

Abstract
Objective
To determine if distinct muscle pathological features exist in scleroderma subjects with weakness.
Methods
This retrospective study included weak scleroderma subjects with muscle biopsies available for review. Biopsies were systematically assessed for individual pathologic features including inflammation, necrosis, fibrosis, and acute neurogenic atrophy. Based on the aggregate individual features, biopsies were assigned a histopathologic category of polymyositis, dermatomyositis, necrotizing myopathy, non-specific myositis, “acute denervation”, “fibrosis only”, or “other”. Clinical data analyzed included autoantibody profiles, scleroderma subtype and disease duration, Medsger muscle severity scores, creatine kinase (CK), electromyography (EMG), and muscle magnetic resonance imaging (MRI).
Results
42 subjects (79% female and 64% diffuse scleroderma) were included in this study. Necrosis (67%), inflammation (48%), acute neurogenic atrophy (48%), and fibrosis (33%) were the most prevalent pathologic features. The presence of fibrosis was strongly associated with anti-PM-Scl antibodies. Histopathologic categories included non-specific myositis (36%), necrotizing myopathy (21%), dermatomyositis (7%), “acute denervation” (7%), “fibrosis only” (7%), and polymyositis (5%). Disease duration of scleroderma at the time of muscle biopsy was shorter in polymyositis than other histopathologic categories. Patients with anti-PM-Scl and Scl-70 antibodies also had a shorter disease duration than those with other auto-antibody profiles.
Conclusion
Non-specific myositis and necrotizing myopathy were the most common histopathologic categories in weak scleroderma subjects. Surprisingly, nearly half of the subjects studied had histological evidence of acute motor denervation (acute neurogenic atrophy); this has not been previously reported. Taken together, these observations suggest that a variety of pathologic mechanisms may underlie the development of myopathy in scleroderma.
Introduction
Systemic sclerosis (SSc) is a systemic rheumatic disease characterized by three main pathologic mechanisms: altered immune function and inflammation, an obliterative vasculopathy and tissue fibrosis. Skeletal muscle involvement in SSc is relatively common and, depending upon the diagnostic criteria applied, occurs in 14-79% of patients with scleroderma (1–6). Muscle involvement in SSc is a poor prognostic feature impacting survival (7) and hasalso been associated with cardiopulmonary complications and even sudden cardiac death (8).
Myopathy in scleroderma can occur as a result of non-autoimmune etiologies such as malnutrition, disuse, or other neuromuscular disorders (9–11). There are also cases of an inflammatory myopathy in scleroderma with pathological features identical to polymyositis or dermatomyositis; these patients are considered to have an overlap syndrome with features of both scleroderma and a primary idiopathic inflammatory myopathy. In one large well-characterized scleroderma cohort, 1.3% (2/1,499) of SSc patients satisfied the 1975 Bohan and Peter diagnostic criteria for definite PM/DM (13). A range of muscle biopsy features have been described including inflammation and prominent necrosis (14,15). One study suggested that fibrosis was the prominent finding in muscle biopsies of weak scleroderma patients (16).
To determine if weak scleroderma subjects exhibit distinct pathologic muscle characteristics, we conducted a retrospective study in our scleroderma cohort using patients identified as having both objective muscle weakness and an available muscle biopsy read at our institution.
Materials and Methods
We reviewed the data on 2830 subjects enrolled in the Johns Hopkins Scleroderma Center database from May 1990 to June 2014. This database includes subjects who meet the 1980 American College of Rheumatology (ACR) criteria for SSc (17), or have 3/5 features of the CREST syndrome, or have all 3 of the following features: Raynaud's phenomenon, nailfold capillary changes and a SSc-specific antibody. Among this cohort, our study population included 42 patients with (i) evidence of myopathy as defined by proximal weakness by an expert at our Center, and (ii) a muscle biopsy reviewed by a muscle pathologist at the Johns Hopkins Neuromuscular Laboratory.
Demographic data including age, sex, race, ethnicity, and scleroderma subtype were obtained. Clinical data for this study included the lowest percent predicted forced vital capacity (FVC) and the lowest percent predicted diffusing capacity of the lungs for carbon monoxide (DLCO) on available pulmonary function tests (PFTs) (18,19), the lowest ejection fraction (EF (%)) and highest right ventricular systolic pressure (RVSP) on echocardiography, the peak recorded modified Rodnan skin score (mRSS), the highest Medsger muscle severity score, and any history of a renal crisis. These most extreme recorded values were selected to provide a composite picture of the severity of the clinical features or phenotype of the study subjects. The disease duration of SSc to the onset of muscle symptoms was calculated from the time of the first recorded non-Raynaud's and Raynaud's phenomenon.
Autoantibody specificities were also obtained from the database; if the data was missing, it was assayed for research purposes. RNA Polymerase 3 was performed by ELISA using commercially available kits (Inova Diagnostics). For anti-PM-Scl, DNA encoding the 100 and 75 kD subunits was used in in vitro transcription/translation reactions per the manufacturer's protocol (Promega). A mix of both the resulting 35S-methionine labeled proteins was used in immunoprecipitations performed with patient sera, and the resulting immunoprecipitates were visualized by fluorography (20).
Additional clinical information such as medication history, co-morbidities, and overlapping rheumatologic diseases at the time of muscle biopsy was obtained from comprehensive medical chart review.
Muscle evaluation of study participants
Muscle weakness was determined by the scleroderma provider at the time of evaluation and categorized using the 5 point Medsger muscle severity score: 0 = no proximal muscle weakness; 1 = mild proximal muscle weakness; 2 = moderate proximal muscle weakness; 3 = severe proximal muscle weakness; and 4 = weakness requiring ambulation aids (20). Electromyography (EMG) with nerve conduction studies (NCS), magnetic resonance imaging (MRI) of affected proximal muscles, and muscle biopsies were obtained as part of routine clinical care and reviewed for this study. The presence of positive sharp waves and fibrillations on EMG in the context of myopathic motor units was defined as an irritable myopathy; myopathic motor units without spontaneous activity were defined as having a non-irritable myopathy (21). Decreased or absent sural nerve responses on NCS was deemed to be electrophysiologic evidence of a sensory neuropathy (22). A myositis-specific protocol on MRI consisted of coronal and axial short tau inversion recovery (STIR) and coronal and axial T1-weighted sequences of proximal muscles. Edema (defined as increased T2 signal) on STIR images and fatty replacement on T1 weighted sequences were read by a radiologist as part of routine clinical care (23–25). All but 3 had EMG/NCS and MRI within 3 months before the muscle biopsy. Two patients had both performed within 3 months after biopsy and one patient had both10 months after the muscle biopsy. Creatine kinase (CK) and aldolase levels were collected from the date closest to the date of muscle biopsy. Highest ever recorded CK and aldolase levels were also obtained from comprehensive medical record review. Institutional review board approval and participants' written informed consent were obtained from each participant.
Muscle Histology
In most cases, muscle biopsies were performed and read at Johns Hopkins. In three instances, muscle biopsy slides were obtained from another institution and then read at Johns Hopkins. To ensure uniform assessment, we excluded samples that were not read at The Johns Hopkins Neuromuscular Pathology Lab. When processed at Hopkins, all frozen sections were stained using H&E, modified Gomori trichrome, adenosine triphosphatase at pH 4.3, 4.6, and 9.4, nicotinomide adenosine dinucleotide -tetrazolium reductase, acid phosphatase, succinic dehydrogenase, cytochrome oxidase, esterase, alkaline phosphatase, Periodic acid-Schiff (PAS), PAS-diastase control, and Congo red. Muscle biopsy slides were read using a predesigned protocol that is used as part of routine care and assessed for the presence or absence of the following individual histologic features: necrotic fibers, inflammation, perifasicular atrophy, fibrosis, angular atrophic esterase-positive fibers (a histopathologic feature of acute neurogenic atrophy), and fiber type grouping (a feature of chronic denervation with re-innervation) (26).
Based on the individual histologic features, most biopsies were assigned to one of 4 well-accepted histopathologic categories of myopathy: (1) polymyositis, (2) dermatomyositis, (3) necrotizing myopathy, (4) or non-specific myositis. The categories were adapted from the European Neuro Muscular Centre (ENMC) histopathologic criteria (28)(Table 1). Some of the muscle biopsies could not be categorized according to this scheme. While not a validated histological subtype, patients with fibrosis as the only analyzed histopathologic feature were categorized as having “fibrosis only”. In cases where positive esterase staining of angular atrophic fibers suggested acute neurogenic atrophy without any other pathological findings, biopsies were categorized as “acute denervation”. If there were other findings such as numerous COX-negative fibers (indicating mitochondrial dysfunction) or non-specific myofiber atrophy in isolation, these were classified as “Other”.
Table 1. Diagnostic Category Definitions (adapted from ENMC criteria (28)).
|
Polymyositis: Muscle biopsy criteria include a or b, and exclude c.
Dermatomyositis: Muscle biopsy criteria include c.
Non-specific myositis: Muscle biopsy criteria include d or e, and exclude all others.
Necrotizing myopathy: Muscle biopsy criteria include f, and exclude all others.
Acute denervation: Muscle biopsy reveals esterase-positive angular atrophic fibers and excludes all others.
Fibrosis only: Muscle biopsy reveals increased connective tissue and excludes all others.
Immunostaining of frozen muscle biopsies
When residual frozen muscle biopsy specimens were available, they were stained with antibodies recognizing CD3, CD4, CD8, CD20, and major histocompatibility complex (MHC I) for the purposes of this study since it is not done for routine clinical care. Muscle biopsies were scored for the presence of CD4, CD8, CD20, and MHC-I by a single neuromuscular pathologist (T.L.) who was aware of the diagnosis of scleroderma associated myopathy.
Statistical Analysis
Statistical analyses were performed using the statistical software, Stata version 12.1 (Stata Corp, College Station, Texas). Continuous variables were reported as a mean value +/- standard deviation. Discrete variables were summarized as proportions. If there was missing data, denominators in proportions included only individuals with data available for that particular variable. Fisher's exact test was used to analyze differences in dichotomous variables. Wilcoxon rank sum test, a non-parametric test, was performed if there were comparisons between two groups with very small sample sizes. Differences between means were examined using the Student's t test for continuous variables. All reported p values are 2-sided with α=0.05.
Results
Clinical characteristics of weak scleroderma subjects
Out of 2830 subjects included in the Johns Hopkins Scleroderma database between 1990 and 2014, 65 had a history of weakness and a muscle biopsy. Forty-two of these had their muscle biopsy read at our institution and constituted the study population. The mean age at the onset of scleroderma was 46 ± 12.5 years, based on their first non-Raynaud's symptom. The mean age at the onset of muscle symptoms was 4 years later at 50 ± 14.2 years of age. 79% of subjects were female and 64% had diffuse scleroderma (Table 2). The most common auto-antibody was anti-RNP (22.5%). The mean maximum RVSP was 48.2 ± 20.9 for the study group. 8/42 subjects (19%) were deceased at the time of our survey. 4/8 subjects died of multisystem disease with myopathy, severe ILD, and pulmonary hypertension thought secondary to scleroderma. The cause of death in the other four subjects was recorded as single organ disease including heart failure (n=1), severe pulmonary hypertension (n=1), respiratory failure from ILD (n=1), and unknown causes (n=1).
Table 2. Clinical characteristics of scleroderma patients with myopathy (N=42).
| Mean age of scleroderma onset (years ± SD) | 46 ± 12.5 |
| Mean age at the onset of muscle weakness (year ± SD) | 50 ± 14.3 |
| Female sex, No (%) | 33 (78.6%) |
| Race, No(%) | |
| Caucasian | 24 (57.1%) |
| African-American | 17 (40.5%) |
| Other | 1 (2.4%) |
| Scleroderma Classification, No.(%) | |
| Limited | 15 (35.7%) |
| Diffuse | 27 (64.3%) |
| Cancer | 1 (2.4%) |
| Death | 8 (19.1%) |
| Renal crisis | 6 (14.3%) |
| Maximum Muscle Severity Score (N=42) | |
| 0 (Full Strength) | 0 (0%) |
| 1 (Mild proximal weaknss) | 26 (61.9%) |
| 2 (Moderate proximal weakness) | 9 (21.4%) |
| 3 (Severe proximal weakness) | 4 (9.5%) |
| Requires ambulation aids | 3 (7.1%) |
| Mean maximum mRSS ± SD (N=40) | 16.5 ± 12.8 |
| Mean maximum RVSP ± SD (N=33) | 48.2 ± 20.9 |
| Mean minimum EF ± SD (N=35) | 53.2 ± 10.1 |
| Mean minimum DLCO ± SD (N=40) | 56.4 ± 24.6 |
| Mean forced vital capacity (FVCˆ)± SD (N=41) | 65 ± 19.1 |
| Auto-antibody status | |
| Anti-nuclear antibody (ANA) (N=40) | |
| ANA negative | 2 (5%) |
| ANA positive titer 1:40-1:160 | 9 (22.5%) |
| ANA Titer ≥ 1:160 | 29 (72.5%) |
| Anti-centromere positive (N=41) | 6 (14.6%) |
| Anti-Scl 70 (N=39) | 4 (10.3%) |
| Anti-RNP (N=40) | 9 (22.5%) |
| Anti-Smith (N=40) | 0 (0%) |
| Anti-Ro (N=32) | 4 (12.5%) |
| Anti-La (N=32) | 0 (0%) |
| Anti-RNA Polymerase III (N=38) | 5 (12%) |
| Anti-PM-Scl (N=41) | 4 (9.8%) |
To determine if the subjects with muscle weakness who had a biopsy were different from those who did not have a biopsy, these two groups were compared. Using the maximum muscle severity score as a surrogate marker of muscle weakness, those who had a score > 1 were considered to be weak. Of the 2830 subjects in the database, 1711 had an available muscle severity score and 405/1711 (23.7%) were weak. Those who had biopsies had a younger mean age of scleroderma onset (49.1 ± 11.6 years vs. 54.4 ± 14.4 years, p=0.007) when compared to those who did not have a biopsy. Another statistically significant difference was a higher CK value (mean CK was 1166 + 2503 vs. 297 + 549, p<0.00001). There was no statistically significant difference in their disease duration, race, sex, disease subtype, maximum Rodnan skin score, or renal crisis.
Muscle weakness developed approximately 4 years after the first non-Raynaud's symptom and approximately 8 years after the onset of Raynaud's phenomenon (Table 3). 74% had an abnormal maximum CK (CK ≥ 200) (Table 3 and Supplemental Table 1). 30/34 (88.2%) had edema on MRI, suggesting ongoing muscle damage at the time of imaging. 10/34 (29.4%) had fatty replacement of muscle tissue on T1 sequences, a finding suggesting chronic and irreversible muscle damage (27). Table S2 includes EMG details from all 39 subjects who underwent this study. Overall, 19/39 (48.7%) had an irritable myopathy and 16/39 (41.3%) had a non-irritable myopathy; myopathic features were more common in proximal than distal muscles.
Table 3. Neuromuscular characteristics of scleroderma patients with myopathy.
| Disease duration of scleroderma(1st non-Raynaud's) | |
| At the time of muscle biopsy (years) | 6.0 ± 7.0 |
| At the onset of muscle weakness (years) | 4.2 ± 7.5 |
| Disease duration of scleroderma (Raynaud's) | |
| At the time of muscle biopsy (years) | 9.4 ± 18.3 |
| At the onset of muscle weakness (years) | 7.9 ± 19.3 |
| Lab values | |
| Elevated maximum CK (CK≥200) (31/42, 73.8%) | 2412 ± 3317 |
| Normal maximum CK (CK<200) (11/42, 26.2%) | 94.4 ± 56.1 |
| MRI Findings N=34 | |
| MRI edema | 30 (88.2%) |
| MRI fatty replacement | 10 (29.4%) |
| EMG results N=39 | |
| Irritable myopathy | 19 (48.7%) |
| Non-irritable myopathy | 16 (41.3%) |
| Normal | 4 (10.3%) |
| Nerve Conduction Results N=39 | |
| Abnormal sural study | 17 (43.5%) |
| Muscle biopsy features | |
| Inflammation (N=42) | 20 (47.6%) |
| Necrosis (N=42) | 28 (66.7%) |
| Fibrosis (N=42) | 14 (33.3%) |
| Acute neurogenic atrophy (N=41) | 20 (47.6%) |
| Histological categories based on aggregate biopsy features | |
| Polymyositis | 2 (4.8%) |
| Dermatomyositis | 3 (7.1%) |
| Non-specific myositis | 15 (35.7%) |
| Fibrosis only | 3 (7.1%) |
| Necrotizing | 9 (21.4%) |
| Acute denervation | 3 (7.1%) |
| Other | 7 (16.6%) |
Inflammation, necrosis, and acute neurogenic atrophy are the predominant histologic muscle biopsy features
Among the individual histologic features analyzed in 42 muscle biopsies, necrosis and inflammation were among the most prevalent, occurring in 67% (n=28) and 48% (n=20) of the biopsies, respectively (Table 3, Figure 1, and Supplemental Table 1). While fibrosis was seen in 33% (n=14) of biopsies, this feature was usually observed in conjunction with inflammation (n=20) and/or necrosis (n=28). Surprisingly, esterase positivity of angular atrophic fibers, a marker of acute neurogenic atrophy on biopsy, was seen in 48% (n=20) of the muscle biopsies. Interestingly, only 3 had evidence of fiber type grouping on ATPase stains, a feature suggesting chronic denervation with reinervation. Each of these also had evidence of acute neurogenic atrophy, suggesting an acute on chronic neurogenic process. As the high prevalence of acute neurogenic atrophy was unexpected, we evaluated 26 available dermatomyositis muscle biopsies stained for esterase read at our institution. Dermatomyositis was chosen because of its relative clinical homogeneity compared to patients with a diagnosis of polymyositis. 4/26 (15%) of dermatomyositis subjects had histologic evidence of acute neurogenic atrophy; this was significantly lower than the prevalence of this feature in scleroderma muscle biopsies (p=0.005).
Figure 1.
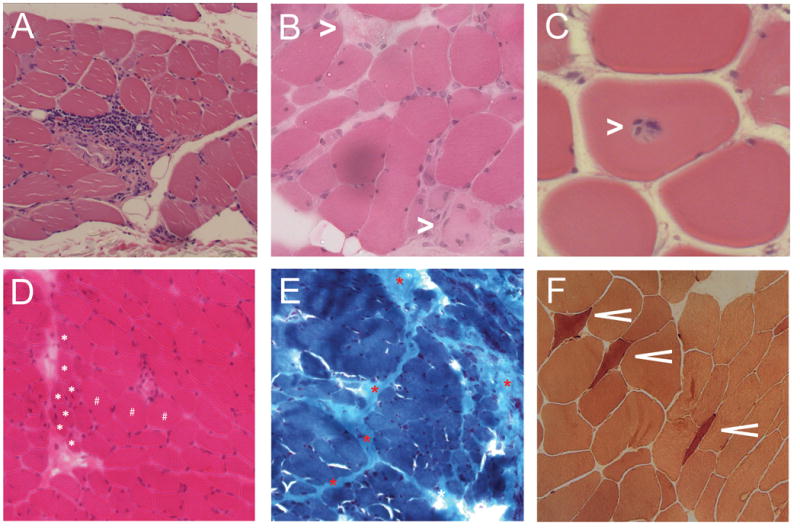
Examples of histologic features from muscle biopsies of weak scleroderma patients. (A) Perivascular inflammation with extension into the endomysium (H&E stain). (B) Necrotic muscle fibers (white asterisks; H&E stain). (C) Focal invasion of an apparently healthy myofiber by lymphocytes (H&E stain). (D) Perifascicular atrophy (white asterisks indicate several atrophic fibers localized to the perifascicular area; H&E stain). (E) Prominent perimysial fibrosis (red asterisks; Gomori trichrome stain). (F) Numerous esterase-positive angular atrophic fibers suggesting an acute neurogenic process (esterase stain).
We sought to determine whether a secondary etiology may have contributed to a necrotizing myopathy. Although 2 subjects had statin exposure, both had non-specific myositis rather than a necrotizing myopathy. In addition, 3 subjects were on hydroxychloroquine at the time of biopsy, but none had the vacuolar changes characteristic of hydroxychloroquine-associated myopathy. Finally, none had major electrolyte disorders or infections requiring hospitalization.
Exposure to potentially neurotoxic medications (e.g. cyclophosphamide, phenytoin, thalidomide, vinca alkaloids, and amiodarone) was obtained from chart review. Three patients had exposure to cyclophosphamide. One patient had this exposure ∼4 months prior to biopsy and the other 2 had exposure more than 2 years prior to biopsy. Evidence of a chronic neurogenic process (i.e, fiber type grouping) was observed in the patient with more recent cyclophosphamide exposure but not in those exposed more than 2 years prior to biopsy.
Clinically relevant co-morbidities to explain the neurogenic biopsy features (e.g., diabetes, Sjogren's, alcoholism, drug abuse, hepatitis, HIV, Lyme disease, or B12 deficiency) were obtained and none had any of these conditions.
Non-specific myositis and necrotizing myopathy are the predominant histopathologic categories
Histopathologic categories were assigned after considering all of the individual histologic features of each biopsy (as described in the Methods section.) The most common histopathologic categories were non-specific myositis (35.7%) and necrotizing myopathy (21.4%) (Table 3). Two subjects had polymyositis, 3 had dermatomyositis, 3 had “fibrosis only”, and 3 had acute denervation. Five had non-specific myofiber atrophy, and 2 had prominent COX negative fibers consistent with mitochondrial damage; these are classified as “other” in Table 2.
39/42 subjects had frozen muscle tissue available for immunostaining. More than half of the biopsies (56%) had sarcolemmal MHC I expression, a characteristic feature commonly seen in patients with autoimmune muscle disease (28). Among the immunostained biopsies, 18% had CD8-positive cells, and 15% had CD4-positive cells. Interestingly, the presence of infiltrating CD3-positive cells and CD8-positive cells associated with necrosis (p=0.02 and p=0.03, respectively). In contrast, CD4 did not associate with necrosis (p=0.28). Increased MHC-I expression associated with fibrosis (p=0.04), but not with inflammation (p=0.06) or necrosis (p=0.27).
Associations of muscle biopsy features and histopathologic categories with CK, EMG, and MRI findings
Inflammation and necrosis were associated with higher CK levels (Table 4). The CK at the time of muscle biopsy for those with inflammation was 1199 ± 3548 while those without inflammation had a CK of 187 ± 1212 (p=0.00006). Similarly, those with necrosis had a CK at biopsy of 915 ± 3187 and those without had a CK of 118 ± 131 (p=0.0001). Irritable myopathy did not associate with muscle biopsy features of inflammation or necrosis but this was not surprising because irritable myopathy is not specific or sensitive for an inflammatory histology (29). Edema or fatty replacement on MRI also did not associate with muscle biopsy features but only 34 had MRI of the muscles completed. Moreover, biopsy was obtained from the biceps or delotid in 35/42 (74%) whereas MRI was performed of the thighs only in 38/42 (90%).
Table 4. CK values (closest to biopsy) and association with histologic features and categories.
| CK if present (Median ± SD) | CK if absent (Median ± SD) | p * | |
|---|---|---|---|
| Histopathologic features | |||
| Inflammation (n=20) | 1199 ± 3548 | 187 ± 1212 | 0.00006 |
| Necrosis (n=28) | 915 ± 3187 | 118 ± 131 | 0.0001 |
| Fibrosis (n=14) | 490 ± 1436 | 304 ± 3212 | 0.96 |
| Acute Neurogenic Atrophy (n=20) | 428 ± 3668 | 223 ± 1530 | 0.49 |
| Histopathologic categories | |||
| Polymyositis (n=2) | 8710 ± 9407 | 554 ± 2173 | 0.06 |
| Dermatomyositis (n=3) | 1760 ± 1095 | 562 ± 3117 | 0.79 |
| Necrotizing myopathy (n=9) | 509 ± 2207 | 830 ± 3208 | 0.47 |
| Non-specific myositis (n=15) | 1467 ± 4108 | 369 ± 1427 | 0.0008 |
| Fibrosis only (n=3) | 168 ± 650 | 662 ± 3105 | 0.29 |
| Acute denervation (n=3) | 172 ± 78.2 | 748 ± 3126 | 0.05 |
The CK of those with non-specific myositis was 1467 ± 4108 versus 369 ± 1427 in those who did not have non-specific myositis (p=0.00008) (Table 4). Among the 15 subjects who had a non-specific myositis, 9 (60%) had an irritable myopathy and 11 (73%) had edema on MRI. Only 1 (6%) had fatty replacement on MRI. Of note, those with acute denervation had lower CK than those without this finding (p=0.05). The other histopathologic subgroups that were analyzed did not have any statistically significant associations with CK, EMG, or MRI findings.
The association of muscle biopsy features with scleroderma clinical features
Interestingly, patients with any fibrosis on muscle biopsy tended to have an increased risk of renal crisis compared to those without any fibrosis (28% vs. 7.1%; p=0.06), though this trend was not statistically significant. There was no association between fibrosis and RNA polymerase III antibodies.
Those who were of the diffuse cutaneous subtype had higher prevalence of acute neurogenic atrophy on muscle biopsy compared to those with the limited cutaneous subtype (p=0.04). Although 44% of the study population had electrophysiological evidence of a sensory neuropathy (i.e., decreased or absent sural amplitude), this finding did not correlate with evidence of neurogenic atrophy on muscle biopsy.
Four patients were positive for anti-PM-Scl antibodies, three of whom had the diffuse cutaneous subtype and one with the limited cutaneous subtype of scleroderma. Compared to PM-Scl negative patients, PM-Scl positive subjects more commonly had some histological evidence of fibrosis (p = 0.004). No anti-PM-Scl positive patient was in the “fibrosis only” category since each had other prominent histological features (e.g. inflammation or necrosis) as well. Anti-centromere, RNA-Polymerase III, Scl-70, and anti-RNP did not associate with muscle histology findings. Moreover, there was no association between the histopathological subgroups and scleroderma subtype, RVSP, DLCO, or FVC. There was also no association between individual histologic features on muscle biopsy and the presence of cancer.
Duration of scleroderma at the time of muscle biopsy is shorter in subjects with polymyositis, PM-Scl, and Scl-70 antibody
Subjects in the polymyositis histopathologic category had dramatically shorter disease duration at the time of muscle biopsy. Among those within this subgroup, the disease duration of scleroderma (defined by first non-Raynaud's symptom) at the time of biopsy was 0.68 ± 0.48 years versus 4.1 ± 7.0 years (p=0.05) for the other histological subgroups (Table 5). Those in the categories of non-specific myositis, “fibrosis only”, necrotizing myopathy, and dermatomyositis did not show different disease duration compared to other histology types. Disease duration was also shorter in those with anti-PM-Scl and Scl-70 antibodies at the time of muscle biopsy compared to those without this antibody (Table 5). In contrast, those with acute denervation (n=3) had a much longer disease duration at the time of biopsy than those without (p=0.04).
Table 5. Disease duration of scleroderma (1st non-Raynaud's symptom to the time of muscle biopsy) and its association with histopathologic categories on muscle biopsy and autoantibodies.
| Disease duration if present (in years) | Disease duration if absent (in years) | p-value | |
|---|---|---|---|
| Polymyositis (n=2) | 0.68 ± 0.48 | 4.1 ± 7.0 | 0.05 |
| Dermatomyositis (n=3) | 4.1 ± 0.34 | 3.8 ± 7.2 | 0.88 |
| Necrotizing myopathy (n=9) | 5.7 ± 5.9 | 3.7 ± 7.3 | 0.29 |
| Non-specific myositis (n=15) | 1.6 ± 3.8 | 4.3 ± 8.2 | 0.07 |
| Fibrosing myopathy (n=3) | 4.0 ± 4.1 | 3.9 ± 7.2 | 0.88 |
| Acute denervation (n=3) | 16.4 ± 1.3 | 3.9 ±6.7 | 0.04 |
| Anti-PM-Scl antibody (n=41) | 1.4 ± 1.6 | 6.6 ± 7.2 | 0.002 |
| Anti-RNP (n=40) | 7.0 ± 11.1 | 5.3 ± 5.1 | 0.67 |
| Anti-RNA polymerase 3 (n=38) | 7.0 ± 2.0 | 5.3 ± 7.1 | 0.27 |
| Anti-centromere (n=41) | 9.1 ± 7.8 | 5.6 ± 6.9 | 0.38 |
| Anti-Scl 70 (n=39) | 2.4 ± 1.7 | 6.1 ± 7.3 | 0.02 |
Discussion
This survey of a large well characterized scleroderma cohort was undertaken to determine if weak scleroderma subjects exhibited distinct muscle histopathologic features. We found that necrosis, inflammation, and acute neurogenic atrophy were the most common individual histologic features in 42 weak scleroderma subjects. The most common histopathologic categories on muscle biopsy were non-specific myositis (38.5%) and a necrotizing myopathy (21.4%). Of note, the presence of a necrotizing myopathy is common in patients with immune-mediated myopathies associated with anti-SRP and anti-HMGCR autoantibodies and has also been reported in the antisynthetase syndrome (30,31). Our findings highlight that a necrotizing muscle biopsy is relatively non-specific and may be found in scleroderma as well as other autoimmune myopathies. Unexpectedly, a high percentage of scleroderma muscle biopsies included angular atrophic fibers that stained positive for esterase (48%). This feature of acute neurogenic atrophy was far less common (15%) in dermatomyositis muscle biopsies. In sclerodema muscle biopsies, acute neurogenic atrophy was often found in the context of inflammation, necrosis, and/or fibrosis. Interestingly, those patients who had acute neurogenic atrophy on biopsy tended to be of the diffuse (80%) rather than limited subtype (20%). Although nerve conduction studies revealed a high prevalence (44%) of sensory neuropathy, there was no correlation of this electrophysiologic feature with the presence of acute neurogenic atrophy on muscle biopsy.
The current study is the first to provide histologic evidence that a neuropathic process may contribute to the underlying pathologic mechanism of weakness in scleroderma subjects with myopathy. Interestingly, Ringel et. al reported that 3/14 (21%) of weak scleroderma patients had electrophysiological evidence of sensorimotor neuropathy, suggesting a combination of neuropathic and myopathic processes in these patients (32). Similarly, Ranque and colleagues found that among weak scleroderma patients who underwent electrophysiological evaluation, 29% had a combination of neuropathic and myopathic findings (13). However, these studies did not include histological evidence of motor denervation on muscle biopsy.
Notably, only 3 patients with evidence of acute denervation had fiber type grouping, a feature seen with chronic denervation followed by reinervation. Fiber type grouping occurs when when a motor axon is destroyed and muscle fibers are reinnervated by a different axon. If, in subjects with scleroderma, damage occurs to the distal nerve terminal and there is subsequent reinnervation by the same nerve fiber, fiber type grouping might not occur. However, this mechanistic explanation is speculative and will require further study to confirm or refute.
Of note, although tissue fibrosis is a well-characterized mechanism of tissue injury in scleroderma, fibrosis in isolation was found in only 3 out of 42 (7.1%) patients. This was unexpected given that a previous study reported that 13/36 (36%) biopsies had fibrosis without inflammation or necrosis (15). This discrepancy may be related to the fact that our patients underwent muscle biopsy while living with active disease. In contrast, some patients in the prior study (19/36 or 52.7%) had muscle tissue examined only at autopsy when fibrosis may more likely be seen due to late stage muscle damage, or did not have clinically relevant muscle disease. It remains to be determined if fibrosis is a primary myopathic process in scleroderma or occurs secondary to chronic muscle damage from necrosis, inflammation, and/or a vasculopathy.
We determined the autoantibody status of most patients included in this study. We found that PM-Scl positive subjects underwent muscle biopsy sooner after the onset of scleroderma compared to patients without this autoantibody. This may indicate that the pathologic insult to the muscles occurred more quickly after scleroderma diagnosis. Anti-PM-Scl patients were also more likely to have evidence of muscle fibrosis compared to other patients, but this was exclusively in the presence of inflammation or necrosis. Of note, 3 of 4 anti-PM-Scl positive subjects had a maximum Medsger muscle severity score of 2 or greater, whereas, the proportion of anti-PM-Scl negative patients with Medsger muscle scores greater than 2 (6/37, 16.2%) was much lower. Taken together, these observations suggest that PM-Scl-positive patients may have unusually rapid onset of particularly severe skeletal muscle involvement soon after the diagnosis of scleroderma. However, our ability to draw conclusions about antibodies and their clinicopathological correlations is limited by the small number of patients studied and retrospective nature of the study.
Immunostaining of muscle biopsies demonstrated that CD 8 cells, CD 4 cells, and CD 20 cells were found in 18%, 15%, and 15% of the 39 muscle biopsies, respectively. This is in agreement with an earlier study of 11 scleroderma muscle biopsies showing that perimysial exudates consisting mostly of CD 8+ T cells and macrophages was the most common type of cellular infiltrate (33). In contrast, findings from another study showed that 3/4 of muscle biopsies in weak scleroderma patients were infiltrated predominantly by CD4-positive T cells (13). Additional immunostaining in our study revealed increased MHC I expression in more than half of the biopsies. Although positive MHC I staining (up to 92%) has been reported by Bhansing and colleagues in scleroderma patients with polymyositis overlap (14), this is the first report of increased MHC I expression in a more diverse collection of weak scleroderma subjects.
This study has several limitations. First, our institution is a tertiary referral center and consequently there may be some referral bias. Second, the retrospective nature of this study precludes drawing conclusions about causal or temporal relationships between the onset of muscle weakness and different histopathologic diagnoses on muscle biopsies. Third, although electrophysiologic and biopsy data suggest that scleroderma associated myopathy patients may also have neuropathic features, this study lacks appropriate control groups needed to determine whether these findings are unique to scleroderma patients with myopathy. We did, however, determine that the prevalence of acute neurogenic atrophy on biopsy was only 15% in dermatomyositis compared to 48% in weak scleroderma patients. Additional studies evaluating nerve function in an unbiased group of scleroderma subjects would be of interest. Finally, while our study population is the largest yet characterized, it is relatively small when attempting to make associations with histological subtypes and other clinical and serological features.
This is the largest analysis of the histologic and clinical features of weak scleroderma patients reported to date. Importantly, this study reveals that myopathy in scleroderma is not uniform, but rather forms a heterogeneous spectrum of disease states. Our study demonstrates that a variety of insults may be contributing to the development of myopathy in scleroderma and emphasizes the need to characterize the specific pathology on muscle biopsy. Future studies should follow to determine if clinical outcomes are impacted following specific targeted therapy.
Supplementary Material
Significance and Innovations.
Non-specific myositis and necrotizing myopathy are the most common histopathologic categories in weak scleroderma patients.
Acute neurogenic atrophy is seen in 48% of muscle biopsies suggesting motor denervation may be a common pathological process in weak scleroderma patients.
Myopathy occurs early after the onset of scleroderma in those with histopathologic diagnoses of polymyositis, and in those with anti-PM-Scl and Scl-70 antibodies.
Acknowledgments
We would like to acknowledge the Scleroderma Research Foundation for their support. We would like to acknowledge Noel Carter for performing the immunostaining of the muscle biopsy slides.
Support: Dr. Paik is supported by the Clinical to Research Transition Award from the Arthritis Foundation. Dr. Casciola-Rosen is supported by NIH grant R56AR062615. Drs. Mammen, Hummers and Casciola-Rosen are supported by funding from the Donald B. and Dorothy L. Stabler Foundation. The Johns Hopkins Rheumatic Disease Research Core Center, where some of the autoantibody assays were performed, is supported by NIH grant P30-AR-053503. The immunostaining of the muscle biopsy slides were supported by The Huayi and Siuling Zhang Discovery Fund. This research was supported [in part] by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health
References
- 1.Mammen AL. Scleroderma: from pathogenesis to comprehensive management. New York, New York: Springer; Myopathy in scleroderma; pp. 525–9. [Google Scholar]
- 2.West SG, Killian PJ, Lawless OJ. Association of myositis and myocarditis in progressive systemic sclerosis. Arthritis Rheum. 1981;24:662–667. doi: 10.1002/art.1780240506. [DOI] [PubMed] [Google Scholar]
- 3.Russell ML, Hanna WM. Ultrastructure of muscle microvasculature in progressive systemic sclerosis: relation to clinical weakness. J Rheumatol. 1983;10:741–747. [PubMed] [Google Scholar]
- 4.Averbuch-Heller L, Steiner I, Abramsky O. Neurologic manifestations of progressive systemic sclerosis. Arch Neurol. 1992;49:1292–1295. doi: 10.1001/archneur.1992.00530360094024. [DOI] [PubMed] [Google Scholar]
- 5.Hietaharju A, Jääskeläinen S, Kalimo H, Hietarinta M. Peripheral neuromuscular manifestations in systemic sclerosis (scleroderma) Muscle Nerve. 1993;16:1204–1212. doi: 10.1002/mus.880161110. [DOI] [PubMed] [Google Scholar]
- 6.Mimura Y, Ihn H, Jinnin M, Asano Y, Yamane K, Tamaki K. Clinical and laboratory features of scleroderma patients developing skeletal myopathy. Clin Rheumatol. 2005;24:99–102. doi: 10.1007/s10067-004-0975-7. [DOI] [PubMed] [Google Scholar]
- 7.Jung M, Bonner A, Hudson M, Baron M, Pope J, on behalf of the Canadian Scleroder Myopathy is a poor prognostic feature in systemic sclerosis: results from the Canadian Scleroderma Research Group (CSRG) cohort. Scand J Rheumatol. 2014;43:217–220. doi: 10.3109/03009742.2013.868512. [DOI] [PubMed] [Google Scholar]
- 8.Follansbee WP, Zerbe TR, Medsger TA. Cardiac and skeletal muscle disease in systemic sclerosis (scleroderma): a high risk association. Am Heart J. 1993;125:194–203. doi: 10.1016/0002-8703(93)90075-k. [DOI] [PubMed] [Google Scholar]
- 9.Fernández-Serna M, Arboleya L, Alonso S, Queiro R, Alperi M. Dropped head syndrome in a patient with scleromyositis. J Clin Rheumatol Pract Rep Rheum Musculoskelet Dis. 2013;19:32–34. doi: 10.1097/RHU.0b013e31827d8778. [DOI] [PubMed] [Google Scholar]
- 10.Zivković SA, Medsger TA. Myasthenia gravis and scleroderma: two cases and a review of the literature. Clin Neurol Neurosurg. 2007;109:388–391. doi: 10.1016/j.clineuro.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Ouhabi H, Bourazza A, Rouimi A, Boutaleb N, Mosseddaq R. Association of amyotrophic lateral sclerosis with scleroderma Study of 2 cases. Rev Neurol (Paris) 1997;153:790–791. [PubMed] [Google Scholar]
- 12.Varga J, Denton C, Wigley FM. Scleroderma: from pathogenesis to comprehnsive management. New York, New York: Springer; 2011. [Google Scholar]
- 13.Ranque B, Authier FJ, Le-Guern V, Pagnoux C, Berezne A, Allanore Y, et al. A descriptive and prognostic study of systemic sclerosis-associated myopathies. Ann Rheum Dis. 2009;68:1474–1477. doi: 10.1136/ard.2008.095919. [DOI] [PubMed] [Google Scholar]
- 14.Bhansing KJ, Lammens M, Knaapen HKA, van Riel PLCM, van Engelen BGM, Vonk MC. Scleroderma-polymyositis overlap syndrome versus idiopathic polymyositis and systemic sclerosis: a descriptive study on clinical features and myopathology. Arthritis Res Ther. 2014;16:R111. doi: 10.1186/ar4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medsger TA, Rodnan GP, Moossy J, Vester JW. Skeletal muscle involvement in progressive systemic sclerosis (scleroderma) Arthritis Rheum. 1968;11:554–568. doi: 10.1002/art.1780110405. [DOI] [PubMed] [Google Scholar]
- 16.Anonymous. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 1980;23:581–590. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 17.Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159:179–187. doi: 10.1164/ajrccm.159.1.9712108. [DOI] [PubMed] [Google Scholar]
- 18.Knudson RJ, Kaltenborn WT, Knudson DE, Burrows B. The single-breath carbon monoxide diffusing capacity. Reference equations derived from a healthy nonsmoking population and effects of hematocrit. Am Rev Respir Dis. 1987;135:805–811. doi: 10.1164/arrd.1987.135.4.805. [DOI] [PubMed] [Google Scholar]
- 19.Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25–34. doi: 10.1016/j.jaad.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Medsger TA, Silman AJ, Steen VD, Black CM, Akesson A, Bacon PA, et al. A disease severity scale for systemic sclerosis: development and testing. J Rheumatol. 1999;26:2159–2167. [PubMed] [Google Scholar]
- 21.Griggs R, Mendell J, Miller R. Evaluation of patients with myopathy. Philadelphia, PA: FA Davis; 1995. Evaluation and Treatment of Myopathies; pp. 17–78. [Google Scholar]
- 22.Veves A, Malik RA, Lye RH, Masson EA, Sharma AK, Schady W, et al. The relationship between sural nerve morphometric findings and measures of peripheral nerve function in mild diabetic neuropathy. Diabet Med J Br Diabet Assoc. 1991;8:917–921. doi: 10.1111/j.1464-5491.1991.tb01530.x. [DOI] [PubMed] [Google Scholar]
- 23.Olsen NJ, Qi J, Park JH. Imaging and skeletal muscle disease. Curr Rheumatol Rep. 2005;7:106–114. doi: 10.1007/s11926-005-0062-3. [DOI] [PubMed] [Google Scholar]
- 24.Napier N, Shortt C, Eustace S. Muscle edema: classification, mechanisms, and interpretation. Semin Musculoskelet Radiol. 2006;10:258–267. doi: 10.1055/s-2007-971997. [DOI] [PubMed] [Google Scholar]
- 25.Young IR, Bydder GM. Magnetic resonance: new approaches to imaging of the musculoskeletal system. Physiol Meas. 2003;24:R1–23. doi: 10.1088/0967-3334/24/4/r01. [DOI] [PubMed] [Google Scholar]
- 26.Thompson SW. Selected Histochemical and histopathological methods. Charkles C Thomas; 1974. [Google Scholar]
- 27.Kuo GP, Carrino JA. Skeletal muscle imaging and inflammatory myopathies. Curr Opin Rheumatol. 2007;19:530–535. doi: 10.1097/BOR.0b013e3282efdc66. [DOI] [PubMed] [Google Scholar]
- 28.Jain A, Sharma MC, Sarkar C, Bhatia R, Singh S, Handa R. Major histocompatibility complex class I and II detection as a diagnostic tool in idiopathic inflammatory myopathies. Arch Pathol Lab Med. 2007;131:1070–1076. doi: 10.5858/2007-131-1070-MHCCIA. [DOI] [PubMed] [Google Scholar]
- 29.Lyu RK, Cornblath DR, Chaudhry V. Incidence of irritable electromyography in inflammatory myopathy. J Clin Neuromuscul Dis. 1999;1:64–67. doi: 10.1097/00131402-199912000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Mehndiratta P, Mehta S, Manjila SV, Kammer GM, Cohen ML, Preston DC. Isolated necrotizing myopathy associated with ANTI-PL12 antibody. Muscle Nerve. 2012;46:282–286. doi: 10.1002/mus.23383. [DOI] [PubMed] [Google Scholar]
- 31.Meyer A, Messer L, Goetz J, Lannes B, Weber JC, Geny B, et al. Immune-mediated necrotizing myopathies are serologically heterogeneous and autoantibodies may predict their clinical phenotype: two cases associated with anti-Pl7 antibodies. Scand J Rheumatol. 2014;43:81–83. doi: 10.3109/03009742.2013.864421. [DOI] [PubMed] [Google Scholar]
- 32.Ringel RA, Brick JE, Brick JF, Gutmann L, Riggs JE. Muscle involvement in the scleroderma syndromes. Arch Intern Med. 1990;150:2550–2552. [PubMed] [Google Scholar]
- 33.Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies I: Quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol. 1984;16:193–208. doi: 10.1002/ana.410160206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.