

Abstract
Sphingosine-1-phosphate (S1P) is a ubiquitous, endogenous small molecule that is synthesized by two isoforms of sphingosine kinase (SphK1 and 2). Intervention of the S1P signaling pathway has attracted significant attention because alteration of S1P levels is linked to several disease states including cancer, fibrosis, and sickle cell disease. While intense investigations have focused on developing SphK1 inhibitors, only a limited number of SphK2-selective agents have been reported. Herein, we report our investigations on the structure-activity relationship studies on the lipophilic tail region of SLR080811, a SphK2-selective inhibitor. Our studies demonstrate that the internal phenyl ring is a key structural feature that is essential in the SLR080811 scaffold. Further, we show the dependence of SphK2 activity and selectivity on alkyl tail length, suggesting a larger lipid binding pocket in SphK2 compared to SphK1.
Keywords: Sphingosine kinase, SphK1, SphK2, Sphingosine-1-phosphate, S1P
Sphingosine 1-phosphate (S1P) is both an intermediate in the catabolism of sphingolipids and an extracellular signaling molecule. The synthesis of S1P in vivo is controlled by two isoforms of sphingosine kinase (SphK1 and SphK2), which phosphorylate sphingosine (Sph) to S1P. S1P is involved in a variety of important intracellular and extracellular functions through a complex network of signaling pathways including G-protein coupled receptors S1P1–5. S1P signaling has been associated with a variety of diseases including cancer, fibrosis, multiple sclerosis, and sickle cell disease.1–4 As a result of its key role in Sph and S1P metabolism, regulation of SphKs has attracted an increasing amount of attention as a therapeutic target. The ability to control SphK function would also aid in the understanding of their in vivo function as well as their effects in the sphingolipid signaling pathway.
Many differences exist between SphK1 and SphK2 including size, cellular localization, and intracellular roles.5,6 While double knockout studies in mice suggests that SphKs are the sole source of S1P, some functional redundancy exists as SphK1 or SphK2 null mice are viable and fertile. Although inhibitor development towards SphK1 has been a focus of intense studies,7 inhibitors of SphK2 are emerging (Figure 1). For example, ABC294640 (Ki = 10 µM) was the first inhibitor with SphK2 activity that has been deployed in a variety of disease models including lupus nephritis, diabetic nephropathy, Crohn’s disease, ulcerative colitis, and osteoarthritis.8,9 However, it was recently reported to inhibit estrogen receptors in breast cancer cells by acting as a partial agonist similar to tamoxifen.10 Another inhibitor, thiazolidine-2,4-dione K145 (Ki = 6.4 µM), which is an analog of sphingosine was recently reported as a selective SphK2 inhibitor.11 K145 was shown to inhibit leukemia cell growth in vitro as well as in a xenograph mouse model.
Figure 1.

Structure of sphingosine kinase 2 inhibitors.
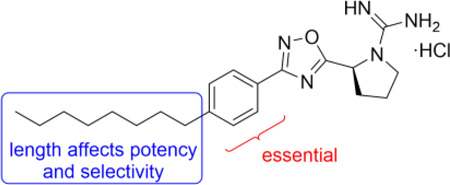
Due to our interest in understanding the in vivo function of SphK2 and the lack of highly potent and selective inhibitors, we focused our studies in developing unique scaffolds to achieve our goals. Our first generation inhibitor, VT-ME6, contained a quaternary ammonium group as a warhead and established that a positively charged moiety is necessary for engaging key amino acid residues in the enzyme binding pocket.13,14 This compound is moderately potent (Ki = 8 µM) and displays three-fold selectivity for SphK2 over SphK1. Subsequent improvement resulted in a scaffold that featured a 1,2,4-oxadiazole linker and guanidine as warhead: SLR080811 possesses a Ki of 13.3 µM and 1.3 µM for SphK1 and SphK2 respectively.15 A significant finding from these studies was that pharmacological inhibition of SphK2 resulted in elevated S1P levels in mice. Further structure-activity relationship studies on the guanidine core revealed that an azetidine-containing derivative SLP1201701 improved the half-life to 8 hrs in mice.16 In this report, we detail our investigations on the tail region of the scaffold (Fig. 2). Our studies demonstrate that the internal phenyl ring is essential to maintain inhibitory activity for SphK2 and that the alkyl tail length has a significant effect on the potency and selectivity towards SphK2.
Figure 2.

Pharmacophore of guanidine-based inhibitors.
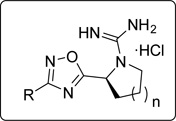
The synthesis of SLR080811 derivatives with varying alkyl length as well as heterocycles attached to the phenyl ring is shown in Schemes 1 and 2. In Scheme 1, 4-iodobenzonitrile was cross-coupled to a series of alkynes or hydroborated intermediates under standard Sonogashira or Suzuki-Miyaura conditions. Subsequent reaction with hydroxylamine afforded amidoximes 2a–e, which were cyclized to 1,2,4-oxadiazoles 3a–f in the presence of HCTU and Boc-L-proline. Deprotection with HCl and reduction of alkynyl groups with tosylhydrazine at refluxing conditions yielded amines 4a–h. To install the guanidine moiety, the amines were treated with DIEA and N,N’-Di-Boc-1H-pyrazole-1-carboxamidine for several days at room temperature and deprotected with HCl to produce the desired derivatives 5a,d,f–h. A similar synthetic strategy was employed to access the remaining phenyl/alkyl derivatives (7c and 7f–g); however, heterocycles 7d–e were obtained via Buchwald-Hartwig coupling conditions as shown in Scheme 2. Similarly, Scheme 3 illustrates the synthesis of various amidopiperazine tail surrogates 10a–d using Buchwald-Hartwig and amide coupling reactions.
Scheme 1.

a.) Alkyne (2 equiv.), TEA (5 equiv.), DMF, PdCl2(PPh3)2 (0.05 equiv.), CuI (0.03 equiv.), 80 °C, 18 h, (72–93%); b.) i. Alkene, 0.5 M 9-BBN, in THF, rt, 12 h; ii. Pd(dppf)Cl2, Cs2CO3, DMF, 70 °C, 18 h, (75–93%); c.) NH2OH·HCl (3 equiv.), TEA (3 equiv.), EtOH, 80 °C, 6 h, (43–95%); d.) Boc-L-Proline (1.4 equiv.), DIEA (1.4 equiv.), HCTU (1.8 equiv.), DMF, 110 °C, 18 h, (25–65%); e.) DME (20 vol/wt), 4-toluenesulfonyl hydrazide (10 equiv.), TEA (5 equiv.), reflux, (67–71%); f.) HCl/MeOH, (35–100%); g.) DIEA (3 equiv.), N,N'-Di-Boc-1H-pyrazole-1-carboxamidine (1.05 equiv.), CH3CN, rt, 3 days, (27–76%).
Scheme 2.

a.) Boc-L-Azetidine (1.4 equiv.), DIEA (1.4 equiv.), HCTU (1.8 equiv.), DMF, 110 °C, 18 h, (63%); b.) Alkyne (2 equiv.), TEA (5 equiv.), DMF, PdCl2(PPh3)2 (0.05 equiv.), CuI (0.03 equiv.), 80 °C, 18 h, (33–57%); c.) Phenylboronic acid (1.3 equiv.), Cs2CO3 (equiv.), DMF, PdCl2(dppf) (0.04 equiv.), 80 °C, 18 h, (91%); d.) Amine, Pd(dba)3, Cs2CO3, PtBu3, toluene, 120 °C, 6 d, (81–83%); e.) DME (20 vol/wt), 4-toluenesulfonyl hydrazide (10 equiv.), TEA (5 equiv.), reflux, (60–71%); f.) HCl/MeOH, (78–96%); g.) DIEA (3 equiv.), N,N'-Di-Boc-1H-pyrazole-1-carboxamidine (1.05 equiv.), CH3CN, rt, 3 days, (43–66%).
Scheme 3.

a.) Piperazine (3 equiv.), Pd2(dba)3 (0.2 equiv.), PtBu3 (0.8 equiv.), Cs2CO3 (1.2 equiv.), toluene, 120 °C, 3 days, (52%); b.) Acid chloride (2.5 equiv.) or benzyl bromide (1 equiv.), TEA (2 equiv.), CH2Cl2, 0 °C—rt, 2 h, (66–88%); c) HCl/MeOH, (76–95%); d) DIEA (3 equiv.), N,N'-Di-Boc-1H-pyrazole-1-carboxamidine (1.05 equiv.), CH3CN, rt, 3 days, (23–74%).
Compounds 14 and 17 were synthesized as shown in Scheme 4. 4-(3-ethoxymethyl)-5-methylbenzyl)benzonitrile 13 was formed in two steps via mono-substitution of 1,3- palladium-catalyzed cross coupling reaction with 4-cyanophenylboronic acid to afford 13. Alternatively, benzonitrile 16 was achieved using sodium benzenesulfonate and 15. Standard oxadiazole formation, guanidylation, and deprotection afforded 14 and 17. Finally, a series of alkyl tails directly linked to the oxadiazole ring were synthesized (Scheme 5). Treatment of alkylbromides with potassium cyanide gave alkylnitriles 19a–c, which were converted to amidoximes 20a–c. Transformation to oxadiazoles 21a–c was effected either by HCTU-mediated cyclization at 110 °C or by two-step coupling/TBAF-catalyzed cyclization, which eventually led to 22a–c.
Scheme 4.

a.) NaH, EtOH, 0 °C—rt, (46%); b.) 4-cyanophenylboronic acid, Pd(PPh3)4 (10 mol%), Na2CO3, THF:H2O, (94%); c.) NH2OH·HCl (3 equiv.), TEA (3 equiv.), EtOH, 80 °C, 6 h, (71–93%); d.) Boc-L-Proline (1.4 equiv.), DIEA (1.4 equiv.), HCTU (1.8 equiv.), DMF, 110 °C, 18 h, (46–82%); e.) HCl/MeOH, (33–91%); f.) DIEA (3 equiv.), N,N'-Di-Boc-1H-pyrazole-1-carboxamidine (1.05 equiv.), CH3CN, rt, 3 days, (53–83%); g.) sodium benzenesulfonate (1.5 equiv.), DMF, 60 °C, 2 h, (89%).
Scheme 5.

a.) KCN (2 equiv.), 9:1 EtOH:H2O, 80 °C, 18 h, (20–93%); b.) NH2OH·HCl (3 equiv.), TEA (3 equiv.), EtOH, 80 °C, 12 h, (53–69%); c.) Boc-L-Proline (1.4 equiv.), DIEA (1.4 equiv.), HCTU (1.8 equiv.), DMF, 110 °C, 18 h, (50%); d.) Boc-L-Proline (1.4 equiv.), DIEA (1.4 equiv.), HCTU (1.8 equiv.), CH2Cl2, rt, 4 h, (57–80%); e.) TBAF (1.0 M, 1 equiv.), THF, rt, 1 h, (93–95%); f.) HCl/MeOH, (66–100%); g) DIEA (3 equiv.), N,N'-Di-Boc-1H-pyrazole-1-carboxamidine (1.05 equiv.), CH3CN, rt, 3 days, (51–71%).
With the library of putative inhibitors synthesized, the inhibitory effects of the compounds were determined for hSphK1 and mSphK2 using a previously published protocol (Table 1).16 Briefly, Sph and cell lysate containing recombinant SphK1 or SphK2 were incubated with or without inhibitor in the presence of γ-[32P]ATP. After 20 minutes, the reaction mixtures were extracted, separated using thin layer chromatography, and quantified using liquid scintillation counting. The kinase inhibition was determined as the amount of 32P-S1P produced as a function of inhibitor concentration. Compounds were screened at 10 µM inhibitor concentrations.
Table 1.
Inhibitory effects of SLR080811 derivatives on SphK1 and SphK2.a
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| % kinase activity b | % kinase activity b | ||||||||||
| entry | R | n | entry | R | n | ||||||
| SphK1 | SphK2 | SphK1 | SphK2 | ||||||||
| 1 | 5a | 1 | 100 ± 2 | 63 ± 1 | 12 | 22b | 1 | 60 ± 1 | 57 ± 1 | ||
| 2 | 7c | 0 | 101 ± 1 | 91 ± 1 | 13 | 22c | 1 | 37 ± 5 | 52 ± 2 | ||
| 3 | 5e | 1 | 78 ± 1 | 70 ± 7 | 14 | 7d | 0 | 90 ± 4 | 94 ± 2 | ||
| 4 | 5f | 1 | 103 ± 2 | 35 ± 3 (76 ± 6) |
15 | 7e | 0 | 97 ± 2 | 90 ± 3 | ||
| 5 | 7f | 0 | 94 ± 2 | 45 ± 2 (86 ± 6) |
16 | 10d | 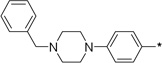 |
1 | 89 ± 2 | 89 ± 3 | |
| 6 | SLR080811 | 1 | 60 ± 1 | 9 ± 4 (44 ± 4) |
17 | 10a | 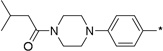 |
1 | 79 ± 2 | 90 ± 2 | |
| 7 | 5g | 1 | 18 ± 9 (64 ± 4) |
9 ± 7 (46 ± 5) |
18 | 10c | 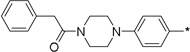 |
1 | 90 ± 2 | 86 ± 2 | |
| 8 | 7g | 0 | 11 ± 4 | 12 ± 2 | 19 | 10b | 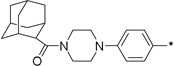 |
1 | 81 ± 28 | 87 ± 3 | |
| 9 | 5h | 1 | 37 ± 2 (82 ± 3) |
15 ± 1 (56 ± 1) |
20 | 14 | 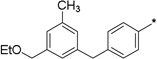 |
1 | 96 ± 1 | 46 ± 9 (99 ± 3) |
|
| 10 | 5d | 1 | 75 ± 1 (88 ± 3) |
16 ± 10 (52 ± 4) |
21 | 17 | 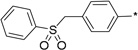 |
1 | 99 ± 1 | 88 ± 2 | |
| 11 | 22a | 1 | 96 ± 1 | 88 ± 1 | |||||||
Values represent percent activity of human SphK1 or mouse SphK2 with 10 and 5 µM Sph, respectively, in the presence of 10 µM inhibitor. Each value is an average of two experiments. Lower SphK activity level indicates better inhibition.
Values in parenthesis indicate compounds assayed at 1 µM.
As shown in Table 1, replacement of the octyl chain of SLR080811 with iodide, phenyl or phenethyl groups did not improve inhibitory activity (entries 1–3). Decreasing or increasing the lipophilic alkyl tail length from hexyl to tetradecyl in two-carbon increments resulted in compounds with similar inhibitory activity as SLR080811 (entries 4–10), although the hexyl chain was slightly less active. In cases where the kinase activity was similar to SLR080811 at 10 µM, rescreening at a more stringent inhibitor concentration (1 µM) was performed: the results indicated that none of these analogs had improved activity compared to SLR080811. We also note that the pyrrolidine and azetidine rings have been shown to have similar potency, but with the advantage of improved in vivo half-life for the azetidine derivatives.16 Interestingly, as the alkyl tail increased to a decyl group, SphK2 selectivity decreased as SphK1 inhibition increased. However, as the chain length increased further to a dodecyl and tetradecyl, inhibition of SphK1 decreased while maintaining SphK2 activity. These results suggest that the lipid binding pocket in SphK2 is much larger than that of SphK1 and is consistent with the prediction based on a crystal structure of SphK1 bound to SphK1 inhibitor PF-543.17 We next investigated the effect of the phenyl substitutent next to the 1,2,4-oxadiazole ring. Removal of this ring while maintaining the overall length of the molecule resulted not only in diminished SphK2 selectivity but also inhibitory activity (entries 11–13). Our data indicate that the phenyl ring is necessary for selectivity and potency using this scaffold
To further determine features of the lipid binding pocket, morpholine and a series of heterocyclic rings were synthesized (entries 14–19). In particular, a piperazine ring is attractive because of increased conformational rigidity as well as an anchor point in which various groups can be appended. Morpholine, N-methyl or N-benzyl piperazine derivatives were inactive. As these substituents are positively charged and the likelihood that the lipid binding pocket is lined with hydrophobic groups, neutral amide versions with increasing steric bulk were tested. Isovaleryl, phenacetyl, and adamantylcarbonyl groups were also inactive. Finally, trisubstituted aryl 14 as well as sulfonate 17 bearing groups, featured in SphK1 inhibitor PF-543, were tested and also found to be poor inhibitors (entries 20–21).18
In summary, a focused library of SphK2-selective inhibitor SLR080811 derivatives that interrogated the lipophilic tail region of the pharmacophore were synthesized. Our studies demonstrate the dependence of SphK2 inhibitory activity on alkyl chain length; the most optimal length includes octyl and decyl substituents, which suggests an ideal ‘head-to-tail’ (positive charge to terminal methyl group) length of approximately 18–21 atoms. Furthermore, our studies provide evidence for the much larger lipophilic binding cavity in SphK2 over SphK1. In the SLR080811 scaffold, the internal phenyl ring appears to be essential for activity and is likely interacting with residues in the kinase binding pocket. These predictions can be aided by a SphK2 crystal structure, which is currently unavailable.
Supplementary Material
Acknowledgments
We acknowledge financial support by NIH (Grants R01 GM104366 and R01 GM067958).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data (characterization data for final compounds) associated with this article can be found, in the online version, at http://dx.doi.org/10
References and notes
- 1.Bigaud M, Guerini D, Billich A, Bassilana F, Brinkmann V. Biochim. Biophys. Acta. 2014;1841:745. doi: 10.1016/j.bbalip.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Takuwa N, Du W, Kaneko E, Okamoto Y, Yoshioka K, Takuwa Y. Am. J. Cancer Res. 2011;1:460. [PMC free article] [PubMed] [Google Scholar]
- 3.Kunkel GT, Maceyka M, Milstien SSS. Nat Rev Drug Discov. 2013;12:688. doi: 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Berka V, Song A, Sun K, Wang W, Zhang W, Ning C, Li C, Zhang Q, Bogdanov M, Alexander DC, Milburn MV, Ahmed MH, Lin H, Idowu M, Zhang J, Kato GJ, Abdulmalik OY, Zhang W, Dowhan W, Kellems RE, Zhang P, Jin J, Safo M, Tsai AL, Juneja HS, Xia Y. J. Clin. Invest. 2014;124:2750. doi: 10.1172/JCI74604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH, Jr, Milstien S, Spiegel S. J. Biol. Chem. 2005;280:37118. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- 6.Neubauer HA, Pitson SM. FEBS J. 2013 doi: 10.1111/febs.12314. [DOI] [PubMed] [Google Scholar]
- 7.Plano D, Amin S, Sharma AK. J. Med. Chem. 2014;57:5509. doi: 10.1021/jm4011687. [DOI] [PubMed] [Google Scholar]
- 8.French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, Upson JJ, Green CL, Keller SN, Smith CD. J. Pharmacol. Exp. Ther. 2010;333:129. doi: 10.1124/jpet.109.163444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neubauer HA, Pitson SM. FEBS J. 2013;280:5317. doi: 10.1111/febs.12314. [DOI] [PubMed] [Google Scholar]
- 10.Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S, Rhodes LV, Ashe HB, Wiese TE, Smith CD, Burow ME, Beckman BS. Endocrinology. 2010;151:5124. doi: 10.1210/en.2010-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu K, Guo TL, Hait NC, Allegood J, Parikh HI, Xu W, Kellogg GE, Grant S, Spiegel S, Zhang S. PloS one. 2013;8:e56471. doi: 10.1371/journal.pone.0056471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santos WL, Lynch KR. ACS Chem. Biol. 2015;10:225. doi: 10.1021/cb5008426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raje MR, Knott K, Kharel Y, Bissel P, Lynch KR, Santos WL. Bioorg. Med. Chem. 2012;20:183. doi: 10.1016/j.bmc.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knott K, Kharel Y, Raje MR, Lynch KR, Santos WL. Bioorg. Med. Chem. Lett. 2012;22:6817. doi: 10.1016/j.bmcl.2012.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kharel Y, Raje M, Gao M, Gellett AM, Tomsig JL, Lynch KR, Santos WL. Biochem. J. 2012;447:149. doi: 10.1042/BJ20120609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patwardhan NNMEA, Raje MR, Gao M, Kharel Y, Tomsig JL, Lynch KR, Santos WL. J. Med. Chem. 2015;58:1879. doi: 10.1021/jm501760d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Knapp S, Pyne NJ, Pyne S, Elkins JM. ACS Med. Chem. Lett. 2014;5:1329. doi: 10.1021/ml5004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schnute ME, McReynolds MD, Kasten T, Yates M, Jerome G, Rains JW, Hall T, Chrencik J, Kraus M, Cronin CN, Saabye M, Highkin MK, Broadus R, Ogawa S, Cukyne K, Zawadzke LE, Peterkin V, Iyanar K, Scholten JA, Wendling J, Fujiwara H, Nemirovskiy O, Wittwer AJ, Nagiec MM. Biochem. J. 2012;444:79. doi: 10.1042/BJ20111929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.