

Abstract
Micro-ribonucleic acids (miRNAs) are a large group of endogenous, tiny, non-coding RNAs consisting of 19–25 nucleotides that regulate gene expression at either the transcriptional or post-transcriptional level by mediating gene silencing in eukaryotes. They are considered to be important regulators that affect growth, development, and response to various stresses in plants. Alexandrium catenella is an important marine toxic phytoplankton species that can cause harmful algal blooms (HABs). To date, identification and function analysis of miRNAs in A. catenella remain largely unexamined. In this study, high-throughput sequencing was performed on A. catenella to identify and quantitatively profile the repertoire of small RNAs from two different growth phases. A total of 38,092,056 and 32,969,156 raw reads were obtained from the two small RNA libraries, respectively. In total, 88 mature miRNAs belonging to 32 miRNA families were identified. Significant differences were found in the member number, expression level of various families, and expression abundance of each member within a family. A total of 15 potentially novel miRNAs were identified. Comparative profiling showed that 12 known miRNAs exhibited differential expression between the lag phase and the logarithmic phase. Real-time quantitative RT-PCR (qPCR) was performed to confirm the expression of two differentially expressed miRNAs that were one up-regulated novel miRNA (aca-miR-3p-456915), and one down-regulated conserved miRNA (tae-miR159a). The expression trend of the qPCR assay was generally consistent with the deep sequencing result. Target predictions of the 12 differentially expressed miRNAs resulted in 1813target genes. Gene ontology (GO) analysis and the Kyoto Encyclopedia of Genes and Genomes pathway database (KEGG) annotations revealed that some miRNAs were associated with growth and developmental processes of the alga. These results provide insights into the roles that miRNAs play in the growth of A. catenella, and they provide the basis for further studies of the molecular mechanisms that underlie bloom growth in red tides species.
Introduction
As a class of biflagellated unicellular microorganisms, the free-swimming dinoflagellates play an important role in the primary production of the marine ecosystem[1]. Inthe past two decades, it has been suggested that marine dinoflagellates, which were previously deemed to be exclusively autotrophic,are actually mixotrophic (i.e., they carry out photosynthesis and ingest prey) [2–4]. Importantly, many dinoflagellate species can produce toxins[5], and they account for 75% of all harmful algal bloom species(HABs).Blooms of toxic dinoflagellates have detrimental effects on the fishing industry, marine environments, and public health[6–8].
Within the dinoflagellate genus Alexandrium, many species can produce neurotoxins called paralytic shellfish toxins (PSTs), which can bring about paralytic shellfish poisoning. In the last few decades, the frequency and intensity of HABs caused by this genus have increased, as has their geographic extent [9]. Alexandrium catenella is widely distributed all over the world and has the ability to produce PSTs [10]. Previous studies of A. catenella focused on transcriptome sequencing and analysis[11], proteomic analysis[12], characterization of genes related to proliferating cells[13], and gene expression in the decline phase of HABs[14] and the effect of different environmental conditions[15], which aimed to elucidate the molecular mechanisms responsible for blooming. However, molecular mechanisms related to the regulation of cell growth during the process of HAB development remain unclear[12, 13]. Moreover, no investigation of micro-ribonucleic acids (miRNAs) and their expression profiles in A. catenella have been reported to date.
miRNAs are a large class of evolutionarily conserved, endogenous, non-coding, single-stranded, tiny (18–25 nucleotide (nt)) RNAs that are ubiquitous in eukaryotes[16]. They play many crucial roles in various biological and cellular processes, such as cell proliferation, differentiation[17], signal transduction[18], and responses to stresses[19] in plants. Mature miRNAs are loaded into the RNA-induced silencing complex, where they can generally guide direct transcript cleavage to regulate gene expression[20]. As a class of master regulators of gene expression, considerable efforts have been made to discover and profile miRNAs from various plant species. In recent decades, Sanger sequencing and computational prediction methods were initially used to identify miRNAs[21, 22].However, some species-specific or specific-expression miRNAs with low abundance are not conservative[23, 24], and traditional methods may not be able to detect these miRNAs.
Next-generation sequencing is not only a rapid and powerful approach to investigating genomes, but it also is a tool that can provide unprecedented coverage depth, which is advantageous for discovering low abundance or novel miRNAs[25]. For many higher organisms, such as Arachishypogaea [26], Oryza sativa[27],and Medicagotruncatula [28], high-throughput sequencing was successfully used after first being used in the model organism Arabidopsis[29]. Recently, miRNAs were found to be expressed in macroalgae (Ectocarpussiliculosus [30], Ulva prolifera [31], and Pyropiayezoensis [32]) and a limited number of unicellular algae (Chlamydomonasreinhardtii[33], Phaeodactylumtricornutum[34], and Symbiodiniummicroadriaticum[35]).At present, a comprehensive profile of the endogenous miRNAs and their regulatory roles in Alexandrium obtained by high-throughput sequencing does not exist,although18 potential miRNAs were screened from expressed sequence tags in Alexandriumtamarense using a computational approach[36].
In this study, two small RNA libraries from different growth stages of A. catenella were constructed and sequenced using an Illumina high-throughput platform. The sequence data were analyzed using an automated, proprietary, bioinformatics analysis pipeline. The aim of this study was to profile differential expression of miRNAs between the two growth phases and to determine their probable roles in the growth of A. catenella. qPCR was conducted to validate the differential expression of identified miRNAs. To our knowledge, this is the first comprehensive report of identification and profiling of miRNAs from Alexandrium by high-throughput Illumina sequencing. Results of this study will provide new insights into the roles of miRNAs in the growth of Alexandrium species and lay the foundation for further study of the molecular regulatory mechanisms involved in HAB development.
Materials and Methods
Sample culture and collection
A. catenella was obtained from the Key Laboratory of Marine Genetics and Breeding, Ministry of Education of China, College of Marine Life Sciences. The stock algal cells were maintained at 20±1°C in f/2 medium(without silicate) with a 12 h/12 h light/dark photocycle[37]. Cool white fluorescent lights with a photon flux density of 30–35 μmol m–2 s–l provided the light. To construct separate small RNA libraries, the cultures were grown into logarithmic phase in f/2 medium (without silicate)and placed in the dark for 48 h[38].Subsequently, the synchronized cells were inoculated into new f/2 medium with an initial concentration of 2×106 cellsL–l. Algal cells at two different growth stages were sampled at 12:00. One sample was the lag phase sample (cells cultured for 2 dinf/2 medium), and the other was the logarithmic phase sample(a mixture of cells cultured for 7 d and 12 d, respectively).Both samples contained 5×106 cells. Because many reports have noted that high concentrations of nitrogen (N), phosphorus (P), and manganese (Mn) promote the growth of A. catenella[39, 40],three nutrient-enhanced cultures(i.e., induced logarithmic phases)with high N, high P, and high Mn were generated and used for the quantitative PCR (qPCR) experiment. The f/2 medium in these cultures was supplied with NaNO3,NaH2PO4⋅2H2OorMnCl2⋅4H2Oat final concentrations of 2.304mmolL–l, 0.144mmolL–l, and 2.730μmol/L–l[11], respectively. Cells (5×106) were collected from each of the induced logarithmic phases of the three nutrient-enhanced cultures. To minimize bacterial contamination, methods described in previous studies were used [41, 42]. Briefly, a 10μm pore size bolting-silk was used to filter every 100mLof the cultures, which was rinsed with 300mL of sterile f/2 medium.
The A. catenella cultures were subjected to the following treatments: Washed cells were suspended in 50 mL of sterile f/2 medium containing 0.05% Tween-80 and 0.01 M EDTA (at 20°C for 30 min), followed by ultrasonication for 1 min (50 w; ultrasonic treatment time of 5 s with an interval of 5 s). Sequentially, lysozyme (0.5 mgmL–l, 20°C for 10 min) and sodium dodecyl sulfonate(SDS) (0.25%, 20°C for 10 min) were added. The algal cells then were washed three times with sterile f/2 medium to remove these reagents. Finally, the cultures were detected using epifluorescence microscopy (Nikon ECLIPSE 80i, Japan) after acridine orange staining (0.01%) for 1 or 2 min[42].Only axenic cultures were used for the following experiments. The 5×106cells of different growth stages were collected by centrifugation for 6 min at 4000g.Thecell pellets in 1.5mL Eppendorf tubes were frozen immediately in liquid nitrogen and stored at –80°C prior to RNA extraction. All samples were collected in triplicate.
Small RNA library construction for Illumina sequencing
Trizolreagent (Invitrogen, Carlsbad, CA,USA) was used to extract total RNA from the lag phase and logarithmic phase samples according to the manufacturer’s instructions, and subsequently DNA was digested with DNase I (TaKaRa, Japan).The quantity and purity of total RNA was analyzed using the NanoDrop 2000 device (Thermo, USA). Only high-quality RNA was selected for subsequent experiments. Approximately 1 μg of total RNA was used to prepare the small RNA(sRNA) library according to the protocol of the TruSeq TM Small RNA Sample Preparation Kit (Illumina).Briefly, fragments of 15–40ntsRNAs were separated by 15% denaturing polyacrylamide gel electrophoresis (PAGE) using aTrackItTM 10 base pair (bp) DNA Ladder (Invitrogen).Synthesis of cDNA using SuperScriptIIReverse Transcriptase (Life Technologies) was carried out after ligating 3’ and 5’RNA adapters with T4 RNA ligase. Subsequently, the sRNAs were subjected to PCR amplification. Final amplification products of approximately 147bp were further purified by 6% PAGE. Finally, an Illumina Hiseq2000 instrument was used to sequence the constructed libraries directly. Sequencing reads could be obtained from the image files generated by the Illumina instrument analyzer.
Bioinformatic processing of Illumina sequencing data
The raw reads were cleaned by masking the low-quality data, adaptor sequence, polyAsequence, and sequences outside of 18–25nt fraction range. The clean reads were processed for miRNA identification with the proprietary in-house software package ACGT101-miR v4.2 (LC Sciences, Houston, TX, USA).Briefly, the filtered reads of 18–25nt were aligned with the Rfam11.0 (ftp.sanger.ac.uk/pub/databases/Rfam)andRepbase 18.02(http://www.girinst.org/) database entries to discard a variety of non-coding sRNA (such as rRNA, tRNA, snRNA, and snoRNA)and repeat sequences (repetitive sequence elements). Subsequently, BLAST searches were used to detect known miRNAs against miRBase 21.0 (http://www.mirbase.org/) with only one mismatch permitted. The priority of the mapped species is listed in S1 Table. To identify potential novel miRNAs, the remaining sequences were further aligned with the transcriptome data to determine their locations. For every sequence mapped to the transcriptome, the sequence of the candidate miRNAs precursor were predicted by RNA fold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) from the adjacent upstream and downstream 60ntsequences in both directions. The following were the default parameters of the RNA fold software: (1) the number of nucleotides in one bulge in the stem was ≤12;(2) the number of bp in the stem region of the predicted hairpin was ≥16;(3) the free energy was ≤ –15 kCalmol–1; (4) the length of the hairpin (up and down stems + terminal loop) was ≥ 50; (5) the length of the hairpin loop was ≤200; (6) the number of nucleotides in one bulge in the mature region was ≤ 4; (7) the number of allowed biased errors in one bulge in the mature region was ≤ 4; (8) the number of allowed biased bulges in the mature region was ≤ 2; (9)the number of allowed errors was ≤ 2; (10)the number of bp in the mature region of the predicted hairpin was ≥12; and (11)the percentage of miRNA in the stem was≥ 80%.A sequence was considered to be a candidate miRNA precursor if it satisfied the criteria according to Meyers[43]. Finally, Mfold software(http://mfold.rna.albany.edu/) was used to fold the secondary structures of miRNA[44].To profile the differential expression of identified miRNAs, the raw copies of miRNAs in the two small RNA libraries were normalized[45]. The Fisher exact test and chi-square test with a Bonferroni correction were used for multiple comparisons between the two libraries. The significance threshold was set at 0.05. The fold-change between the two different growth phases was calculated as:
If an miRNA satisfied |log2fold-change|>1 and P< 0.05, it was considered to be a differentially expressed miRNA. It was considered to be significant expression when P<0.01 and |log2fold-change|>1[46].
Target prediction for differentially expressed miRNAs and function annotation
To further understand the function of the differentially expressed miRNAs, computational target prediction algorithms (TargetScan5.0 and miRanda 3.3a) were used to predict the miRNA binding sites (potential target genes) [47]. All target gene candidates were mapped to gene ontology (GO) terms in Gene Ontology (http://www.geneontology.org/), and the gene numbers in each term were calculated. To identify significantly enriched GO terms, a hypergeometric test was utilized to compare the target gene candidates with the reference gene background following the formula for P-value[48]:
N represents the number of all genes with GO annotation, n is the number of differentially expressed target gene candidates in N, M is the number of all genes annotated to a certain GO term, and m is the number of potential target genes in M. Fisher’s exact test also was used to determine the P-value. GO terms with a P-valueless than the threshold value0.05 were considered to be significantly enriched. The Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg) was used to analyze metabolic pathway assignments. The formula and threshold value for estimating significantly enriched metabolic pathways and signal transduction pathways were the same as those used in the GO analysis. However, the meanings of the letters in the formula for KEGG analysis were as follows: N represents the number of all genes with KEGG annotation; n is the number of differentially expressed target gene candidates in N, M is the number of all genes annotated to a certain pathway, and m is the number of target gene candidates in M.
Quantitative real-time PCR of miRNA
To validate the existence of A. catenella miRNAs identified by the high-throughput sequencing and their expression trends, two miRNAs were selected. Poly(A)-tailed qPCR [49] was conducted using an Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems Ltd, USA).In addition to the lag phase and logarithmic phase samples inf/2 medium, the three induced logarithmic phases (high N, high P, and high Mn cultures)were also analyzed byqPCR. Algal cells of different growth stages and conditions were collected. Each sample contained 5×106cells. The small RNAs (<200 nt) were extracted from the samples following the manufacturer's instructions for the RNAiso for Small RNA kit(TaKaRa).The integrity and quality of sRNAs was evaluated by 3% agarose gel electrophoresis and the NanoDrop 2000 (Thermo, USA).Equal amounts of high-quality sRNAs (OD 260/280 ranging from 1.9 to 2.1) were reverse transcribed to cDNA. Each reverse transcriptase assay consisted of 10 μL of 2× miRNA Reaction Buffer Mix, 2μL of 0.1%BSA, 2μLof miRNA PrimeScript® RT Enzyme Mix, approximately 1μg of total RNA, and 5μL of RNase-free ddH2O.The resulting cDNA products were stored at −20°C. The cDNA was subsequently used for qPCR with miRNA-specific forward primers, which were designed based on mature miRNA sequences, and universal reverse primer, which was provided in the kit. Appropriate adjustments were made by adding several guanine (G)or cytosine(C)residues to the 5’ terminus of the miRNA sequence to obtain mature miRNATm values of about 60°C.All primers used are listed in S2 Table. The qPCR reaction mixture consisted of SYBR® Premix Ex Taq™II(10 μl), 10mM specific forward primer(0.4 μl), 10mMuniversal reverse primer(0.4 μl), RNase-free H2O(7.2 μl), and cDNA (approximately 2 μl) according to the manufacturer's instructions of the SYBR® PrimeScriptx™miRNA RT-PCR Kit (TaKaRa, Japan). The 5.8s RNA gene of A. catenella was chosen to normalize the expression levels of miRNAs. Reactions for three technical replicates and three biological replicates were prepared. The program for the miRNA qPCR was 95°C for 30s and 40 cycles of 95°C for 5 s, 60°C for 34 s. The comparative threshold (2–ΔΔCt) method was used to assess the relative expression levels. Data for mean Ct value and means ±SE (standard error) were calculated for each miRNA. A paired t-test was performed to identify statistically significant differences. P<0.05 was deemed to be statistically significant.
Results
Construction of small RNA libraries and high-throughput sequencing data analysis
To identify miRNAs involved in the growth of the A. catenella, Illumina deep sequencing technology was used on two small RNA libraries that were constructed from two different developmental life stages, namely the lag phase(cultured for 2d) and the logarithmic phase (mix of cultures grown for 7 d and 12 d, respectively). A total of 38,092,056(representing 9,311,258 unique reads) and 32,969,156(representing 7,713,621 unique reads) raw reads (SRP055721) were obtained from the lag phase and the logarithmic phase libraries, respectively. Subsequently, the sequence data were further refined to16,527,710 (43.39% of the raw reads) and 13,324,372(40.41% of the raw reads)clean reads of 18–25ntfor the lag phase and logarithmic phase libraries (Table 1). Among the unique reads,3,230,369(34.69% of the unique reads in the lag phase library) and 2,411,293 (31.26% of the unique reads in the logarithmic phase)were found to be similar to miRNAs. The unique read ratios of the miRNA candidates in the respective libraries were 34.69% and 31.26%.Several other types of non-coding RNAs, including tRNA, rRNA, snRNAs, and snoRNAs, were identified by alignment with the Rfam database. The number and proportion of different categories of small RNAs are given in Table 2.When the length distribution of 18–25nt silencing small RNAs in A. catenella was investigated (Fig 1),the most abundant reads fell into the 24nt fraction, which accounted for 15.96% and16.65%of two libraries, respectively, followed by the 22nt (15.52%, 16.21%), 25 nt(15.58%, 16.15%), and 23nt(15.72% and 16.12%)fractions.
Table 1. Data statistics of small RNAs produced by Illumina deep sequencing.
| Category | The lag phase | The logarithmic phase | ||
|---|---|---|---|---|
| Total small RNAs | Unique small RNAs | Total small RNAs | Unique small RNAs | |
| Raw reads | 38,092,056 | 9,311,258 | 32,969,156 | 7,713,621 |
| 3ADT&length filter | 19,081,207 | 5,670,799 | 17,749,062 | 4,952,497 |
| Junk reads | 205,372 | 61,494 | 213,241 | 52,070 |
| Rfam | 2,203,115 | 284,387 | 1,623,385 | 246,854 |
| Repeats | 106,331 | 93,061 | 84,599 | 74,144 |
| Clean reads | 16,527,710 | 3,230,369 | 13,324,372 | 2,411,293 |
Table 2. Distribution of small RNAs among different RNA categories in A. catenella by mapping to Rfam database.
| Category | The lag phase | The logarithmic phase | ||
|---|---|---|---|---|
| Total small RNAs | Unique small RNAs | Total small RNAs | Unique small RNAs | |
| rRNA | 1,617,108(4.25℅) | 135,768(0.36℅) | 1,197,675(3.63℅) | 123,465(0.37℅) |
| tRNA | 70,911(0.19℅) | 23,706(0.06℅) | 58,754(0.18℅) | 24,125(0.07℅) |
| snoRNA | 42,572(0.11℅) | 30,220(0.08℅) | 31,274(0.09℅) | 23,939(0.07℅) |
| snRNA | 59,056(0.16℅) | 23,287(0.06℅) | 46,852(0.14℅) | 18,316(0.06℅) |
| other Rfam RNA 1 | 413,468(1.09℅) | 71,406(0.19℅) | 288,830(0.88℅) | 57,009(0.17℅) |
1snoRNA:small nucleolar RNA; snRNA: small nuclear RNA; other Rfam RNA: riboswitch, ribozyme, splicing and so on.
Fig 1. Length distribution and abundance of sequenced small RNA sequences in A. catenella libraries.
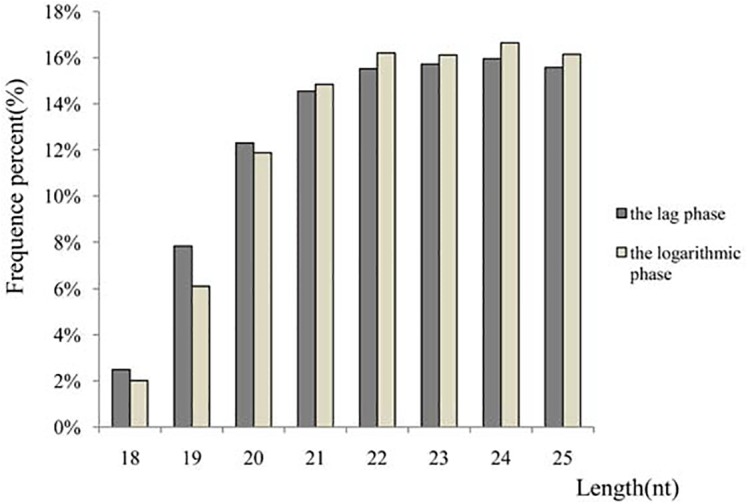
Among the small RNAs in the range of 18–25 nt, the most abundant size was 24 nt, followed by 22 nt and 25 nt.
Identification and analysis of known miRNAs in A. catenella
To identify the expression of known miRNAs in A. catenella,3,230,369 unique sRNA sequences in the lag phase and 2,411,293 unique sRNA sequences in the logarithmic phase were aligned against known miRNAs and miRNA precursors of selected species(S1 Table) in miRBase (ftp://mirbase.org/pub/mirbase/CURRENT/21.0), with a maximum of one mismatch allowed. Based on the sequence similarity,88 mature miRNAs belonging to 32 miRNA families were identified in the two libraries. Among them, 62 known co-expressed miRNAs were present, and 83 miRNA precursors were also detected. he Venn diagram in Fig 2 displays the distribution of the 88known mature miRNAs between the two libraries.
Fig 2. Venn diagram of identified known miRNAs in the two libraries.
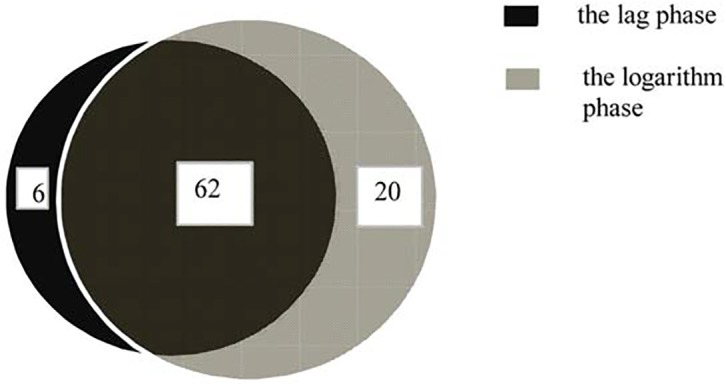
The diagram not only shows the numbers of miRNA that were expressed in the lag phase and the logarithmic phase preferentially but also the co-expressed miRNAs in both phases.
The sequences obtained by high-throughput sequencing made it possible to compare not only the relative abundance of various miRNA families but also that of different members within an miRNA family. Fig 3 illustrates the expression level of miRNA families with more than two copies. Among them, miR166 exhibited the highest abundance in both libraries, with 1076 and 3738 copies respectively. The expression of miR396 ranked second in the lag phase with 202copies, and the expression level of miR156 ranked second with 713 copies in the logarithmic phase. Some families (miR171, miR393, and miR390) had an expression level lower than 10 copies in both phases, whereas each of the following five miRNAs were expressed abundantly(>100 copies) in both phases: miR166, miR396, miR156, miR168, and miR159. These dominantly expressed miRNAs may play important roles in cell growth duringthe two different stages. Overall, these results indicate that the expression level of different families varied greatly.
Fig 3. Expression abundance of miRNA families in two growth phases in A. catenella.
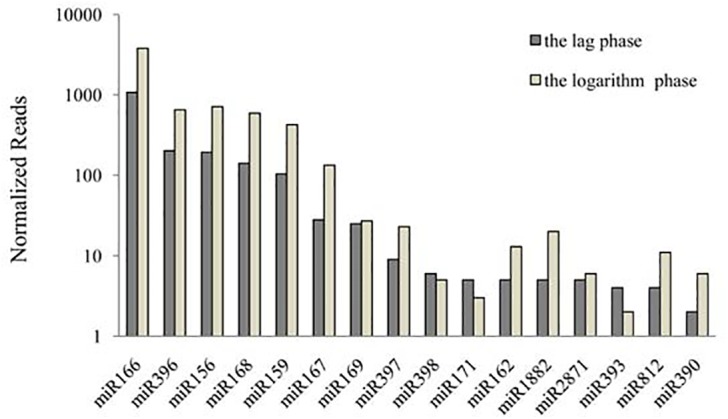
X-axis represents the known miRNA families, Y-axis represents the normalized expression level of each miRNA family.
Within a given miRNA family, the relative abundance of different members varied widely. For example, miR166a exhibited extremely high expression (1851 copies), whereas miR166m had only 3copies in the log phase library. Similarly, in the miR396 family, miR396a expressed 650 copiesbutmiR396e expressed only 3copies. This result implies that largely divergent functions may exist within one miRNA family, which was previously reported in chicken [50].
The number of members present in different miRNA families may be indicative of the diversity of A. catenella miRNA families. Fig 4 shows all of the miRNA families that contained more than two members. Among them, miR169 had the most members (11), miR182 and miR166 ranked second, whereas many families only had one member. Similar results were found in Arabidopsis thaliana and Caragana intermedia; these studies suggested that the size of miRNA families might be indicative of their function[51, 52].
Fig 4. miRNA families with more than two members.
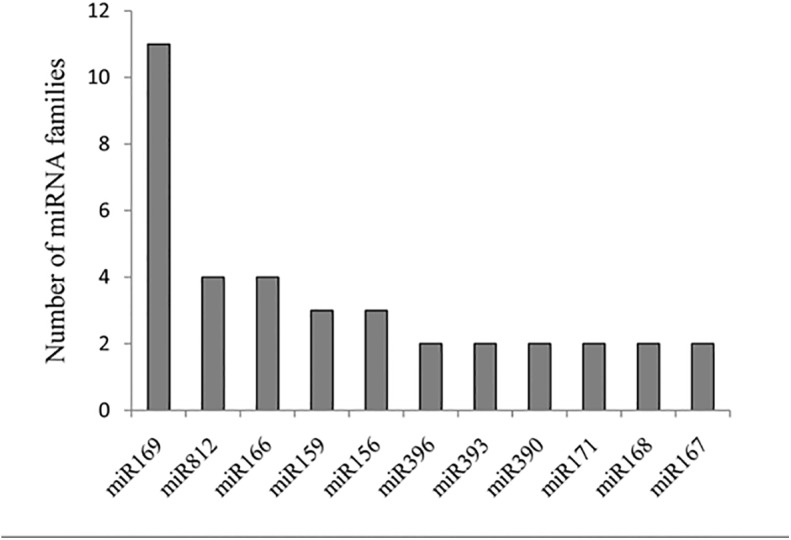
Studies have shown that miRNAs are highly conserved among various organisms. The identified miRNAs in the A. catenella libraries were further compared with those of other selected species, and the frequency of occurrence in those species was determined (Fig 5). As expected, miRNAs in A. catenella were identified in many higher plants, including Glycine max, O. sativa, Populustrichocarpa, and Malusdomestica. Only miR1158 and miR1160 were shared by C. reinhardtii, which might be attributable to the limited miRNA information for algal organisms in miRBase. These results indicate that miRNAs are highly conserved across species.
Fig 5. The occurrence of A. catenella miRNAs that appeared also in other selected species.
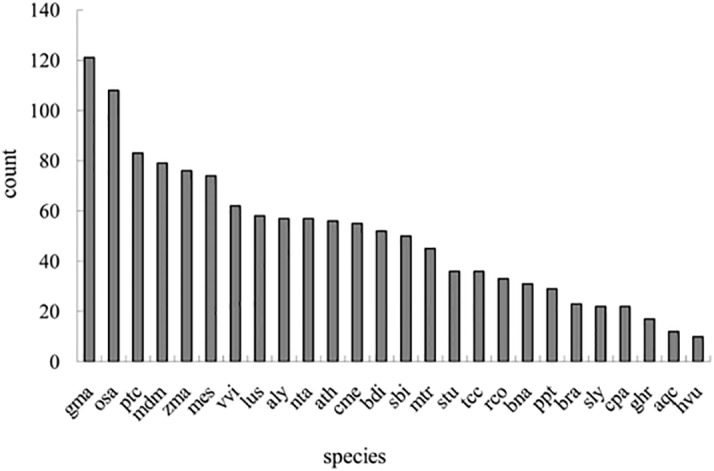
Gma: Glycine max, osa: Oryza sativa, ptc: Populus trichocarpa, mdm: Malus domestica, zma: Zea mays, mes: Manihot esculenta, vvi: Vitis vinifera, lus: Linum usitatissimum, aly: Arabidopsis lyrata, nta: Nicotiana tabacum, ath: Arabidopsis thaliana, cme: Cucumis melo, bdi: Brachypodium distachyon, sbi: Sorghum bicolor, mtr: Medicago truncatula, stu: Solanum tuberosum, tcc: Theobroma cacao, rco: Ricinus communis, bna: Brassica napus, ppt: Physcomitrella patens, bra: Brachypodium distachyon, sly: Solanum lycopersicum, cpa: Carica papaya, ghr: Gossypium hirsutum, aqc: Aquilegia caerulea, hvu: Hordeum vulgare, Gma: Glycine max, osa: Oryza sativa, ptc: Populus trichocarpa, mdm: Malus domestica, zma: Zea mays, mes: Manihot esculenta, vvi: Vitis vinifera, lus: Linum usitatissimum, aly: Arabidopsis lyrata, nta: Nicotiana tabacum, ath: Arabidopsis thaliana, cme: Cucumis melo, bdi: Brachypodium distachyon, sbi: Sorghum bicolor, mtr: Medicago truncatula, stu: Solanum tuberosum, tcc: Theobroma cacao, rco: Ricinus communis, bna: Brassica napus, ppt: Physcomitrella patens, bra: Brachypodium distachyon, sly: Solanum lycopersicum, cpa: Carica papaya, ghr: Gossypium hirsutum, aqc: Aquilegia caerulea, hvu: Hordeum vulgare.
It has been reported that the first residue at the 5’terminus of the vast majority of 20–23 nt small RNAs is predominantly uridine (U) in the plant kingdom[53]. Studies of Arabidopsis have indicated that the incorporation of sRNAs into Argonaute proteins(located in silencing complexes) mainly relies on a 5' terminal U[35]. Accordingly, the 5’nucleotides of the 88 identified known miRNAs were analyzed (Fig 6). The first residue of the 20 nt small RNA was either U and adenine (A). The 22 nt small RNAs had a bias for U and C. Small RNAs of 21 nt started with U, C, or A. The base at the first position of all 23 nt small RNAs was U exclusively. Thus, U was dominant in the first position of 20–23 nt small RNAs, which is consistent with previous studies of other organisms, such as S.microadriaticum, Physcomitrella patensprotonema, Arabidopsis, and Triticumaestivum L [35, 54–56].These results provide support for the existence of miRNA functionality in A. catenella[55].
Fig 6. First nucleotide bias of known miRNAs in A. catenella.
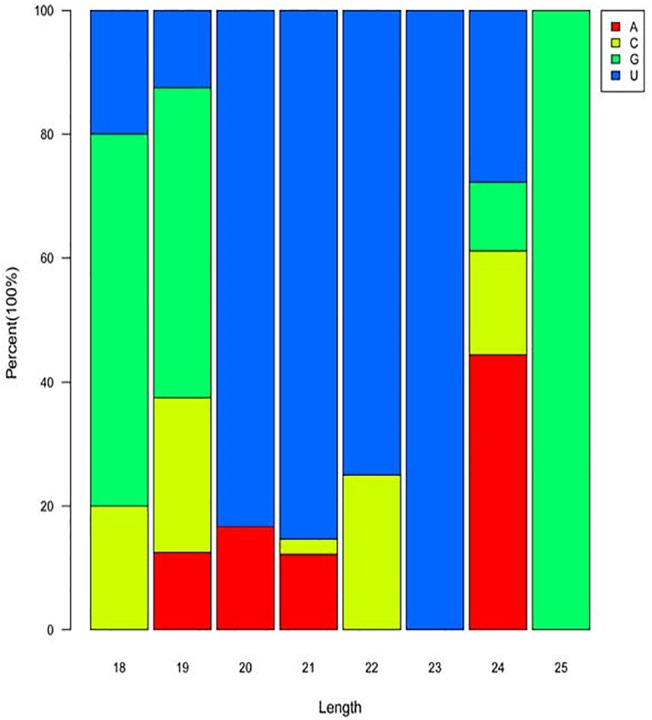
The figure indicates the first nucleotide of 18–25 nt miRNAs. U had the greatest frequency among miRNAs of 20–23 nt.
Prediction of novel A. catenella-specific miRNAs
Following the identification of conserved miRNAs, sequences without similarity to known miRNAs in miRBase were further examined to uncover additional specific miRNAs in A. catenella. After mapping to the transcriptome information, 15 potentially new miRNAs in the two libraries fulfilled the criteria established for identification of miRNA of higher eukaryotes in compliance with Meyers et al.[43]. These novel candidates mainly ranged from 20 to 24nt(Table 3). A primary criterion for distinguishing miRNA from siRNA and piRNA is that the flanking sequences of miRNA are capable of folding back into a hairpin structure. The secondary hairpin structures of the precursors were predicted by RNA fold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). The precursors of all 15 candidate miRNAs could fold into the typical hairpin structure(S1 Fig–S15 Fig). The average length of precursor sequences(approximately 123nt) was shorter than the average length of known miRNAs (163nt). The minimum free folding energies of these predicted novel miRNA precursors ranged from −46kcalmol–1 to −132 kcalmol–1(average,−78.32kcalmol–1)according to the RNAfold analysis. These results are in line with the values of other miRNA precursors reported previously(−68.2kcalmol–1 in S.microadriaticum, −72.4kcalmol–1 in T.aestivum, and −70kcalmol–1 in O.sativa[35, 56, 57]),but they are lower than the free folding energies of other non-coding RNAs such as tRNAs or rRNAs[58].
Table 3. Novel miRNAs predicted in A. catenella.
| Name | Sequence | length | Precursor length | GC% | dG |
|---|---|---|---|---|---|
| aca-miR43924-5p | GCGGAGGAAAAGAAACTAAC | 20 | 96 | 52.50 | -93.20 |
| aca- miR4542336-3p | GTTCCGCTTTCTCGTCGC | 18 | 96 | 52.50 | -90.30 |
| aca- miR35336-5p | CGGACTTGCCATTCCTAGCCT | 21 | 187 | 42.10 | -98.10 |
| aca- miR311414-3p | AAAGGCTAGGAATTGCAATTC | 21 | 187 | 42.10 | -98.10 |
| aca- miR129519-3p-1 | CCATCAGCTGTTGCCATGTCGT | 22 | 123 | 53.50 | -73.90 |
| aca- miR129519-3p-2 | CCATCAGCTGTTGCCATGTCGT | 22 | 175 | 52.80 | -132.30 |
| aca- miR312912-5p | AAAAGGCTGGGAATTGCAATTC | 22 | 122 | 55.70 | -73.20 |
| aca- miR5257085-3p | GAAGGGCTGGAGATTGCAACT | 21 | 122 | 55.70 | -73.20 |
| aca- miR456915-3p | CAAAATGGGCGGCAAGAAAGGCT | 23 | 103 | 55.70 | -67.80 |
| aca- miR342562-5p | TTTTTTAAAAGGCTGGGAAT | 20 | 56 | 38.70 | -33.30 |
| aca- miR126213-3p | CAACGAAAGGTTATCTTCCTGGAT | 24 | 119 | 37.70 | -59.30 |
| aca- miR879241-3p | CGGAGGAAAAGAAACTAACA | 20 | 96 | 42.40 | -46 |
| aca- miR254217-3p | CTCAGTCCTTGGGCTGGTTGCTCT | 24 | 129 | 57.50 | -101.20 |
| aca- miR5619838-5p | GCCGCTGATAGAGGGCCTGCAACCT | 25 | 146 | 65.10 | -98.30 |
| aca- miR190576-3p | CGAGCCATTCAAAGTCTGGACTTTG | 25 | 95 | 46.90 | -51.60 |
Differentially expressed miRNAs in two different life stages of A. catenella
To identify miRNAs related to the growth of A. catenella, the normalized expression of miRNAs in the two libraries were compared. Twelve known miRNAs were expressed differently in the two phases (Table 4). Among them osa-miR2876-3p_R+1 was preferentially expressed in the lag phase and zma-miR529-5p was only expressed in the logarithmic phase. The other 10 miRNAs were expressed in both libraries. Compared with the lag phase cultures, four miRNAs were significantly down-regulated and eight miRNAs were up-regulated in the logarithmic phase algae. Of the12 sequences, the majority ranged from a 1- to 2-fold difference, and only four miRNAs showed greater than 2-fold differences between the two growth stages.
Table 4. Twelve differentially expressed miRNAs in two different growth phases in A. catenella.
| miRNA name | log2 (fold change) | Pvalue (chi square_2x2) | up/down |
|---|---|---|---|
| osa-miR168a-5p | -2.06 | 0.0044 | down |
| tae-miR159a | -2.05 | 0.0202 | down |
| aca- miR43924-5p | -1.11 | 0.0000 | down |
| zma-miR529-5p | -∞ | 0.0452 | down |
| osa-miR2876-3p_R+1 | +∞ | 0.0027 | up |
| rgl-miR5139 | 1.12 | 0.0060 | up |
| aca- miR456915-3p | 1.21 | 0.0003 | up |
| sbi-miR169c | 1.23 | 0.0294 | up |
| stu-miR169a-5p_R+1 | 1.23 | 0.0294 | up |
| zma-miR169f-5p | 1.23 | 0.0294 | up |
| stu-miR171b-3p | 1.38 | 0.0248 | up |
| bdi-miR7732-3p_L-1_1ss11GC | 1.79 | 0.0055 | up |
log2 (fold change):log2 (the normalized expression in the lag phase /the normalized expression in the logarithmic phase); R+1 means the miRNA_seq (detected) is one base more than known rep_miRSeq on the right side; L-1 means the miRNA_seq (detected) is one base less than known rep_miRSeq on the left side; 1ss11GC means one substitution (ss), where G was substituted by C at position 11.
Some of the miRNA families were expressed solely in the lag phase or the logarithmic phase. For example, miR1862 (with one copy), miR1507, and miR947 were expressed exclusively in the lag stage, whereas miR535, miR820, and miR529 were only expressed in the logarithmic stage. These significant result ssuggested that these families may have potentially important effects on the initial and exponential growth stages, respectively.
qPCR was performed to confirm the expression of the miRNAs identified by the high-throughput sequence approach. The two selected miRNAs included one up-regulated novel miRNA(aca-miR-3p-456915) and one down-regulated conserved miRNA (tae-miR159a).Fig 7 illustrates the relative expression levels oftae-miR159a. Its expression in the logarithmic phase and in the logarithmic phases of the three nutrient-enhanced cultures were all significantly higher than those in the lag phase (P<0.01). The |log2fold change|oftae-miR159awas 2.05 as determined by high-throughput sequencing. The expression trend of the qPCR assay was generally consistent with the deep sequencing results. For aca-miR-3p-456915,expressionsin the high P and high Mn induced logarithmic phases were both significantly lower than those in the lag phase (P<0.05), whereas its expression in the logarithmic phase was slightly higher than that in the lag phase (Fig 8).
Fig 7. qPCR validation of the differentially expressedtae-miR159a.

Fig 8. qPCR validation of the differentially expressedaca-miR-3p-456915.
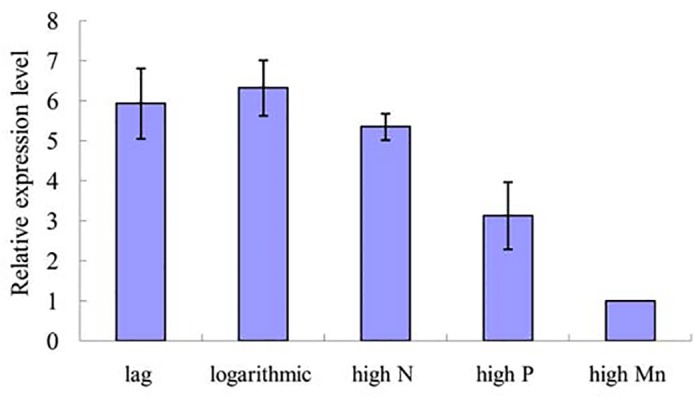
Normalized expression level (2–ΔΔCT) of tae-miR159a and aca-miR-3p-456915 under different growth phases and conditions.
Prediction of potential targets for differentially expressed miRNAs and function annotation
To understand the specific functions of the 12 differentially expressed miRNAs during the growth of A. catenella, potential target genes were predicted by integrating two software programs (TargetScan5.0 and miRanda3.3a), and1813 potential target genes were identified (S3 Table).GO enrichment and KEGG pathway annotation were then applied to these targets. On the basis of three GO ontologies, 656target genes were primarily classified into 412 enriched GO terms. Among these, 38 GO terms were significantly enriched. The following GO terms were the most significantly enriched:protein binding, ATP binding, catalytic activity, membrane in cellular component, and the oxidation-reduction process (Fig 9).
Fig 9. Partial gene ontology (GO) classification annotated for predicted target genes of 12 differentially expressed miRNAs.

The figure shows partial GO enrichment of the 1813 predicted target genes in three GO ontologies: biological processes, cellular component and molecular function.
KEGG pathway analysis of all predicted potential targets was then performed to reveal the enriched metabolic pathways that were regulated by differentially expressed miRNAs in A. catenella. In total,175 KEGG pathways were identified, with 13 pathways significantly enriched (Table 5). Biosynthesis of unsaturated fatty acids was enriched extremely significantly (P<0.01). Other very significantly enriched pathways were the TGF-beta signaling pathway, phenylalanine, tyrosine, and tryptophan biosynthesis, the Ca2+ signaling pathway, and the Wnt signaling pathway. These results suggest that the genes targeted by differentially expressed miRNAs might be involved in the growth processes of A. catenella.
Table 5. The significantly enriched KEGG pathways of target genes for 12 differentially expressed miRNAs.
| Pathway ID | Pathway | target gene number | P-value of Fisher's Exact Test |
|---|---|---|---|
| ko01040 | Biosynthesis of unsaturated fatty acids | 5 | 0.0037 |
| ko04350 | TGF-beta signaling pathway | 4 | 0.0155 |
| ko00400 | Phenylalanine, tyrosine and tryptophan biosynthesis | 4 | 0.0169 |
| ko04020 | Calcium signaling pathway | 6 | 0.0225 |
| ko04310 | Wnt signaling pathway | 6 | 0.0247 |
| ko00592 | alpha-Linolenic acid metabolism | 2 | 0.0255 |
| ko02040 | Flagellar assembly | 1 | 0.0255 |
| ko04111 | Cell cycle—yeast | 8 | 0.0356 |
| ko00260 | Glycine, serine and threonine metabolism | 7 | 0.0364 |
| ko00380 | Tryptophan metabolism | 7 | 0.0378 |
| ko05210 | Colorectal cancer | 3 | 0.0437 |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 6 | 0.0460 |
| ko04910 | Insulin signaling pathway | 10 | 0.0486 |
Discussion
Sequence of small RNA libraries of A. catenella
The high-throughput Illumina deep sequencing platform offers a powerful approach for profiling small RNAs, and it is efficient for discovering miRNAs in various organisms. In the present study, two smallRNA cDNA libraries were constructed, and detailed miRNA profiles were obtained for the lag phase library and the logarithmic phase library using this method. Altogether, 38,092,056 and 32,969,156 unfiltered reads, respectively, representing 9,311,258 and 7,713,621unique reads were acquired. These results confirmed the existence of miRNAs in this unicellular dinoflagellate. These miRNAs also exhibited an unexpected complexity in the small RNA regulation mechanism of A. catenella.
To the best of our knowledge, this is the first report of comprehensive identification and expression profiling of miRNAs of A. catenella in different developmental life stages. The 24nt small RNA sequences were dominant, which is in accordance with data for typical higher plants such as A. thaliana[23],A. hypogaea[26],and O. sativa[27]. The 22nt sequences were next in dominance, followed by the25 and 23nt sequences. In P.tricornutum, small RNAs of 22nt were the most common[34], and the same was found for aquatic animals such as Paralichthysolivaceus[59]and Cynoglossussemilaevis[44]. In S.microadriaticum(a dinoflagellate symbiont of reef-building corals) [35]and U.prolifera[31], the most frequent read was 25nt;however, the 21nt sequences ranked first in the unicellular green alga C.reinhardtii[33]and the marine red alga P.yezoensis[32]. It has been reported that small RNAs of 24ntwereabsent in C.reinhardtii[33] and the plant P.patens[54],which is different from the scenario in many higher plants [23, 26, 27]. These reports suggested that small RNAs have multiple distribution patterns in various algal species.
The kinds of enzymes that participated in sample processing were responsible for the length of small RNAs. For instance, 24nt molecules can be generated by Dicer-like protein (DCL) DCL3 and 21nt small RNAs can be processed by DCL1, whereas in animals the 22nt fraction require the RNase III endonuclease Drosha [60–62]. In plants, the most abundant size are always 24 nt, while in animal kingdoms, that are 22 nt. In A. catenella, 24 nt size is the most abundant, followed by 22 nt. Therefore, these typical length distributions of sRNAs that found in the plant and animal kingdoms were unsuitable for A. catenella. The characteristic length distribution of small RNAs in A. catenella suggests that lower level unicellular algae might have different kinds of RNA processing enzymes when compared to the plants and animals. It is likely that both the typical DCL as found in plants and the classic Dicer as in animals are used to process miRNA in A. catenella, which is consistent with the dual animal and plant behavior of this alga [63].Previous studies have shown that the Dicer-like enzymes in U.prolifera have a sequence similar to that found in higher organisms [31]. Thus, studies are needed to investigate whether the enzymes in A. catenella are Dicer, DCL, or other enzymes that have sequence homology with them. Additionally more work is necessary to elucidate their exact roles.
The roles of identified miRNAs in different growth stages
Eighty-eight miRNAs belonging to 32 miRNA families were identified and their expression profiles were further analyzed. Our results showed that the member number, the expression level of various families, and the expression level of each member within one family were significantly different. Two highly conserved miRNA families,miR167 and miR396, were abundant in both libraries.miR167 exerts a key role in roots and shoots by controlling the transcriptional response to auxin signals, including growth, division, and differentiation of cells of the plant [17]. A study of A.thaliana indicated that miR396 targeted transcription factor AtGRF, which participates in the regulation of cell division and differentiation in leaf development[26].Based on these results, we propose that highly expressed miR167 and miR396 may be related to the rapid growth of the algal cell.
Targets of differentially expressed miRNAs
After prediction of the target genes of differentially expressed miRNAs, KEGG annotation of these potential targets was conducted to gain information about gene regulation during different growth periods in A. catenella. The differentially expressed miRNAs are likely related to growth of the unicellular alga.miR159appears to target MYB (v-myb avian myeloblastosis viral oncogene homolog) transcription factors, which have important roles in responding to hormones; this premise has been validated experimentally in previous study[64]. Another report on Arabidopsis indicated that the expression level of miR159can be enhanced by gibberellins [65].In the tomato plant, researchers reported that miR159 targeted 1-amino cyclopropane carboxylic acid (ACC) synthase, which plays crucial roles in fruit ripening[66]. In our study, miR159a targeted many functional enzymes that are crucial in multiple metabolic pathways, such as fatty acid synthase subunit beta, cysteine synthase A, cytochrome c oxidase cbb3-type subunit I, and various dehydrogenases and transferases. Due to the lack of MYB transcription factor genes and ACC synthase in the transcriptome that was referenced during target prediction, our results are not identical with those of other reports. Therefore, we suggested that159a might target different genes to ultimately regulate the growth of A. catenella. qPCR results indicated that the expression of miR159a was dramatically up-regulated in the exponential growth phase and the exponential phases of the three nutrient-enhanced cultures which were consistent with the deep sequencing results. (Fig 7). These results also provide evidence that high-throughput sequencing is a reliable method to identifying differentially expressed miRNAs indifferent growth phases of A. catenella. The expression of target genes is regulated negatively by miRNA. Therefore, we propose that miR159with higher expression levels in the logarithmic phase might play an important part in the rapid growth of A. catenella during red tide blooms.
Our results indicated that replication licensing factor (RLF) and the cell division cycle gene (cdc15)in the cell cycle were the target genes of miR171b. As a critical initiation factor, RLF combines with chromatin when the karyotheca ruptures at M phase so that the cell cycle can progress to S phase [67, 68].Previous research showed that RLF is involved in the precise replication during every single cell cycle by holding back re-replication of chromosomal DNA[69]. Meanwhile, the cdc gene codes a cyclin-dependent protein that directly takes part in cell division and regulation of the cell cycle[70].In our study, miR171b was up-regulated in the logarithmic phase; thus, we suggest that miR171b might carry out an important regulatory function in the rapid growth of A. catenella by controlling precise replication during the cell cycle and cell division.
In addition, miR169 (miR169a, miR169c, and miR169f) targeted calmodulin-dependent protein kinase II (CaMKII), which is an important member of the ubiquitous calcium ion(Ca2+) signaling pathway. Many reports have noted that CaMKII might be related to the cell cycle and rapid cellular metabolism[71]. To date, CaMK homologs have been successfully isolated from O.sativa, Maluspumila, Liliumbrownieand Nicotianatabacum [72–75]. In maize, two CaMK homologs of MCK1and MCK2 were identified and validated experimentally to be accumulated, particularly in plant parts undergoing rapid growth such as the apical meristem, root cap, and flower primordium [76]. Similar results have been found in O. sativa [72].In animals, researchers have found that CaMKII can promote the proliferation and growth of the cell by facilitating the replication of the centriole. At the same time, G2 in the cell cycle can be arrested by the use of CaMKII inhibitor[77,71]. Therefore, we speculated that CaMKII might play a major role in regulating the cell cycle and the rapid growth of the logarithmic phase in single-celled dinoflagellates. In A. catenella, CaM was shown to activate CaMKII, which provided evidence of its regulatory function [78]. These results suggest that miR169, which was found to be up-regulated in the logarithmic phase, might have a significant role in the blooming growth of A. catenella. However, much more research is needed tov alidate the target genes of CaMKII and to elucidate how miR169exerts its effect on growth in dinoflagellates.
In this study,aca-miR-3p-456915 was one of the predicted potentially novel miRNAs that expressed differentially in the two different growth phases (down-regulated in the exponential phase), and this finding was further validated by qPCR (Fig 8). KEGG pathway annotation was conducted on the target genes of aca-miR-3p-456915. These target genes were mainly enriched in the metabolic processes related to DNA replication and cell division, such as biosynthesis of unsaturated fatty acids, mismatch repair, biosynthesis of purine and pyrimidine, metabolism of various amino acids, and oxidative phosphorylation. Reports have shown that the proteins that take part in DNA replication and tRNA synthesis had a higher expression level in the logarithmic phase compared to the lag phase in A. catenella[12]. Thus,aca-miR-3p-456915might affect the rapid growth of algal cells during the logarithmic phase by providing the materials and energy necessary for speedy proliferation of the cell.
Conclusions
The present report is the first to identify and profile the expression pattern of miRNA in A. catenella through high-throughput sequencing. Eighty-eight conserved miRNAs belonging to 32 families and 15 novel miRNAs were found. Twelve miRNAs were identified that were differentially expressed in the lag phase and the logarithmic phase. Two of the differentially expressed miRNAs were verified by qPCR. Target predictions of the 12 differentially expressed miRNAs resulted in 1813 target genes. GO and KEGG annotation revealed highly enriched GO terms and metabolic pathways that were related to the growth of toxic A. catenella. miRNAs may play important roles in the rapid growth of A. catenella by the regulation of DNA replication and the cell cycle. These findings provide new insights into understanding the molecular mechanisms involved in regulation of algal cell growth.
Supporting Information
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
All the aligned species in the miRBase and their priority are listed. Chlamydomonas reinhardii (cre) was the preferentially aligned species.
(DOCX)
The specific forward primersof 5.8s, aca-miR-3p-456915 and tae-miR159a are listed.
(DOCX)
In total, 1813 target genes were predicted.
(DOCX)
Data Availability
Data are available in the NCBI Sequence Read Archive (SRA) database with accession number SRP055721. All other relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by The National Natural Science Foundation of China (41176098), and Shandong Provincial Natural Science Foundation, China (ZR2011DZ002). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Sournia A. Red tide and toxic marine phytoplankton of the world ocean: an inquiry into biodiversity Lavoisier, Paris(France) 1995; 103–112. [Google Scholar]
- 2. Stoecker DK. Mixotrophy among Dinoflagellates1.Journal of Eukaryotic Microbiology. 1999;46: 397–401. [Google Scholar]
- 3. Burkholder JM, Glibert PM and Skelton HM. Mixotrophy, a major mode of nutrition for harmful algal species in eutrophic waters. Harmful Algae. 2008;8: 77–93. [Google Scholar]
- 4. Schnepf E and Elbrachter M. Nutritional strategies in dinoflagellates: a review with emphasis on cell biological aspects. European journal of protistology. 1992;28: 3–24. 10.1016/S0932-4739(11)80315-9 [DOI] [PubMed] [Google Scholar]
- 5. Toulza E, Shin MS, Blanc G, Audic S, Laabir M, Collos Y, et al. Gene expression in proliferating cells of the dinoflagellate Alexandrium catenella (Dinophyceae). Applied and environmental microbiology. 2010; 76: 4521–4529. 10.1128/AEM.02345-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schnepf E and Winter S. A microtubular basket in the armoured dinoflagellate Prorocentrum micans (Dinophyceae). Archiv für Protistenkunde. 1990; 138: 89–91. [Google Scholar]
- 7. Burkholder JM. Implications of harmful microalgae and heterotrophic dinoflagellates in management of sustainable marine fisheries. Ecological applications. 1998; 8: S37–S62. [Google Scholar]
- 8. Matsuyama Y. Harmful effect of dinoflagellate Heterocapsa circularisquama on shellfish aquaculture in Japan. Japan Agricultural Research Quarterly. 1999;33: 283–294. [Google Scholar]
- 9. Hallegraeff GM, Anderson DM, Cembella AD. Manual on harmful marine microalgae Unesco; 2003; 25–49. [Google Scholar]
- 10. Uribe P, Fuentes D, Valdes J, Shmaryahu A, Zuniqa A, Holmes D, et al. Preparation and Analysis of an Expressed Sequence Tag Library from the Toxic Dinoflagellate Alexandrium catenella . Marine Biotechnology. 2008;10:692–700. 10.1007/s10126-008-9107-8 [DOI] [PubMed] [Google Scholar]
- 11. Zhang S, Sui ZH, Chang LP, Kang KH, Ma JH, Kong FN, et al. Transcriptome de novo assembly sequencing and analysis of the toxic dinoflagellate Alexandrium catenella using the Illumina platform. Gene. 2014;537: 285–293. 10.1016/j.gene.2013.12.041 [DOI] [PubMed] [Google Scholar]
- 12. Wang DZ, Lin L, Wang MH, Li C and Hong HS. Proteomic analysis of a toxic dinoflagellate Alexandrium catenella under different growth phases and conditions. Chinese Science Bulletin. 2012;57:3328–3341. [Google Scholar]
- 13. Huang J, Liang S, Sui ZH, Mao YX, Guo H. Cloning and characterization of proliferating cell nuclear antigen gene of Alexandrium catenella (Dinoflagellate) with respect to cell growth. Acta Oceanologica Sinica. 2010;29: 90–96. [Google Scholar]
- 14. Zhong J, Sui ZH, Wang CY, Ding HY, Zhou W, Zhang Y. Screening of growth decline related genes from Alexandrium catenella . Acta Ecol. Sin.2012;32: 431–437. [Google Scholar]
- 15. Wang CY, Sui ZH, Mao YX, Wen RB. Screening of environment factors responsive genes via suppressive subtractive hybridization technique from Alexandrium catenella . Periodical of Ocean University of China. 2009;39: 127–132. [Google Scholar]
- 16. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. cell. 2004;116: 281–297. [DOI] [PubMed] [Google Scholar]
- 17. Kidner CA, Martienssen RA. The developmental role of microRNA in plants. Current opinion in plant biology. 2005; 8: 38–44. [DOI] [PubMed] [Google Scholar]
- 18. Inui M, Martello G and Piccolo S. MicroRNA control of signal transduction. Nature reviews Molecular cell biology. 2010;11: 252–263. 10.1038/nrm2868 [DOI] [PubMed] [Google Scholar]
- 19. Qin YR, Duan ZX, Xia XL, Yin WL. Expression profiles of precursor and mature microRNAs under dehydration and high salinity shock in Populus euphratica . Plant cell reports. 2011;30: 1893–1907. 10.1007/s00299-011-1096-9 [DOI] [PubMed] [Google Scholar]
- 20. Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol. 2006;57: 19–53. [DOI] [PubMed] [Google Scholar]
- 21. Bonnet E, Wuyts J, Rouzé P, Van de Peer Y. Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101: 11511–11516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pilcher RLR, Moxon S, Pakseresht N, Moulton V, Manning K, Seymour G, et al. Identification of novel small RNAs in tomato (Solanum lycopersicum). Planta. 2007; 226: 709–717. [DOI] [PubMed] [Google Scholar]
- 23. Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana . Genes & development. 2006; 20: 3407–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Allen E, Xie ZX, Gustafson AM, Sung GH, Spatafora JW, Carrington JC. Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana . Nature genetics. 2004;36: 1282–1290. [DOI] [PubMed] [Google Scholar]
- 25. Liu SP, Li D, Li QB, Zhao P, Xiang ZH, Xia QY. MicroRNAs of Bombyx mori identified by Solexa sequencing. BMC genomics. 2010;11:148 10.1186/1471-2164-11-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chi XY, Yang QL, Chen XP, Wang JY, Pan LJ, Chen MN, et al. Identification and characterization of microRNAs from peanut (Arachis hypogaeaL.) by high-throughput sequencing. PloS one. 2011; 6: e27530 10.1371/journal.pone.0027530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sunkar R, Zhou XF, Zheng Y, Zhang WX, Zhu JK. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC plant biology. 2008; 8: 25 10.1186/1471-2229-8-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szittya G, Moxon S, Santos DM, Jing R, Fevereiro MP, Moulton V, et al. High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC genomics. 2008; 9: 593 10.1186/1471-2164-9-593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PloS one. 2007; 2: e219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Billoud B, Nehr Z, Le Bail AL, Charrier B. Computational prediction and experimental validation of microRNAs in the brown alga Ectocarpus siliculosus . Nucleic acids research. 2013; 42:417–429. 10.1093/nar/gkt856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang AY, Wang GC, He LW, Niu JF, Zhang BY. Characterization of small RNAs from Ulva prolifera by high-throughput sequencing and bioinformatics analysis. Chinese Science Bulletin. 2011; 56: 2916–2921. [Google Scholar]
- 32. Liang CW, Zhang XW, Zou J, Xu D, Su F, Ye NH. Identification of miRNA from Porphyra yezoensis by high-throughput sequencing and bioinformatics analysis. PloS one. 2010;5: e10698 10.1371/journal.pone.0010698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao T, Li GL, Mi SJ, Li S, Hannon GJ, Wang XJ. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii . Genes & development. 2007; 21: 1190–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang AY, He LW, Wang GC. Identification and characterization of microRNAs from Phaeodactylum tricornutum by high-throughput sequencing and bioinformatics analysis. BMC genomics. 2011;12: 337 10.1186/1471-2164-12-337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baumgarten S, Bayer T, Aranda M, Liew YJ, Carr A, Micklem G. Integrating microRNA and mRNA expression profiling in Symbiodinium microadriaticum, a dinoflagellate symbiont of reef-building corals. BMC genomics. 2013;14: 704 10.1186/1471-2164-14-704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao DH, Qiu LM, Hou ZH, Zhang QC, Wu JM, Gao Q, et al. Computational Identification of MicroRNAs from the Expressed Sequence Tags of Toxic Dinoflagellate Alexandrium Tamarense . Evolutionary bioinformatics. 2013;9: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guillard RR. Culture of phytoplankton for feeding marine invertebrates Culture of marine invertebrate animals. Springer; 1975; pp 29–60. [Google Scholar]
- 38. Galleron C. Synchronizationofthemarine dinoflagellate amphidinium carteriindense clutures. Journal of Phycology. 1976;12(1): 69–73. [Google Scholar]
- 39. Matsuda A, Nishijima T, Fukami K. Effects of nitrogenous and phosphorus nutrients on the growth of toxic dinoflagellate Alexandrium catenella . Nippon Suisan Gakkaishi. 1999;65: 847–855. [Google Scholar]
- 40. Brand LE, Sunda WG, Guillard RR. Limitation of marine phytoplankton reproductive rates by zinc, manganese, and iron. Limnology and oceanography. 1983;28: 1182–1198. [Google Scholar]
- 41. Su QJ, Yang XR, Zheng TL, Hong HS. An efficient method to obtain axenic cultures of Alexandrium tamarense—a PSP-producing dinoflagellate. Journal of microbiological methods. 2007;69: 425–430. [DOI] [PubMed] [Google Scholar]
- 42. Hobbie JE, Daley RJ, Jasper S. Use of nuclepore filters for counting bacteria by fluorescence microscopy. Applied and environmental microbiology. 1977;33: 1225–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Mardis ER, et al. Criteria for annotation of plant MicroRNAs. The Plant Cell. 2008;20: 3186–3190. 10.1105/tpc.108.064311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic acids research. 2003;31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. D’haene B, Mestdagh P, Hellemans J, Vandesompele J. miRNA expression profiling: from reference genes to global mean normalization Next-Generation MicroRNA Expression Profiling Technology. Springer; 2012;pp. 261–272. [DOI] [PubMed] [Google Scholar]
- 46. Audic S, Claverie JM. The significance of digital gene expression profiles. Genome research. 1997;7: 986–995. [DOI] [PubMed] [Google Scholar]
- 47. Yang G, Yang L, Zhao Z, Wang JJ, Zhang XB. Signature miRNAs involved in the innate immunity of invertebrates. PloS one. 2012;7: e39015 10.1371/journal.pone.0039015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sha ZX, Gong GY, Wang SL, Lu Y, Wang L, Wang QL, et al. Identification and characterization of Cynoglossus semilaevi microRNA response to Vibrio anguillarum infection through high-throughput sequencing. Developmental & Comparative Immunology. 2014;44: 59–69. [DOI] [PubMed] [Google Scholar]
- 49. Fuj H, Chiou TJ, Lin SI, Aung K, Zhu JK. A miRNA Involved in Phosphate-Starvation Response in Arabidopsis . Current Biology. 2005;15: 2038–2043. [DOI] [PubMed] [Google Scholar]
- 50. Kang L, Cui XX, Zhang YJ, Yang CH, Jiang YL. Identification of miRNAs associated with sexual maturity in chicken ovary by Illumina small RNA deep sequencing. BMC genomics. 2013;14: 352 10.1186/1471-2164-14-352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang B, Pan X, Cannon CH, Cobb GP, Anderson TA. Conservation and divergence of plant microRNA genes. The Plant Journal. 2006;46: 243–259. [DOI] [PubMed] [Google Scholar]
- 52. Zhu JF, Li WF, Yang WH, Qi LW, Han SY. Identification of microRNAs in Caragana intermedia by highthroughput sequencing and expression analysis of 12 microRNAs and their targets under salt stress. Plant Cell Reports. 2013;32:1339–1349. 10.1007/s00299-013-1446-x [DOI] [PubMed] [Google Scholar]
- 53. Bartel B, Bartel DP. MicroRNAs: at the root of plant development? Plant Physiology. 2003;132: 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arazi T, Talmor-Neiman M, Stav R, Riese M, Huijser P, Ni FR, et al. Cloning and characterization of microRNAs from moss. The Plant Journal. 2005;43: 837–848. [DOI] [PubMed] [Google Scholar]
- 55. Mi S, Cai T, Hu Y, Chen Y, Hodges E, Li L. Sorting of Small RNAs into Arabidopsis Argonaute Complexes Is Directed by the 5′ Terminal Nucleotide. cell. 2008;133: 116–127. 10.1016/j.cell.2008.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Su C, Yang XZ, Gao SQ, Tang YM, Zhao CP, Li L. Identification and characterization of a subset of microRNAs in wheat (Triticum aestivum L.). Genomics. 2014;103: 298–307. 10.1016/j.ygeno.2014.03.002 [DOI] [PubMed] [Google Scholar]
- 57. Morin RD, Aksay G, Dolgosheina E, Ebhardt HA, Magrini V, Mardis ER, et al. Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa . Genome Research. 2008;18: 571–584. 10.1101/gr.6897308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bonnet E, Wuyts J, Rouzé P, Van de Peer Y. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics. 2004;20: 2911–2917. [DOI] [PubMed] [Google Scholar]
- 59. Fu Y, Shi Z, Wu M, Zhang J, Jia L, Chen XW. Identification and differential expression of microRNAs during metamorphosis of the Japanese flounder (Paralichthys olivaceus). PloS one. 2011;6: e22957 10.1371/journal.pone.0022957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vazquez F. Arabidopsis endogenous small RNAs: highways and byways. Trends in plant science. 2006;11: 460–468. [DOI] [PubMed] [Google Scholar]
- 61. Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, Zilberman D, et al. Genetic and functional diversification of small RNA pathways in plants. PLoS biology. 2004;2: e104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ou JT, Meng QG, Li Y, Xiu YJ, Du J, Gu W, et al. Identification and comparative analysis of the Eriocheir sinensis microRNA transcriptome response to Spiroplasma eriocheirisinfection using a deep sequencing approach. Fish & shellfish immunology. 2012;32: 345–352. [DOI] [PubMed] [Google Scholar]
- 63. Jeong HJ, Du Yoo Y, Kim JS, Seong KA, Kang NS, Kim TH. Growth, feeding and ecological roles of the mixotrophic and heterotrophic dinoflagellates in marine planktonic food webs. Ocean science journal. 2010;45: 65–91. [Google Scholar]
- 64. Millar AA, Gubler F. The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. The Plant Cell. 2005;17: 705–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Achard P, Herr A, Baulcombe DC, Harberd NP. Modulation of floral development by a gibberellin-regulated microRNA. Development. 2004;131: 3357–3365. [DOI] [PubMed] [Google Scholar]
- 66. Zuo JH, Wang YX, Liu HP, Ma YZ, Ju Z, Zhai BQ, et al. MicroRNAs in tomato plants. Science China Life Sciences 54: 599–605. 10.1007/s11427-011-4188-4 [DOI] [PubMed] [Google Scholar]
- 67. Mahbubani HM, Chong JP, Chevalier S, Thommes P, Blow JJ. Cell cycle regulation of the replication licensing system: involvement of a Cdk-dependent inhibitor. The Journal of cell biology. 1997;136: 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Alberts B, Bray D, Lewis J, Raff M, Roberts K, et al. 1994. Molecular biology of the cell Garland, New York: 139–194. [Google Scholar]
- 69. Thömmes P, Kubota Y, Takisawa H, Blow JJ. The RLF-M component of the replication licensing system forms complexes containing all six MCM/P1 polypeptides. The EMBO Journal. 1997;16: 3312–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Weinert TA, Hartwell LH. Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics. 1993;134: 63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Means AR. Calcium, calmodulin and cell cycle regulation. FEBS letters. 1994;347: 1–4. [DOI] [PubMed] [Google Scholar]
- 72. Zhang L, Liu BF, Liang SP, Jones RL, Lu YT. Molecular and biochemical characterization of a calcium/calmodulin-binding protein kinase from rice. Biochem J. 2002;368: 145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Watillon B, Kettmann R, Boxus P, Burny A. A calcium/calmodulin-binding serine/threonine protein kinase homologous to the mammalian type II calcium/calmodulin-dependent protein kinase is expressed in plant cells. Plant Physiology. 1993;101: 1381–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Patil S, Takezawa D, Poovaiah BW. Chimeric plant calcium/calmodulin-dependent protein kinase gene with a neural visinin-like calcium-binding domain. Proceedings of the National Academy of Sciences. 1995;92: 4897–4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Liu ZH, Xia M, Poovaiah BW. Chimeric calcium/calmodulin-dependent protein kinase in tobacco: differential regulation by calmodulin isoforms. Plant molecular biology. 1998;38: 889–897. [DOI] [PubMed] [Google Scholar]
- 76. Wang L, Liang S, Lu YT. Characterization, physical location and expression of the genes encoding calcium/calmodulin-dependent protein kinases in maize (Zea mays L.). Planta. 2001;213: 556–564. [DOI] [PubMed] [Google Scholar]
- 77. Zhao L, Lu R, Yao Z. The roles of Ca2+/ calmodulin-dependent kinases in cell proliferation.Chinese Journal of Cell Biology. 2007;29: 331–335. [Google Scholar]
- 78. Wen RB, Sui ZH, Bao ZM, Zhou W, Wang CY. Isolation and characterization of calmodulin gene of Alexandrium catenella (Dinoflagellate) and its performance in cell growth and heat stress. Journal of Ocean University of China;13: 290–296. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
All the aligned species in the miRBase and their priority are listed. Chlamydomonas reinhardii (cre) was the preferentially aligned species.
(DOCX)
The specific forward primersof 5.8s, aca-miR-3p-456915 and tae-miR159a are listed.
(DOCX)
In total, 1813 target genes were predicted.
(DOCX)
Data Availability Statement
Data are available in the NCBI Sequence Read Archive (SRA) database with accession number SRP055721. All other relevant data are within the paper and its Supporting Information files.