

Abstract
Hypocalcemia and hyperphosphatemia are encountered in idiopathic hypoparathyroidism (IHP) and pseudohypoparathyroidism type Ib (PHP1B). In contrast to PHP1B, which is caused by resistance toward parathyroid hormone (PTH), the genetic defects leading to IHP impair production of this important regulator of mineral ion homeostasis. So far, only five PTH mutations were shown to cause IHP, each of which is located in the hormone’s pre-pro leader segment and thus impair hormone secretion. In three siblings affected by IHP, we now identified a homozygous arginine-to-cysteine mutation at position 25 (R25C) of the mature PTH(1-84) polypeptide; heterozygous family members are healthy. Depending on the assay used for evaluating these patients, plasma PTH levels were either low or profoundly elevated, thus leading to ambiguities regarding the underlying diagnosis, namely IHP or PHP1B. Consistent with increased PTH levels, recombinant [Cys25]PTH(1-84) and wild-type PTH(1-84) were secreted equally well by transfected COS-7 cells. However, synthetic [Cys25]PTH(1-34) was found to have a lower binding affinity for the PTH receptor type-1 (PTH1R) than PTH(1-34) and consequently a lower efficiency for stimulating cAMP formation in cells expressing this receptor. Consistent with these in vitro findings, long-term infusion of [Cys25]PTH(1-34) resulted only in minimal calcemic and phosphaturic responses, despite readily detectable levels of [Cys25]PTH(1-34) in plasma. The mineral ion abnormalities observed in the three IHP patients are thus most likely caused by the inherited homozygous missense PTH mutation, which reduces bioactivity of the secreted hormone. Based on these findings, screening for PTH(1-84) mutations should be considered when clinical and laboratory findings are consistent with PHP1B, but GNAS methylation changes have been excluded. Differentiating between IHP and PHP1B has considerable implications for genetic counseling, therapy, and long-term outcome because treatment of IHP patients with inappropriately high doses of active vitamin D and calcium can contribute to development of nephrocalcinosis and chronic kidney disease.
Keywords: IDIOPATHIC HYPOPARATHYROIDISM, PSEUDOHYPOPARATHYROIDISM TYPE IB, PTH(1-84) MUTATION
Introduction
Idiopathic hypoparathyroidism (IHP), namely hypocalcemia and hyperphosphatemia without additional developmental defects, can be the result of inadequate production of bioactive PTH. Molecular causes of IHP include activating mutations in the calcium-sensing receptor (CaSR), which lead to a reduction in PTH secretion by the parathyroid glands(1–3) and, less frequently, mutations in the parathyroid-specific transcription factor GCM2(4–10) or in GNA11, which encodes Gα11, one of the G protein α-subunits that mediate signaling at the CaSR.(11–13) Other well-defined causes of IHP are five different mutations in the PTH gene itself that are all located in the hormone’s prepro leader segment and thus impair hormone synthesis or secretion.(3,14–17)
Hypocalcemia and hyperphosphatemia are also observed in patients affected by pseudohypoparathyroidism type Ia (PHP1A) or pseudohypoparathyroidism type Ib (PHP1B). Both PHP variants are caused by mutations on the maternal allele of GNAS, a complex genetic locus on chromosome 20q13.3 that undergoes parent-specific methylation at multiple sites and gives rise to several different transcripts.(18–21) Although the promoter giving rise to transcripts encoding the alpha-subunit of the stimulatory G protein (Gαs) is not methylated, this critically important effector at the PTH receptor type-1 (PTH1R) and numerous other G protein-coupled receptors appears to be derived in the proximal renal tubular cells and few other tissues predominantly from the maternal allele.(22–24) Thus, maternal mutations that impair biological activity of Gαs protein itself cause PHP1A,(19–21) whereas maternal deletions that affect GNAS methylation and hence Gαs transcription cause PHP1B.(25–32) However, most PHP1B cases with GNAS methylation changes are sporadic,(33–38) and only those affected by paternal uniparental disomy involving the chromosome 20q13 region have thus far been defined at the molecular level.(28,39–42)
In contrast to IHP, PHP1A and PHP1B are both associated with elevated circulating levels of bioactive PTH.(18–21) PHP1A patients are readily recognized by skeletal and developmental abnormalities that are referred to as Albright’s hereditary osteodystrophy (AHO), ie, features that are only rarely observed in patients affected by PHP1B.(29,43–45) Because obvious physical abnormalities are usually lacking, individuals affected by PHP1B are often not diagnosed until the second decade of life when symptomatic hypocalcemia develops.(34) Testing for changes in GNAS methylation patterns, which establish PHP1B as the underlying defect, is not available in most clinical laboratories. As a result, measurement of the blood PTH concentration is the diagnostic test usually performed to discriminate between PHP1B and IHP because the two conditions are typically associated with high and low PTH levels, respectively.
Differentiating between PHP1B and IHP is critical because very different therapeutic interventions need to be followed for both disorders. In PHP1B, PTH-resistance is limited to the proximal renal tubules; the usually profound PTH elevations encountered in this disorder can thus readily increase bone resorption, thereby mobilizing calcium from the skeleton. Increased PTH levels, furthermore, enhance the rate of calcium reabsorption at the distal tubules.(46,47) This lowers urinary calcium excretion and allows treatment of PHP1B patients with oral doses of calcium and activated vitamin D that are high enough to normalize blood calcium concentration. In IHP, on the other hand, where bioactive PTH levels are low, urinary calcium excretion is inappropriately normal or elevated, such that overly aggressive treatment with calcium and activated vitamin D can increase the risk for nephrocalcinosis and chronic kidney disease.(48)
We now report a Korean family with three members showing biochemical findings consistent with either PHP1B or IHP, in that hypocalcemia and hyperphosphatemia were present with either considerably elevated PTH levels, as measured by one immunometric PTH assay, or with low-normal PTH levels, as measured by two other such assays. Genetic analyses revealed that all three patients carry a novel, homozygous PTH mutation, which reduces hormonal bioactivity in vitro and in vivo, and dramatically impairs detection of the mutant hormone in some immunometric PTH assays. Our findings highlight that it can be difficult to discriminate between IHP and PHP1B on the basis of immunoreactive PTH levels alone.
Materials and Methods
Peptide synthesis and quantification
Human PTH(1-34), [Cys25]PTH(1-34), and [Nle8,21, Tyr34] rat PTH(1-34) (rPTH(1-34)) were synthesized by the MGH Biopolymer Core facility, as described.(49) Peptide quality was verified by analytical HPLC and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry; peptide concentrations in stock solutions were established by optical absorbance at 280 nM (NanoDrop Spectrophotometer, Thermo Scientific, Wilmington, DE, USA).
Enzyme-linked immunometric sandwich assays (ELISA) for PTH detection
PTH levels were measured in EDTA plasma with two commercially available ELISA kits that use the same polyclonal goat anti-PTH capture antibody affinity-purified with immobilized human PTH(39-84). For detection, two different HRP-conjugated anti-PTH antibodies were used that had been affinity-purified using either immobilized human PTH(1-3) or PTH(13-34), thus measuring only full-length PTH (PTH(1-84) assay) or a combination of full-length and N-truncated PTH (ntPTH assay), respectively (Immutopics, Inc., San Clemente, CA, USA).(50) The ADVIA Centaur assay for PTH measurement (Siemens Healthcare Diagnostics, Tarrytown, NY, USA) (PTH-S) also employs a capture antibody affinity-purified with immobilized PTH(39-84), whereas the detection antibody was affinity-purified with immobilized PTH(1-34).(51) PTH analogs infused into mice were measured by an ELISA specific for human PTH(1-34);(52) cross-reactivity with [Cys25]PTH(1-34) was 36.4 ± 3.6% (n =3); peptide concentrations were read off the respective standard curves.
Plasma CTX measurement
C-terminal collagen cross-links (CTX) concentration was measured by ELISA (Immunodiagnostic Systems, Fountain Hills, AZ, USA) using plasma collected from mice 4 days after starting the infusions.
Ellsworth-Howard test
After an overnight fast, patients began drinking 250 mL/h and urine was collected for 1 hour before and hourly after sc injection of recombinant human PTH(1-34) (40 μg; Forteo, Lilly, Indianapolis, IN, USA) for measurement of cAMP, phosphate, and creatinine.
Molecular genetic studies
Informed consent was obtained from participants after study approval by the Institutional Review Board of Gachon University Gil Medical Center, Incheon, South Korea (GIRBA2151). Genomic DNA was extracted by standard methods from EDTA blood. Sanger sequencing of PTH, CASR, and GCM2, and whole exome sequencing were performed as described.(10,12) Animal studies were approved by the MGH Institutional Animal Care and Use Committee (IACUC) (2004N000205).
PTH expression vectors and mutagenesis
The Cys25 mutation was introduced into a plasmid encoding wild-type, preproPTH (PTH(1-84)), as described,(7) to generate the plasmid encoding [Cys25]PTH(1-84).
Cell culture and functional assays
COS-7 cells were cultured and transiently transfected with plasmids encoding wild-type PTH(1-84) or the [Cys25]PTH(1-84) mutant, as described.(7) Forty-eight hours after transfection, PTH levels were measured in conditioned medium using ntPTH and PTH(1-84) assays. Competition binding assays were performed using membranes prepared from GP-2.3 cells, which are HEK-293 cells stably expressing the wild-type human PTH1R and the Glosensor cAMP reporter,(53) and 125I-rPTH(1-34) radioligand tracer, as described.(54) cAMP assays were performed in SGS-72 cells, which are SaOS-2 cells stably transfected to express the Glosensor cAMP reporter using an Envision (PerkinElmer, Waltham, MA, USA) plate reader to measure intracellular cAMP-dependent, luciferase-based luminescence, as described.(55) Signaling via the PLC/IP3 pathway was assessed using GP-2.3 cells; the cells were challenged with PTH(1-34) or [Cys25]PTH(1-34) (30 minutes, 37°C), and the resulting intracellular inositol monophosphate (IP1) levels were measured using the IP-One Tb assay kit (CisBio, Bedford, MA, USA).
In vivo testing of PTH(1-34) and [Cys25]PTH(1-34)
Acute injection
PTH(1-34) or [Cys25]PTH(1-34) were diluted in 0.05% Tween 80, 10 mM citric acid, and 150 mM NaCl (pH 5.0), and injected iv at 80 μg/kg into 9-week-old male C57BL6 mice; control mice received only vehicle. Plasma and urine cAMP were measured, as described,(55) before and 15 minutes after peptide or vehicle injection. Blood ionized calcium was determined 2 hours after injection of peptide or vehicle.
PTH analog infusions
PTH(1-34) or [Cys25]PTH(1-34) (40 or 120 μg/kg/d) were infused, as described,(52) for 4 days by Alzet osmotic minipumps (Model 1002, DURECT Corporation, Cupertino, CA, USA) into 7-week-old male C57BL6 mice; control animals received vehicle. Both peptides were diluted in a solution of 150 mM NaCl, 1 mM HCl, and 2% heat-inactivated mouse serum. Blood ionized calcium and plasma phosphate levels were determined before and daily after starting the infusion.
To determine the biological activity of circulating PTH(1-34) or [Cys25]PTH(1-34), pooled plasma collected from mice infused with either ligand was incubated with GP2 cells (stable hPTH1R expression) and cAMP-dependent increases in luminescence were measured, as described.(55)
For some experiments, heparinized plasma was collected before and daily after minipump implantation. Furthermore, aliquots of the peptide solution in each minipump were recovered before implantation and stored at −80°C. Four days after implantation into an interscapular subcutaneous pocket, aliquots were obtained from each pump and stored at −80°C; aliquots from pumps loaded with vehicle served as controls. All samples were evaluated in the same assays.
Quantitative real-time PCR (qPCR)
RNA was TRIzol extracted from kidneys, 4 days after infusion with either vehicle or PTH analogs. After reverse-transcription using Superscript VILO cDNA Synthesis Kit (Invitrogen, Carlsbad, CA, USA), gene expression by qPCR was assessed using a SYBR Green PCR Kit (Invitrogen). Relative expression for each gene was calculated by the 2−ΔΔCT method with Gapdh for normalization.
Statistical analyses
In vitro data
Data were processed using Excel 2008 (Microsoft Corp., Redmond, WA, USA) and Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA). Aggregate data are expressed as mean ± SEM. Curves were fit to the data using a four-parameter, no-linear regression function. Statistical analyses were performed using the Student’s t test (two-tailed, unequal variances).
In vivo data
All values are expressed as mean ± SEM. Statistical analyses were performed using unpaired, 2-tailed Student’s t tests for comparisons between 2 groups and 1-way ANOVA and Fisher’s protected least significant difference tests for comparison of more than 2 groups.
Results
Investigated patients
The Korean proband (Fig. 1A, individual II-3) reported herein was first diagnosed with hypocalcemia in his early teens when he presented with transient muscle cramps and sudden loss of consciousness; seizures were noted in his late twenties. After reevaluation at age 55 years because of persistent hypocalcemia and hyperphosphatemia, the dose of calcium carbonate was increased and treatment with Rocaltrol was started. One of the proband’s younger twin sisters (II-5) was stillborn at term, and the surviving twin (II-6) suffers from severe mental retardation, most likely from perinatal complications. Sisters II-8 and II-10 exhibited severe hypocalcemia but were otherwise asymptomatic when evaluated for the current study at ages 50 and 47 years, respectively (Table 1). The parents had no clinical or laboratory evidence for hypocalcemia and were not knowingly consanguineous. However, their ancestors have lived for generations on the same remote Yellow Sea island, which raised the possibility that the hypocalcemia observed in affected family members was caused by a recessive mutation.
Fig. 1.
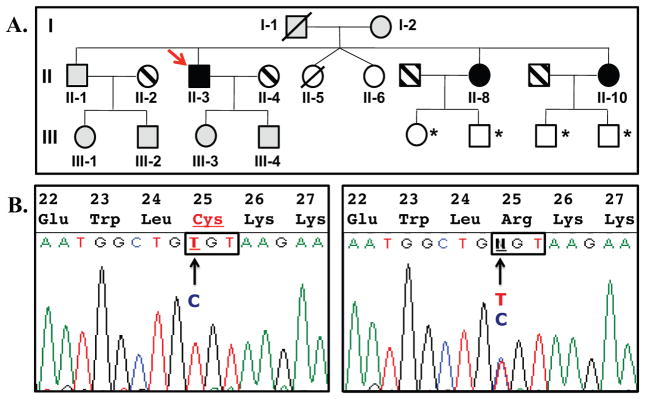
Pedigree of the investigated family and partial nucleotide sequence of PTH for the proband and his mother. (A) The proband (II-3) and his two affected sisters (II-8 and II-10) are carriers of a homozygous PTH mutation (black symbols); their mother (I-2), the brother (II-1), the proband’s son (III-4) and daughter (III-3), as well as niece (III-1) and nephew (III-2) are carriers of the heterozygous mutation (gray symbols). Striped symbols =unrelated spouses; white symbols = not available for testing. Note that the deceased father of the proband (I-1) is predicted to have been a heterozygous carrier. All heterozygous carriers of the PTH mutation, who were available for testing, showed no evidence for biochemical or radiographic abnormalities, indicating that hypocalcemia follows in this family an autosomal recessive trait. *Healthy, not tested;/, deceased. (B) Nucleotide sequence analyses of the gene encoding PTH revealed for the proband II-3 a homozygous c.166C>T mutation (left panel), which replaces arginine at position 25 with cysteine ([R25C]PTH) (partial nucleotide sequence and amino acid sequence are shown; codon 25 is boxed). His healthy mother I-2 is heterozygous for the mutation (right panel).
Table 1.
Age, Sex, Genetic, and Laboratory Findings for the Different Family Members Investigated for This Study
| Family member | Age (years)/sex | PTH R25C |
Ca | P | iCa | 25(OH) Vit D |
1,25(OH) 2 Vit D |
24-h urine Ca |
PTH-S | ntPTH | PTH(1-84) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference range | 8.2~10.8 (mg/dL) | 2.5~4.7 (mg/dL) | 1.05~1.35 (mmol/L) | 4.8~52.8 (ng/mL) | 25.1~66.1 (pg/mL) | 100~300 (mg/d) | 10~65 (pg/mL) | 10~65 (pg/mL) | 9–39 (pg/mL) | ||
| I-2 | 81/F | Het | 9.2 | 4.2 | 1.22 | 102.0 | 34.2 | ||||
| II-1 | 59/M | Het | 9.1 | 4.2 | 1.2 | 92.5 | 40.7 | ||||
| II-2 | 57/F | WT | 9.2 | 4.1 | 1.18 | 96.0 | 25.5 | ||||
| II-3 | 55/M | Hom | 4.8 | 8.1 | 0.35 | 17.1 | 30.7 | 15.2 | <5 | ||
| II-4 | 55/F | WT | 9.3 | 3.8 | 1.26 | 19.0 | 65.5 | 60.0 | 33.2 | ||
| II-8 | 50/F | Hom | 7.1 | 4.8 | 0.93 | 16.4 | 26.2 | 98.8 | 89.7 | 7.6 | 1455 |
| II-10 | 47/F | Hom | 6.5 | 6.2 | 0.74 | 17.3 | 23.9 | 26.4 | 24.1 | 4.6 | 679 |
| III-1 | 32/F | Het | 9.3 | 4.2 | 1.24 | 97.5 | 27.6 | ||||
| III-2 | 30/M | Het | 9.2 | 4.1 | 1.22 | 104.5 | 32.6 | ||||
| III-3 | 28/F | Het | 9.4 | 4.1 | 1.28 | 17.2 | 32.7 | 89.0 | 25.1 | ||
| III-4 | 26/M | Het | 9.7 | 4.4 | 1.25 | 17.7 | 50.2 | 95.7 | 29.8 |
Measurement of PTH and PTH-stimulated cAMP and phosphate excretion
The proband’s blood PTH levels were found to be inadequately low, relative to the profound hypocalcemia, when first evaluated by the “PTH-S” assay at the age of 55 years (Table 1). The PTH levels of the two affected yet untreated sisters, II-8 and II-10, were subsequently measured by the “PTH-S” assay and two additional commercially available kits (Fig. 2). Both were found to have normal or slightly elevated PTH levels in the “PTH-S” assay, subnormal levels in the “ntPTH” assay, and extremely elevated levels in the “PTH(1-84)” assay. Administration of synthetic PTH(1-34) increased urinary cAMP excretion in the three affected family members to the same extent as in healthy controls(56) (34.8 ± 10-fold over baseline at 60 minutes postinjection; mean ± SEM), and the peptide, furthermore, induced a normal increase in urinary phosphate excretion (4.34 ± 1.5-fold over baseline at 120 minutes postinjection). Thus, PTH1R-mediated Gαs signaling and downregulation of phosphate reabsorption in renal proximal tubules were intact.
Fig. 2.
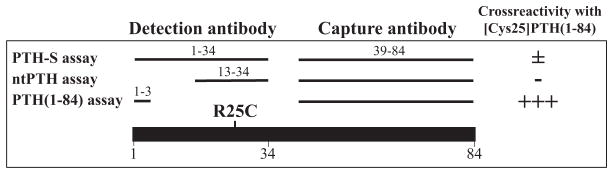
Schematic representation of the PTH fragments used for affinity purification of the antibodies employed in different PTH assays. For ligand capture, all three PTH assays use similar antibodies that were affinity-purified with immobilized PTH(39-84). The detection antibodies were affinity-purified either with immobilized PTH(1-34) (PTH-S assay), PTH(13-34) (ntPTH assay), or PTH(1-3) (PTH(1-84) assay). Overall cross-reactivity with [Cys25] PTH(1-84) in the three assays is indicated by − and +.
Genetic analyses
Nucleotide sequencing of II-3, II-8, and II-10 excluded mutations in CaSR and GCM2 but revealed a homozygous nucleotide transition in PTH exon 3 (c.166C>T), which had previously been identified in a single unrelated individual as a heterozygous variant (rs199955107).(57) The unaffected mother as well as available unaffected siblings and children were each heterozygous for the same nucleotide change. The identified PTH mutation results in substitution of arginine-25 by cysteine, ie, an amino acid residue known to interact with the receptor’s ligand-binding region(54) (Fig. 1B). Whole exome sequencing of DNA samples from the three affected siblings did not identify any other genetic variant that could be disease-causing (data not shown).
Efficient secretion of the mutant PTH from transfected COS-7 cells
To assess whether the identified mutation alters hormone secretion, we measured PTH levels in conditioned medium from COS-7 cells transfected with plasmids encoding either wild-type PTH(1-84) or [Cys25]PTH(1-84). Although the ntPTH assay detected markedly lower levels of the Cys25 mutant than the wild-type hormone (30.9 ± 23 and 2285 ± 277 pg/mL, respectively), the PTH(1-84) assay detected equal levels of both (1812 ± 175 and 1812 ± 94 pg/mL, respectively). Thus, the mutant hormone was efficiently secreted into the medium, and, moreover, it reacted poorly in the ntPTH assay. These findings are consistent with the divergent PTH levels measured with both assays in the two affected sisters (Table 1).
In vitro biological properties of the PTH mutant
The mutant peptide [Cys25]PTH(1-34) was approximately threefold weaker than PTH(1-34) for binding to the PTH1R in membranes and for stimulating cAMP formation in a human osteoblast-derived SaOS-2 cell line transfected to stably express a glosensor cAMP reporter (Fig. 3A, B; Table 2). In time-course experiments, PTH(1-34) and [Cys25]PTH(1-34) induced comparable increases in cAMP over the first 10 minutes after ligand addition, but then, after washout of unbound ligand, the cAMP signal in cells treated with [Cys25]PTH(1-34) decayed faster than that in cells treated with PTH(1-34) (Fig. 3C). No significant effect of the mutation on signaling via the PLC/IP3 pathway was detected (Table 2). Overall, the effects of the Arg25-->Cys mutation on PTH hormone action as observed in these binding and cAMP signaling studies are consistent with residue 25 of the ligand being situated in or near the portion of the ligand that forms critical contacts with the amino-terminal extracellular domain (ECD) portion of the receptor (Fig. 3D).(58)
Fig. 3.

Effects of the Cys25 mutation on binding to and activation of the PTH1R. (A) Binding of PTH(1-34) and [Cys25]PTH(1-34) to the human PTH1R was assessed by competition methods in membranes prepared from GP-2.3 cells (HEK-293 cells stably expressing the hPTH1R) using 125I-[Nle8,21, Tyr34] ratPTH(1-34)NH2 as tracer radioligand. (B) Ligand potency for cAMP signaling was assessed in SGS-72 cells, which are SaOS-2-derived cells that stably express the luciferase-based cAMP reporter. The cells were preloaded with luciferin, treated with varying concentrations of PTH(1-34) or [Cys25]PTH(1-34). Peak luminescence responses, occurring 10 to 15 minutes after ligand addition, were measured in a plate reader and plotted versus ligand concentration. (C) cAMP signaling before and after ligand wash-out was also assessed in SGS-72 cells. The cells were preloaded with luciferin, then treated with PTH(1-34) or [Cys25]PTH(1-34) (0.3 nM) and luminescence was measured for 16 minutes; the cells were then removed from the plate reader, rinsed twice to remove unbound ligand, and after replenishing with buffer containing luciferin, luminescence was measured for an additional 120 minutes. The gap in the x-axis indicates the time of wash-out. Data are means (±SEM) of 4 (A, B) or 5 (C) experiments, each performed in duplicate. Curve fitting parameters for dose-response data are presented in Table 2. (D) Hypothetical model of PTH(1-34) bound to the PTH1R. The receptor is colored gray and the ligand is colored yellow, except for Arg25, which is colored red. The model was constructed using the crystal structure of the PTH1R amino-terminal extracellular domain (ECD) in complex with the PTH(15-34) fragment (protein data base file 3C4M(58)) and that of the transmembrane domain region (TMD) of the related CRFR1 (PDB file 4K5Y(66)), which was used as a template for the PTH1R1 TMD region. The PTH(1-14) segment, in a helical conformation, was docked to the TMD region, and the ECD•PTH(15-34) and TMD•PTH(1-14) components were aligned manually; several residues at the extracellular ends of helices II and III of the TMD region were removed to permit better views of the ligand.
Table 2.
Effects of the Cys25 Mutation on PTH1R Binding, cAMP, and IP3 Signaling
| Binding to PTH1Ra
|
cAMP signalingb
|
PLC/IP signalingc
|
|||
|---|---|---|---|---|---|
| IC50 (nM)d | EC50 (nM)d | Max. (cps)e | EC50 (nM)d | Max. (nanomole/well)e | |
| PTH(1-34) | 4.33 ± 0.07 | 0.095 ± 0.015 | 178 ± 8 | 857 ± 165 | 1252 ± 297 |
| [Cys25]PTH(1-34) | 12.3 ± 0.2 | 0.270 ± 0.02 | 186 ± 8 | 265 ± 14 | 1036 ± 167 |
| p =0.0007f | p =0.002f | p =0.5f | p =0.07f | p =0.6f | |
Competition binding assays were performed in membranes prepared from GP-2.3 cells (HEK293-derived cells stably expressing Glosensor cAMP reporter and hPTH1R) using 125I-PTH(1-34) as tracer radioligand (n = 4).
cAMP dose-response assays were performed in SGS-72 cells (SaOS-2-derived cells stably expressing the Glosensor cAMP reporter, n = 4).
PLC/IP3 assays were performed in GP-2.3 cells using the Cisbio IP1 assay kit (n = 3).
Potency terms for binding (IC50) and signaling (EC50) are expressed as nM ligand concentrations.
Maximum (Max.) values indicate the peak response as luminesence counts per second (cps, cAMP-glosensor) or nanomole IP1 (PLC/IP3); the basal value for cAMP was 5930 ± 469 cps and that for IP1 was 28 ± 4 nanomole/well).
p = statistical comparison to PTH(1-34).
Acute injection and continuous infusion of wild-type and mutant PTH(1-34)
Single injection of high-dose PTH(1-34) and [Cys25]PTH(1-34) into mice led to comparable increases in plasma and urine cAMP levels, and both peptides had a similar calcemic effect (Table 3). However, although mice continuously infused for 4 days with PTH(1-34) via Alzet osmotic minipumps showed significant increases in blood ionized calcium, infusion of [Cys25]PTH(1-34) led to significantly smaller increases in blood ionized calcium levels (Fig. 4A–D). Differences in stability of PTH(1-34) and [Cys25]PTH(1-34) within the infusion pumps were excluded because the potencies of both peptides were similar when recovered from the pumps 4 days after implantation (Supplemental Fig. S1). Consistent with a lower bioactivity of the mutant peptide, plasma samples obtained from mice on days 1–2 and 3–4 (pooled from 2 consecutive days) were inactive for stimulating a cAMP response when applied to HEK293 cells stably expressing the PTH1R (hPTH1R/glosensor cells). In contrast, plasma samples obtained from mice infused with PTH(1-34) stimulated a robust cAMP response (Fig. 5A). The lack of cAMP-stimulating activity in plasma from the [Cys25]PTH(1-34)-infused mice was not owing to a more rapid rate of disappearance of the peptide from the circulation because [Cys25]PTH(1-34) was readily detectable in plasma by immunoassay (Fig. 5B).
Table 3.
In Vivo Response in Mice to an Acute Challenge With Either PTH(1-34) or [Cys25]PTH(1-34)
| Plasma cAMP (pmol/mL)
|
Urine cAMP (pmol/mg creat)
|
Blood Ca2+ (mM)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Pre-injection | 15 minutes post | t test (versus vehicle) | Pre-injection | 15 minutes post | t test (versus vehicle) | Pre-injection | 2 hours post | t test (versus vehicle) | |
| PTH(1-34) | 47 ± 2.8 | 178 ± 9.0 | 0.000 | 44 ± 1.8 | 291 ± 61.1 | 0.004 | 1.23 ± 0.01 | 1.30 ± 0.01 | 0.010 |
| [Cys25]PTH(1-34) | 47 ± 2.9 | 164 ± 28.4 | 0.001 | 39 ± 1.7 | 224 ± 63.1 | 0.130 | 1.24 ± 0.01 | 1.32 ± 0.02 | 0.003 |
| vehicle | 44 ± 1.7 | 28 ± 3.4 | 44 ± 2.9 | 40.2 ± 14.0 | 1.23 ± 0.01 | 1.22 ± 0.02 | |||
Plasma and urine cAMP levels 15 minutes after injection of wild-type PTH(1-34), [Cys25]PTH(1-34), or vehicle (80 μg/kg BW, n = 4–6/group); blood ionized calcium levels 2 hours after injection. Plasma cAMP, PTH(1-34) versus [Cys25]PTH(1-34): p = 0.660; Urine cAMP, PTH(1-34) versus [Cys25]PTH(1-34): p = 0.786; Blood Ca2+, PTH(1-34) versus [Cys25]PTH(1-34): p = 0.279.
Fig. 4.

Effect of continuous PTH infusion on blood ionized calcium and serum phosphate concentration. Male C57BL/6J mice at the age of 7 weeks received continuous infusions of vehicle or PTH analogs at a dose of 40 μg/kg/d (A, C) or 120 μg/kg/d (B, D) for 4 days. Infusion of PTH(1-34) resulted in a sustained increase in blood ionized calcium from 1.18 ± 0.012 to 1.53 ± 0.055 mmol/L and from 1.20 ± 0.013 to 2.52 ± 0.159 mmol/L, respectively. In contrast, infusion of [Cys25]PTH(1-34) revealed much smaller calcium increases from 1.18 ± 0.012 to 1.24 ± 0.012 mmol/L and from 1.20 ± 0.013 to 1.36 ± 0.053 mmol/L, respectively. Likewise, in comparison to PTH(1-34), both doses of the infused [Cys25]PTH(1-34) led to a severely blunted hypophosphatemic effect.
Fig. 5.

Bioactivity and immunoreactivity of infused PTH analogs. Seven-week-old male mice received continuous infusions of vehicle (black bars) or PTH(1-34) (blue bars) and [Cys25]PTH(1-34) (red bars) at a dose of 120 μg/kg/d for 4 days (n =6–8 per group). (A) Plasma was pooled from days 1–2 and 3–4 after starting infusion of PTH(1-34) or [Cys25]PTH(1-34) to assess in vitro the bioactivity of each PTH analog in the circulation. (B) Measurement of the PTH concentrations in plasma of mice infused with vehicle, PTH(1-34), or [Cys25]PTH(1-34) (n = 6–8 for each condition; plasma was collected daily after starting the infusions).
The 4-day infusion of [Cys25]PTH(1-34), compared with PTH(1-34), also resulted in a much lower bone-resorption response, as reflected by a much smaller increase in C-terminal collagen cross-links in the plasma (vehicle, 25.8 ± 2.6 ng/mL; PTH(1-34), 58.3 ± 4.6 ng/mL; [Cys25]PTH(1-34), 33.1 ± 2.7 ng/mL; p <0.01; n =6). Likewise, [Cys25]PTH(1-34) infusion resulted in a much smaller increase in the levels of mRNA for CYP27B (1α-hydroxylase) in the kidney (levels relative to GAPDH mRNA: vehicle, 1.0 ± 0.41; PTH(1-34), 48.9 ± 0.5; [Cys25]PTH(1-34), 1.7 ± 0.5) (Fig. 6). Similarly blunted responses were observed for the effects of infused [Cys25] PTH(1-34) on mRNA transcripts for CYP24A1, Npt2a, Npt2c, PTH1R, and TRPV5.
Fig. 6.

Assessment of gene expression in the kidneys by quantitative real-time PCR (qPCR). Seven-week-old male mice received continuous infusion of vehicle or PTH(1-34) or [Cys25]PTH(1-34) at doses of 120 μg/kg/d for 4 days (n =6–8 per group). Primer sequences used for qPCR amplification are as follows (forward and reverse, respectively): Cyp27B (1α-hydroxylase), 5′-aagtcactgtccagagcgctg-3′ and 5′-gttgtccagagttccagcata-3′; Cyp24A1 (24-hydroxylase, 5′-tcaatgaggtcttggctgatt-3′ and 5′-aaggtcagggcttcttcctct-3′; Npt2a, 5′-ccttcacaagactcatcatcc-3′ and 5′-atggtggtgtttgcaaggctg-3′; Npt2c, 5′-cttggaagagggaggtacaga-3′ and 5′-aagcagagctgaggatgtcca-3′; Pth1r, 5′-gcactgcacgcgcaactaca-3′ and 5′-acctgcgcgatgatatgcaac-3′; Trpv5, 5′-ctccatacttggtcacagagt-3′ and 5′-gtagatgaggttgtgtgaact-3′; and Gapdh, 5′-tggagtggtgtcttcactact-3′ and 5′-aagcagttggtggtgcaggat-3′. Data represent mean ± SEM over control levels (vehicle-infused mice); note that differences in the scale of the y-axis for the left and right panel. a: p <0.01 (PTH(1-34) versus vehicle); b: p <0.01 (PTH(1-34) versus [Cys25]PTH(1-34)).
Discussion
The term isolated hypoparathyroidism (IHP) refers to a heterogeneous group of disorders characterized by hypocalcemia, hyperphosphatemia, and reduced levels of bioactive PTH in the circulation. A small fraction of IHP cases have been resolved at the molecular level, in that affected individuals often carry activating mutations in the CaSR,(1,2,9) but loss- and gain-of-function mutations in GCM2(4–10) and gain-of-function mutations in Gα11(11–13) have also been reported. IHP can also be caused by mutations in PTH, but such cases are rare, as only 5 PTH mutations have been reported thus far. The identified mutations all occur in exon 2 of the PTH gene, which encodes the prepro leader sequence of the hormone, thus leading to inadequate secretion of the mature PTH(1-84) polypeptide.(3,14–17)
In a family from a geographically isolated island off the Korean coast with three members affected by IHP, we identified the first germline PTH mutation that changes an amino acid residue in the bioactive portion of PTH(1-84) rather than in the prepro leader sequence as for all five other cases.(3,14–17) The homozygous missense mutation c.166C>T changes the conserved arginine residue at position 25 to a cysteine. Measurements of PTH levels in the blood of the affected individuals varied widely, depending on the immunometric PTH assay that was used. Thus, the proband’s two affected sisters, who were naïve to treatment with calcium and vitamin D when investigated, had PTH levels that were below the normal range (ntPTH assay) or only moderately above normal (PTH-S assay), which is consistent with a diagnosis of hypoparathyroidism, whereas PTH levels measured with the PTH(1-84) assay were dramatically elevated, which is consistent with a diagnosis of pseudohypoparathyroidism (Table 1). It appears likely that the identified missense mutation lowers the affinity of the detection antibodies used by the ntPTH and the PTH-S assay because these were affinity-purified with immobilized PTH(13-34) and PTH(1-34), respectively (Fig. 2). In contrast, measurements by the PTH(1-84) assay are less likely to be influenced by the Cys25 mutation because this assay uses an antibody that was affinity-purified using immobilized PTH(1-3)(50) and hence allowed detection of substantially elevated PTH levels. Similar results would be expected with other widely used assays that measure only full-length PTH,(59–61) thus mistakenly leading in these hypocalcemic patients to the diagnosis of PHP1B as the underlying disease.
The [Cys25]PTH(1-34) mutant was about threefold less potent than control PTH(1-34) for stimulation of the formation of cAMP in an SaOS-2-derived osteoblastic cell line. Although the maximum signaling efficacy attained by the mutant peptide was comparable to that attained with PTH(1-34), the response induced by the mutant peptide was less sustained than that of the control peptide, as shown by kinetic wash-out assays. These effects on cAMP signaling could largely be explained by a reduction in affinity with which the mutant peptide binds to the PTH1R, as revealed by competition binding assays.
Consistent with the similar maximal levels of cAMP formation observed for both peptides in the in vitro cAMP assays, iv injection of high-dose [Cys25]PTH(1-34) or PTH(1-34) into wild-type mice induced comparable elevations in plasma and urinary cAMP levels as well as in blood ionized calcium levels. However, when the peptides were continuously infused into mice at a dose of 40 μg/kg/d, the calcemic and phosphaturic responses induced by [Cys25]PTH(1-34) were markedly reduced compared with those induced by PTH(1-34). Even when infused at a dose of 120 μg/kg/d, [Cys25]PTH(1-34) elicited only a moderate calcemic response and was thus still less effective than PTH(1-34) infused at the lower dose of 40 μg/kg/d. The reduced calcemic activity observed for the infused Cys25 mutant was not because of a loss of peptide activity in the implanted infusion device because samples recovered from the minipumps at the end of the experiment induced cAMP signaling responses in PTH1R-expressing cells that were consistent with the initial starting concentration of the peptide (Supplemental Fig. S1). Moreover, the blood levels of the mutant peptide, as determined by an immunometric PTH(1-34) assay, were comparable to those of PTH(1-34); this finding suggests that the rates of clearance of the two peptides from the circulation were similar. Overall, the data obtained from our infusion studies indicate that the Cys25 mutant PTH peptide has an in vivo activity that is ≈3-fold lower than that of control PTH(1-34) and are thus consistent with data from in vitro studies showing reduced PTH1R binding and cAMP signaling of [Cys25]PTH(1-34).
Consistent with the interpretation that the mutant hormone is not cleared from the circulation at accelerated rates, the blood levels of [Cys25]PTH(1-84) in the two affected individuals, who were naïve to treatment, were dramatically elevated, relative to the normal range of endogenous PTH. The elevated blood levels of [Cys25]PTH(1-84) in the patients, as determined with the PTH(1-84) assay, are also consistent with a lack of an effect of the mutation on secretion from the parathyroid gland, as suggested by our findings on the secretion of the mutant hormone from transiently transfected COS-7 cells. Our in vitro and in vivo data thus indicate that the mutant PTH is efficiently secreted from the parathyroid gland but has a reduced capacity to bind to and activate the PTH1R.
The identified Arg25>Cys mutation is located within the α-helical portion of PTH that mediates critical binding interactions with the PTH1R (Fig. 3D).(54,58) Indeed, the mode of binding, which involves interaction of the amphipathic α-helix ligand domain with a central groove within the receptor’s extracellular domain, is used by multiple family B-GPCRs.(62) The precise mechanistic impact that the Cys25 mutation has on the overall binding interaction and subsequently on signaling in target cells, as mediated at the plasma membrane or potentially from within the endosomal compartment,(63) remains to be determined. In any case, it is now clear that a relatively moderate reduction in the binding affinity of [Cys25]PTH(1-84) for the receptor can lead to substantial biological consequences, as shown by the chronic hypocalcemia occurring in these IHP patients. That the Cys25 mutation did not completely abolish interaction with the PTH1R likely explains why the patients were able to maintain blood calcium at levels that are high enough to avoid even more profound hypocalcemia. On the other hand, it remains puzzling that the parathyroid glands in the affected individuals did not produce/secrete even greater amounts of the mutant PTH such that the blood calcium levels could be maintained fully within the normal range.(64)
In summary, a homozygous PTH mutation was identified as a novel cause of autosomal recessive IHP. The R25C change does not affect hormonal processing or secretion from the parathyroid glands, but rather reduces binding of the mature PTH(1-84) to the PTH1R and hence impairs activity in the two main target tissues, kidney and bone. Increased production of the mutant PTH likely prevented symptomatic hypocalcemia for years (proband) or even decades (affected sisters). Importantly, elevated circulating levels of the mutant hormone were readily detectable by the PTH(1-84) immunometric assay but not by the ntPTH and S-PTH assays. As a result, distinguishing between IHP and PHP1B, disorders of PTH-resistance and PTH-deficiency, respectively, was not straightforward. Mutations such as Cys25 affecting the bioactive portion of PTH should, therefore, be considered in patients suspected of having PHP1B or IHP, particularly if no abnormality in GNAS methylation is found. Differentiating between IHP and PHP1B has considerable implications for genetic counseling, therapy, and long-term outcome because treatment of IHP patients with inappropriately high doses of active vitamin D and calcium can contribute to the development of nephrocalcinosis and chronic kidney disease.(48) Likewise, PTH mutations that prevent detection by immunoassay, yet do not dramatically alter the hormone’s biological activity, such as the truncation mutation observed in a patient with hyperparathyroidism,(65) should be excluded in cases of hypercalcemia, hypophosphatemia, and unexpectedly low or normal PTH levels.
Supplementary Material
Acknowledgments
The authors thank all family members for their participation and Professors Jaesang Kim, John Lew, Ung-il Chung, Sung-Kil Lim, Jean-Pierre Vilardaga, and Henry Kronenberg for their insightful advice. Supported by the Basic Science Research Program, National Research Foundation of Korea (NRF-2013R1A1A1A05005629 to SL) and by grants from the National Institute of Diabetes and Digestive and Kidney Disease (R01-DK46718 to HJ; P01-DK11794, project I to TJG and project IV to HJ; and R01-DK100584 to MM).
Footnotes
Disclosures
All authors state that they have no conflicts of interest.
Authors’ roles: SL identified and evaluated the index case and his family. SL, SMK, and HSY performed the initial sequence analyses of candidate genes. SL, MM, and HJ designed and/or performed subsequent clinical and laboratory investigations, and assessed the presence or absence of the PTH mutation in the entire family. JG and MO performed in vivo studies in mice. AK synthesized, purified, and quantified synthetic PTH peptides. TJG, TD, and MM performed in vitro experiments. SL drafted the first version of the manuscript. TJG and HJ directed the entire project and wrote the final version of the manuscript.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81:239–97. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- 2.Hannan FM, Nesbit MA, Zhang C, et al. Identification of 70 calcium-sensing receptor mutations in hyper- and hypo-calcaemic patients: evidence for clustering of extracellular domain mutations at calcium-binding sites. Hum Mol Genet. 2012;21:2768–78. doi: 10.1093/hmg/dds105. [DOI] [PubMed] [Google Scholar]
- 3.Tomar N, Gupta N, Goswami R. Calcium-sensing receptor auto antibodies and idiopathic hypoparathyroidism. J Clin Endocrinol Metab. 2013;98:3884–91. doi: 10.1210/jc.2013-2158. [DOI] [PubMed] [Google Scholar]
- 4.Ding C, Buckingham B, Levine MA. Familial isolated hypoparathyroidism caused by a mutation in the gene for the transcription factor GCMB. J Clin Invest. 2001;108:1215–20. doi: 10.1172/JCI13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baumber L, Tufarelli C, Patel S, et al. Identification of a novel mutation disrupting the DNA binding activity of GCM2 in autosomal recessive familial isolated hypoparathyroidism. J Med Genet. 2005;42:443–8. doi: 10.1136/jmg.2004.026898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomee C, Schubert SW, Parma J, et al. GCMB mutation in familial isolated hypoparathyroidism with residual secretion of parathyroid hormone. J Clin Endocrinol Metab. 2005;90:2487–92. doi: 10.1210/jc.2004-2450. [DOI] [PubMed] [Google Scholar]
- 7.Mannstadt M, Bertrand G, Muresan M, et al. Dominant-negative GCMB mutations cause an autosomal dominant form of hypoparathyroidism. J Clin Endocrinol Metab. 2008;93:3568–76. doi: 10.1210/jc.2007-2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirczuk SM, Bowl MR, Nesbit MA, et al. A missense glial cells missing homolog B (GCMB) mutation, Asn502His, causes autosomal dominant hypoparathyroidism. J Clin Endocrinol Metab. 2010;95:3512–6. doi: 10.1210/jc.2009-2532. [DOI] [PubMed] [Google Scholar]
- 9.Tomar N, Bora H, Singh R, et al. Presence and significance of a R110W mutation in the DNA-binding domain of GCM2 gene in patients with isolated hypoparathyroidism and their family members. Eur J Endocrinol. 2010;162:407–21. doi: 10.1530/EJE-09-0303. [DOI] [PubMed] [Google Scholar]
- 10.Yi HS, Eom YS, Park Ie B, et al. Identification and characterization of C106R, a novel mutation in the DNA-binding domain of GCMB, in a family with autosomal-dominant hypoparathyroidism. Clin Endocrinol (Oxf) 2012;76:625–33. doi: 10.1111/j.1365-2265.2011.04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nesbit MA, Hannan FM, Howles SA, et al. Mutations affecting G-protein subunit alpha11 in hypercalcemia and hypocalcemia. N Engl J Med. 2013;368:2476–86. doi: 10.1056/NEJMoa1300253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mannstadt M, Harris M, Bravenboer B, et al. Germline mutations affecting Galpha11 in hypoparathyroidism. N Engl J Med. 2013;368:2532–4. doi: 10.1056/NEJMc1300278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li D, Opas EE, Tuluc F, et al. Autosomal dominant hypoparathyroidism caused by germline mutation in GN A11: phenotypic and molecular characterization. J Clin Endocrinol Metab. 2014;99:E1774–83. doi: 10.1210/jc.2014-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnold A, Horst SA, Gardella TJ, Baba H, Levine MA, Kronenberg HM. Mutation of the signal peptide-encoding region of the preproparathyroid hormone gene in familial isolated hypoparathyroidism. J Clin Invest. 1990;86:1084–7. doi: 10.1172/JCI114811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parkinson D, Thakker R. A donor splice site mutation in the parathyroid hormone gene is associated with autosomal recessive hypoparathyroidism. Nat Genet. 1992;1:149–53. doi: 10.1038/ng0592-149. [DOI] [PubMed] [Google Scholar]
- 16.Sunthornthepvarakul T, Churesigaew S, Ngowngarmratana S. A novel mutation of the signal peptide of the preproparathyroid hormone gene associated with autosomal recessive familial isolated hypoparathyroidism. J Clin Endocrinol Metab. 1999;84:3792–6. doi: 10.1210/jcem.84.10.6070. [DOI] [PubMed] [Google Scholar]
- 17.Ertl DA, Stary S, Streubel B, Raimann A, Haeusler G. A novel homozygous mutation in the parathyroid hormone gene (PTH) in a girl with isolated hypoparathyroidism. Bone. 2012;51:629–32. doi: 10.1016/j.bone.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 18.Jan de Beur S, Levine M. Pseudohypoparathyroidism: clinical, biochemical, and molecular features. In: Bilezikian JP, Markus R, Levine MA, editors. The parathyroids: basic and clinical concepts. New York: Academic Press; 2001. pp. 807–25. [Google Scholar]
- 19.Weinstein L, Yu S, Warner D, Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001;22:675–705. doi: 10.1210/edrv.22.5.0439. [DOI] [PubMed] [Google Scholar]
- 20.Bastepe M, Jüppner H. Pseudohypoparathyroidism, Albright’s hereditary osteodystrophy, and progressive osseous heteroplasia: disorders caused by inactivating GNAS mutations. In: DeGroot LJ, Jameson JL, editors. Endocrinology. 6. Philadelphia, PA: W.B. Saunders Company; 2010. pp. 1223–35. [Google Scholar]
- 21.Mantovani G. Clinical review: pseudohypoparathyroidism: diagnosis and treatment. J Clin Endocrinol Metab. 2011;96:3020–30. doi: 10.1210/jc.2011-1048. [DOI] [PubMed] [Google Scholar]
- 22.Weinstein LS, Xie T, Qasem A, Wang J, Chen M. The role of GNAS and other imprinted genes in the development of obesity. Int J Obes (Lond) 2010;34:6–17. doi: 10.1038/ijo.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mantovani G, Ballare E, Giammona E, Beck-Peccoz P, Spada A. The gsalpha gene: predominant maternal origin of transcription in human thyroid gland and gonads. J Clin Endocrinol Metab. 2002;87:4736–40. doi: 10.1210/jc.2002-020183. [DOI] [PubMed] [Google Scholar]
- 24.Turan S, Fernández-Rebollo E, Aydin C, et al. Postnatal establishment of allelic Gαs silencing as a plausible explanation for delayed onset of parathyroid hormone resistance owing to heterozygous galphas disruption. J Bone Miner Res. 2014;29:749–60. doi: 10.1002/jbmr.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bastepe M, Fröhlich LF, Hendy GN, et al. Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J Clin Invest. 2003;112:1255–63. doi: 10.1172/JCI19159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linglart A, Gensure RC, Olney RC, Jüppner H, Bastepe M. A novel STX16 deletion in autosomal dominant pseudohypoparathyroidism type Ib redefines the boundaries of a cis-acting imprinting control element of GNAS. Am J Hum Genet. 2005;76:804–14. doi: 10.1086/429932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chillambhi S, Turan S, Hwang DY, Chen HC, Jüppner H, Bastepe M. Deletion of the noncoding GNAS antisense transcript causes pseudohypoparathyroidism type Ib and biparental defects of GNAS methylation in cis. J Clin Endocrinol Metab. 2010;95:3993–4002. doi: 10.1210/jc.2009-2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin HY, Lee BH, Choi JH, et al. Clinical characterization and identification of two novel mutations of the GNAS gene in patients with pseudohypoparathyroidism and pseudopseudohypoparathyroidism. Clin Endocrinol (Oxf) 2011;75:207–13. doi: 10.1111/j.1365-2265.2011.04026.x. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez J, Perera E, Jan de Beur S, et al. Madelung-like deformity in pseudohypoparathyroidism type 1b. J Clin Endocrinol Metab. 2011;96:E1507–11. doi: 10.1210/jc.2011-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turan S, Ignatius J, Moilanen JS, et al. De novo STX16 deletions: an infrequent cause of pseudohypoparathyroidism type Ib that should be excluded in sporadic cases. J Clin Endocrinol Metab. 2012;97:E2314–9. doi: 10.1210/jc.2012-2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richard N, Abeguile G, Coudray N, et al. A new deletion ablating NESP55 causes loss of maternal imprint of A/B GNAS and autosomal dominant pseudohypoparathyroidism type Ib. J Clin Endocrinol Metab. 2012;97:E863–7. doi: 10.1210/jc.2011-2804. [DOI] [PubMed] [Google Scholar]
- 32.Elli FM, de Sanctis L, Peverelli E, et al. Autosomal dominant pseudohypoparathyroidism type Ib: a novel inherited deletion ablating STX16 causes loss of imprinting at the A/B DMR. J Clin Endocrinol Metab. 2014:jc20133704. doi: 10.1210/jc.2013-3704. [DOI] [PubMed] [Google Scholar]
- 33.Mantovani G, Bondioni S, Linglart A, et al. Genetic analysis and evaluation of resistance to thyrotropin and growth hormone-releasing hormone in pseudohypoparathyroidism type Ib. J Clin Endocrinol Metab. 2007;92:3738–42. doi: 10.1210/jc.2007-0869. [DOI] [PubMed] [Google Scholar]
- 34.Linglart A, Bastepe M, Jüppner H. Similar clinical and laboratory findings in patients with symptomatic autosomal dominant and sporadic pseudohypoparathyroidism type Ib despite different epigenetic changes at the GNAS locus. Clin Endocrinol (Oxf) 2007;67:822–31. doi: 10.1111/j.1365-2265.2007.02969.x. [DOI] [PubMed] [Google Scholar]
- 35.Mantovani G, de Sanctis L, Barbieri AM, et al. Pseudohypoparathyroidism and GNAS epigenetic defects: clinical evaluation of Albright hereditary osteodystrophy and molecular analysis in 40 patients. J Clin Endocrinol Metab. 2010;95:651–8. doi: 10.1210/jc.2009-0176. [DOI] [PubMed] [Google Scholar]
- 36.Fernández-Rebollo E, Pérez de Nanclares G, Lecumberri B, et al. Exclusion of the GNAS locus in PHP-Ib patients with broad GNAS methylation changes: evidence for an autosomal recessive form of PHP-Ib? J Bone Miner Res. 2011;26:1854–63. doi: 10.1002/jbmr.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maupetit-Mehouas S, Mariot V, Reynes C, et al. Quantification of the methylation at the GNAS locus identifies subtypes of sporadic pseudohypoparathyroidism type Ib. J Med Genet. 2011;48:55–63. doi: 10.1136/jmg.2010.081356. [DOI] [PubMed] [Google Scholar]
- 38.Elli FM, de Sanctis L, Bollati V, et al. Quantitative analysis of methylation defects and correlation with clinical characteristics in patients with pseudohypoparathyroidism type I and GNAS epigenetic alterations. J Clin Endocrinol Metab. 2014;99:E508–17. doi: 10.1210/jc.2013-3086. [DOI] [PubMed] [Google Scholar]
- 39.Bastepe M, Lane AH, Jüppner H. Paternal uniparental isodisomy of chromosome 20q (patUPD20q)—and the resulting changes in GNAS1 methylation—as a plausible cause of pseudohypoparathyroidism. Am J Hum Genet. 2001;68:1283–9. doi: 10.1086/320117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bastepe M, Altug-Teber O, Agarwal C, Oberfield SE, Bonin M, Jüppner H. Paternal uniparental isodisomy of the entire chromosome 20 as a molecular cause of pseudohypoparathyroidism type Ib (PHP-Ib) Bone. 2011;48:659–62. doi: 10.1016/j.bone.2010.10.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernández-Rebollo E, Lecumberri B, Garin I, et al. New mechanisms involved in paternal 20q disomy associated with pseudohypoparathyroidism. Eur J Endocrinol. 2011;163:953–62. doi: 10.1530/EJE-10-0435. [DOI] [PubMed] [Google Scholar]
- 42.Dixit A, Chandler KE, Lever M, et al. Pseudohypoparathyroidism type 1b due to paternal uniparental disomy of chromosome 20q. J Clin Endocrinol Metab. 2013;98:E103–8. doi: 10.1210/jc.2012-2639. [DOI] [PubMed] [Google Scholar]
- 43.Pérez de Nanclares G, Fernández-Rebollo E, Santin I, et al. Epigenetic defects of GNAS in patients with pseudohypoparathyroidism and mild features of Albright’s hereditary osteodystrophy. J Clin Endocrinol Metab. 2007;92:2370–3. doi: 10.1210/jc.2006-2287. [DOI] [PubMed] [Google Scholar]
- 44.Unluturk U, Harmanci A, Babaoglu M, et al. Molecular diagnosis and clinical characterization of pseudohypoparathyroidism type-Ib in a patient with mild Albright’s hereditary osteodystrophy-like features, epileptic seizures, and defective renal handling of uric acid. Am J Med Sci. 2008;336:84–90. doi: 10.1097/MAJ.0b013e31815b218f. [DOI] [PubMed] [Google Scholar]
- 45.Mariot V, Maupetit-Mehouas S, Sinding C, Kottler ML, Linglart A. A maternal epimutation of GNAS leads to Albright osteodystrophy and parathyroid hormone resistance. J Clin Endocrinol Metab. 2008;93:661–5. doi: 10.1210/jc.2007-0927. [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto M, Takuwa Y, Masuko S, Ogata E. Effects of endogenous and exogenous parathyroid hormone on tubular reabsorption of calcium in pseudohypoparathyroidism. J Clin Endocrinol Metab. 1988;66:618–25. doi: 10.1210/jcem-66-3-618. [DOI] [PubMed] [Google Scholar]
- 47.Kruse K, Kracht U, Wohlfart K, Kruse U. Biochemical markers of bone turnover, intact serum parathyroid hormone and renal calcium excretion in patients with pseudohypoparathyroidism and hypoparathyroidism before and during vitamin D treatment. Eur J Pediatr. 1989;148:535–9. doi: 10.1007/BF00441552. [DOI] [PubMed] [Google Scholar]
- 48.Mitchell DM, Regan S, Cooley MR, et al. Long-term follow-up of patients with hypoparathyroidism. J Clin Endocrinol Metab. 2012;97:4507–14. doi: 10.1210/jc.2012-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimizu M, Carter PH, Khatri A, Potts JT, Jr, Gardella TJ. Enhanced activity in parathyroid hormone (1-14) and (1-11): novel peptides for probing the ligand-receptor interaction. Endocrinology. 2001;142:3068–74. doi: 10.1210/endo.142.7.8253. [DOI] [PubMed] [Google Scholar]
- 50.Salusky IB, Goodman WG, Kuizon BD, et al. Similar predictive value of bone turnover using first- and second-generation immunometric PTH assays in pediatric patients treated with peritoneal dialysis. Kidney Int. 2003;63:1801–8. doi: 10.1046/j.1523-1755.2003.00915.x. [DOI] [PubMed] [Google Scholar]
- 51.Tan K, Ong L, Sethi SK, Saw S. Comparison of the Elecsys PTH(1-84) assay with four contemporary second generation intact PTH assays and association with other biomarkers in chronic kidney disease patients. Clin Biochem. 2013;46:781–6. doi: 10.1016/j.clinbiochem.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 52.Guo J, Song L, Liu M, et al. Activation of a non-cAMP/PKA signaling pathway downstream of the PTH/PTHrP receptor is essential for a sustained hypophosphatemic response to PTH infusion in male mice. Endocrinology. 2013;154:1680–9. doi: 10.1210/en.2012-2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheloha RW, Maeda A, Dean T, Gardella TJ, Gellman SH. Backbone modification of a polypeptide drug alters duration of action in vivo. Nat Biotech. 2014;32:653–5. doi: 10.1038/nbt.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dean T, Khatri A, Potetinova Z, Willick GE, Gardella TJ. Role of amino acid side chains in region 17-31 of parathyroid hormone (PTH) in binding to the PTH receptor. J Biol Chem. 2006;281:32485–95. doi: 10.1074/jbc.M606179200. [DOI] [PubMed] [Google Scholar]
- 55.Maeda A, Okazaki M, Baron DM, et al. Critical role of parathyroid hormone (PTH) receptor-1 phosphorylation in regulating acute responses to PTH. Proc Natl Acad Sci USA. 2013;110:5864–9. doi: 10.1073/pnas.1301674110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linglart A, Menguy C, Couvineau A, et al. Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. N Engl J Med. 2011;364:2218–26. doi: 10.1056/NEJMoa1012717. [DOI] [PubMed] [Google Scholar]
- 57.Biesecker LG, Mullikin JC, Facio FM, et al. The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–74. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pioszak AA, Parker NR, Gardella TJ, Xu HE. Structural basis for parathyroid hormone-related protein binding to the parathyroid hormone receptor and design of conformation-selective peptides. J Biol Chem. 2009;284:28382–91. doi: 10.1074/jbc.M109.022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bouillon R, Coopmans W, Degroote D, Radoux D, Eliard P. Immunoradiometric assay of parathyrin with polyclonal and monoclonal region-specific antibodies. Clin Chem. 1990;36:271–6. [PubMed] [Google Scholar]
- 60.John M, Goodman W, Gao P, Cantor T, Salusky I, Jüppner H. A novel immunoradiometric assay detects full-length human PTH but not amino-terminally truncated fragments: implications for PTH measurements in renal failure. J Clin Endocrinol Metab. 1999;84:4287–90. doi: 10.1210/jcem.84.11.6236. [DOI] [PubMed] [Google Scholar]
- 61.Fournier A, Solal M, Oprisiu R, et al. Optimal range of plasma concentration of true 1-84 parathyroid hormone in patients on maintenance dialysis. J Clin Endocrinol Metab. 2001;86:1840–2. doi: 10.1210/jcem.86.4.7436-8. [DOI] [PubMed] [Google Scholar]
- 62.Couvineau A, Laburthe M. The family B1 GPCR: structural aspects and interaction with accessory proteins. Curr Drug Targets. 2012;13:103–15. doi: 10.2174/138945012798868434. [DOI] [PubMed] [Google Scholar]
- 63.Vilardaga JP, Gardella TJ, Wehbi VL, Feinstein TN. Non-canonical signaling of the PTH receptor. Trends Pharmacol Sci. 2012;33:423–31. doi: 10.1016/j.tips.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brown EM. Control of parathyroid hormone secretion by its key physiological regulators. In: Bilezikian J, editor. The parathyroids: basic and clinical concepts. San Diego: Academic Press; 2015. pp. 101–18. [Google Scholar]
- 65.Au AY, McDonald K, Gill A, et al. PTH mutation with primary hyperparathyroidism and undetectable intact PTH. N Engl J Med. 2008;359:1184–6. doi: 10.1056/NEJMc0802570. [DOI] [PubMed] [Google Scholar]
- 66.Hollenstein K, Kean J, Bortolato A, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–43. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.