the Immune Transcriptomic Subclass
Demonstrating the clinical relevance of molecular classification requires interpretation in the context of existing clinical practice. As a proof of concept, we addressed the clinical relevance and potential application of the observation that the “immune” transcriptomic subclass was associated with improved post-accession survival of patients with regional metastatic melanoma.
Although tumor-infiltrating lymphocytes have been associated with favorable prognosis in primary melanoma (Azimi et al., 2012), such an association has not been investigated in regional disease. To assess whether our transcriptomic classification of melanoma captures the biology of tumor-associated lymphocytes, we complemented the clinicopathological annotation provided by tissue source sites with a standardized pathology review of frozen section slides by TCGA Analysis Working Group (AWG) dermatopathologists (see Author Contributions); the density and distribution of melanoma-associated lymphocytes were used to derive a “lymphocyte score” (LScore), a semiquantitative measure of the number of lymphocytes in a sample (see Supplemental Experimental Procedures). Additional histopathological parameters included percent tumor content, percent necrotic tissue, and amount of melanin pigment. Melanomas from regional or distant lymph nodes showed significantly higher LScore than tumors from other tissues (Wilcoxon rank-sum test, p = 5.6e–8; Figure 5C). Among the subgroup of regional metastatic melanomas, elevated LScore was significantly associated with prolonged post-accession survival (Figure 5D), corroborating prior observations that tumor-associated lymphocytes are a favorable prognostic factor in melanoma (Bogunovic et al., 2009; Mihm et al., 1996). Remarkably, there was a striking concordance between high LScore (3–6) and assignment to the immune subclass (Figure S6A) (Fisher’s exact test, p < 1e–12).
Next, we asked whether transcriptomic features that defined the “immune” cluster are seen at the protein level by RPPA. In particular, we focused on two immune-related proteins, LCK and SYK, non-receptor tyrosine kinases commonly associated with T- and B-lymphocyte signaling. Interestingly, unsupervised clustering of RPPA data revealed that LCK and SYK are highly expressed in a subset of samples (Figure S5C) that are enriched with tumors in the transcriptomic immune subclass and/or that have high LScores (Figure 5E). However, high LCK, but not SYK, protein expression was also strongly associated with favorable post-accession survival of patients with regionally metastatic tumors (Figure 5F and data not shown). Tumors with high LScores tended to be assigned to the transcriptomic immune subclass and also express elevated levels of LCK protein (Figures S6A and S6B). These three characteristics overlapped considerably, and a combination of the three predicted mela-noma outcome more accurately than did any one of the features alone (log-rank, p = 8.0e–6, post-accession survival in regionally metastatic tumors; Figure S6C). This observation is consistent with the hypothesis that the three reflect unique (although overlapping) biological characteristics, each of which confers favorable outcomes in melanoma.
Finally, recognizing that unsupervised cluster analysis of a transcriptomic profile is not readily applicable to clinical practice, we tested the hypothesis that a bivariate model of LScore and LCK protein expression level offers a comparable prognostic prediction. Indeed, tumors with high LScore and high LCK expression were associated with significantly improved post-accession survival compared with those having low LScore and low LCK expression (log-rank p = 7.9–5, hazard ratio = 5.5, tumors with both high LScore and LCK versus both scores low; Figure S6D). Multivariable Cox proportional hazard regression also demonstrated that both LScore and LCK expression have independent predictive value in the two-factor model (Figure S6E). Overall, this integrative analysis suggests that a combination of LCK protein expression and pathologists’ scoring of tumor-infiltrating lymphocytes may be more prognostic for patients with nodal metastases than assessment of tumor-infiltrating lymphocytes alone.
DISCUSSION
We propose here that cutaneous melanomas can be divided into four genomic subtypes, designated BRAF, RAS (N/H/K), NF1, and Triple-WT. Such a genomic classification provides a framework for exploring how additional molecular alterations may explain observed biological and clinical differences among the subtypes. It also provides signposts for identification of drugable targets and predictive biomarkers, as well as potentially useful guidance for decisions about therapy.
Based on evidence that (1) BRAF/RAS (N/H/K) mutant melanomas are driven, at least in part, by MAPK signaling (Hodis et al., 2012; Krauthammer et al., 2012); (2) melanomas lacking NF1 expression are dependent on MAPK signaling and respond to MAPK inhibitors (Maertens et al., 2013; Nissan et al., 2014); and (3) there are clinicopathologic and molecular differences among melanomas that do not have hot-spot mutations in BRAF/RAS but differ with respect to NF1 mutation status, melanoma joins two other RTK/RAS-driven solid tumor types (GBM and lung adenocarcinoma) analyzed by the TCGA, among which a subset of these cancers has loss-of-function NF1 mutations (TCGA, 2008, 2014b).
We suggest that significantly mutated genes and other molecular alterations identified here, combined with previously described melanoma-associated genes, are likely to have important implications for prognosis and therapy (Table 1). For example, we postulate that patients with BRAF wild-type, NF1 mutant melanomas respond to MEK and/or ERK inhibitors (Maertens et al., 2013; Nissan et al., 2014; Whittaker et al., 2013), supported by cell line studies that demonstrate that at least some NF1 mutant cell lines respond to MEK inhibitors (Ranzani et al., 2015). In the setting of frequently co-occurring NF1 and ARID2 mutations, synthetic lethal strategies targeting chromatin modifiers represent a rational area for pre-clinical research (Helming et al., 2014). In addition to therapeutic strategies currently under clinical development, melanomas with RAS (N/H/K) mutations, frequently concurrent with PPP6C hot-spot mutations, may provide therapeutic opportunities for combinatorial treatment strategies that include Aurora kinase inhibition (Gold et al., 2014). Previous studies have shown frequent co-occurrence of BRAF mutations and PTEN mutations or deletions (Tsao et al., 2012). Here, we showed a higher frequency of amplifications and overexpression of AKT3 in RAS, NF1, and Triple-WT melanomas, which may provide additional biomarkers to support the use of combination MEK and PI(3)K/AKT/mTOR pathway inhibitors in such subtypes. In addition, mutations in PIK3CA (E545K, H1047L) and AKT1/3 (E17K) in BRAF, as well as RAS (N/H/K) mutant melanoma (Table S2E), may serve as biomarkers that predict response to the above-mentioned targeted therapies.
Candidate driver events in Triple-WT melanomas provide opportunities for pre-clinical and clinical efforts to effectively target these molecular aberrations. These include KIT mutations/amplifications, co-amplified RTKs, PDGFRA and KDR (VEGFR2), and even rare GNAQ Q209P (n = 1) and GNA11 Q209L (n = 2) mutations (sample IDs: TCGA-ER-A3ES, TCGA-ER-A3ET, and TCGA-ER-A2NF)—the latter of which, interestingly, co-occur with hot-spot SF3B1 R625H mutations (n = 2 for co-occurrence with GNA11/Q hot-spot mutations) in our cutaneous melanoma cohort, but not BAP1 mutations, which are frequently found in metastatic uveal melanoma (Field and Harbour, 2014). Although GNAQ and GNA11 hot-spot mutations are common in uveal melanomas, they have also been reported in blue nevi and primary melanocytic neoplasms of the central nervous system (Küsters-Vandevelde et al., 2010). Our classification supports the use of imatinib and dasatinib to treat patients with KIT-mutated/amplified cutaneous melanomas (Carvajal et al., 2011; Hodi et al., 2008; Lutzky et al., 2008; Terheyden et al., 2010) and consideration of combination therapies with sorafenib, crenolanib, regorafenib, and pazopanib to target co-amplified RTKs, PDGFRA, and KDR (VEGFR2). Triple-WT melanomas with amplifications of MDM2 and overexpression of BCL2 may respond to inhibitors such as AMG 232, nutlin-3, and BH3 mimetics, currently in preclinical or clinical development in melanoma. Such agents may also be beneficial for patients with wild-type TP53 across the genetic subtypes (Frederick et al., 2014; Ji et al., 2013; Sun et al., 2014). Other potentially actionable mutations include recurrent IDH1 R132 (~6%) and EZH2 Y641 mutations (<1%) (Table S2E).
Overall, approximately half of all cases were assigned to the “immune” subtype. Interestingly, the response rate to inhibitors of the PD-1/PD-L1 pathway is approximately one-third (Brahmer et al., 2012; Hamid et al., 2013; Topalian et al., 2012). In our study, expression of both PD-1 and PD-L1 was significantly higher in “immune” compared to each of the two other groups (Figure S6F), similar to a recent report showing that pre-existing CD8+ T cells distinctly located at the invasive tumor margin are associated with immunohistochemical expression of PD-1 and PD-L1, and was also predictive of response to pembrolizumab (Tumeh et al., 2014). However, it is important to emphasize that our data do not prove that the immune subtype represents a population responsive to immunotherapies.
We show that immune infiltration is statistically correlated with more favorable prognosis, irrespective of genomic subtype. The lack of a genomic correlation with outcome provides a plausible molecular explanation for the lack of observed preferential anti-tumor responses in clinical trials employing immune checkpoint blockade, at least in relation to BRAF status (Ascierto et al., 2014; Robert et al., 2014). Nonetheless, despite these data, the question of whether specific mutated melanoma antigens are responsible for differences in the degree of tumor infiltration by lymphocytes is an area of active investigation (Robbins et al., 2013; Snyder et al., 2014). Our combined RPPA analysis, including exploration of LCK and SYK proteins that are associated with T cell and B cell signaling effectors, respectively, suggests that T cell, but not B cell, signaling has prognostic significance. This relevance of T cells, and in particular effector CD8+ T cells, is congruent with clinical benefit seen with high-dose bolus IL-2, a T cell growth factor used as a therapeutic agent for advanced melanoma (McArthur and Ribas, 2013).
Among the cohort of patients in this study with advanced stage III disease (Balch et al., 2010), high lymphocytic score and immune-associated gene expression was associated with prolonged post-accession survival, potentially reflecting a clinical benefit of immunotherapies for stage III melanoma patients (Eggermont et al., 2008; Kirkwood et al., 1996). Such markers should be considered for further evaluation and potential integration into future AJCC staging systems and associated prognostic models, as well as for exploration as a potential predictor of response to adjuvant therapies for stage III disease.
EXPERIMENTAL PROCEDURES
Patients and Biospecimens
Eligible patients had a diagnosis of either primary or metastatic cutaneous melanoma or metastatic melanoma of unknown primary (Balch et al., 2009; Dasgupta et al., 1963), but no previous systemic therapy (except that adjuvant interferon-α ≥90 days prior was permitted); the site from which the bio-specimen was collected could not have been previously treated at any time with radiotherapy. Biospecimens from resected primary and/or metastatic melanomas were obtained from patients with appropriate informed consent and institutional review board or ethics board approval. Biospecimens were classified as either primary or metastatic based on the available clinical and pathological information. Independent pathological review confirmed that each biospecimen was consistent with melanoma. As specimens were required to have sufficient mass and quality for downstream molecular analyses, those from advanced primary and/or metastatic tumors were over-represented. The complete methodology for patient eligibility, clinical and pathological data elements, biospecimen acquisition, and molecular analyte extraction is described in the Supplemental Experimental Procedures.
Data Generation
Data from at least one platform were available for 333 patients. The data types included: (1) clinical, (2) whole-exome sequencing, (3) DNA copy-number and single-nucleotide polymorphism array, (4) whole-genome sequencing, (5) RNA-sequencing data, (6) DNA methylation, (7) reverse-phase protein array, and (8) microRNA sequencing. Details of data generation and analyses are described in the Supplemental Experimental Procedures. All data sets are available through the Cancer Genome Atlas (TCGA) data portal (https://tcga-data.nci.nih.gov/tcga).
Whole-Genome and Exome-Sequencing Data Analysis
Whole-exome sequencing was performed as previously described (TCGA, 2012). Exome capture was performed using the Agilent Sure-Select Human All Exon v2.0, 44 Mb kit, followed by 2 × 76 bp paired-end sequencing on the Illumina HiSeq platform. Read alignment and processing were performed using BWA and the Picard and Firehose pipelines at the Broad Institute. For each file, Picard generates a single BAM file that includes reads, calibrated quantities, and alignments to the genome. The Firehose pipeline performs quality control, local realignment, mutation calling, small insertion and deletion identification, and coverage calculations, among other analyses. Complete details of the pipeline can be found online at http://www.broadinstitute.org/cancer/cga. Whole-genome sequencing methods are described in detail in the Supplemental Experimental Procedures.
RNA-Sequencing Data Analysis
Total RNA was converted to mRNA libraries using the lllumina mRNA TruSeq kit, following the manufacturer’s directions. Libraries were sequenced on the Illumina HiSeq 2000 as previously described (TCGA, 2012). Read mapping, gene expression quantitation, and identification of fusion transcripts are described in the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Represents the largest integrative analysis of cutaneous melanoma (331 patients)
Establishes a framework for melanoma genomic classification: BRAF, RAS, NF1, and Triple-WT
Identifies additional subtypes that may benefit from MAPK-and RTK-targeted therapies
Multi-dimensional analyses identify immune signatures associated with improved survival
Acknowledgments
We thank all patients and families who contributed to this study. We are grateful to Chris Gunter for manuscript editing and Ina Felau and Margi Sheth for project management. This article is dedicated to the memory of Donald L. Morton, M.D., a pioneer in melanoma oncology, who passed away on January 10, 2014. This study was supported by NIH grants: U54 HG003273, U54 HG003067, U54 HG003079, U24 CA143799, U24 CA143835, U24 CA143840, U24 CA143843, U24 CA143845, U24 CA143848, U24 CA143858, U24 CA143866, U24 CA143867, U24 CA143882, U24 CA143883, U24 CA144025, and P30 CA016672.
A.D.C. and M.M. receive research funding from Bayer AG. M.M. is a founder of, equity holder in, and consultant for Foundation Medicine, a next-generation sequencing-based cancer diagnostics company. L.A.G. received a commercial research grant from Novartis and is a consultant/advisory board member for Novartis, Foundation Medicine, and Boehringer Ingelheim. L.A.G. also has equity interest in Foundation Medicine. D.J.W. is a consultant for Zymo Research Corporation, which distributes commercially available products for DNA methylation-based experiments. Zymo Research neither supported this work nor has an interest in the outcome of this research. O.P and O.V. are co-founders and shareholders of Cureline, Inc., which received a contract payment from the NIH for this work. J.E.G. is an advisory board member for Merck.
CONSORTIA
The members of The Cancer Genome Atlas Research Network for this project are Rehan Akbani, Kadir C. Akdemir, B. Arman Aksoy, Monique Albert, Adrian Ally, Samirkumar B. Amin, Harindra Arachchi, Arshi Arora, J. Todd Auman, Brenda Ayala, Julien Baboud, Miruna Balasundaram, Saianand Balu, Nandita Barnabas, John Bartlett, Pam Bartlett, Boris C. Bastian, Stephen B. Baylin, Madhusmita Behera, Dmitry Belyaev, Christopher Benz, Brady Bernard, Rameen Beroukhim, Natalie Bir, Aaron D. Black, Tom Bodenheimer, Lori Boice, Genevieve M. Boland, Riccardo Bono, Moiz S. Bootwalla, Marcus Bosenberg, Jay Bowen, Reanne Bowlby, Christopher A. Bristow, Laura Brockway-Lunardi, Denise Brooks, Jakub Brzezinski, Wiam Bshara, Elizabeth Buda, William R. Burns, Yaron S.N. Butterfield, Michael Button, Tiffany Calderone, Giancarlo Antonini Cappellini, Candace Carter, Scott L. Carter, Lynn Cherney, Andrew D. Cherniack, Aaron Chevalier, Lynda Chin, Juok Cho, Raymond J. Cho, Yoon-La Choi, Andy Chu, Sudha Chudamani, Kristian Cibulskis, Giovanni Ciriello, Amanda Clarke, Stephen Coons, Leslie Cope, Daniel Crain, Erin Curley, Ludmila Danilova, Stefania D’Atri, Tanja Davidsen, Michael A. Davies, Keith A. Delman, John A. Demchok, Qixia A. Deng, Yonathan Lissanu Deribe, Noreen Dhalla, Rajiv Dhir, Daniel DiCara, Michael Dinikin, Michael Dubina, J. Stephen Ebrom, Sophie Egea, Greg Eley, Jay Engel, Jennifer M. Eschbacher, Konstantin V. Fedosenko, Ina Felau, Timothy Fennell, Martin L. Ferguson, Sheila Fisher, Keith T. Flaherty, Scott Frazer, Jessica Frick, Victoria Fulidou, Stacey B. Gabriel, Jianjiong Gao, Johanna Gardner, Levi A. Garraway, Julie M. Gastier-Foster, Carmelo Gaudioso, Nils Gehlenborg, Giannicola Genovese, Mark Gerken, Jeffrey E. Gershenwald, Gad Getz, Carmen Gomez-Fernandez, Thomas Gribbin, Jonna Grimsby, Benjamin Gross, Ranabir Guin, Tony Gutschner, Angela Hadjipanayis, Ruth Halaban, Benjamin Hanf, David Haussler, Lauren E. Haydu, D. Neil Hayes, Nicholas K. Hayward, David I. Heiman, Lynn Her-bert, James G. Herman, Peter Hersey, Katherine A. Hoadley, Eran Hodis, Robert A. Holt, Dave SB Hoon, Susan Hoppough, Alan P. Hoyle, Franklin W. Huang, Mei Huang, Sharon Huang, Carolyn M. Hutter, Matthew Ibbs, Lisa Iype, Anders Jacobsen, Valerie Jakrot, Alyssa Janning, William R. Jeck, Stuart R. Jefferys, Mark A. Jensen, Corbin D. Jones, Steven J.M. Jones, Zhenlin Ju, Hojabr Kakavand, Hyojin Kang, Richard F. Kefford, Fadlo R. Khuri, Jaegil Kim, John M. Kirkwood, Joachim Klode, Anil Korkut, Konstanty Korski, Michael Krauthammer, Raju Kucherlapati, Lawrence N. Kwong, Witold Kycler, Marc La-danyi, Phillip H. Lai, Peter W. Laird, Eric Lander, Michael S. Lawrence, Alexander J. Lazar, Radoslaw Łaźniak, Darlene Lee, Jeffrey E. Lee, Junehawk Lee, Kenneth Lee, Semin Lee, William Lee, Ewa Leporowska, Kristen M. Leraas, Haiyan I. Li, Tara M. Lichtenberg, Lee Lichtenstein, Pei Lin, Shiyun Ling, Jia Liu, Ouida Liu, Wenbin Liu, Georgina V. Long, Yiling Lu, Singer Ma, Yussanne Ma, Andrzej Mackiewicz, Harshad S. Mahadeshwar, Jared Malke, David Mallery, Georgy M. Manikhas, Graham J. Mann, Marco A. Marra, Brenna Matejka, Michael Mayo, Sousan Mehrabi, Shaowu Meng, Matthew Meyerson, Piotr A. Mieczkowski, John P. Miller, Martin L. Miller, Gordon B. Mills, Fedor Moiseenko, Richard A. Moore, Scott Morris, Carl Morrison, Donald L. Morton, Stergios Moschos, Lisle E. Mose, Florian L. Muller, Andrew J. Mungall, Dawid Murawa, Pawel Murawa, Bradley A. Murray, Luigi Nezi, Sam Ng, Dana Nicholson, Michael S. Noble, Adeboye Osunkoya, Taofeek K. Owonikoko, Bradley A. Ozenberger, Elena Pagani, Oxana V. Paklina, Angeliki Pantazi, Michael Parfenov, Jeremy Parfitt, Peter J. Park, Woong-Yang Park, Joel S. Parker, Francesca Passarelli, Robert Penny, Charles M. Perou, Todd D. Pihl, Olga Potapova, Victor G. Prieto, Alexei Protopopov, Michael J. Quinn, Amie Radenbaugh, Kunal Rai, Suresh S. Ramalingam, Ayush T. Raman, Nilsa C. Ramirez, Ricardo Ramirez, Uma Rao, W. Kimryn Rathmell, Xiaojia Ren, Sheila M. Reynolds, Jeffrey Roach, A. Gordon Robertson, Merrick I. Ross, Jason Roszik, Giandomenico Russo, Gordon Saksena, Charles Saller, Yardena Samuels, Chris Sander, Cindy Sander, George Sandusky, Netty Santoso, Melissa Saul, Robyn PM Saw, Dirk Schadendorf, Jacqueline E. Schein, Nikolaus Schultz, Steven E. Schumacher, Charles Schwallier, Richard A. Scolyer, Jonathan Seidman, Pedamallu Chandra Sekhar, Harmanjatinder S. Sekhon, Yasin Senbabaoglu, Sahil Seth, Kerwin F. Shannon, Samantha Sharpe, Norman E. Sharpless, Kenna R. Mills Shaw, Candace Shelton, Troy Shelton, Ronglai Shen, Margi Sheth, Yan Shi, Carolyn J Shiau, Ilya Shmulevich, Gabriel L. Sica, Janae V. Simons, Rileen Sinha, Payal Sipahimalani, Heidi J. Sofia, Matthew G. Soloway, Xingzhi Song, Carrie Sougnez, Andrew J. Spillane, Arkadiusz Spycha1a, Jonathan R. Stretch, Joshua Stuart, Wiktoria M. Suchorska, Antje Sucker, S. Onur Sumer, Yichao Sun, Maria Synott, Barbara Tabak, Teresa R. Tabler, Angela Tam, Donghui Tan, Jiabin Tang, Roy Tarnuzzer, Katherine Tarvin, Honorata Tatka, Barry S. Taylor, Marek Teresiak, Nina Thiessen, John F. Thompson, Leigh Thorne, Vesteinn Thorsson, Jeffrey M. Trent, Timothy J. Triche, Jr., Kenneth Y. Tsai, Peiling Tsou, David J. Van Den Berg, Eliezer M. Van Allen, Umadevi Veluvolu, Roeland G. Verhaak, Douglas Voet, Olga Voronina, Vonn Walter, Jessica S. Walton, Yunhu Wan, Yuling Wang, Zhining Wang, Scot Waring, Ian R. Watson, Nils Weinhold, John N. Weinstein, Daniel J. Weisenberger, Peter White, Matthew D. Wilkerson, James S. Wilmott, Lisa Wise, Maciej Wiznerowicz, Scott E. Woodman, Chang-Jiun Wu, Chia-Chin Wu, Junyuan Wu, Ye Wu, Ruibin Xi, Andrew Wei Xu, Da Yang, Liming Yang, Lixing Yang, Travis I. Zack, Jean C. Zenklusen, Hailei Zhang, Jianhua Zhang, Wei Zhang, Xiaobei Zhao, Jingchun Zhu, Kelsey Zhu, Lisa Zimmer, Erik Zmuda, and Lihua Zou.
Footnotes
AUTHOR CONTRIBUTIONS
The Cancer Genome Atlas Research Network contributed collectively to this study. Biospecimens were provided by the tissue source sites and processed by the Biospecimen Core Resource. Data generation and analyses were performed by the genome-sequencing centers, cancer genome-characterization centers, and genome data analysis centers. All data were released through the Data Coordinating Center. The NCI and NHGRI project teams coordinated project activities. Individual contributions of TCGA investigators are detailed in the Supplemental Information.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, four tables, and one data file and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2015.05.044.
References
- Andersen LB, Fountain JW, Gutmann DH, Tarlé SA, Glover TW, Dracopoli NC, Housman DE, Collins FS. Mutations in the neuro-fibromatosis 1 gene in sporadic malignant melanoma cell lines. Nat Genet. 1993;3:118–121. doi: 10.1038/ng0293-118. [DOI] [PubMed] [Google Scholar]
- Ascierto PA, Simeone E, Sileni VC, Pigozzo J, Maio M, Altomonte M, Del Vecchio M, Di Guardo L, Marchetti P, Ridolfi R, et al. Clinical experience with ipilimumab 3 mg/kg: real-world efficacy and safety data from an expanded access programme cohort. J Transl Med. 2014;12:116. doi: 10.1186/1479-5876-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthy SW, Saw RP, Thompson JF. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J Clin Oncol. 2012;30:2678–2683. doi: 10.1200/JCO.2011.37.8539. [DOI] [PubMed] [Google Scholar]
- Baade P, Meng X, Youlden D, Aitken J, Youl P. Time trends and latitudinal differences in melanoma thickness distribution in Australia, 1990-2006. Int J Cancer. 2012;130:170–178. doi: 10.1002/ijc.25996. [DOI] [PubMed] [Google Scholar]
- Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Ding S, Byrd DR, Cascinelli N, Cochran AJ, Coit DG, Eggermont AM, et al. Multivariate analysis of prognostic factors among 2,313 patients with stage III melanoma: comparison of nodal micrometastases versus macrometastases. J Clin Oncol. 2010;28:2452–2459. doi: 10.1200/JCO.2009.27.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–506. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogunovic D, O’Neill DW, Belitskaya-Levy I, Vacic V, Yu YL, Adams S, Darvishian F, Berman R, Shapiro R, Pavlick AC, et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc Natl Acad Sci USA. 2009;106:20429–20434. doi: 10.1073/pnas.0905139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash DE. UV signature mutations. Photochem Photobiol. 2015;91:15–26. doi: 10.1111/php.12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, Panageas KS, Busam KJ, Chmielowski B, Lutzky J, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305:2327–2334. doi: 10.1001/jama.2011.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscione VD, Weinstock MA. Melanoma thickness trends in the United States, 1988-2006. J Invest Dermatol. 2010;130:793–797. doi: 10.1038/jid.2009.328. [DOI] [PubMed] [Google Scholar]
- Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- Dasgupta T, Bowden L, Berg JW. Malignant Melanoma of Unknown Primary Origin. Surg Gynecol Obstet. 1963;117:341–345. [PubMed] [Google Scholar]
- Draper GJ, Sanders BM, Kingston JE. Second primary neoplasms in patients with retinoblastoma. Br J Cancer. 1986;53:661–671. doi: 10.1038/bjc.1986.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton-Regester K, Gartner JJ, Emmanuel R, Qutob N, Davies MA, Gershenwald JE, Robinson W, Robinson S, Rosenberg SA, Scolyer RA, et al. A highly recurrent RPS27 5′ UTR mutation in melanoma. Oncotarget. 2014;5:2912–2917. doi: 10.18632/oncotarget.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont AM, Suciu S, Santinami M, Testori A, Kruit WH, Marsden J, Punt CJ, Salès F, Gore M, Mackie R, et al. EORTC Melanoma Group. Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: final results of EORTC 18991, a randomised phase III trial. Lancet. 2008;372:117–126. doi: 10.1016/S0140-6736(08)61033-8. [DOI] [PubMed] [Google Scholar]
- Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, Patterson JW, Slingluff CL., Jr Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012;72:1070–1080. doi: 10.1158/0008-5472.CAN-11-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MG, Harbour JW. Recent developments in prognostic and predictive testing in uveal melanoma. Curr Opin Ophthalmol. 2014;25:234–239. doi: 10.1097/ICU.0000000000000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick DT, Salas Fragomeni RA, Schalck A, Ferreiro-Neira I, Hoff T, Cooper ZA, Haq R, Panka DJ, Kwong LN, Davies MA, et al. Clinical profiling of BCL-2 family members in the setting of BRAF inhibition offers a rationale for targeting de novo resistance using BH3 mimetics. PLoS ONE. 2014;9:e101286. doi: 10.1371/journal.pone.0101286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershenwald JE, Ross MI. Sentinel-lymph-node biopsy for cutaneous melanoma. N Engl J Med. 2011;364:1738–1745. doi: 10.1056/NEJMct1002967. [DOI] [PubMed] [Google Scholar]
- Gold HL, Wengrod J, de Miera EV, Wang D, Fleming N, Sikkema L, Kirchhoff T, Hochman T, Goldberg JD, Osman I, Gardner LB. PP6C hotspot mutations in melanoma display sensitivity to Aurora kinase inhibition. Mol Cancer Res. 2014;12:433–439. doi: 10.1158/1541-7786.MCR-13-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ, et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med. 2014;20:251–254. doi: 10.1038/nm.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, Friedlander P, Corless CL, Heinrich MC, Mac Rae S, Kruse A, Jagannathan J, Van den Abbeele AD, Velazquez EF, Demetri GD, Fisher DE. Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol. 2008;26:2046–2051. doi: 10.1200/JCO.2007.14.0707. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman SS, Rayhan DJ, Hazany S, Kolodney MS. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J Invest Dermatol. 2014;134:213–220. doi: 10.1038/jid.2013.276. [DOI] [PubMed] [Google Scholar]
- Ji Z, Kumar R, Taylor M, Rajadurai A, Marzuka-Alcalá A, Chen YE, Njauw CN, Flaherty K, Jönsson G, Tsao H. Vemurafenib synergizes with nutlin-3 to deplete survivin and suppresses melanoma viability and tumor growth. Clin Cancer Res. 2013;19:4383–4391. doi: 10.1158/1078-0432.CCR-13-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14:7–17. doi: 10.1200/JCO.1996.14.1.7. [DOI] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küsters-Vandevelde HV, Klaasen A, Küsters B, Groenen PJ, van Engenvan Grunsven IA, van Dijk MR, Reifenberger G, Wesseling P, Blokx WA. Activating mutations of the GNAQ gene: a frequent event in primary melanocytic neoplasms of the central nervous system. Acta Neuropathol. 2010;119:317–323. doi: 10.1007/s00401-009-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Siva-chenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez GY, Reitman ZJ, Solomon D, Waldman T, Bigner DD, McLendon RE, Rosenberg SA, Samuels Y, Yan H. IDH1(R132) mutation identified in one human melanoma metastasis, but not correlated with metastases to the brain. Biochem Biophys Res Commun. 2010;398:585–587. doi: 10.1016/j.bbrc.2010.06.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzky J, Bauer J, Bastian BC. Dose-dependent, complete response to imatinib of a metastatic mucosal melanoma with a K642E KIT mutation. Pigment Cell Melanoma Res. 2008;21:492–493. doi: 10.1111/j.1755-148X.2008.00475.x. [DOI] [PubMed] [Google Scholar]
- Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter S, Flaherty K, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013;3:338–349. doi: 10.1158/2159-8290.CD-12-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann GJ, Pupo GM, Campain AE, Carter CD, Schramm SJ, Pianova S, Gerega SK, De Silva C, Lai K, Wilmott JS, et al. BRAF mutation, NRAS mutation, and the absence of an immune-related expressed gene profile predict poor outcome in patients with stage III melanoma. J Invest Dermatol. 2013;133:509–517. doi: 10.1038/jid.2012.283. [DOI] [PubMed] [Google Scholar]
- McArthur GA, Ribas A. Targeting oncogenic drivers and the immune system in melanoma. J Clin Oncol. 2013;31:499–506. doi: 10.1200/JCO.2012.45.5568. [DOI] [PubMed] [Google Scholar]
- Mihm MC, Jr, Clemente CG, Cascinelli N. Tumor infiltrating lymphocytes in lymph node melanoma metastases: a histopathologic prognostic indicator and an expression of local immune response. Lab Invest. 1996;74:43–47. [PubMed] [Google Scholar]
- Morton DL, Thompson JF, Cochran AJ, Mozzillo N, Nieweg OE, Roses DF, Hoekstra HJ, Karakousis CP, Puleo CA, Coventry BJ, et al. MSLT Group. Final trial report of sentinel-node biopsy versus nodal observation in melanoma. N Engl J Med. 2014;370:599–609. doi: 10.1056/NEJMoa1310460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, Harshman K, Guipponi M, Bukach O, Zoete V, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2012;44:133–139. doi: 10.1038/ng.1026. [DOI] [PubMed] [Google Scholar]
- Nissan MH, Pratilas CA, Jones AM, Ramirez R, Won H, Liu C, Tiwari S, Kong L, Hanrahan AJ, Yao Z, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74:2340–2350. doi: 10.1158/0008-5472.CAN-13-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Cancer Genome Atlas Research Network. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Rakosy Z, Ecsedi S, Toth R, Vizkeleti L, Hernandez-Vargas H, Lazar V, Emri G, Szatmari I, Herceg Z, Adany R, Balazs M. Integrative genomics identifies gene signature associated with melanoma ulceration. PLoS ONE. 2013;8:e54958. doi: 10.1371/journal.pone.0054958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranzani M, Alifrangis C, Perna D, Dutton-Regester K, Pritchard A, Wong K, Rashid M, Robles-Espinoza CD, Hayward NK, McDermott U, et al. BRAF/NRAS wild-type melanoma, NF1 status and sensitivity to trametinib. Pigment Cell Melanoma Res. 2015;28:117–119. doi: 10.1111/pcmr.12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19:747–752. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- Santa Cruz DJ, Hamilton PD, Klos DJ, Fernandez-Pol JA. Differential expression of metallopanstimulin/S27 ribosomal protein in melano-cytic lesions of the skin. J Cutan Pathol. 1997;24:533–542. doi: 10.1111/j.1600-0560.1997.tb01457.x. [DOI] [PubMed] [Google Scholar]
- Shields JM, Thomas NE, Cregger M, Berger AJ, Leslie M, Torrice C, Hao H, Penland S, Arbiser J, Scott G, et al. Lack of extracellular signal-regulated kinase mitogen-activated protein kinase signaling shows a new type of melanoma. Cancer Res. 2007;67:1502–1512. doi: 10.1158/0008-5472.CAN-06-3311. [DOI] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, Canon J, Chen A, Chen X, Chow D, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem. 2014;57:1454–1472. doi: 10.1021/jm401753e. [DOI] [PubMed] [Google Scholar]
- TCGA (Cancer Genome Atlas Research Network) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA (Cancer Genome Atlas Research Network) Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA (Cancer Genome Atlas Research Network) Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014a;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA (Cancer Genome Atlas Research Network) Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014b;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terheyden P, Houben R, Pajouh P, Thorns C, Zillikens D, Becker JC. Response to imatinib mesylate depends on the presence of the V559A-mutated KIT oncogene. J Invest Dermatol. 2010;130:314–316. doi: 10.1038/jid.2009.197. [DOI] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Chin L, Garraway LA, Fisher DE. Melanoma: from mutations to medicine. Genes Dev. 2012;26:1131–1155. doi: 10.1101/gad.191999.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. Dermatologic Cooperative Oncology Group of Germany (DeCOG) The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson IR, Li L, Cabeceiras PK, Mahdavi M, Gutschner T, Genovese G, Wang G, Fang Z, Tepper JM, Stemke-Hale K, et al. The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res. 2014;74:4845–4852. doi: 10.1158/0008-5472.CAN-14-1232-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, Schadendorf D, Root DE, Garraway LA. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3:350–362. doi: 10.1158/2159-8290.CD-12-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnepenninckx V, Lazar V, Michiels S, Dessen P, Stas M, Alonso SR, Avril MF, Ortiz Romero PL, Robert T, Balacescu O, et al. Melanoma Group of the European Organization for Research and Treatment of Cancer. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J Natl Cancer Inst. 2006;98:472–482. doi: 10.1093/jnci/djj103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.