

Abstract
The genetic and molecular mechanisms that initiate and maintain pituitary tumorigenesis are poorly understood. Nonfunctioning tumors of the gonadotrope lineage represent 35% of all tumors; are usually macroadenomas, often resulting in hypopituitarism; and have no medical treatments. Using expression microarrays combined with whole-genome copy number screens on individual human tumors, we identified the mammalian sterile-20-like kinase (MST4) transcript, which was amplified within chromosome Xq26.2 in one tumor and up-regulated in all gonadotrope tumor samples. MST4 mRNA and protein were consistently overexpressed in human tumors compared with normal pituitaries. To mimic the pituitary tumor microenvironment, a hypoxia model using LβT2 murine gonadotrope cells was created to examine the functional role of the kinase. During long-term hypoxia, MST4 expression increased colony formation in a soft agar assay and rates of cell proliferation by activating p38 MAPK and AKT. Under short-term severe hypoxic stress, MST4 decreased the rates of apoptosis via p38 MAPK, AKT, hypoxia-inducible factor-1, and its cell-specific downstream targets. Analysis of MST4 mutants confirmed the importance of the kinase sequence but not the regulatory C terminus for its functional effects. Together these data identify the MST4 kinase as a novel candidate to mediate human pituitary tumorigenesis in a hypoxic environment and position it as a potential therapeutic target.
Pituitary tumors are among the most common intracranial neoplasms, with 1 per 10 000 persons/year seeking clinical evaluation, but with 15%–20% of the population demonstrating pituitary tumors at autopsy (1, 2). Tumors may present with hormone overproduction such as in patients with acromegaly due to excess GH, Cushing's disease due to excess adrenocorticotropin or amenorrhea, or hypogonadism due to elevated prolactin in patients with prolactinomas (3, 4). Gonadotrope pituitary tumors, on the other hand, were initially called nonfunctioning because they were thought to be more clinically silent. These tumors are more common in men than women, often presenting with hypogonadism, headaches, and visual disturbances that can progress to blindness (4, 5).
Gonadotrope tumors, representing 35% of pituitary tumors, are identified by protein expression of FSH, LH, and/or α-subunit by immunohistochemistry. The tumors are usually macroadenomas (>1 cm) and can lead to compromise in normal pituitary function and clinical hypopituitarism (5). Local invasion into adjacent structures and dura occurs in approximately 50% of patients, leading to increased risk of residual tumor regrowth in approximately 30% after transsphenoidal surgical resection with a need for additional surgery or radiation treatment (6).
Although monoclonal in nature, the underlying pathogenesis of pituitary tumors is poorly understood due to a limited access to human tissue, lack of human cell lines, and lack of animal models (7, 8). Bone morphogenetic and retinoic acid inducible neuronal protein-3, a highly up-regulated gonadotrope-specific mitochondrial protein, and epidermal growth factor receptor-associated protein-8 (EPS8), a docking protein downstream of growth factor receptors, have been shown to promote pituitary gonadotrope cell proliferation and/or survival in response to growth factor withdrawal (9, 10). Several tumor suppressor genes downstream of p53 in gonadotrope tumors have been identified, including MEG3, GADD45β, GADD45γ, RPRM, and ZAC1 (11–15). These are down-regulated in gonadotrope tumors, and their functional relevance is under active investigation. To date, there are no animal models of gonadotrope pituitary tumors, and most animal models in which tumors have been induced do not replicate the genetic or genomic defects observed in human tumor samples (16).
Prior studies by our group and others have used expression microarray profiling of individual human tumor samples to identify and characterize candidate genes involved in pituitary tumor promotion or maintenance (9, 10, 13, 14). A combinatorial approach of techniques that link genomic aberrations with transcriptional changes has recently been useful for the identification of important pathways involved in tumorigenesis (17–19); thus, we performed copy number variation microarrays together with gene expression microarray profiling of human gonadotrope tumors and normal pituitaries. A deletion of most of chromosome X (ChrX), but with a small amplification at region of chromosome Xq26.2 was identified in a single tumor specimen. The mammalian Ste20-like kinase 4 (MST4) gene was localized to this amplified region, and the transcript was consistently up-regulated in all human gonadotrope tumor samples compared with normal pituitary from autopsies; thus, it was selected for further investigation.
Sterile 20 (Ste20) is a serine threonine kinase identified in yeast, upstream of MAPK kinase, involved in pheromone signaling (20, 21). Subsequently, several mammalian Ste20-like kinases sharing the homology with the yeast Ste20 were identified and grouped into two structurally distinct families: the p21-activated kinase family and the germinal center kinase (GCK) family (20). The GCK group is further divided into 8 groups (GCKI to GCKVIII) and defined by the presence of a kinase domain at the NH2 terminus, a regulatory region in the C terminal and lack of GTPase binding domains for domain organization and structure, respectively (22–24). The GCKIII subfamily contains MST3, MST4, and SOK1 (Ste20-like oxidant stress response kinase 1), which share almost 90% amino acid identity in the kinase domain but less than 20% in the C-terminal domain (20, 25). These three members are implicated in a broad array of cellular responses. MST3 is proapoptotic and SOK1 has a putative role in cell migration (26–28). Less is known about MST4, but it appears to play diverse roles in a cell-specific fashion (25).
Because pituitary adenomas have lower regional oxygen saturation than that of the surrounding tissues (29, 30) and the other two GCKIII family members, SOK1 and MST3, are activated by hypoxia to regulate cell movement and/or survival (27, 31), we asked whether MST4 plays a role in response to a hypoxic microenvironment. Here we observed under a hypoxic microenvironment that MST4 promoted colony formation in soft agar via effects on proliferation and survival in the LβT2 cells. We determined that the p38MAPK, AKT, and hypoxia inducible factor-1 (HIF-1) pathways are downstream effectors of the MST4 kinase and confirmed the importance of the MST4 kinase sequence for the functional effects. Together these data identify MST4 as a novel intracellular kinase in human pituitary tumors that responds to a hypoxic microenvironment to promote tumorigenesis and support future efforts to target it with small molecule inhibitors as a new medical therapy for human pituitary tumors.
Materials and Methods
Characterization of human pituitary tumors and normal pituitary
Pituitary tumor samples were obtained from patients at the University of Colorado Hospital at the time of transsphenoidal surgery after informed consent. Portions of the specimens were immediately placed in RNA Later (QIAGEN) at the time of resection, dissected, and stored at −80°C. Tumors were classified as chromophobic tissue, immunostaining positively for FSH, LH, and α-subunit. Normal pituitary glands, were obtained at autopsy within 2–18 hours of death from University of Colorado Denver Pathology Department. Demonstration of RNA and protein integrity of all samples was confirmed as previously described (9, 13, 14). The University of Colorado Pituitary Program has a tissue bank containing more than 400 pituitary tumor samples and 100 normal pituitary samples.
Microarray and copy number variation analysis
The DNA microarray was performed as previously reported (9, 13, 14). To ensure intact RNA, total RNA integrity was verified using the Agilent 2100 bioanalyzer in the University of Colorado Health Science Center Cancer Center Array Core. For copy number variation analysis, genomic DNA was prepared, labeled, and hybridized to Affymetrix Genome-Wide Human SNP 6.0 Arrays according to the manufacturer's instructions. For copy number variation analysis, genomic DNA was restriction digested, PCR amplified, fragmented, labeled, and hybridized to Affymetrix Genome-Wide Human SNP 6.0 Arrays according to the manufacturer's instructions. Data files were analyzed using Partek Genomics Suite software. Copy number was assessed using the 794 GenomeWide SNP 6.0 HapMap samples available from the International HapMap Project as the copy number baseline and the Partek segmentation algorithm to define chromosomal regions of amplifications/deletions. For each detected region amplified or deleted, visual inspection for genes was performed and compared with a gene expression data set from the microarrays to detect candidates that were either amplified and up-regulated or conversely deleted and down-regulated.
Reagents
The identity of amino acid sequence between human and murine MST4 is 98% as assessed by protein sequence analysis (National Center for Biotechnology Information protein blast tool). Thus, mMst4 was created from pCMV.Sport6-mMst4 and inserted into pcDNA3 vectors (Open Biosystems). Mst4 mutants of K53E, T178A, and δC were generated by using a mutagenesis kit (Agilent Technologies). SB203580 was from Tocris Bioscience. PD98059 and LY294002 were purchased from EMD Millipore.
Immunoblot analysis and immunohistochemistry
The immunoblotting was performed as previously described (14). Protein concentrations in tumor or cell lysates were quantified by a bicinchoninic acid assay (Pierce). Equal amounts of proteins were separated by SDS-PAGE and blotted to polyvinyl difluoride membranes using the mini transblotter system (Bio-Rad Laboratories). After blocking, the membranes were incubated with primary antibodies at 4°C overnight. Antibodies against mouse and human AKT, ERK, p38, MST4, phospho-AKT, phospho-ERK, phospho-p38, and antihuman glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies (Cell Signaling Technology) were used at 1:1000 dilutions. Antihuman and mouse HIF-1 was used at 1:500 dilutions (BD Biosciences). Antimouse β-tubulin (Abcam) was used at 1:2000 dilutions. The membranes were washed and then incubated with horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences) for 1 hour at room temperature, and proteins were visualized by enhanced chemiluminescence according to the manufacturer's protocol (Pierce). For immunohistochemistry, tissue samples were deparaffinized and rehydrated and then soaked in a 10-mM citrate buffer (pH 6.0) and incubated in a pressure cooker for 10 minutes. Sections were incubated in 3% H2O2, blocked with 5% normal horse serum for 1 hour, and then incubated with the antihuman MST4 antibody or IgG control (BD Biosciences; 1:500 dilutions) overnight at 4°C. After washing, the samples were incubated with the biotinylated goat antimouse IgG and then with streptavidin-peroxidase complex each for 30 minutes. After three washes, the peroxidase-binding sites were demonstrated by the diaminobenzidine method.
RNA preparation and RT-PCR
Total RNA was extracted from tissues or cells using TRIzol reagent according to the manufacturer's protocol (Invitrogen), and RNA (0.5 μg) was reversed transcribed using a Thermo Verso cDNA kit (Fisher Scientific). The semiquantitative RT-PCR was conducted on tumor and normal pituitary cDNA to analyze the genes of human MST4 and GAPDH. The primers used in RT-PCR are listed in Supplemental Table 1.
Real-time PCR was performed with a Power SYBR Green master mix kit (Applied Biosystems) using an ABI PRISM 7300 real-time recycler (Applied Biosystems). The primer sequences used in real-time PCR analysis are listed in Supplemental Table 1. Primers of Glut-4 (QT01044946) and Aldolase (QT00291753) were purchased from QIAGEN. All samples were run in triplicate.
Cell culture
LβT2 gonadotrope cells from P. Mellon (University of California, San Diego, San Diego, California) were cultured as previously described (32). These cells, immortalized with simian virus 40 T-antigen, are the only functional gonadotrope cell lines available. The cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (HyClone), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37ºC in humidified 5% CO2. LβT2 stable transfectants including vector pcDNA3, MST4 wild-type, and MST4 mutants were generated using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol (Gemini). The selection of stably overexpressing pcDNA3, MST4, and MST4 mutant cells were generated from the population of clones under geneticin selection (Invitrogen; 600 μg/mL).
Soft agar assays
Soft agar assays were performed as previously described (13). Cells were loaded at a concentration of 4 × 104 cells/well in 0.35% agar and incubated for 18 hours under normoxic conditions before hypoxia (5% O2) treatment. After 14 days of chronic hypoxia, colonies were counted in triplicate plates and photographed at ×2 using an Olympus microscope BX51 mounted Microfire digital camera.
Proliferation assays
To assess proliferation, 5000 cells were plated in the 24-well plate with coverslips in growth medium and incubated under normoxia for 24 hours and then placed under hypoxia (5% O2) for up to 14 days or for 3 days under 1% O2 for studies using signaling pathway inhibitors. For 5-bromo-2′-deoxyuridine (BrdU) staining, the growing LβT2 transfectants were incubated with BrdU (Sigma-Aldrich; 10 μM) under 5% O2 for 24 hours. Cells were rinsed in PBS three times and fixed in 4% paraformaldehyde for 20 minutes at room temperature, and then the cells were treated with 0.3% Triton X-100 in PBS for an additional 15 minutes at room temperature to increase cellular permeability. After blocking by preincubation with 10% bovine serum in PBS at room temperature for 30 minutes, 1 μL of anti-BrdU (BD Pharmingen) with 100 μL of 0.5% Tween 20 in PBS was applied and incubated in a humidified chamber at room temperature for 1 hour. After three washes with PBS, the cells were incubated with rhodamine-conjugated goat antimouse IgG for 30 minutes. After three washes with PBS, cells were incubated for 6 minutes in 0.5 μg/mL of diamidino-2-phenylindole in methanol. The cells were washed with water and a coverslip was applied using Flo-Texx mounting medium prior to microscopic examination. In addition, cells were counted at indicated time points and viability was determined by trypan blue exclusion.
Apoptosis assays in response to hypoxic microenvironment
Terminal deoxynucleotidyl transferase deoxyuridine 5-triphosphate nick end labeling (TUNEL) was performed to analyze apoptosis. Briefly, cells were fixed with 4% paraformaldehyde for 25 minutes at 4°C. After three washes with PBS, cells were permeabilized with 0.2% Triton X-100 in PBS for 5 minutes at room temperature. TUNEL reaction mixture was obtained by adding terminal deoxynucleotidyl transferase to nucleotide mixture as instructed by the manufacturer's manual (Promega). The cells were then incubated with 50 μL of the TUNEL reaction mixture in a humidified chamber at 37°C for 1 hour. After rinsing with PBS, cell nuclei were visualized using 0.5 μg/mL 4′,6′-diamino-2-phenylindole. The green fluorescence of apoptotic cells was detected by fluorescent microscopy (Nikon Elipse E600).
LβT2 transfectants were exposed to 20% O2 or 1% O2 for 17 hours, and then rates of cell death were detected by nuclear fluorescence staining by Hoechst 33258. Briefly, cells were fixed with 1% paraformaldehyde at room temperature followed by treatment of 70% ethanol (in 100 mM glycine buffer) for 20 minutes at −20°C. Cells were washed and incubated with Hoechst 33258 (8 μg/mL; Life Technologies) and observed under a fluorescence microscope (Axiovert 200 Zeiss). Rates of cell death were quantified by counting the number of cells with condensed nuclei.
Silencing of HIF-1
LβT2 transfectants with decreased HIF-1 expression were generated by lentiviral-mediated short hairpin RNA (shRNA) expression. Briefly, the pLKO.1 lentiviral vectors targeting mouse Hif-1a were purchased from Sigma-Aldrich (TRCN 0000232223). Nontargeting shRNA (SHC002; Sigma) was used as a control. Cells were infected with the virus at a desired multiplicity of infection of 10:1, and Hif-1 mRNA was analyzed 48 hours after infection. The on-target control rescue was generated by overexpression of an insensitive HIF-1 construct in LβT2 transfectants with HIF-1 silencing. Briefly, the HIF-1 expression construct or control vector was transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Real-time PCR was performed to analyze Hif-1 mRNA expression 24 hours after transfection.
Luciferase assay
Conventional luciferase assays were performed. Individual plates of LβT2 transfectants (1 × 106 cells/well) were transfected with 1.5 μg HRE plasmid (number 26731; Addgene) and 0.5 μg of Renilla constitutive reporter plasmid. Twenty-four hours after the transfection, cells were trypsinized and replated at equal cell numbers to ensure equal and comparable transfection efficiency in 96-well plates. Twenty-four hours after replating, one set of plates was placed under a hypoxia condition (1% O2) for 17 hours, whereas a parallel set was maintained under normoxia (20% O2). Cells were collected and analyzed for luciferase activity using a dual-luciferase assay kit (Promega) according to the manufacturer's instructions. For analysis, the experimental reporter was normalized to Renilla constitutive reporter to control for differences in transfection efficiency.
Statistical analysis
Data are presented as means ± SEM from three or more separate experiments. P value calculations were conducted using an unpaired Student's t test for two-group comparison or ANOVA (with Bonferroni posttest analysis for multiple comparisons). All data were analyzed and presented by using GraphPad Prism software (version 5.0; GraphPad Software Inc).
Study approval
All studies involving human subjects were performed with approval from the Institutional Review Board of the University of Colorado School of Medicine.
Results
Gene expression microarray analysis
Microarray based expression profiling of 14 individual human gonadotrope pituitary tumors and nine normal pituitary samples was used to characterize candidates involved in pituitary tumorigenesis as previously described (9, 10, 13, 14). This analysis revealed 1911 unique genes expressed more than 2.0-fold (gonadotrope tumor compared with normal pituitary). Among these, 1121 genes were down-regulated, whereas 790 genes were upregulated in gonadotrope tumor vs normal pituitary samples.
Analysis of copy number aberrations
To identify genetic alterations in human pituitary tumor samples, copy number variation profiling was performed on 10 individual gonadotrope pituitary tumors. Overall, 446 copy number variations were observed, including 110 amplifications and 336 deletions.
Identification of Xq26.2 amplification
Genomic copy number variation analysis detected that chromosome X was almost entirely deleted with the exception of the amplification of region ChrXq26.2 in one tumor sample (case 119). This was a tumor (see magnetic resonance image, Figure 1A) in a patient with a highly invasive gonadotrope adenoma, characterized by cavernous, bony invasion, and suprasellar extension. The amplification of the segment of Xq26.2 was confirmed and corresponded to 2.6 copies of region ChrX q26.2 in case 119, compared with two copies in female tumors and normal pituitary samples and one copy number in the male tumor sample analyzed (Figure 1B).
Figure 1.
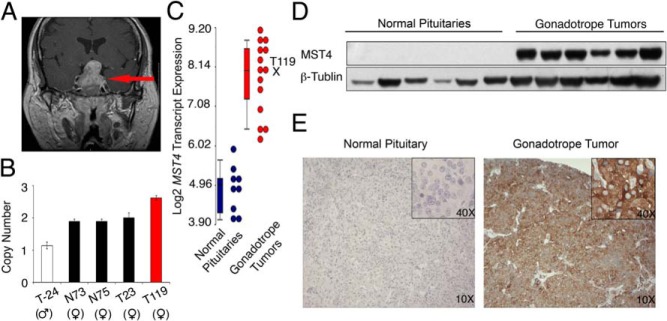
The MST4 kinase is up-regulated in human gonadotrope pituitary tumors. A, Magnetic resonance imaging of the gonadotrope tumor with genomic amplification of region containing MST4 (red arrow). B, MST4 copy number was increased in the tumor. Quantitative PCR was performed using a Taqman probe in case 119 and male and female tumor (T) and normal (N) pituitary samples. C, MST4 transcript levels are amplified in gonadotrope tumors compared with normal pituitary samples (see Materials and Methods for details). D, MST4 protein levels are increased in gonadotrope tumors and low to absent in normal pituitaries. Lysates of human tumor samples and normal pituitary were analyzed by immunoblot using antibodies against human MST4 and GAPDH. E, MST4 protein expression by immunohistochemistry is up-regulated in gonadotrope tumors. Immunohistochemistry was performed on paraffin sections of human normal pituitary and gonadotrope tumor samples.
The MST4 transcript is up-regulated in gonadotrope pituitary tumors
Our prior data set of gene expression profiling (13, 14) was queried for genes localizing to this region, ChrX q26.2. Four genes were detected in this locus: MST4, LOC286467 (an unknown gene), FRMD7 (FERM domain containing 7), and RAP2c (member of RAS oncogene family) (Supplemental Figure 1A). Among these, there was no transcript expression of LOC286467 or FRMD7 in the tumor samples and no differential expression of RAP2c between tumors and normal tissue. In contrast, the MST4 transcript was increased 6.3-fold in the pituitary tumors compared with normal pituitary (Figure 1C). Consistent up-regulation of MST4 mRNA levels was confirmed in a panel of gonadotrope pituitary tumor samples using semiquantitative PCR (Supplemental Figure 1B).
MST4 protein levels are up-regulated in tumors
MST4 protein levels were assessed by immunoblots and demonstrated that the MST4 protein was consistently expressed in gonadotrope tumors and not detected in normal pituitary samples (Figure 1D). Immunohistochemical studies then confirmed that MST4 was abundantly expressed in the cytoplasm of gonadotrope tumors and was very low or absent in normal pituitary samples (Figure 1E).
MST4 enables colony formation and promotes proliferation in pituitary cells under long-term hypoxic stress
In the absence of human pituitary gonadotrope cell lines, functional studies were performed in LβT2 mouse gonadotrope pituitary cells (32). Endogenous Mst4 mRNA as well as MST4 protein levels are low to undetectable in LβT2 cells (data not shown). In Figure 2B (insert), mouse MST4 was stably overexpressed, and its levels similar to that in human tumors confirmed by immunoblot. Given that a primary role of the Ste20-like kinases in other systems is to regulate cell growth and survival (31, 33) and pituitary tumors have lower oxygen saturation (29, 30), we asked whether MST4 acts as a tumorigenic signal in pituitary gonadotrope cells under a hypoxic microenvironment. To mimic a chronic hypoxic milieu, LβT2-MST4 or vector controls were grown in soft agar under a normoxic (20% O2) or moderate hypoxic conditions (5% O2) for 14 days (Figure 2, A and B). MST4 and control cells showed similar number of colonies under normoxic conditions. In contrast, under chronic hypoxic stress, maintained numbers of colonies expressing MST4 were observed (369 ± 8.15 compared with 247 ± 12.67 colonies per well, respectively, a 1.5-fold increase, P = .02, Figure 2B). These data suggest that the MST4 kinase expression enables colony formation of pituitary gonadotrope cells in a hypoxic microenvironment.
Figure 2.

MST4 overexpression enables tumor colony formation and promotes proliferation under long-term hypoxia. A, Photomicrograph of soft agar colony growth in vector and MST4 stable transfectants under normoxia (20% O2) or hypoxia (5% O2) for 14 days. B, MST4 was overexpressed in LβT2 pituitary cells and protein expression detected by immunoblot (insert). MST4 overexpression increased colony formation under hypoxia. Number of colonies in vector control and MST4 transfectants were counted at 14 days. Colony numbers of transfectants cultured under 5% O2 were compared with the transfectants cultured under 20% O2 (n = 3). C, Images of the BrdU staining of LβT2 transfectants grown under normoxic or hypoxic conditions for 14 days. LβT2 MST4 and control transfectants were grown under 20% or 5% O2, and cell proliferation was determined by BrdU staining of nuclei (white arrows). Bar, 20 μm. D, MST4 increased rates of cell proliferation under chronic hypoxia. The proliferation rate of LβT2 transfectants was analyzed from 1 to 14 days and was expressed as percentage of BrdU-positive cells to total cells (n = 3. E, p38MAPK and AKT are hyperactivated in gonadotrope pituitary tumors and LBT2-MST4 transfectants. Levels of endogenous kinases (total p38MAPK, ERK, and AKT) and their phosphorylated forms (P-p38MAPK, P-ERK, and P-AKT) were analyzed by immunoblot in individual human gonadotrope tumor samples compared with normal pituitary (left panel), and the same analysis was performed in LβT2-MST4 transfectants compared with vector control cells (right panel) (n = 3). F and G, p38MAPK and AKT are activated during MST4-induced proliferation. After exposure to hypoxia (1% O2 for 3 d), cell proliferation was assessed by BrdU staining of nuclei in the presence or absence of a p38MAPK inhibitor (SB203580, 10 μM), AKT inhibitor (LY294002, 10 μM), or MEK inhibitor (PD98059, 30 μM), respectively. F, Images represent BrdU-positive cells of MST4 transfectants after the inhibitor treatment. Bar, 20 μm. G, The proliferation rate was expressed as BrdU-positive cells to total cells (n = 3). Values represent mean ± SEM of three independent experiments. *, P = .02, **, P < .01, MST4 cells compared with vector control cells; #, P < .01, inhibitor-treated MST4 cells compared with DMSO vehicle treatment.
To further explore the mechanisms underlying MST4-induced tumorigenic effects, we examined cell proliferation by BrdU staining under normoxia (20% O2) or chronic hypoxia (5% O2) for 14 days (Figure 2C). Under normoxic conditions, MST4 and control transfectants showed similar rates of proliferation (Figure 2D). However, MST4 transfectants showed increased rates of proliferation compared with controls under chronic hypoxia (Figure 2D; 2.60-fold at 7 d, P < .001; 2.35-fold at 9 d, P = .002; 2.27-fold at 11 d, P = .002; 2.16-fold at 14 d, P = .001). In addition, MST4 transfectants showed increased cell numbers as assessed by cell counts compared with control cells (1.64-fold at 9 d, P = .003; 1.72-fold at 11 d, P = .001; 1.82-fold at 14 d, P = .001, Supplemental Figure 2A). Together these data suggest that the increased MST4 expression in human pituitary tumors may serve the role of increasing cellular proliferation, thus aiding in tumor progression.
MST4 regulates cell proliferation via p38 MAPK and AKT signaling cascades
In prior work, MST family members have been shown to regulate cell growth, transformation, and cell death via modulation of diverse intracellular signaling pathways (20). To identify cell specific MST4-dependent pathways in gonadotrope tumors, we first asked which intracellular signaling pathways are activated in human pituitary tumor samples and whether this pattern was replicated in LβT2-MST4 transfectants. Gonadotrope tumors displayed an up-regulation of active phospho-p38MAPK, phospho-ERK, and phospho-AKT compared with normal pituitary samples (Figure 2E, left panel). In the LβT2-MST4 transfectants (Figure 2E, right panel), phospho-ERK was constitutively high at baseline and not increased further with MST4 overexpression. In contrast, a robust activation of p38MAPK and AKT was detected with stable overexpression of MST4, similar to that in human tumor samples. These data demonstrate that the AKT and p38MAPK signaling pathways are increased in both human tumors and MST4 pituitary gonadotrope cell transfectants and suggested their potential importance in transducing the downstream effects of the kinase.
To investigate whether p38MAPK and phosphatidylinositol 3-kinase/AKT pathways were downstream effectors of MST4 actions to promote proliferation, LβT2 transfectants were incubated in the absence or presence of inhibitors specific for each pathway, and BrdU incorporation assays were performed for 3 days under a standard model of severe hypoxic stress (1% O2) (Figure 2F). As shown in Figure 2G, MST4 transfectants proliferated at a higher rate than control cells (3 d, 5.2-fold, P < .001). However, in the presence of the p38MAPK inhibitor (SB203580) or the AKT inhibitor (LY294002), MST4 transfectants lost their proliferative advantage, displaying rates of cell proliferation similar to vector controls (1.2-fold in the presence of SB203580, P = .15; 1.18-fold in the presence of LY294002, P = .18). MST4 cells incubated with the MAPK kinase (MEK [Mitogen-activated protein kinase kinase]) inhibitor (PD98059), however, maintained increased rates of proliferation (5.1-fold increase compared with vector controls, P = .03), similar to dimethylsulfoxide (DMSO)-treated MST4 cells.
To confirm these data, cell proliferation was assessed by cell counts (Supplemental Figure 2B). Similar to studies with BrdU incorporation, the MST4 transfectants showed a higher proliferation rate compared with controls (2.1-fold, P = .032). In the presence of the p38MAPK inhibitor (SB203580) or the AKT inhibitor (LY294002), MST4 induced cell proliferation was significantly inhibited back to levels in vector controls (1.33-fold in the presence of SB203580, P = .16 and 1.2-fold in the presence of LY294002, P = .23). In contrast, MST4 cells incubated in the presence of the MEK inhibitor (PD98059) to block ERK MAPK, maintained increased rates of proliferation (1.52-fold compared with controls, P = .03). Together these data suggest that p38MAPK and AKT, but not ERK MAPK, are downstream effectors of the MST4 kinase to promote pituitary gonadotrope cell proliferation.
MST4 decreases rates of cell death in response to hypoxia
Based on studies showing that hypoxia modifies a vast range of cellular functions that allow tumor cells to not only proliferate but to augment survival (34, 35), we hypothesized that MST4 may protect pituitary gonadotrope cells from hypoxic stress-induced apoptosis. Rates of gonadotrope cell death were assessed in models of chronic and acute hypoxic stress. Under long-term hypoxia (5% O2 for 14 d, Supplemental Figure 2C), cell death was measured by trypan blue exclusion. MST4 expression resulted in decreased rates of cell death in response to chronic hypoxia (1.32-fold at 11 d, P = .01; 1.50-fold at 14 d, P = .001).
To further address the survival effect observed, cells were exposed to a standard severe hypoxia model (1% O2) overnight, and cell death was measured by TUNEL (Figure 3A). An increase rate of apoptosis was detected in vector cells in a time-dependent manner in response to hypoxic stress (8.9-fold at 7 h, P < .001; 9.8-fold at 17 h, P < .001). In contrast, MST4 cells were less susceptible to hypoxia-induced apoptosis compared with vector controls (1.37-fold at 4 h, P = .06; 2.92-fold at 7 h, P < .001; 3.1-fold at 17 h, P < .001, Figure 3B). Cell viability was also assessed by Hoechst staining of nuclear condensation (Supplemental Figure 3, A and B), which confirmed the antiapoptotic effects of MST4 with exposure to hypoxia. Rates of cell death increased with exposure to hypoxia in control cells (5.11-fold at 7 h, P < .001; 8.2-fold at 17 h, P < .001). In contrast, MST4 transfectants showed decreased rates of apoptosis (2.9-fold at 7 h, P = .007; 3.0-fold at 17 h, P = .003). Together these data support the hypothesis that the MST4 kinase allows pituitary gonadotrope cells to respond to a hypoxic stress with decrease in rates of apoptosis to promote and/or maintain tumorigenesis.
Figure 3.

MST4 protects pituitary cells from hypoxia-induced apoptosis. A, Overexpression of MST4 decreases hypoxia-induced apoptosis. MST4 or control cells were exposed to 1% O2 for 17 hours, and apoptosis was assessed by TUNEL staining (white arrows). Bar, 20 μm. B, Rates of apoptosis were expressed as percentages of TUNEL-positive cells to total cells (n = 3). C, p38MAPK and AKT mediate protective effects of MST4. In the presence of SB203580 (10 μM), LY294002 (10 μM), and PD98059 (30 μM), cell death was measured by TUNEL after exposure to 1% O2 for 17 hours. In the upper panel, images represent TUNEL positive staining, and the rates of apoptosis were expressed as percentages of TUNEL cells to total cells (n = 3). Bar, 20 μm. **, P < .01, MST4 cells compared with vector control cells; †, P < .001, rates of apoptosis of vector cells at 7 and 17 hours of hypoxia compared with 0 hours, respectively; #, P < .01, inhibitor-treated MST4 cells compared with DMSO vehicle treatment. DAPI, 4′,6′-diamino-2-phenylindole.
To investigate whether similar intracellular signaling pathways mediated the ability of MST4 to confer survival advantage under hypoxic stress, cells were incubated with pathway specific inhibitors in the presence of severe hypoxia (1% O2 overnight), and apoptosis was assessed by TUNEL and Hoechst staining of nuclear condensation. As shown in Figure 3, C and D, decreased rates of apoptosis were detected in MST4 cells in response to hypoxia compared with vector cells (2.6-fold at 17 h, P = .01) by TUNEL staining. Inhibition of p38MAPK (SB203580) or AKT (LY294002) abolished the MST4 protective effects on cell survival with rates of cell death similar to that of vector controls (0.97-fold in the presence of SB203580, P = .89 and 0.95-fold in the presence of LY294002, P = .8, compared with vector controls, respectively). However, incubation with the ERK pathway inhibitor had no effect on MST4 actions to protect from hypoxia-induced apoptosis. Similarly, using Hoechst staining of nuclear condensation, MST4 cells displayed decreased rates of hypoxia-induced apoptosis compared with vector controls (2.83-fold, P = .006, Supplemental Figure 3C). Incubation with p38MAPK (SB203580) and AKT (LY294002) inhibitors diminished the antiapoptotic effects of MST4 (1.0-fold in the presence of SB203580, P = .76 and 0.8-fold in the presence of LY294002, P = .09, compared with vector controls, respectively), whereas inhibition of ERK pathway did not (2.6-fold compared with vector controls, P = .002). Thus, both MST4-dependent proliferative and survival actions under hypoxic stress are dependent on the p38 MAPK and AKT but not the ERK MAPK signaling pathways.
HIF-1 mediates MST4 protected pituitary cell survival and proliferation under the hypoxic condition
Hypoxic stress triggers the stabilization of the hypoxia inducible factor proteins that then translocate to the nucleus to activate cell specific downstream events (36). Compared with controls, gonadotrope pituitary cells expressing MST4 had increased levels of HIF-1 protein in the presence of 1% O2 (Figure 4A and Supplemental Figure 4A). Considering the protumorigenic role of HIFs, we hypothesized that HIF-1 played a key role in mediating MST4 effects of protection from apoptotic cell death during hypoxic stress. HIF-1 blockade using lentiviral Hif-1 shRNA (70% inhibition, Supplemental Figure 4, B and C) resulted in a loss of the ability of MST4 to protect cells from apoptosis under hypoxic stress as assessed by TUNEL (P < .001, Figure 4B) and Hoechst staining (P < .001, Supplemental Figure 4D). Similarly, HIF-1 inactivation by shRNA treatment abolished the MST4-induced cell proliferation as analyzed by BrdU staining (1.2-fold compared with vector controls, P = .12, Figure 4C). The selectivity of the HIF-1 silencing was confirmed by a rescue experiment in which a HIF-1 construct insensitive to Hif-1 shRNA was overexpressed (Supplemental Figure 4E). In these studies, HIF-1 silencing in the presence of the Hif-1 insensitive construct retained the MST4 protected survival (compare Supplemental Figure 4D with 4F) and proliferative effects (compare Figure 4C with Supplemental Figure 4G). Together these data confirm that HIF-1 serves as a downstream effector of the MST4 kinase.
Figure 4.

MST4 protection of pituitary cell survival requires HIF-1. A, MST4 overexpression up-regulates HIF-1 protein during hypoxia. After exposure to hypoxia (1% O2 for 0, 4, 7 and 17 h), HIF-1α protein expression was measured by immunoblot. B and C, Inactivation of HIF-1 impaired MST4-protected cell survival and proliferation. MST4 or vector cells were treated with scrambled or Hif-1 shRNA for 48 hours. B, TUNEL staining was performed to assess the rates of apoptosis after exposure to 1% O2 for 17 hours, and the rates of apoptosis were expressed as a percentage to TUNEL positive cells to total cells (n = 3). C, BrdU staining was used to analyze cell proliferation after 1% O2 treatment for 3 days, and the rates of proliferation were expressed as a percentage of BrdU-positive cells to total cells (n = 3). D, MST4 increases HIF-1 activity in response to hypoxia. The MST4 or vector cells were transfected with HRE-luciferase reporter constructs. After 24 hours of transfection, cells were subjected to normoxia (20% O2) or hypoxia (1% O2) for 17 hours. HRE luciferase values were detected and normalized to Renilla luciferase values (n = 5 independent experiments). E, and F, Glut1 and Ldh mRNA expression are increased in MST4 transfectants under hypoxia. mRNA levels of Glut1 and Ldh were detected by quantitative PCR in LβT2 transfectants under normoxia (20% O2) or hypoxia (1% O2).*, P < .01 and **, P < .001, MST4 cells compared with vector control cells; #, P < .001, treated MST4 cells compared with nontreated cells; †, P < .001, vector cells during hypoxia compared with normoxia.
HIF-1 translocates to the nucleus to activate nuclear targets containing a hypoxic response element. To confirm that the MST4 kinase directs nuclear HIF-1 activity, a HIF-1 target assay was performed in which a luciferase construct expressing three copies of the hypoxic response element was transiently transfected into pituitary cells and luciferase activity was assessed. Compared with controls, MST4 transfectants showed a hyperactivation of the luciferase reporter activity in response to hypoxia (2.0-fold increase, P < .001, Figure 4D).
To test the possibility that activation of p38MAPK or AKT might be upstream of the increase in HIF-1 activity, the LβT2 transfectants were treated with either SB203580 or LY294002 prior to exposure to hypoxia (1% O2). As shown in Supplemental Figure 4H, neither inhibitor altered HIF-1 activity in the MST4 transfectants in response to the hypoxic stimulation, indicating that HIF-1 activation is not directly downstream of the p38MAPK and AKT pathways in the mediation of the MST4 kinase effects.
Because there are more than a hundred reported nuclear promoters activated by HIF-1 (37), as an initial screen, we tested the effects of MST4 to modulate a panel of HIF-1 target genes during hypoxia (Figure 4, E and F). Under short-term severe hypoxia, MST4 transfectants displayed increased Glut1 and Ldh mRNA expression compared with vector cells (2.1-fold, P = .03, and 2.6-fold, P = .01, respectively), and this effect was diminished by inactivation of HIF-1 (0.96-fold compared with vector controls, P = .2, and 0.82-fold compared with vector controls, P = .12, respectively; Supplemental Figure 4, I and J). In contrast, MST4 overexpression had no significant effects on a panel of other HIF-1 target genes (Supplemental Figure 5). Together these data suggest that the MST4 kinase signals via HIF-1 and gonadotrope cell-specific nuclear targets to promote tumor cell survival during hypoxic stress.
MST4 kinase-induced protection from hypoxia-induced death requires the kinase but not the regulatory C terminus
Because the MST kinases have multiple domains that may act as docking sites for different protein-protein interactions as well as domains that mediate kinase function (25), we next dissected the regions of MST4 that promote its functional effects in pituitary gonadotrope cells. MST4 mutants, including K53E, a kinase dead mutant, T178A, which results in a loss of autophosphorylation and a C-terminus deletion (δC) that removes the major docking domains, were tested for their ability to confer a survival advantage in response to hypoxic stress (Figure 5A). Semiquantitative PCR confirmed the expression of wild-type and mutant Mst4 (Figure 5B and Supplemental Figure 6A), and their protein expression was determined by immunoblot analysis (Supplemental Figure 6B). Transfectants expressing wild-type MST4 or mutant MST4 proteins were exposed to hypoxia (1% O2 overnight) and rates of apoptosis assessed by TUNEL labeling of apoptotic cells. MST4 wild-type cells showed a decreased number of apoptotic cells compared with vector controls (2.6-fold, P = .001, Figure 5C). Mutation of the kinase (K53E) or the autoactivation site (T178A) abolished the protective effects (P = .005 and P = .002, respectively) back to the rates of cell death observed in controls. Similar results were observed measuring cell viability by Hoechst staining (P = .02 and P = .01, respectively; Supplemental Figure 6C). In contrast, the C-terminal mutant (δC), with an intact kinase domain but lacking the docking or regulatory domain, displayed effects similar to MST4 wild-type, with decreased rates of hypoxia-induced apoptosis (2.5-fold compared with vector control, P = .001; Figure 5C), and cell viability (2.89-fold compared with vector control, P < .001; Supplemental Figure 6C). These data support the importance of the kinase in mediating the effects of MST4 to protect pituitary gonadotrope cells from hypoxia-induced cell death.
Figure 5.
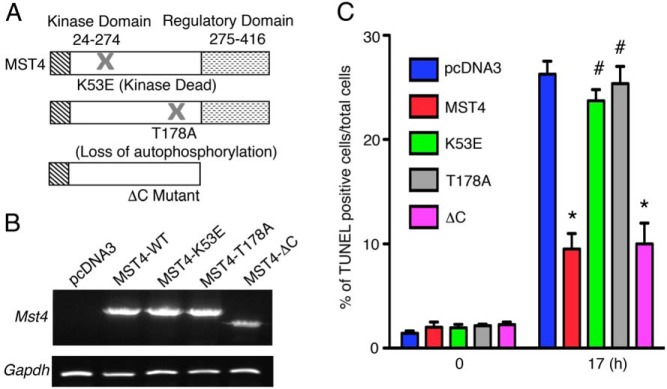
The functional effects of MST4 are dependent on its kinase domain. A, Structure of wild-type and MST4 mutants. B, MST4 wild-type and mutants (K53E, T178A, and δC) were stably overexpressed in LβT2 pituitary cells, and mRNA levels were detected by semiquantitative PCR. C, The MST4 kinase sequence modulates MST4-induced protective effects against cell death. Hypoxia-induced apoptosis (1% O2 for 17 h) was measured by TUNEL staining in LβT2 transfectants. Rates of apoptosis were expressed as percentage of TUNEL-positive cells to total cells (n = 4). *, P < .001, MST4 wild-type and δC compared with vector control; #, P < .01, MST4 mutants of K53E and T178A compared with wild type.
Discussion
Using copy number variation analysis and DNA microarray profiling of individual human gonadotrope pituitary tumors and normal pituitaries, we identified the MST4 kinase as a genetically and genomically dysregulated gene. Whereas only 1 of 10 tumors in our screen had a specific genomic amplification, the MST4 transcript, RNA, and protein levels were consistently high in all tested human gonadotrope tumor samples, suggesting its potential role as a novel kinase to target. In functional studies, MST4 promoted pituitary tumorigenesis inducing colony formation in soft agar by modulating cell proliferation and survival in response to a hypoxic microenvironment. Both proliferation and antiapoptotic effects of MST4 were dependent on the downstream activation of p38 MAPK and AKT, similar to some of the activated signaling pathways observed in human tumor samples. Activation of HIF-1 and its downstream nuclear targets were also critical for MST4 action. Together these data suggest that the MST4 kinase and its downstream signal transduction pathways are novel candidates involved in human gonadotrope pituitary tumor initiation, maintenance, and/or progression.
Whereas MST4 was consistently up-regulated in pituitary tumors, there were no differences in the transcript levels of the other family members, MST3 or SOK1, in tumors compared with normal pituitary samples (data not shown). Although we identified a mutation causing amplification of the chromosomal region containing MST4 in one tumor, all tumors displayed up-regulation of the MST4 transcript, suggesting that MST4 may be dysregulated by multiple mechanisms in pituitary tumorigenesis. The role of MST4 in tumor development or maintenance has not been extensively explored. High expression is observed in a variety of malignancies, including prostate cancer and malignant pleural mesothelioma (33, 38). Functional roles have been observed in tumor cell lines through overexpression experiments, suggesting roles in proliferation, colony formation, and transformation. Prior to our studies, only one report showed that MST4 increased anchorage independent growth of prostate cells and tumor formation in nude mice (33). However, MST4 is proapoptotic in other systems such as breast cancer (39), implying that MST4 serves a diversity of roles and cell specific functions.
Our data suggest that MST4 plays a role in cellular transformation and progression under a hypoxic microenvironment, which is known to be observed in human pituitary tumors (29, 30, 40). Tumor cells respond to hypoxic stress by overcoming nutrient deprivation or escape from the microenvironment by proliferation and invasion (35). Responding to hypoxia stress, pituitary cells overexpressing MST4 showed an increased rate of growth and increased survival. Prior studies suggested that in embryonic kidney cells, cerebral cavernous modulator-3 modulates MST4 function to enhance cell growth and shuttles MST4 from the Golgi to the plasma membrane to interact with ezrin/radixin/moesin proteins to promote cell survival (41, 42). We have not detected any interaction with cerebral cavernous modulator-3 or shuttling of MST4 to activate ezrin/radixin/moesin proteins in response to hypoxia in pituitary gonadotrope cells (our unpublished observations). Thus, further studies are warranted to identify the upstream activators of MST4.
How is the signal transduced from the MST4 kinase to the downstream events in response to a hypoxic microenvironment? Earlier studies suggested that MST kinases activate multiple signaling pathways, including p38, c-Jun N-terminal kinase, ERK MAPKs, and AKT, in a cell-specific fashion (reviewed in reference 20). Initial reports suggested that MST4 did not activate ERK, c-Jun N-terminal kinase, or p38 MAPK in human embryonic kidney 293 cells but activated ERK via mitogen-activated protein/ERK kinase in Phoenix cells (43, 44). Our studies show that human gonadotrope pituitary tumors display activated p38MAPK and AKT, and functional studies indicate that both pathways are required for the MST4 effects on proliferation and survival during hypoxia in gonadotrope pituitary cells. In some systems, p38MAPK is implicated as a negative regulator of cell proliferation; conversely, in gonadotrope pituitary cells and other model systems (45), the effects of p38MAPK are to promote cell proliferation and tumorigenesis.
Hypoxia signaling via HIFs leads to multiple cellular alterations during tumorigenesis (34, 35), including protection from hypoxia or nutrient deprivation-induced apoptosis (46). Our studies suggest that HIF-1 is a critical downstream component in the MST4 signaling pathway but is independent of p38MAPK and AKT pathways. HIF-1 activates a diverse set of genes during hypoxic stress (37). Our initial screen of candidate targets identified that the Glut1 and Ldh genes are downstream of MST4 and HIF-1. The constitutive expression of glucose transporter 1 would be expected to contribute to the Warburg effect to promote tumor cell growth as an adaptive response to hypoxia (47). Further studies with a nonbiased screen will be needed to identify other pituitary specific targets.
Recent structure analysis showed the MST4 kinase is localized to the N-terminal domain, whereas the C-terminal domain mediates heterodimerization with other docking proteins (43, 48–50). Analysis of site specific MST4 mutants (K53E and T178A) suggests that the MST4 actions to promote cell proliferation and survival in response to a hypoxic stress in pituitary gonadotrope cells are critically dependent on the N-terminal kinase and autophosphorylation sites. The C-terminal domain is dispensable, indicating that MST4 mediates its protective effects independent of a requirement for crosstalk with other docking proteins (49, 50).
Our focus in these studies was to identify novel therapeutic targets in human gonadotrope pituitary tumors in which there are currently no medical therapies. Preliminary data suggest that MST4 is also variably expressed in other human pituitary tumor types (our unpublished observations). Thus, further studies are warranted to address whether it plays a more general role in human pituitary tumor development and/or progression in a hypoxic microenvironment. Our results support future research efforts to use small molecules and kinase inhibitor screens to test whether MST4 is a viable therapeutic target in human pituitary tumors.
Acknowledgments
We thank Maozhen Tian for initiating the project; Taylor S. Mills, Smita Salian-Mehta, and Holger Elzschig for reviewing the manuscript; and Francisco La Rosa for help with photography. The LβT2 cells were provided by Dr Pamela Mellon (University of California, San Diego, California). The human HIF-1 construct was provided by Dr Louise Glover (University of Colorado, Aurora, Colorado).
This work was supported by Veterans Affairs Merit Review Award 001 (to M.E.W.), NIH K12CA086913-12 (to K.K.V.), University of Colorado Cancer Center Support Grant P30-CA046934, and the Cancer Center Genomics Core.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BrdU
- 5-bromo-2′-deoxyuridine
- ChrX
- chromosome X
- DMSO
- dimethylsulfoxide
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GCK
- germinal center kinase
- HIF-1
- hypoxia inducible factor-1
- MEK
- MAPK kinase
- MST
- mammalian Ste20-like kinase
- shRNA
- short hairpin RNA
- Ste20
- sterile 20
- TUNEL
- terminal deoxynucleotidyl transferase deoxyuridine 5-triphosphate nick end labeling.
References
- 1. Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004;101:613–619. [DOI] [PubMed] [Google Scholar]
- 2. Melmed S. Update in pituitary disease. J Clin Endocrinol Metab. 2008;93:331–338. [DOI] [PubMed] [Google Scholar]
- 3. Melmed S. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest. 2003;112:1603–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rogers A, Karavitaki N, Wass JA. Diagnosis and management of prolactinomas and non-functioning pituitary adenomas. BMJ. 2014;349:g5390. [DOI] [PubMed] [Google Scholar]
- 5. Molitch ME. Nonfunctioning pituitary tumors. Handb Clin Neurol. 2014;124:167–184. [DOI] [PubMed] [Google Scholar]
- 6. Freda PU, Beckers AM, Katznelson L, et al. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Herman V, Fagin J, Gonsky R, Kovacs K, Melmed S. Clonal origin of pituitary adenomas. J Clin Endocrinol Metab. 1990;71:1427–1433. [DOI] [PubMed] [Google Scholar]
- 8. Alexander JM, Biller BM, Bikkal H, Zervas NT, Arnold A, Klibanski A. Clinically nonfunctioning pituitary tumors are monoclonal in origin. J Clin Invest. 1990;86:336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shorts-Cary L, Xu M, Ertel J, et al. Bone morphogenetic protein and retinoic acid-inducible neural specific protein-3 is expressed in gonadotrope cell pituitary adenomas and induces proliferation, migration, and invasion. Endocrinology. 2007;148:967–975. [DOI] [PubMed] [Google Scholar]
- 10. Xu M, Shorts-Cary L, Knox AJ, Kleinsmidt-DeMasters B, Lillehei K, Wierman ME. Epidermal growth factor receptor pathway substrate 8 is overexpressed in human pituitary tumors: role in proliferation and survival. Endocrinology. 2009;150:2064–2071. [DOI] [PubMed] [Google Scholar]
- 11. Zhou Y, Zhang X, Klibanski A. MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol. 2012;48:R45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang X, Sun H, Danila DC, et al. Loss of expression of GADD45 γ, a growth inhibitory gene, in human pituitary adenomas: implications for tumorigenesis. J Clin Endocrinol Metab. 2002;87:1262–1267. [DOI] [PubMed] [Google Scholar]
- 13. Michaelis KA, Knox AJ, Xu M, et al. Identification of growth arrest and DNA-damage-inducible gene β (GADD45β) as a novel tumor suppressor in pituitary gonadotrope tumors. Endocrinology. 2011;152:3603–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu M, Knox AJ, Michaelis KA, et al. Reprimo (RPRM) is a novel tumor suppressor in pituitary tumors and regulates survival, proliferation, and tumorigenicity. Endocrinology. 2012;153:2963–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pagotto U, Arzberger T, Theodoropoulou M, et al. The expression of the antiproliferative gene ZAC is lost or highly reduced in nonfunctioning pituitary adenomas. Cancer Res. 2000;60:6794–6799. [PubMed] [Google Scholar]
- 16. Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol. 2011;7:257–266. [DOI] [PubMed] [Google Scholar]
- 17. Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Riddick G, Fine HA. Integration and analysis of genome-scale data from gliomas. Nat Rev Neurol. 2011;7:439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carlsson E, Ranki A, Sipila L, et al. Potential role of a navigator gene NAV3 in colorectal cancer. Br J Cancer. 2012;106:517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ling P, Lu TJ, Yuan CJ, Lai MD. Biosignaling of mammalian Ste20-related kinases. Cell Signal. 2008;20:1237–1247. [DOI] [PubMed] [Google Scholar]
- 21. Leberer E, Dignard D, Harcus D, Thomas DY, Whiteway M. The protein kinase homologue Ste20p is required to link the yeast pheromone response G-protein βγ subunits to downstream signalling components. EMBO J. 1992;11:4815–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dan I, Watanabe NM, Kusumi A. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 2001;11:220–230. [DOI] [PubMed] [Google Scholar]
- 23. Pombo CM, Force T, Kyriakis J, Nogueira E, Fidalgo M, Zalvide J. The GCK II and III subfamilies of the STE20 group kinases. Front Biosci. 2007;12:850–859. [DOI] [PubMed] [Google Scholar]
- 24. Strange K, Denton J, Nehrke K. Ste20-type kinases: evolutionarily conserved regulators of ion transport and cell volume. Physiology (Bethesda). 2006;21:61–68. [DOI] [PubMed] [Google Scholar]
- 25. Sugden PH, McGuffin LJ, Clerk A. SOcK, MiSTs, MASK and STicKs: the GCKIII (germinal centre kinase III) kinases and their heterologous protein-protein interactions. Biochem J. 2013;454:13–30. [DOI] [PubMed] [Google Scholar]
- 26. Lin CY, Wu HY, Wang PL, Yuan CJ. Mammalian Ste20-like protein kinase 3 induces a caspase-independent apoptotic pathway. Int J Biochem Cell Biol. 2010;42:98–105. [DOI] [PubMed] [Google Scholar]
- 27. Wu HY, Lin CY, Chen TC, Pan ST, Yuan CJ. Mammalian Ste20-like protein kinase 3 plays a role in hypoxia-induced apoptosis of trophoblast cell line 3A-sub-E. Int J Biochem Cell Biol. 2011;43:742–750. [DOI] [PubMed] [Google Scholar]
- 28. Chen XD, Cho CY. Downregulation of SOK1 promotes the migration of MCF-7 cells. Biochem Biophys Res Commun. 2011;407:389–392. [DOI] [PubMed] [Google Scholar]
- 29. Kristof RA, Aliashkevich AF, Hans V, et al. The regional oxygen saturation of pituitary adenomas is lower than that of the pituitary gland: microspectrophotometric study with potential clinical implications. Neurosurgery. 2003;53:880–885; discussion 885–886. [DOI] [PubMed] [Google Scholar]
- 30. Vidal S, Scheithauer BW, Kovacs K. Vascularity in nontumorous human pituitaries and incidental microadenomas: a morphometric study. Endocr Pathol. 2000;11:215–227. [DOI] [PubMed] [Google Scholar]
- 31. Nogueira E, Fidalgo M, Molnar A, et al. SOK1 translocates from the Golgi to the nucleus upon chemical anoxia and induces apoptotic cell death. J Biol Chem. 2008;283:16248–16258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thomas P, Mellon PL, Turgeon J, Waring DW. The LβT2 clonal gonadotrope: a model for single cell studies of endocrine cell secretion. Endocrinology. 1996;137:2979–2989. [DOI] [PubMed] [Google Scholar]
- 33. Sung V, Luo W, Qian D, Lee I, Jallal B, Gishizky M. The Ste20 kinase MST4 plays a role in prostate cancer progression. Cancer Res. 2003;63:3356–3363. [PubMed] [Google Scholar]
- 34. Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. [DOI] [PubMed] [Google Scholar]
- 35. Vaupel P, Harrison L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist 2004;9(suppl 5):4–9. [DOI] [PubMed] [Google Scholar]
- 36. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123:3664–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies hypoxia inducible factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009;37:4587–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gordon GJ, Rockwell GN, Jensen RV, et al. Identification of novel candidate oncogenes and tumor suppressors in malignant pleural mesothelioma using large-scale transcriptional profiling. Am J Pathol. 2005;166:1827–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dan I, Ong SE, Watanabe NM, et al. Cloning of MASK, a novel member of the mammalian germinal center kinase III subfamily, with apoptosis-inducing properties. J Biol Chem. 2002;277:5929–5939. [DOI] [PubMed] [Google Scholar]
- 40. Vidal S, Horvath E, Kovacs K, Kuroki T, Lloyd RV, Scheithauer BW. Expression of hypoxia-inducible factor-1α (HIF-1α) in pituitary tumours. Histol Histopathol. 2003;18:679–686. [DOI] [PubMed] [Google Scholar]
- 41. Fidalgo M, Guerrero A, Fraile M, Iglesias C, Pombo CM, Zalvide J. Adaptor protein cerebral cavernous malformation 3 (CCM3) mediates phosphorylation of the cytoskeletal proteins ezrin/radixin/moesin by mammalian Ste20–4 to protect cells from oxidative stress. J Biol Chem. 2012;287:11556–11565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ma X, Zhao H, Shan J, et al. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol Biol Cell. 2007;18:1965–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qian Z, Lin C, Espinosa R, LeBeau M, Rosner MR. Cloning and characterization of MST4, a novel Ste20-like kinase. J Biol Chem. 2001;276:22439–22445. [DOI] [PubMed] [Google Scholar]
- 44. Lin JL, Chen HC, Fang HI, Robinson D, Kung HJ, Shih HM. MST4, a new Ste20-related kinase that mediates cell growth and transformation via modulating ERK pathway. Oncogene. 2001;20:6559–6569. [DOI] [PubMed] [Google Scholar]
- 45. Han J, Sun P. The pathways to tumor suppression via route p38. Trends Biochem Sci. 2007;32:364–371. [DOI] [PubMed] [Google Scholar]
- 46. Parks SK, Chiche J, Pouyssegur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer. 2013;13:611–623. [DOI] [PubMed] [Google Scholar]
- 47. Munoz-Pinedo C, El Mjiyad N, Ricci JE. Cancer metabolism: current perspectives and future directions. Cell Death Dis. 2012;3:e248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Preisinger C, Short B, De Corte V, et al. YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14–3-3ζ. J Cell Biol. 2004;164:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shi Z, Jiao S, Zhang Z, et al. Structure of the MST4 in complex with MO25 provides insights into its activation mechanism. Structure. 2013;21:449–461. [DOI] [PubMed] [Google Scholar]
- 50. Zhang M, Dong L, Shi Z, et al. Structural mechanism of CCM3 heterodimerization with GCKIII kinases. Structure. 2013;21:680–688. [DOI] [PubMed] [Google Scholar]