

Abstract
Human induced pluripotent stem cells (hiPSCs) are a type of pluripotent stem cells artificially derived from an adult somatic cell (typically human fibroblast) by forced expression of specific genes. In recent years, different feeders like inactivated mouse embryonic fibroblasts (MEFs), human dermal fibroblasts (HDFs), and feeder free system have commonly been used for supporting the culture of stem cells in undifferentiated state. In the present work, the culture of hiPSCs and their characterizations on BD Matrigel (feeder-and serum-free system), MEF and HDF feeders using cell culture methods and molecular techniques were evaluated and compared. The isolated HDFs from foreskin samples were reprogrammed to hiPSCs using gene delivery system. Then, the pluripotency ability of hiPSCs cultured on each layer was determined by teratoma formation and immunohistochemical staining. After EBs generation the expression level of three germ layers genes were evaluated by Q-real-time PCR. Also, the cytogenetic stability of hiPSCs cultured on each condition was analyzed by karyotyping and comet assay. Then, the presence of pluripotency antigens were confirmed by Immunocytochemistry (ICC) test and alkaline phosphatase staining. This study were showed culturing of hiPSCs on BD Matrigel, MEF and HDF feeders had normal morphology and could maintain in undifferentiated state for prolonged expansion. The hiPSCs cultured in each system had normal karyotype without any chromosomal abnormalities and the DNA lesions were not observed by comet assay. Moreover, up-regulation in three germ layers genes in cultured hiPSCs on each layer (same to ESCs) compare to normal HDFs were observed (p < 0.05). The findings of the present work were showed in stem cells culturing especially hiPSCs both MEF and HDF feeders as well as feeder free system like Matrigel are proper despite benefits and disadvantages. Although, MEFs is suitable for supporting of stem cell culturing but it can animal pathogens transferring and inducing immune response. Furthermore, HDFs have homologous source with hiPSCs and can be used as feeder instead of MEF but in therapeutic approaches the cells contamination is a problem. So, this study were suggested feeder free culturing of hiPSCs on Matrigel in supplemented media (without using MEF conditioned medium) resolves these problems and could prepare easy applications of hiPSCs in therapeutic approaches of regenerative medicine such as stem-cell therapy and somatic cell nuclear in further researches.
Keywords: hiPSCs, MEF, HDF, BD Matrigel matrix ICC, Karyotyping, Comet assay, IHC
Introduction
Human induced pluripotent stem cells (hiPSCs) are capable of unlimited reproduction and have a potential to differentiate into the variety of cell types (Park et al. 2008). HiPSCs could be obtained by reprogramming of adult somatic cells such as human dermal fibroblasts (HDFs) using defined factors (Takahashi and Yamanaka 2006; Takahashi et al. 2007; Bayart and Cohen-Haguenauer 2013). The properties of hiPSCs are similar to human embryonic stem cells (hESCs) and the potentially of them have major impact on cell therapy and these cells are useful for future studies of stem cell biology as well as regenerative medicine (Aguilar-Gallardo et al. 2010; Robinton and Daley 2012; Jung et al. 2012). Mouse embryonic fibroblasts (MEFs) and HDFs cell lines are used as a feeder layer to support long-term undifferentiating of iPSCs and ESCs in vitro (Richards et al. 2002; Cheng et al. 2003). The advantages of MEFs feeder are secretion of essential factors for undifferentiated growth of stem cells like iPSCs and ESCs in long-term period. MEF feeder and FBS supplementation in culture media could increase the immune response and the risk of exposure of iPSCs to exogenous antigens and zoonotic animal pathogens such as retrovirus (Jung et al. 2012; Ghasemi-Dehkordi et al. 2014). Furthermore, separations of iPSCs from MEFs in therapeutic aims are difficult. The MEF feeder could be replace by HDF feeder cells or matrix. HDF feeder could be used for iPSCs culture and provides suitable base to support the stem cells. HDF feeder in low passages (P 1–3) could decrease the immune response, easy handling, removing the contamination with animal pathogens, and maintains for long-term periods in the culture (Richards et al. 2002; Tecirlioglu et al. 2010; Bisson et al. 2013; Ghasemi-Dehkordi et al. 2014).
In recent years, feeder-free culture such as BD Matrigel™ using MEF conditioned medium has been reported to be efficient for culture of stem cells. Matrigel is a complex protein mixture containing components such as collagens, laminin, and proteoglycans used as support in the feeder free stem cell culture for long-term. However, Matrigels have an animal origin and using conditioned medium (CM) in therapeutic approaches is important and could increase the risk of animal pathogens transmission (Chen et al. 2014; Ghasemi-Dehkordi et al. 2014).
The culture of stem cells on feeder free systems (like Matrigel matrix) and feeder cell lines (such as HDF and MEF) have advantages and disadvantages. In the present study, the characterizations of hiPSCs on feeder-and serum-free culture (BD Matrigel) were compared to MEF and HDF feeder layers.
Materials and methods
HDFs isolation and preparation
In this study, the healthy male neonatals were subjected to obtain foreskin samples from Kashani Hospital (Shahrekord City, southwest Iran) and specimens were transferred to Cellular and Molecular Research Center. The foreskin tissue was kept in 70 % ethanol for 2 min and then was dissected and washed with phosphate-buffered saline (PBS) and was centrifuged at 1200 rpm for 5 min. The supernatant was discarded and the tissue was enzymatically digested using 0.25 % Trypsin/EDTA (Gibco Cat no. 15090–046) solution, 100 U/mL collagenase type IV, and 100 μg/mL of DNase (both Sigma-Aldrich, Poole, UK), for 20 min. The enzyme activity was neutralized with equal volume of Dulbecco’s modified Eagle’s medium (DMEM) containing 10 % fetal bovine serum (FBS) and cell suspension was passed through a Mesh filter (BD Falcon, 9340329) and centrifuged at 1200 rpm for 5 min. Then, the single cells were cultured in DMEM including 10 % FBS, and 1 % penicillin/streptomycin antibiotics (all Gibco, Grand Island, NY, USA) at 37 °C in a 5 % CO2 atmosphere.
Preparing of plasmids
The TetO-FUW-OSKM and Oct4-GFP (transgenic reporter and contain green fluorescent protein used as a positive control) plasmids and psPAX2 and pMD2.G vectors (used as a lentivirus packaging and producing viral particles) were taken from the Rudolf Jaenisch laboratory and Tronolab (Switzerland), respectively. The plasmids were transformed separately in E. coli strain Top10F (Invitrogen) using T/A Cloning Kit (Fermentas, Germany) according to the manufacturerʼs protocol. The plasmids were extracted from bacterial cells using Plasmid Mini Kit (BIONEER, South Korea) and after confirmation the plasmids by restriction enzymes digestion and PCR techniques the E. coli cells contain each plasmid were cultured for large scale. The viral plasmids were purified from bacterial cells using the QIAGEN Plasmid Maxi Kit (Qiagen) according to the manufacturerʼs instructions and were used for lentiviruses production.
Culture of HEK-293 T cells for lentiviruses production and concentration
The HEK-293 T cells were obtained from American Type Culture Collection (ATCC, Rockville, MD) and were cultured in 10 cm2 plates (Techno Plastic Products (TPP), 93100) containing DMEM with FBS (10 %) and after cell confluence reach to 70–80 % were transfected with lentiviruses based on the calcium phosphate precipitation principle to producing viruses (King et al. 1995; Kipreos et al. 1996). In brief, a 22.5 μg of TetO-FUW-OSKM lentiviral vector (Rudolf Jaenischʼs lab) containing OCT4, SOX2, C-MYC, and KLF4 genes, 14.6 μg of gag-pol expression plasmid (psPAX, Tronolab), and 7.9 μg VSV-G envelop plasmid (pMDG2, Tronolab) in 50 μL of CaCl2 2.5 mM and 450 μL of 2X HBS (140 mM NaCl, 1.5 mM Na2HPO4, 50 mM 4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES, (Sigma-Aldrich, St. Louis, MO, cat. no. H7006) pH = 7.05) was added and mixed gently and then incubated at room temperature (RT) for 20 min. Furthermore, in another group the eGFP-expressing vector (Tronolab) was used as a positive control instead of TETO-FUW-OSKM. The mixture was added drop-wise onto the HEK-293 T cells. After 48–72 h of transfection the supernatant containing lentiviruses were collected and centrifuged at 4000 rpm to remove cell debris, and then were passed through 0.45 μm cellulose acetate filters. Viral supernatants were concentrated by ultracentrifugation at 50,000 × g for 2 h to make virus stocks.
Transduction of HDFs with TETO-FUW-OSKM and Oct4-GFP lentiviral vectors
HDFs at passage 2 were seed in 6 wells plate (5 × 106) and were transduced three times with concentrated TETO-FUW-OSKM lentiviral vector and eGFP lentiviruses (positive control), separately. After 12 h the culture media was changed by fresh medium. Forty-eight hours after transduction, eGFP positive fluorescent cells in each group were detected by fluorescent microscope (Nikon eclipse E600, Japan).
MEF isolation for feeder cell preparation
Female mice at days 12–14 of gestation were used for MEF isolation. All mice were anesthetized with chloroform and killed by cervical dislocation and embryos were removed from the uterus and were collected in the transfer media containing DMEM and 1 % penicillin/streptomycin antibiotics. The embryos were maintained in ethanol for 1 min and were washed with PBS. Then, the fetal liver, head, and residual limbs were removed. The samples were spliced into smaller size and washed with PBS again and centrifuged for 5 min at 1200 rpm and tissues were digested for 15 min using 0.05 % Trypsin-EDTA (1X). The enzyme activity were naturalized with FBS and cell suspension were transferred in DMEM containing 10 % FBS and cultured in an incubator at 37 °C in a 5 % CO2 atmosphere. After 7 days, the cells were prepared for first passage. The supernatant medium was discarded and washed with PBS and the 0.05 % Trypsin-EDTA was added to the plate and incubated for 2 min. After cells detached the enzyme was neutralized by FBS and the suspension was centrifuged for 5 min at 1200 rpm. Then, the cells were transferred to a new flask. After 2 to 4 passages the cells proliferation were inactivated with 4 μg/mL of mitomycin C (MMC; Sigma-Aldrich, St. Louis, MO) for 2 h at 37 °C before seeding and used as feeder layer for growing hiPSCs.
Culture of hiPSCs on MEF feeder
The reprogrammed HDFs were transferred on inactivated MEF feeder (2.4 × 104/cm2) for generation of hiPS colonies. The undifferentiated state cells were grown in DMEM/F-12 (Invitrogen, cat no. 11330) supplemented with 20 % knockout serum replacement (SR, Gibco), 100 M nonessential amino acids (Gibco, 11140), 2 mM glutamine (Invitrogen, 35050), 50 U/mL penicillin, 50 g/mL streptomycin, 10−4 M β-mercaptoethanol (Sigma, St Louis, MO), and 20 ng/mL human basic fibroblast growth factor (hbFGF, PeproTech, RockyHill, NJ, USA, 100-18B) in 25 T cell culture flask (Techno Plastic Products (TPP, 93100)). The culture medium was changed every day and cultures were passaged upon colony density were reached 70 to 80 % and incubated at 37 °C in a 5 % CO2 atmosphere. Cell proliferation and characterization were checked at high passages (passage 15).
HiPSCs culture on HDF feeder
The isolated HDFs in low passage (P 1–3) were seeded in six wells plate (2.4 × 104/cm2) and mitotically inactivated with mitomycin C (Sigma-Aldrich; cat. no. M-4287) via protocol mentioned above for supporting the culture of hiPSCs. The culture medium was changed every day and when iPSC colonies reached to 70–80 % confluence, the cells were passaged and maintained at 37 °C in a 5 % CO2 incubator and cultured for long term period (passage 12).
Culture of hiPSCs on BD Matrigel matrix (feeder-free system)
For coating of Matrigel (BD Biosciences, Bedford, MA), 500 μL of BD Matrigel was transferred into each well of six wells plate on ice and spread by rocking gently. The plates were then incubated at 37 °C for 2 h to allow the BD Matrigel to polymerize into gel. Then, the suspensions of reprogrammed HDFs (2.4 × 104/cm2) were transferred onto coated palates with BD Matrigel matrix in DMEM media containing 10 % FBS and the plate was returned into incubator. After 48 h the media was replaced by supplemented DMEM/F-12 (including supplemented factors as mentioned above) and was changed every day for formation of hiPSC colonies. The iPSCs culture were maintained and followed for 10 passages.
Reverse transcription polymerase chain reaction (RT-PCR)
Total RNA from hiPSCs that cultured on MEF and HDF feeders as well as feeder free system (BD Matrigel matrix) were extracted using Biozol (BioFlux Kit Bioer Technology, BSC51M1) according to the manufactureʼs instructions. The quality of extracted RNA was measured by NanoDrop ND- 1000 (Peqlab, Erlangen, Germany) at both 260 and 280 nm according to the method described by Sambrook and Russell (Sambrook and Russell 2001). One μg of RNA samples from each group was used for cDNA synthesize by PrimeScriptTM RT Reagent Kit (Takara Bio Inc, Product Code: RR037A) according to manufacturerʼs instruction (incubated at 37 °C for 15 min for reverse transcription), and at 85 °C for 5 s for inactivation. The expression of genes responsible for pluripotency of hiPSCs including Oct-4, Sox-2, Nonog, Rex-1, hTERT8, FGF4, and AP genes using specific oligonucleotide primers were determined by RT-PCR technique and the sequences were analyzed by basic local alignment search tool (BLAST) in GenBank data (Table 1). Furthermore, the GAPDH primers were used as internal control of reaction as described previously (Pakzad et al. 2010). The PCR reaction was performed in a total volume of 25 μL containing 2 mM MgCl2, 200 μM dNTP mix, 2.5 μL of 10X PCR buffer (all Fermentas, Germany), 1 μg of template cDNA, 0.25 μM of each primer, and 1 unit of Taq DNA polymerase (Roche Applied Science, Germany). The negative control contained all reagents without DNA. The PCR were performed in a Gradient Palm Cycler (Corbett Research, Australia) with the following parameters: initial denaturation at 94 °C for 5 min; followed by 35 cycles at 94 °C for 30 s, annealing at 63 °C (for Oct-4), 65 °C (for FGF4), and 66 °C (for Sox-2, Nonog, Rex-1, and hTERT8 genes) for 30 s, extension at 72 °C for 30 s, and final extension (72 °C for 5 min) was done at the end of the amplification. Ten μL of amplified cDNA were applied to the 3 % agarose gel electrophoresis and constant voltage of 80 V for 30 min was used for products separation. The 50 bp DNA ladder (Fermentas, Germany) was used as a molecular weight marker to determine the length of the amplified fragments. After electrophoresis, the gel was stained with ethidium bromide and examined under UV light and photograph was obtained in UVIdoc gel documentation system (Uvitec, UK).
Table 1.
The sequence of primers used for gene amplification of hiPSCs
| Gene | Primers name | Sequence | Product length (bp) | Accession number |
|---|---|---|---|---|
| Sox-2 | SOX2-F SOX2-R |
5′-ACAGATGCAACCGATGCACC-3′ 5′-TGGAGTTGTACTGCAGGGCG-3′ |
53 | NM_003106 |
| Oct-4 | OCT4-F OCT4-R |
5′-TCGCCGTGTCAATCATTTTC-3′ 5′-AACACCCCGAAAAGACGAG-3′ |
91 | NM_004064 |
| Nonog | NANOG-F NANOG-R |
5′-CCAAAGGCAAACAACCCACTT-3′ 5′-CGGGACCTTGTCTTCCTTTTT-3′ |
62 | NM_001297698 |
| Rex-1 | REX1-F REX1-R |
5′-CCCACAGTCCATCCTTACAGAGTT-3′ 5′-GGGACTTTGCCCCCAAAC-3′ |
67 | XM_005259599 |
| hTERT8 | TERT8-F TERT8-R |
5′-CGGAGACCACGTTTCAAAAGA-3′ 5′-TTTGCAACTTGCTCCAGACACT-3′ |
66 | NM_001193376 |
| FGF4 | FGF4-F FGF4-R |
5′-AGTACCCCGGCATGTTCATC-3′ 5′-CGGTTCCCCTTCTTGGTCTT-3′ |
58 | XM_006718475 |
| GAPDH | GAPDH-F GAPDH-R |
5′-CTCATTTCCTGGTATGACAACGA-3′ 5′-TTCCTCTTGTGCTCTTGCTG-3′ |
121 | NM_001256799 |
Immunocytochemistry (ICC)
ICC test was performed on a number of 1 × 103 hiPSCs cultured on BD Matrigel matrix, MEF and HDF feeder layers. The hiPSC colonies were detached from bottom of flask by mechanically and enzymatically (using collagenase type-IV (Gibco, 17104–019) for 3 min) then were cultured in 24-well plates. The ICC coverslips were placed and coated by gelatin 1 day before the test. After adhering of the colonies, hiPSCs were washed twice with PBS and were fixed with 4 % paraformaldehyde in PBS for 10 min. After that the cells were permeabilized with 0.2 % Triton X-100 in PBS for 15 min in RT and were washed with PBS three times for 5 min. Then, the cells were blocked in 5 % BSA (Bovine serum albumin) for 1 h. The blocking solution were discarded and primary antibody diluted in a BSA 5 % solution were added overnight at 4 °C and then were washed with PBS three times for 5 min again. The primary antibodies included anti-Nanog (0.2 μM; Abcam, ChIP Grade, ab21624), anti-Oct4 (1 μM; Abcam, ChIP Grade, ab19857), anti-SOX2 (1 μM; Abcam, ab97959), anti-SSEA-4 [MC813] (1 μM; Abcam, ab16287). Then, secondary antibodies were added and incubated at RT for 1 h. Finally, the cells were incubated with DAPI solution (nuclei localization) for 2 min and were washed with PBS two times and were evaluated visualized under the fluorescent microscope.
Alkaline phosphatase (AP) staining
Alkaline phosphatase staining was performed using the Leukocyte Alkaline Phosphatase Kit (catalog number: 86R-1KT, Sigma-Aldrich) to evaluate an AP-expressing hiPSC colony. Briefly, hiPSCs were fixed in 2 % paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for 30 min at RT before staining according to manufacturer’s protocol.
Karyotype analysis
The media of cultured iPSCs (1 × 103 cells) on BD Matrigel matrix, MEF and HDF feeder layers in low passages (P 1–3) were replaced by media containing 0.1 μg/mL Karyomax Colcemid solution (Cat. no. 15212–012. Invitrogen, Carlsbad, CA, USA) for 6 h to arrest the cells in metaphase for analyze and evaluation the size, shape, and number of chromosomes. Then, the colonies were detached from the 6 wells plate either mechanically or using collagenase type-IV and then the cells were centrifuged at 1200 rpm for 5 min. After washing in PBS (two times), the cells were gently resuspended in a hypotonic (75 mM) KCl solution and incubated at 37 °C for 20 min. Then, cells were centrifuged at 900 rpm for 10 min at RT and resuspended in 1 mL of Carnoy’s fixative (methanol/acetic acid, 3:1) for two times and in each time the cells were centrifuged at 900 rpm for 10 min. Finally, the pellet was resuspended in a small volume (200 μL/106 cells) of the same cold methanol: acetic acid solution and the cells were spread onto glass slides and allowed to dry (incubating at 75 °C for 3 h) and stained with KaryoMAX Giemsa Stain (Invitrogen). After washing the slides with H2O, routine karyotype analysis was applied in a minimum of 10 chromosome spreads per each slide.
Comet assay
The alkaline comet assay was performed on cultured iPSCs on BD Matrigel matrix, MEF and HDF feeder layers in low passages (P 1–3) according to standard protocol described by McKelvey-Martin with some modifications (McKelvey-Martin et al. 1998). The hydrogen peroxide (H2O2) treatment (to create DNA damages) and healthy normal HDFs were used as controls of comet assay. The 150 × 103 cells/mL of each group were seeded in 3 cm2 Petri dish containing DMEM media 10 % FBS, 2 mM glutamine, and 1 % (100 μg/mL) penicillin/streptomycin antibiotics and were incubate at 37 °C in a 5 % CO2 atmosphere for 2 h. For inducing the DNA lesions and massive single-stranded DNA damages in HDFs the media was replaced by DMEM containing 50 μM H2O2 for 60 min and used as an alkaline comet assay positive control.
The slides of comet assay were prepared by washing with methanol and were heated to remove the proteins. The dakin microscope slides were covered with 250–300 μL of 1 % normal melting point agarose (NMA) (Carlsbad, Ca, USA) that prepared in PBS at 50 °C and were allowed to be fully frosted. For solidify the agarose, coverslips was placed on top and the slides were kept on ice. Then, the cells were resuspended in 80 μL of 0.1 % low melting point agarose (LMA) incubated at 37 °C and were placed directly on a slide. After that the coverslips were gently removed and the cell suspension was pipette rapidly onto the first agarose layer and the coverslips were replaced on top and the slides were maintained on ice quickly to solidify the agarose. The coverslips were gently removed again and the third layer of 1 % NMA (250–300 μL) was placed on slides. As soon as agarose was solidified, the slides were immersed in freshly prepared cold lysis solution (2.5 M NaCl, 100 mM Na2EDTA, and 10 mM Tris, pH = 10, with 1 % Triton X-100 and 10 % DMSO were added just before use) at 4 °C for 12 h to cell lysis and disperse the components. Then, the slides were placed in a horizontal gel electrophoresis tank. The electrophoresis tank was filled with fresh and ice-cold electrophoresis buffer (300 mM NaOH, 1 mM Na2EDTA, pH > 13) and slides were left in the alkaline buffer for 30 min for unwinding the DNA strands. Electrophoresis was performed at 25 V (0.66 V/cm) or 300 mA for 30 min at RT and the slides were drained and placed on a tray and flooded slowly with three changes of neutralization buffer (0.4 M Tris, pH = 7.5) for 5 min to remove alkali and detergents. All slides were rinsed in neutral buffer and the gel and its contents were fixed with ethanol 95 % for 5 min. The slides were stained with 2 μg/mL of ethidium bromide and placed into humidified chamber at 4 °C. Finally, the slides were visualized under UV light and duplicate slides for each treatment were prepared. The parameters of alkaline comet assay including head area, tail area, head DNA, tail DNA, tail length, comet length, and tail moment in cultured hiPSCs on BD Matrigel matrix, MEF and HDF feeder layers as well as normal HDFs and treated cells with H2O2 were evaluated by CaspLab (Comet Assay Software Project) software version 1.0.0.
Teratoma formation in mice
For analysis of pluripotency of iPSCs in vivo the hiPSCs cultured on BD Matrigel matrix, MEF and HDF feeder layers were injected separately at amount of 1 × 106 cells suspended in PBS into dorsal flanks of immunodeficient (SCID) mice previously anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg). Seven weeks after injection tumor was developed. Then, the tumor was harvested and fixed in 10 % neutral buffered formaldehyde, and embedded in paraffin and sectioned. Finally, the tumor was analyzed for various differentiated cell types. The hematoxylin and eosin (H&E) and immunohistochemical stain using mouse anti-human E-cadherin antibody (Abcam, Cambridge,UK) were used for histopathological analysis of teratomas generated from hiPSCs cultured on each condition.
Formation of embryoid bodies (EBs)
EB formation is an early stage in the differentiation process of iPSCs. For generation of EBs, the hiPSC colonies that cultured in three groups (BD Matrigel matrix, MEF and HDF feeder layers) were dissociated into smaller clusters (# 10–20 cells) using collagenase IV (1 mg/mL) for 5 min followed by Trypsin–EDTA 0.05 % solution (Sigma, T4799) for 2 min at 37 °C and were centrifuged at 1200 rpm for 5 min. The hiPSC clusters of each group were plated in 6-well non-treated-plate (JET BIOFIL®) in 2 mL aggregation media, consisting of Iscove’s Modified Dulbecco’s Medium (IMDM) (Gibco, Invitrogen) supplemented with 20 % fetal bovine serum (FBS), 100 M non-essential amino acids, 2 mM glutamine, 50 U/mL penicillin, 50 g/mL streptomycin (Invitrogen, Grand Island, NY), 10−4 M β-mercaptoethanol (Sigma, St Louis, MO), 10 ng/mL BMP-4 (PeproTech, RockyHill, NJ, USA), and 50 g/mL ascorbic acid (Sigma). The plates were incubated in a humidified atmosphere at 37 °C. Twenty-four hours later, the EB formation was checked by inverted phase contrast microscope (Nikon, ECLIPSE TS100).
Quantitative real-time PCR
Total RNA was extracted from EB colonies generated from cultured hiPSCs on BD Matrigel matrix, MEF and HDF feeder layers according to the protocol mentioned above. RNA yield and its purity were quantified by NanoDrop® and the cDNA was synthesized. The specific oligonucleotide primers used in the present study for gene amplification were designed by Gene Runner version 3.05 (Hastings Software, Inc.) and Oligo Primer Analysis software version 7 and after that the sequences were analyzed using BLAST in GenBank data (Table 2). The quantitative real-time PCR (real-time Q-PCR) was performed to evaluate the expression level of three germ layers genes including msh homeobox 1 (MSX1) and Brachyury T for mesoderm, paired box 6 (PAX6) and microtubule-associated protein 2 (MAP2) for ectoderm, and alpha-fetoprotein (AFP) and KRT8 or keratin 8 (CK8) for endoderm in EBs that created from cultured hiPSCs in each layers compare to these genes in control cells (hESC line obtained from Royan Institute, Tehran, Iran) and HDFs. The real-time Q-PCR was carried out with SYBR Green master mix (Roche Applied Science, Indianapolis, IN, USA) in 10 μL reaction involved: 5 μL of SYBR Green master mix, 2.5 nM concentration of each designed forward and reverse primers, and 60 ng/μL of cDNA sample. The micro-tubes were placed into Rotorgene-3000 real-time thermal cycler (Corbett, Australia). The real-time Q-PCR program was consist of an initial denaturation step (95 °C for 10 min), 45 cycles including denaturation at 95 °C (10 s), primer annealing (20 s) at 63, 60, 58, 62, 60 and 60 °C for MSX1, Brachyury T, PAX6, MAP2, AFP, and CK8 genes, respectively, and extension (20 s) at 72 °C. The expression level of three germ layers genes were measured in triplicate for each EB formed from hiPSCs cultured in three conditions. The relative quantification of genes expression in each group compared with these genes in hESC and HDFs were determined using the 2-∆Ct method and the fold-difference of expression levels of genes were calculated and compared in cycle of threshold values (Ct). In final the melting curve was generated immediately after amplification by holding the reaction mixture at 95 °C for 60 s, and then lowering the temperature to 45 °C at a transition rate of 0.1 °C/s and maintained for 120 s. Then, the samples were heated slowly at a transition rate of 0.05 to 80 °C with continuous collection of fluorescence at 640 nm.
Table 2.
The primers sequence used for investigate the expression level of expression level of three germ layers genes of EBs by real-time PCR
| Primers name | Sequences | Germ layer | Products length (bp) | GenBank accession number |
|---|---|---|---|---|
| MSX1-F MSX1-R |
5′-AGCGCAGAGAAATCGGTGTC-3′ 5′-ATTCATCTTCCTGGCTGGC-3′ |
Mesoderm | 124 | NM_002448 |
| Brachyury T-F Brachyury T-R |
5′-CCTCGAATCCACATAGTGAGAG-3′ 5′-AAGAGCTGTGATCTCCTCGT-3′ |
117 | NM_001270484 | |
| PAX6-F PAX6-R |
5′-ACACACATTAACACACTTGAGC-3′ 5′-TGCTGTTGTTGCTTGAAGAC-3′ |
Ectoderm | 158 | NM_000280 |
| MAP2-F MAP2-R |
5′-AGGAAGGGAAGAGCATGTAAG-3′ 5′-ACACCACTTCCTCAACCACAG-3′ |
190 | NM_001039538 | |
| AFP-F AFP-R |
5′-AGGAGATGTGCTGGATTGTC-3′ 5′-ACATTGACCACGTTCCAGC-3′ |
Endoderm | 127 | NM_001134 |
| CK8-F CK8-R |
5′-ATGAGATCAATAAGCGTACAGAG-3′ 5′-TAGCTGCCTGAGGAAGTTGA-3′ |
140 | NM_001256282 |
Ethical approval
In this study, the protocols of MEF and HDF isolation were approved by the Regional Research Ethical Committee of Shahrekord University of Medical Sciences (Grant numbers: 90-12-12, 91-10-5, 91-11-4, and 91-11-6). The informed consent forms were filled out by the healthy male newborns parents before collection of foreskin samples. Male newborns with cytogenetic instabilities such as Downʼs syndrome were excluded from the study.
Statistical analysis
All date were collected in Statistics programs for the Social Sciences software, version 20 (SPSS, Inc., Chicago, IL, USA) and differences between genes expression of hiPSCs in each group were compared with internal control using analysis of variance (ANOVA). All experiments in each group were repeated at least three times. The alkaline comet assay parameters were evaluated by CaspLab software version 1.0.0 and the mean difference between groups was calculated by ANOVA statistical method in SPSS software. In this study, the p-value of ≤0.05 was considered statistically significant and graphs were prepared using Graph Pad Prism version 5.01 (2003, San Diego, CA) software.
Results
HDFs preparation
Isolated HDFs from neonatal human foreskin after 7 days were ready for first passage and cells in second passage were used for lentivirus transduction to generate hiPSCs (Fig. 1a).
Fig. 1.
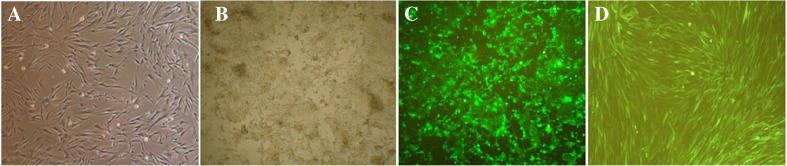
a Isolated HDFs from neonatal human foreskin in passage 2, HEK-293T cells transfected with lentiviral plasmid expressing eGFP after 72 h observed in b normal light and c fluorescence, and d HDFs transducing by eGFP lentiviruses after 72 h
HEK-293 T cell transfection and virus production
The HEK-293 T cells were transfected with a FUW-OSKM plasmid together psPAX, and pMDG2 for producing lentiviral vectors and in positive control Oct4 expressing eGFP was used by CaPO4 precipitation method. After 48 to 72 h the lentivirus particles were produced successfully with high efficiency of eGFP expressing in transfected HEK-293 T cell at a multiplicity of infection (MOI) = 10 and supernatant of cells contained lentiviruses were collected and concentrated by ultra centrifuge at 26000 rpm for 2 h (Fig. 1b and c). The titer of produced viruses was measured by HIV AB & Ag ELISA Kit (DIA.PRO, Milan, Italy) and the data are shown in Table 3.
Table 3.
The results of ELISA test for confirmation of lentiviruses production
| Sample | Blank | Negative control | Positive control | Calibrator | OSKM | Oct4-GFP |
|---|---|---|---|---|---|---|
| Optical density (OD) | 0.045 | 0.811 | 3.021 | 0.934 | 3.072 | 3.067 |
Transduction of HDFs
The HDFs were transduced by TETO-FUW-OSKM lentiviral vector in three times and for positive control eGFP lentiviruses was used (Fig. 1d). After successfully transduction the total RNA was extracted from transduced HDFs and was used for cDNA synthesis to evaluate the target genes.
HiPSCs morphology on BD Matrigel matrix, MEF and HDF feeder layers
Six days after transduction, the transduced HDFs were detached from 6 wells plate and reprogrammed cells were cultured on BD Matrigel matrix, MEF and HDF feeder layers in iPS media (supplemented DMEM/F-12) to generate hiPSC colonies. The hiPSCs morphology was checked every day and after 9–14 days the normal shape of hiPSCs on each condition were observed and let to create colonies (Fig. 2). After that hiPSC colonies were picked up by enzymatic and mechanical methods and sub-cultured again on each feeder and BD Matrigel to produce sufficient colonies for using in further examination.
Fig. 2.

The cultures of hiPSCs on a MEF feeder, b HDF feeder, and c BD Matrigel matrix (as a feeder free condition)
Conventional reverse transcriptase PCR analysis
The pluripotency genes of hiPSCs including Oct-4, Sox-2, Nonog, Rex-1, hTERT8, FGF4, and AP genes using specific oligonucleotide primers were amplified by reverse transcriptase PCR and the products of each group were evaluated on 3 % agarose gel electrophoresis (Fig. 3).
Fig. 3.

The agarose gel electrophoresis of amplified cDNA samples of pluripotency genes of cultured iPSCs on different layers
Characterization of hiPSCs
For characterization of hiPSCs AP staining, ICC test, karyotyping and comet assay were conducted on cultured hiPSC colonies on different layers. The ICC test was used for determination of the specific antigens in surface of hiPSCs using primary antibodies including anti-Nanog, anti-Oct4, anti-SOX2, and anti-SSEA-4. The presence of these antigens was confirmed and specific binding of antigen antibodies was established (Fig. 4). The hiPSC colonies were stained positive for alkaline phosphatase and the morphology typical of human ESCs were showed (Fig. 4).
Fig. 4.

The ICC test on hiPSC colonies cultured on a BD Matrigel matrix, b MEF and c HDF feeders, and their alkaline phosphatase positive test, respectively
G-banding was conducted to determine the karyotype of hiPSCs cultured on feeder free system (BD Matrigel), MEF and HDF feeders. A number of 20 karyotypes per each slide were analyzed and cultured hiPSCs on each condition in low passages (P 1–3) had normal karyotype (20/20, 46 chromosomes, XY) without any abnormalities including deletions, insertions, dislocation, and duplications (Fig. 5)
Fig. 5.

The karyotype of hiPSCs cultured on a feeder free system (BD Matrigel), b MEF and c HDF feeders without cytogenetic abnormalities
After alkaline comet assay the damaged DNA and DNA single-strand breaks were not observed in cultured hiPSCs on BD Matrigel matrix, MEF and HDF feeders in low passages (Fig. 6a–c) compare to positive control (H2O2) and normal HDFs (Fig. 6d and e). The mean values of alkaline comet assay parameters including head area, tail area, head DNA, tail DNA, tail length, comet length, and tail moment of cultured hiPSCs in each condition compared with normal HDFs and positive control (cells treated by H2O2) were calculated by CaspLab software version 1.0.0 to investigate the cytogenetic status (Table 4). The relationship and analysis of variance between parameters in each group were evaluated by ANOVA (Analysis of variance) test (Table 5) and the p-value less than 0.05 were considered significant. The comparison of analysis of variance of alkaline comet assay parameters involving head area, tail area, head DNA%, tail DNA%, tail length, comet length, and tail moment by ANOVA test between hiPSCs cultured on each layer compared with normal HDFs were not statistically significant (p > 0.05) but compared with positive control (H2O2 treatment) were more significantly different (p < 0.05).
Fig. 6.

The alkaline comet assay of hiPSCs cultured on a MEF feeder, b HDF feeder, and c feeder free system (BD Matrigel) in low passage compare to d H2O2 group (positive control) and e normal HDFs
Table 4.
The mean value of alkaline comet assay parameters of cultured hiPSCs on Matrigel matrix, MEF and HDF feeders compare to normal HDFs and positive control (cells treated by H2O2)
| Parameters | Healthy HDFs (normal cells) No. of cells (100) | hiPSCs cultured on MEF feeder No. of cells (80) | hiPSCs cultured on HDF feeder No. of cells (80) | hiPSCs cultured on BD Matrigel No. of cells (80) | HDFs exposed by H2O2 (positive control) No. of cells (100) |
|---|---|---|---|---|---|
| Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | |
| Head area | 1512.29 ± 725.854 | 1149.2 ± 369.104 | 1080.69 ± 310.2 | 1149.2 ± 369.104 | 1602.66 ± 1511.712 |
| Tail area | 102.75 ± 99.158 | 61.99 ± 72.656 | 63.25 ± 68.44 | 65.6 ± 74.362 | 4779.36 ± 1752.768 |
| Head DNA | 209.271 ± 101.533 | 108.539 ± 47.005 | 112.44 ± 52.2 | 106.66 ± 45.1 | 171.149 ± 198.411 |
| Tail DNA | 3.631 ± 4.137 | 1.512 ± 2.256 | 1.4 ± 1.924 | 1.62 ± 2.34 | 363.916 ± 178.368 |
| Head DNA% | 98.257 ± 1.8 | 98.765 ± 1.421 | 100.56 ± 1.22 | 97.6 ± 1.35 | 29.178 ± 7.69 |
| Tail DNA% | 1.743 ± 1.8 | 1.235 ± 1.421 | 1.325 ± 1.22 | 1.415 ± 1.35 | 70.8 ± 7.69 |
| Tail length | 3.44 ± 0.978 | 3.71 ± 1.436 | 4.2 ± 2.02 | 3.892 ± 1.65 | 80.59 ± 20.66 |
| Comet length | 48.46 ± 9.784 | 43.07 ± 6.574 | 45.66 ± 7.78 | 40.58 ± 4.66 | 124.68 ± 25.733 |
| Tail moment | 0.068 ± 0.0802 | 0.056 ± 0.087 | 0.12 ± 0.11 | 0.09 ± 0.099 | 57.84 ± 18.75 |
Head Area Area of the comet head in pixels, Tail Area Area of the comet tail in pixels, Head DNA Amount of DNA in the comet head, Tail DNA Amount of DNA in the comet tail, Tail Length Length of the comet tail measured from right border of head area to end of tail (in pixels), Comet Length Length of the entire comet from left border of head area to end of tail (in pixels), Tail Moment Tail DNA% x Tail Length ([percent of DNA in the tail] x [tail length]), SD Std. Deviation
Table 5.
The relationship of alkaline comet assay parameters between cultured hiPSCs on MEF, HDF and BD Matrigel matrix compare with normal healthy cells and H2O2 (positive control) analyzed by ANOVA test
| Parameters | hiPSCs cultured on MEF feeder compare with positive control (H2O2 treatment) | hiPSCs cultured on MEF feeder compare with normal cells (healthy HDFs) | ||
| Mean difference ± SE | p-value | Mean difference ± SE | p-value | |
| Head area | 471.599* ± 121.78 | 0.011 | 616.65 ± 58.234 | 0.7 |
| Tail area | −4583.101* ± 190.087 | 0.000 | 687.36 ± 86.258 | 0.12 |
| Head DNA% | 64.372* ± 15.987 | 0.007 | 55.26 ± 7.36 | 0.000 |
| Tail DNA% | −69.181* ± 0.85 | 0.000 | 54.66 ± 3.65 | 0.08 |
| Tail length | −75.521* ± 2.464 | 0.000 | 112.3 ± 10.45 | 0.32 |
| Comet length | −80.41* ± 2.856 | 0.000 | 92.41 ± 4.875 | 0.24 |
| Tail moment | −86.43* ± 2.093 | 0.000 | 75.36 ± 24.23 | 0.000 |
| hiPSCs cultured on HDF feeder compare with positive control (H2O2 treatment) | hiPSCs cultured on HDF feeder compare with normal cells (healthy HDFs) | |||
| Mean difference ± SE | p-value | Mean difference ± SE | p-value | |
| Head Area | 556.112* ± 98.236 | 0.02 | 487.25 ± 38.24 | 0.602 |
| Tail Area | −6659.25* ± 150.3 | 0.000 | 587.89 ± 45.2 | 0.84 |
| Head DNA% | 78.36* ± 0.85 | 0.000 | 240 ± 58.3 | 0.000 |
| Tail DNA% | −85.7* ± 0.722 | 0.032 | 64.36 ± 4.22 | 0.212 |
| Tail Length | −84.38* ± 1.215 | 0.000 | 148 ± 24.684 | 0.000 |
| Comet length | −100.2* ± 2.3 | 0.000 | 88.24 ± 4.98 | 0.264 |
| Tail moment | −90.245* ± 1.66 | 0.000 | 105.7 ± 20.2 | 0.000 |
| hiPSCs cultured on BD Matrigel (feeder free) compare with positive control (H2O2 treatment) | hiPSCs cultured on BD Matrigel (feeder free) compare with normal cells (healthy HDFs) | |||
| Mean difference ± SE | p-value | Mean difference ± SE | p-value | |
| Head area | 541.268* ± 109.122 | 0.022 | 652.8 ± 140.6 | 0.000 |
| Tail area | −5212.25* ± 112.65 | 0.000 | 420.8 ± 78.2 | 0.405 |
| Head DNA% | 72.3* ± 0.854 | 0.000 | 452 ± 112.3 | 0.000 |
| Tail DNA% | −84.9* ± 0.692 | 0.000 | 64.36 ± 10.2 | 0.000 |
| Tail length | −92.24* ± 1.5 | 0.000 | 187.54 ± 24.684 | 0.000 |
| Comet length | −87.134* ± 1.96 | 0.000 | 194.32 ± 14.54 | 0.2 |
| Tail moment | −85.124* ± 1.44 | 0.000 | 144.5 ± 42 | 0.000 |
*The mean difference is significant at p < 0.05
SE Std. Error
Histopathology of teratomas
Teratoma was performed to investigate the pluripotency of cultured hiPSCs on Matrigel matrix, MEF and HDF feeder layers by injection of cells into dorsal flanks of SCID mice and was confirmed in more than one set of mouse models. Six to nine weeks post-injection, teratomas were recovered from the SCID mice. The results were showed developing of tumors in each group (an example is presented in Fig. 7 left). The tissue expansion was prepared with the microtome and histological analysis was defined by microscopic morphology by H&E stained images of teratomas derived from cultured hiPSCs on MEF and HDF feeders as well as feeder free condition and were showed multi-differentiated tissues and three germ layers including gut-like epithelial tissues (endoderm), striated muscle (mesoderm), cartilage (mesoderm), neural tissues (ectoderm), and keratin-containing epidermal tissues (ectoderm) and were demonstrated the pluripotency of hiPSCs cultured on each condition (Fig. 7). Finally, immunohistochemical stain (IHC) were demonstrated robust expression of human E-cadherin antibody (as a marker of undifferentiated stage) on the surface of generated teratomas from hiPSCs (Fig. 7).
Fig. 7.

Histopathology assay of teratomas The teratoma formation of hiPSCs (left) by injection amount of 1 × 106 cells suspended in PBS into dorsal flanks of immunodeficient (SCID) mice and the tissue expansion from teratoma in vivo (right) by H&E staining and immunohistochemical staining (after three times repeated) using human E-cadherin (a-e: three germ layers including neural tissues (ectoderm), gut-like epithelial tissues (endoderm), striated muscle (mesoderm), cartilage (mesoderm), and keratin-containing epidermal tissues (ectoderm), respectively were observed. f-j: IHC staining demonstrates robust expression of human E-cadherin antibody on the surface of each germ layer of generated teratomas)
EB generation
The cultured hiPSCs on BD Matrigel matrix, MEF and HDF feeder layers were transferred to the EB media (without bFGF and a rock inhibitor). After 4 days of culture, EB colonies were created in 24 wells ultra low attachment plate. Following 15 days the EBs growth were checked by invert microscope in all groups (Fig. 8) and were used for further molecular assays.
Fig. 8.

The EB formation from hiPSCs cultured on a MEF feeder, b HDF feeder, and c feeder free system (BD Matrigel matrix)
Real-time PCR analyze
The analyze of quantitative real-time PCR were showed that the expression level of three germ layers genes in EBs derived from cultured hiPSCs on MEF and HDF feeders and BD Matrigel compare to hESCs and HDFs including MSX1 (0.007878256, 0.006589552, and 0.006624884 vs. 0.007238562 and 0.002188755), Brachyury T (0.007495524, 0.006525632, and 0.006533204 vs. 0.007055842 and 0.002122865), PAX6 (0.007765866, 0.006432568, and 0.006586224 vs. 0.006895521 and 0.002212536), MAP2 (0.007622585, 0.006325487, and 0.006632585 vs. 0.006624698 and 0.002388755), AFP (0.007565422, 0.006523642, and 0.006722055 vs. 0.007122365 and 0.002243844), and CK8 (0.007822004, 0.006400885, and 0.006418965 vs. 0.006635287 and 0.002105586) genes, respectively (Fig. 9). These results were indicated these genes were up-regulated in derived EBs from cultured hiPSCs in different layers compare to HDFs and were statistically significant (p < 0.05), while the expression level were closest to hESCs and were not significant (p > 0.05).
Fig. 9.

Comparison the mean difference (a 95% confidence interval (CI) were used for the difference) of the expression level of three germ layers genes (mesoderm, ectoderm, and endoderm) in EBs derived from hiPSCs that cultured on each layer, hESCs and HDFs
Discussion
In the present study, the cultures of hiPSCs were characterized and compared on feeder-serum-free system (Matrigel matrix), MEF and HDF feeders. The cytogenetic stability of hiPSCs cultured on each layer was evaluated by karyotyping and comet assay. All cells had normal karyotype without any abnormalities and DNA lesions. The specific binding of antigen antibodies including anti-Nanog, anti-Oct4, anti-SOX2, and anti-SSEA-4 was established and expression of these antigens was confirmed. The alkaline phosphatase test on hiPSC colonies was positive and the morphology of cells typically same to the human ESCs. The pluripotency of hiPSCs were investigated by teratoma formation and preparing the tissues expansion. The immunohistochemical stain using anti-human E-cadherin antibody was down on tissues expansion and confirmed that hiPSCs in each condition had ability to maintain in undifferentiated stage. After EB generation the expression level of three germ layers genes were determined by real-time PCR. Similar to ESCs, up-regulation of these genes were observed in hiPSCs cultured on different layers compare to HDFs. According to the results of this study, cultured hiPSCs on these layers could maintain for long term-period with normal morphology, expressed pluripotency genes and generated three germ layers including ectoderm, mesoderm and endoderm. However, for therapeutic purposes each layer has its advantages and disadvantages. Although, MEF and HDFs are good feeders for supporting the hiPSCs and hESCs (Hovatta et al. 2003; Takahashi et al. 2007), but using Matrigel matrix as a xeno-feeder free system without conditioned medium could decrease immune interaction and inhibit the animal pathogens transfer. Because using conditioned medium collected from MEFs instead of iPS media supplemented with growth factors could increase the risk of above problems.
Previous study demonstrated that iPSCs can be cultured in similar conditions used for hESCs on MEF and foreskin fibroblasts as feeders (Takahashi and Yamanaka 2006; Ellerström et al. 2006; Takahashi et al. 2007; Crook et al. 2007). Amit and co-workers indicated the important advantage of foreskin feeder is its ability to continuously culture for more than 42 passages (Amit et al. 2003). In addition, HDF feeder has been used for derivation and continued undifferentiated growth of hESCs, successfully (Hovatta et al. 2003). HDF is not only the most frequently used human feeders, but also it can be replace by xeno-free culture system like Matrigel and these culture systems promote animal-free conditions for culturing iPSCs (Kleinman and Martin 2005). Therefore, the ideal culture of iPSCs can achieve by combination of an animal-free matrix and both serum-free and animal-free medium. Denham et al. introduced a new feeder-free technique for expanding the culture of hESC and iPS for rapid amplification in many cell biology methods (Denham et al. 2009). In another study SNL fibroblast feeder layers were used to support the derivation and maintenance of hiPSCs but it was shown that they could transfer the zoonotic pathogens and inducing immune response (Pan et al. 2010). Sun and colleagues cultured iPSCs generated from adult human adipose stem cells (hASCs) on feeder free system and showed hiPSCs can be readily generated under feeder-free conditions like BD Matrigel as an ideal autologous source of cells for generation of individual-specific iPSCs (Sun et al. 2009). Unger and co-workers demonstrated iPSCs derived from human fetal fibroblasts could be established and maintained on isogenic feeder cells (Unger et al. 2009), and their findings are similar to the results of the present study. Xu et al. cultured hESCs on Matrigel or laminin in medium conditioned by MEF and showed that cells retain fundamental characteristics of hESCs in this culture system and are suitable for scaleup production (Xu et al. 2001). Another study suggests that autologous fibroblasts can be not only a source for iPSCs but also be feeder layers and it is an important step toward the establishment of clinical grade iPSCs (Takahashi et al. 2009). The present study, also pointed out this potential of HDFs. In recent research by Choi et al. they indicated that feeder-free culture system provides a simple approach for maintaining spermatogonial stem cells (SSCs, also called germline stem cells) in vitro and studying the basic biology of these cells (Choi et al. 2014). In a review study by Ghasemi-Dehkordi and colleagues they demonstrated that the culture of these cells on feeder free systems like Matrigel matrix in conditioned media supplemented with essential growth factors is useful for avoiding the stem cell contamination (Ghasemi-Dehkordi et al. 2014). In the present work the characterization and cytogenetic stability of hiPSCs cultured on Matrigel matrix, MEF and HDFs feeder for long term period in vivo were compare. This study suggested that hiPSCs application for therapeutic aims and regenerative medicine are more suitable using feeder free systems like Matrigel in supplemented media (without using MEF conditioned media).
Conclusions
In this study, hiPSCs were cultured on BD Matrigel, MEF and HDF feeder layers in undifferentiated state for long term period with normal growth and morphology. Although MEF provides a powerful supportive feeder for growth of hiPSC with normal proliferation, but this feeder layer expose hiPSCs to many possible infectious agents and unknown factors from animal sources. Therefore, in therapeutic approaches this feeder is not appropriate as it can potentially activate immune response and increase the risk of cell contamination. Moreover, compare to MEF, HDF is a more suitable feeder for hiPSCs culture because this cell line is cytogenetically more stable and it is ideal homologue cell line for hiPSCs. Therefore, using isogenic feeder is more benefit for therapeutic goals compare to autologous feeders like MEF. In addition, it is difficult to interpret the results obtained from hiPSC-MEF or hiPSC-HDF co-culture during different interventions. Also, in xeno-free culture system using MEF conditioned media is routine but it can only prevent cell contamination and the problem of animal infection and immune response still remains. So, according to this study hiPSCs can culture on BD Matrigel as a feeder layer-free and serum-free environment in iPS media supplemented with essential factors for long-term period and this xeno-free culture system could eliminate these problems and present an appropriate layer for culturing stem cells for therapeutic aims in regenerative medicine.
Acknowledgments
We would like to thank Rudolf Jaenisch laboratory and Tronolab for providing the plasmids. This study was supported by research Grants: 90-12-12, 91-10-5, 91-11-4, 91-11-6, and 91-12-13 of Shahrekord University of Medical Sciences, Shahrekord, Iran. The study team would like to gratefully acknowledge to the staffs of Cellular and Molecular Research Center for their sincere cooperation.
Abbreviations
- HDFs
Human dermal fibroblasts
- hiPSC
Human induced pluripotent stem cell
- hESCs
Human embryonic stem cells
- ICC
Immunocytochemistry
- IHC
Immunohistochemical
References
- Aguilar-Gallardo C, Poo M, Gomez E, Galan A, Sanchez E, Marques-Mari A, Ruiz V, Medrano J, Riboldi M, Valbuena D, Simon C. Derivation, characterization, differentiation, and registration of seven human embryonic stem cell lines (VAL-3, −4, −5, −6 M, −7, −8, and −9) on human feeder. In Vitro Cell Dev Biol Anim. 2010;46(3–4):317–326. doi: 10.1007/s11626-010-9285-3. [DOI] [PubMed] [Google Scholar]
- Amit M, Margulets V, Segev H, Shariki C, Laevsky I, Coleman R, Itskovitz-Eldor J. Human feeder layers for human embryonic stem cells. Biol Reprod. 2003;68:2150–2156. doi: 10.1095/biolreprod.102.012583. [DOI] [PubMed] [Google Scholar]
- Bayart E, Cohen-Haguenauer O. Technological overview of iPS induction from human adult somatic cells. Curr Gene Ther. 2013;13(2):73–92. doi: 10.2174/1566523211313020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson F, Rochefort É, Lavoie A, Larouche D, Zaniolo K, Simard-Bisson C, Damour O, Auger FA, Guérin SL, Germain L. Irradiated human dermal fibroblasts are as efficient as mouse fibroblasts as a feeder layer to improve human epidermal cell culture lifespan. Int J Mol Sci. 2013;14(3):4684–4704. doi: 10.3390/ijms14034684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KG, Mallon BS, McKay RDG, Robey PG. Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell. 2014;14(1):13–26. doi: 10.1016/j.stem.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Hammond H, Ye Z, Zhan X, Dravid G. Human adult marrow cells support prolonged expansion of human embryonic stem cells in culture. Stem Cells. 2003;21:131–142. doi: 10.1634/stemcells.21-2-131. [DOI] [PubMed] [Google Scholar]
- Choi NY, Park YS, Ryu JS, Lee HJ, Araúzo-Bravo MJ, Ko K, Han DW, Schöler HR, Ko K. A novel feeder-free culture system for expansion of mouse spermatogonial stem cells. Mol Cell. 2014;37(6):473–479. doi: 10.14348/molcells.2014.0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook JM, Peura TT, Kravets L, Bosman AG, Buzzard JJ, Horne R, Hentze H, Dunn NR, Zweigerdt R, Chua F, Upshall A, Colman A. The generation of six clinical-grade human embryonic stem cell lines. Cell Stem Cell. 2007;1:490–494. doi: 10.1016/j.stem.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Denham M, Leung J, Tay C, Wong RCB, Donovan P, Dottori M, Pébay A. A new feeder-free technique to expand human embryonic stem cells and induced pluripotent stem cells. Open Stem Cell J. 2009;1:76–82. doi: 10.2174/1876893800901010076. [DOI] [Google Scholar]
- Ellerström C, Strehl R, Moya K, Andersson K, Bergh C, Lundin K, Hyllner J, Semb H. Derivation of a xeno-free human embryonic stem cell line. Stem Cells. 2006;24:2170–2176. doi: 10.1634/stemcells.2006-0130. [DOI] [PubMed] [Google Scholar]
- Ghasemi-Dehkordi P, Allahbakhshian-Farsani M, Abdian N, Mirzaeian A, Hashemzadeh-Chaleshtori M, Jafari-Ghahfarokhi H. Effects of feeder layers, culture media, conditional media, growth factors, and passages number on stem cell optimization. Proc Natl Acad Sci India Sect B Biol Sci. 2014 [Google Scholar]
- Hovatta O, Mikkola M, Gertow K, Strömberg A, Inzunza J, Hreinsson J, Rozell B, Blennow E, Andäng M, Ahrlund‐Richter L. A culture system using human foreskin fibroblasts as feeder cells allows production of human embryonic stem cells. Hum Reprod. 2003;18(7):1404–1409. doi: 10.1093/humrep/deg290. [DOI] [PubMed] [Google Scholar]
- Jung YW, Hysolli E, Kim KY, Tanaka Y, Park IH. Human induced pluripotent stem cells and neurodegenerative disease: prospects for novel therapies. Curr Opin Neurol. 2012;25(2):125–130. doi: 10.1097/WCO.0b013e3283518226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Peters J-M, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM. cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell. 1996;85:829–839. doi: 10.1016/S0092-8674(00)81267-2. [DOI] [PubMed] [Google Scholar]
- Kleinman HK, Martin GR. Matrigel: basement membrane matrix with biological activity. Semin Cancer Biol. 2005;15(5):378–386. doi: 10.1016/j.semcancer.2005.05.004. [DOI] [PubMed] [Google Scholar]
- McKelvey-Martin VJ, Ho ETS, McKeown SR, Johnston SR, McCarthy PJ, Rajab NF, Downes CS. Emerging applications of the single cell gel electrophoresis (Comet) assay. I. Management of invasive transitional cell human bladder carcinoma. II. Fluorescent in situ hybridization Comets for the identification of damaged and repaired DNA sequences in individual cells. Mutagen. 1998;13:1–8. doi: 10.1093/mutage/13.1.1. [DOI] [PubMed] [Google Scholar]
- Pakzad M, Totonchi M, Taei A, Seifinejad A, Hassani SN, Baharvand H. Presence of a ROCK inhibitor in extracellular matrix supports more undifferentiated growth of feeder-free human embryonic and induced pluripotent stem cells upon passaging. Stem Cell Rev Rep. 2010;6(1):96–107. doi: 10.1007/s12015-009-9103-z. [DOI] [PubMed] [Google Scholar]
- Pan C, Hicks A, Guan X, Chen H, Bishop CE. SNL fibroblast feeder layers support derivation and maintenance of human induced pluripotent stem cells. J Genet Genomics. 2010;37(4):241–248. doi: 10.1016/S1673-8527(09)60042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451(7175):141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Richards M, Fong CY, Chan WK, Wong PC, Bongso A. Human feeders support prolonged undifferentiated growth of human inner cell masses and embryonic stem cells. Nat Biotechnol. 2002;20:933–936. doi: 10.1038/nbt726. [DOI] [PubMed] [Google Scholar]
- Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481(7381):295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001. pp. 148–190. [Google Scholar]
- Sun N, Panetta NJ, Gupta DM, Wilson KD, Lee A, Jia F, Hu S, Cherry AM, Robbins RC, Longaker MT. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. PNAS. 2009;106(37):15720–15725. doi: 10.1073/pnas.0908450106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fi broblast cultures by defi ned factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fi broblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Narita M, Yokura M, Ichisaka T, Yamanaka S. Human induced pluripotent stem cells on autologous feeders. PLoS ONE. 2009;4(12):e8067. doi: 10.1371/journal.pone.0008067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecirlioglu RT, Nguyen L, Koh K, Trounson AO, Michalska AE. Derivation and maintenance of human embryonic stem cell line on human adult skin fi broblast feeder cells in serum replacement medium. In Vitro Cell Dev Biol Anim. 2010;46(3–4):231–235. doi: 10.1007/s11626-010-9278-2. [DOI] [PubMed] [Google Scholar]
- Unger C, Gao S, Cohen M, Jaconi M, Bergstrom R, Holm F, Galan A, Sanchez E, Irion O, Dubuisson JB. Immortalized human skin fibroblast feeder cells support growth and maintenance of both human embryonic and induced pluripotent stem cells. Hum Reprod. 2009;24:2567–2581. doi: 10.1093/humrep/dep232. [DOI] [PubMed] [Google Scholar]
- Xu C, Inokuma MS, Denham J, Golds K, Kundu P, Gold JD, Carpenter MK. Feeder-free growth of undifferentiated human embryonic stem cells. Nat Biotechnol. 2001;19:971–974. doi: 10.1038/nbt1001-971. [DOI] [PubMed] [Google Scholar]