

Abstract
Background and Objectives
Oral enzalutamide (160 mg once daily) is approved for the treatment of metastatic castration-resistant prostate cancer (mCRPC). This article describes the pharmacokinetics of enzalutamide and its active metabolite N-desmethyl enzalutamide.
Methods
Results are reported from five clinical studies.
Results
In a dose-escalation study (n = 140), enzalutamide half-life was 5.8 days, steady state was achieved by day 28, accumulation was 8.3-fold, exposure was approximately dose proportional from 30–360 mg/day, and intersubject variability was ≤30 %. In a mass balance study (n = 6), enzalutamide was primarily eliminated by hepatic metabolism. Renal excretion was an insignificant elimination pathway for enzalutamide and N-desmethyl enzalutamide. In a food-effect study (n = 60), food did not have a meaningful effect on area under the plasma concentration–time curve (AUC) of enzalutamide or N-desmethyl enzalutamide, and in an hepatic impairment study, AUC of the sum of enzalutamide plus N-desmethyl enzalutamide was similar in men with mild (n = 6) or moderate (n = 8) impairment (Child–Pugh Class A and B) versus men with normal hepatic function (n = 14). In a phase III trial, an exposure-response analysis of steady-state predose (trough) concentrations (Ctrough) versus overall survival (n = 1103) showed that active treatment Ctrough quartiles for 160 mg/day were uniformly beneficial relative to placebo, and no threshold of Ctrough was associated with a statistically significant better response.
Conclusions
Enzalutamide has predictable pharmacokinetics, with low intersubject variability. Similar efficacy was observed in patients across the concentration/exposure range associated with a fixed oral dose of enzalutamide 160 mg/day.
Electronic supplementary material
The online version of this article (doi:10.1007/s40262-015-0271-5) contains supplementary material, which is available to authorized users.
Key Points
| This article summarizes data from several trials in order to provide clinical investigators with an understanding of the pharmacokinetics of enzalutamide. Collectively, the results show that enzalutamide has a half-life of 5.8 days, achieves steady state by day 28, accumulates 8.3-fold with once-daily dosing, shows approximate dose proportionality from 30–360 mg/day, and has ≤30 % intersubject variability. |
| In addition, enzalutamide is primarily eliminated by hepatic metabolism, while renal excretion is an insignificant elimination pathway for enzalutamide and its active metabolite, N-desmethyl enzalutamide. Exposure to enzalutamide active moieties is not affected by food or baseline mild or moderate hepatic impairment. |
| In an exposure-response analysis of overall survival in patients with mCRPC, active treatment C trough quartile groups for a fixed dose of 160 mg/day were uniformly beneficial relative to placebo, and there was no specific threshold of plasma concentrations in patients receiving enzalutamide that was associated with achieving a statistically significant better response; therefore, similar efficacy was observed in patients across the concentration/exposure range associated with a fixed oral dose of enzalutamide 160 mg/day. |
Introduction
Enzalutamide acts on multiple steps in the androgen receptor (AR) signaling pathway. It has been shown to competitively inhibit androgen binding to the AR, inhibit AR nuclear translocation, and inhibit AR interaction with DNA [1]. Enzalutamide decreased proliferation and induced cell death of prostate cancer cells in vitro and decreased tumor volume in a mouse prostate cancer xenograft model.
Enzalutamide (Xtandi®, Astellas Pharma US, Inc., Northbrook, IL, USA, and Medivation Inc., San Francisco, CA, USA) is approved in more than 40 countries for the treatment of patients with metastatic prostate cancer that has progressed on androgen deprivation therapy (gonadotropin-releasing hormone therapy or bilateral orchiectomy), a disease setting that is also defined as metastatic castration-resistant prostate cancer (mCRPC). The efficacy and safety of enzalutamide were assessed in randomized, placebo-controlled, multicenter, phase III clinical trials (AFFIRM and PREVAIL) [2, 3].
Enzalutamide (Fig. 1) is a small molecule with no ionizable groups at biologically relevant pH; therefore, enzalutamide solubility is not affected by pH over the physiological range. Enzalutamide exhibits limited aqueous solubility (≤2.0 μg/mL at relevant pH range), high permeability across Caco-2 monolayers (mean apparent permeability coefficient ≥31 × 10−6 cm/sec), and is not a substrate for P-glycoprotein (data on file, Medivation, Inc., 2012). Given its low solubility and high permeability, enzalutamide is considered a Biopharmaceutics Classification System (BCS) [4] class 2 drug substance.
Fig. 1.

Structures of enzalutamide, N-desmethyl enzalutamide, and the carboxylic acid metabolite. The asterisk indicates the position of the 14C atom in the radiolabeled molecule that was used in the mass balance and biotransformation study
The commercial product is a liquid-filled capsule of enzalutamide fully dissolved in caprylocaproyl polyoxylglycerides. This presentation was used throughout the development program. The approved dose is 160 mg (4 × 40 mg capsules) administered orally once daily with or without food.
Two major metabolites have been identified in human plasma, N-desmethyl enzalutamide and a carboxylic acid derivative (Fig. 1). Based on in vitro assays, N-desmethyl enzalutamide is an active metabolite that is thought to contribute to pharmacologic effects because it demonstrates key primary pharmacodynamics of similar potency to enzalutamide. In contrast, the carboxylic acid metabolite is inactive. At steady state, N-desmethyl enzalutamide circulates at approximately the same plasma concentration as enzalutamide, while the carboxylic acid metabolite is approximately 25 % lower.
The pharmacokinetics of enzalutamide were characterized in studies in patients with mCRPC, healthy male volunteers, and male subjects with impaired hepatic function. This article summarizes the data in order to provide clinical investigators with an understanding of the pharmacokinetics of enzalutamide.
Methods
Study Population
Pharmacokinetic results from clinical studies are summarized in this report: two studies in patients with mCRPC (n = 934), two in healthy male volunteers (n = 66), and one in healthy male volunteers (n = 14) and male subjects with impaired hepatic function (n = 14) (Table 1). The two studies in patients were assigned the following ClinicalTrials.gov registry numbers: NCT00510718 (dose-escalation study [5]) and NCT00974311 (phase III efficacy and safety study, AFFIRM [2]), while one of the studies in healthy volunteers was assigned the ClinicalTrials.gov registry number NCT01911715 (mass balance and biotransformation study) and the study in healthy volunteers and subjects with impaired hepatic function was assigned the registry number NCT01901133. The food-effect study in healthy volunteers was not assigned a registry number. All studies were conducted in accordance with the Declaration of Helsinki and with the approval of the appropriate local ethics committees. Informed consent was obtained from all subjects before study entry. Key eligibility criteria are summarized in Table 1, and the disease-specific criteria for the patient studies are described elsewhere [2, 5].
Table 1.
Overview of clinical studies and studies of enzalutamide in healthy volunteers
| Study | Population | Key eligibility criteria | Dosea and food intake |
|---|---|---|---|
| Dose escalation | 140 patients with mCRPC | Progressive castration-resistant prostate cancer [5] and ECOG performance status grade of 0–1 (2 was allowed if due to bone pain) | Dose escalation: enzalutamide 30, 60, 150, 240, 360, 480 (as 240 mg bid), and 600 (as 300 mg bid) mg qd; for the single dose, drug was taken in the clinic with breakfast (patient could provide his own breakfast), and for the multiple-dose period, food intake was not controlled |
| Mass balance and biotransformation | 6 healthy males | Healthy subjects, aged 18–55 years, BMI ≥18.5 and ≤30.0 kg/m2 inclusive | 14C-enzalutamide 160 mg (100 µCi) single dose under fasting conditionsb |
| Food effect | 60 healthy males under fed (n = 30) or fasted (n = 30) conditions | Healthy subjects, aged 18–55 years, body weight >50 kg, BMI 18–30 kg/m2 inclusive | Enzalutamide 160 mg single dose under fastedb or fed conditions (high-fat, high-calorie meal) |
| Hepatic impairment | 28 males: 6 with mild hepatic impairment, 8 with moderate hepatic impairment, and 14 with normal hepatic functionc | Aged 18–69 years, BMI ≥18.5 and ≤34.0 kg/m2 inclusive; criteria for mild and moderate hepatic impairment were based on Child–Pugh classification [9] | Enzalutamide 160 mg single dose under fasting conditionsb |
| Phase III efficacy and safety (AFFIRM) | 1199 patients with mCRPC (randomized 2:1 active-to-placebo) | Progressive castration-resistant prostate cancer [2] previously treated with one or two prior chemotherapy regimens, at least one of which was docetaxel-based. ECOG performance status grade of 0–2 | Enzalutamide 160 mg qd; food intake was not controlled |
bid twice daily, BMI body mass index, ECOG Eastern Cooperative Oncology Group, mCRPC metastatic castration-resistant prostate cancer, qd once daily
aAll clinical trials used the same drug product presentation as the commercial product (Xtandi®): a liquid-filled capsule of enzalutamide fully dissolved in caprylocaproyl polyoxylglycerides
bNo caloric intake for at least 10 h predose and 4 h postdose
cA total of 32 subjects were evaluated for pharmacokinetics in this study; however, the pharmacokinetic comparisons were based on 28 subjects due to the exclusion of four subjects (i.e. two pairs of subjects) in the mild hepatic impairment arms because of a >15 % difference in BMI between the matched pairs
Pharmacokinetic and Analytical Methods
Pharmacokinetic blood samples were collected with dipotassium ethylenediaminetetraacetic acid (K2 EDTA) as the anticoagulant. Validated bioanalytical methods based on high-performance liquid chromatography with tandem mass spectrometry (LC–MS/MS) were used to analyze plasma, plasma dialysate, and urine samples for concentrations of enzalutamide and the two major human metabolites. The bioanalytical methods were validated in accordance with US FDA guidance [6]. In the method that supported plasma pharmacokinetic determinations in the phase I dose-escalation (first-in-man) trial, enzalutamide was analyzed over the concentration range of 0.002–5.0 µg/mL [electronic supplementary material (ESM) 1]. In the method that supported all subsequent studies, enzalutamide, N-desmethyl enzalutamide, and the carboxylic acid metabolite were simultaneously measured over the range of 0.02–50 µg/mL for each analyte [7].
Plasma concentrations within the validated concentration range were used to calculate pharmacokinetics parameters, which were estimated by non-compartmental methods using WinNonlin® (Pharsight Corporation, Palo Alto, CA, USA) and applicable complimentary software, such as SAS® (SAS Institute, Cary, NC, USA) and Microsoft Excel® (Microsoft Corporation, Redmond, WA, USA). The estimated pharmacokinetics parameters included maximum plasma concentration (Cmax), time to Cmax (tmax), trough (predose) plasma concentration (Ctrough), area under the plasma concentration–time curve (AUC) from time zero to the last quantifiable concentration (AUClast), AUC for one 24-h dosing interval at steady state (AUCτ), AUC from time zero to 24 h after administration of a single dose (AUC24h), AUC from time zero to infinity (AUC∞), terminal half-life (t½), apparent oral clearance (CL/F), and apparent volume of distribution during the terminal phase (Vz/F).
Study Design
Dose-Escalation Study
A phase I, open-label, dose-escalation safety and pharmacokinetics study of enzalutamide was performed in 140 patients with mCRPC [5]. Two patient populations were enrolled: chemotherapy-naïve patients (n = 65) and post-chemotherapy patients (n = 75). Patients received enzalutamide orally at 30, 60, 150, 240, 360, 480 (as 240 mg twice daily), and 600 mg (as 300 mg twice daily), depending on dosing cohort.
Full pharmacokinetic profiles for enzalutamide were collected from three or six patients per dose level during the single-dose period (with a full pharmacokinetics profile over an approximate 6-day period) and from all patients during the multiple-dose period (with a full pharmacokinetics profile on day 84 over a 24-h dosing interval). The pharmacokinetic sampling times during the single-dose period were as follows: predose and 0.5, 1, 2, 4, 6, 24, 48, 72, 96, and 120 h postdose. The pharmacokinetic sampling times during the multiple-dose period were as follows: predose on days 1, 3, 7, 28, 56, and 84, and postdose on day 84 at 0.5, 1, 2, 4, 6, 8, 12, and 24 h. In addition, predose Ctrough samples were collected from all patients on days 112, 140, and 168, and approximately every 4 weeks thereafter until discontinuation of enzalutamide. For the single-dose period, enzalutamide was taken in the clinic with breakfast. For the multiple-dose period, food intake was not controlled.
N-desmethyl enzalutamide and the carboxylic acid metabolite were not measured in this study because this was the first study in humans and the relative concentrations of these metabolites were not yet known.
Dose proportionality was assessed by a power model involving regression of logarithmically transformed AUCτ versus dose. A statistical test for dose proportionality was applied by determining if the slope was wholly contained within the 90 % confidence intervals (CI) of 0.800–1.25 and the p value for goodness of fit was ≤0.05.
Mass Balance and Biotransformation Study
A phase I, open-label, one-period, single-dose study in six healthy male subjects was performed to evaluate pharmacokinetics, metabolism, and excretion. On day 1, subjects received a single oral dose of enzalutamide 160 mg (4 × 40 mg capsules) plus an additional liquid-filled capsule containing a tracer dose of 14C-labeled enzalutamide (100 μCi) under fasting conditions. The radiochemical purity of 14C-enzalutamide was ≥99.7 %. Whole blood, plasma, urine, and feces were collected through day 77 postdose, and subjects were confined to the clinic up to day 11. After discharge, subjects visited the clinic for overnight stays (2 consecutive nights) on days 13–15, 20–22, 27–29, 34–36, 48–50, 62–64, and 76–78. During these visits, blood samples were taken, and urine and feces were collected for 24 h.
Details regarding the collection times for blood, plasma, urine, and feces, as well as methods used to calculate excretion parameters, can be found in ESM 2.
Food-Effect Study
A phase I, open-label, randomized, single-dose, parallel design, food-effect study was performed in healthy male subjects. The parallel design was considered appropriate because the t½ of enzalutamide is long and intersubject variability in pharmacokinetics is low.
Subjects were randomized to receive a single oral dose of enzalutamide 160 mg (4 × 40 mg capsules) under fasting conditions (a minimum 10 h fast from food) or under fed conditions (n = 30 subjects per treatment arm). In the fed condition, subjects consumed a standard FDA high-fat, high-calorie breakfast containing 800–1000 calories, with approximately 50 % of this caloric content as fat [8] (the breakdown was approximately 150 calories from protein, 250 calories from carbohydrate, and 500–600 calories from fat). Blood samples for plasma pharmacokinetics were collected predose on day 1 and postdose through day 42 at the following times: on day 1 at 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, and 12 h, on day 2 at 0 and 12 h, and on days 3, 5, 7, 14, 21, 28, 35, and 42 at 0 h. These samples were analyzed for enzalutamide, N-desmethyl enzalutamide, and the carboxylic acid metabolite. The primary food-effect comparison was based on enzalutamide.
The food-effect analysis involved parallel group comparisons in which the fed condition (test) was compared with the fasted condition (reference). An analysis of variance was performed on the natural logarithmically transformed parameters AUClast, AUC∞, and Cmax using a mixed-effect model with food effect as the only factor.
Hepatic Impairment Study
A phase I, open-label, two-arm study was performed to determine the effect of mild and moderate hepatic impairment on the pharmacokinetics, safety, and tolerability of a single oral 160 mg dose of enzalutamide. Hepatic function was classified by the Child–Pugh system, as described by the FDA [9]. Arm A consisted of six subjects with mild hepatic impairment (Child–Pugh A, arm A1) and six matched control subjects with normal hepatic function (arm A2). Arm B consisted of eight subjects with moderate hepatic impairment (Child–Pugh B, arm B1) and eight control subjects with normal hepatic function (arm B2). The control subjects were individually matched for age (±5 years) and body mass index (BMI) (±15 %).
Each subject received a single oral dose of enzalutamide 160 mg, administered under fasting conditions. Blood samples for plasma pharmacokinetics were collected predose on day 1 and postdose through day 50 at the following times: on day 1 at 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, and 12 h; on days 2, 24, and 36 at 0 and 12 h; and on days 3, 4, 6, 8, 12, 15,19, 22, 26, 29, 36, 43, and 50 at 0 h. These samples were analyzed for enzalutamide, N-desmethyl enzalutamide, and the carboxylic acid metabolite. Plasma protein binding samples were collected at approximately 2, 96, and 120 h postdose, which corresponded to the expected tmax for enzalutamide, the carboxylic acid metabolite, and N-desmethyl enzalutamide, respectively. The unbound fractions were determined by a validated method based on equilibrium dialysis.
Pharmacokinetic parameters were expressed in terms of total concentrations and (as appropriate) unbound concentrations. Statistical analyses compared subjects with mild hepatic impairment (arm A1, test) versus matched controls (arm A2, reference) and subjects with moderate hepatic impairment (arm B1, test) versus matched controls (arm B2, reference). The primary parameters of interest were the logarithmically transformed AUC∞ and Cmax. If the data did not permit estimation of AUC∞, then AUClast was used. All observations for test and reference were included in the statistical analysis, which used the MIXED procedure in SAS® with an unstructured covariance matrix. Treatment arm (A, B) and treatment group (test, reference) were included as fixed effects, and subject pair was included as a random effect. The least-square means (LSMs) of the primary parameters for each treatment group/arm combination were estimated, and a 90 % CI was constructed around the difference between the LSMs of test and reference for each arm.
Phase III Efficacy and Safety Study in Patients
The phase III AFFIRM trial [2] was a multinational, randomized, double-blind, placebo-controlled efficacy and safety study of oral enzalutamide in 1199 patients with mCRPC whose disease was progressing after one or two prior chemotherapy regimens, one of which was docetaxel-based. All patients continued androgen deprivation therapy. Patients were allowed, but not required, to take glucocorticoids. Patients were randomized 2:1 to receive enzalutamide 160 mg/day or matching placebo. Randomization was stratified by Eastern Cooperative Oncology Group Performance Status grade at baseline.
Predose Ctrough samples were collected from all patients at weeks 1, 2, 5, 9, 13, and 25, and every 12 weeks thereafter to assess plasma concentrations of enzalutamide, N-desmethyl enzalutamide, and the carboxylic acid metabolite. Based on t½ estimates, enzalutamide was expected to be at steady state by week 5, and the metabolites were expected to be at steady state by week 9.
An exposure–response analysis was performed to assess relationships between plasma exposure of the sum of enzalutamide plus N-desmethyl enzalutamide versus the efficacy endpoint of overall survival. The purpose of this analysis was to look for trends in the data that might shed light on the suitability of a fixed oral dose of 160 mg/day. The source data, endpoint derivation, and stratification were as described previously [2], and the intent-to-treat population was as described in ESM 3. At the time of the data cutoff for the analysis for overall survival, the median time on study drug was 8.3 months for enzalutamide and 3.0 months for placebo. Exposure for each patient was defined as his mean steady-state Ctrough value (i.e. the mean concentration for all predose samples that were collected after at least 56 days of consecutive dosing at a particular dose of enzalutamide). The Ctrough values were evaluated as continuous and discrete parameters. When evaluated as discrete parameters, the Ctrough values were classified into quartiles that divided the derived non-zero exposure parameters into four approximately equal groups, from lowest exposure quartile (Q1) to highest exposure quartile (Q4), after sorting by rank order. Exposure categories included the placebo‐randomized patients as one category and four categories (quartiles) of exposure for the enzalutamide‐randomized patients. Patients randomized to placebo were assigned a Ctrough value of zero.
Log‐rank tests for homogeneity assessed differences between Kaplan–Meier exposure category curves, and pairwise log‐rank tests compared exposure categories with one another. In addition, a Cox proportional hazard analysis of Ctrough as a continuous variable assessed the association between exposure and event risk. Where a statistically significant slope was observed, pairwise Cox proportional hazard estimates compared exposure categories. Statistical significance was assigned using a nominal two‐sided type I error rate of 5 %, with no adjustments made for multiplicity.
Results
Participants
Demographics and baseline characteristics of subjects in the five studies are summarized in Table 2.
Table 2.
Demographic and baseline characteristics of subjects
| Characteristic/statistic | Dose escalation | Mass balance | Food effect | Hepatic impairment | Phase III | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fasted | Fed | Mildly impaired | Matched control | Moderately impaired | Matched control | Active | Placebo | |||
| Male sex [n (%)] | 140 (100) | 6 (100) | 30 (100) | 30 (100) | 8 (100) | 8 (100) | 8 (100) | 9 (100) | 800 (100) | 399 (100) |
| Race [n (%)] | ||||||||||
| White | 135 (96) | 5 (83) | 25 (83) | 23 (77) | 8 (100) | 8 (100) | 8 (100) | 9 (100) | 745 (93) | 366 (92) |
| Other | 5 (4) | 1 (17) | 5 (17) | 7 (23) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 55 (7) | 33 (8) |
| Ethnicity [n (%)] | ||||||||||
| NHL | 134 (96) | 6 (100) | 18 (60) | 24 (80) | 8 (100) | 8 (100) | 8 (100) | 9 (100) | 768 (96) | 376 (94) |
| HL | 5 (4) | 0 (0) | 12 (40) | 6 (20) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 325 (4) | 23 (6) |
| Age, years | ||||||||||
| Median | 68 | 24 | 28 | 31 | 39 | 43 | 52 | 54 | 69 | 69 |
| Range | 44–93 | 21–38 | 19–42 | 19–55 | 24–63 | 26–59 | 36–67 | 37–63 | 41–92 | 49–89 |
| Actual body weight, kg [mean (SD)] | 93.9 (17.2) | 75.5 (10.6) | 79.0 (12.7) | 78.7 (7.4) | 87.2 (19.5) | 87.4 (10.0) | 88.8 (15.6) | 90.8 (9.5) | 84.2 (14.5) | 85.0 (16.6) |
| BMI, kg/m2 [mean (SD)] | ND | 23.3 (2.3) | 25.6 (3.1) | 25.2 (2.4) | 28.5 (4.5) | 27.1 (2.8) | 27.8 (3.5) | 27.6 (2.3) | ND | ND |
BMI body mass index, HL Hispanic or Latino, ND not determined, NHL not Hispanic or Latino, SD standard deviation
Dose-Escalation Study
In the single-dose period, enzalutamide was absorbed rapidly after a single oral dose, with a median tmax of 1 h (range 0.4–4.0 h) (Table 3). The t½, CL/F, and Vz/F values were not affected by dose size.
Table 3.
Pharmacokinetic parameters of enzalutamide in castration-resistant prostate cancer patients after a single oral dose
| Study | Dose (mg) | Subjects (n) | C max (μg/mL)a | t max (h)b | AUC24 (μg·h/mL)a | AUC∞ (μg·h/mL)a | t ½ (days)a | CL/F (L/h)a | V z/F (L)a |
|---|---|---|---|---|---|---|---|---|---|
| Dose escalationc | 30 | 3 | 0.4 ± 0.1 | 2.0 [0.4–4.0] | 5.5 ± 0.7 | 54 ± 21 | 6.9 ± 2.9 | 0.61 ± 0.22 | 133 ± 20 |
| 60 | 3 | 1.7 ± 0.5 | 0.5 [0.5–1.0] | 15.6 ± 0.5 | 94 ± 17 | 4.2 ± 1.3 | 0.66 ± 0.13 | 91 ± 14 | |
| 150 | 3 | 3.4 ± 0.8 | 0.5 [0.5–2.0] | 38.9 ± 8.4 | 334 ± 50 | 6.0 ± 1.5 | 0.46 ± 0.06 | 92 ± 12 | |
| 240 | 3 | 5.3 ± 0.9 | 1.0 [0.6–1.0] | 62.1 ± 23.9 | 474 ± 138 | 5.8 ± 0.9 | 0.54 ± 0.18 | 112 ± 56 | |
| 360 | 6 | 7.1 ± 2.5 | 1.0 [0.5–2.2] | 80.5 ± 14.9 | 715 ± 122 | 6.2 ± 1.1 | 0.52 ± 0.09 | 109 ± 18 | |
| 480d | 6 | 6.8 ± 3.5 | 1.5 [0.5–2.1] | ND | 1010 ± 378 | 6.0 ± 2.9 | 0.54 ± 0.23 | 101 ± 30 | |
| 600d | 3 | 5.2 ± 1.0 | 1.0 [1.0–2.0] | ND | 896 ± 270 | 5.4 ± 1.6 | 0.72 ± 0.26 | 127 ± 11 | |
| All doses combined | 27 | NC | 1.0 [0.4–4.0] | NC | NC | 5.8 ± 1.6 | 0.56 ± 0.17 | 110 ± 32 | |
AUC ∞ area under the plasma concentration–time curve from time zero to infinity, AUC 24 area under the plasma concentration–time curve from time zero to 24 h after administration of a single dose, CL/F apparent oral clearance, C max maximum plasma concentration, NC not calculated, ND not determined, t ½ half-life, t max time to reach Cmax, V z /F apparent volume of distribution during the terminal phase
aValues are expressed as mean ± standard deviation
bValues are expressed as median [range]
cDose-escalation study, single-dose period [5]
d480 and 600 mg were administered as a divided dose (240 and 300 mg twice daily, respectively), with approximately 12 h between the two doses
In the multiple-dose period, enzalutamide was absorbed rapidly on day 84, with the median tmax ranging from 0.00 to 2.07 h (Table 4), which was similar to the median tmax in the single-dose period. The mean CL/F was 0.61 L/h, also similar to the CL/F during the single-dose period. In general, dosing for 1 month was required to reach steady state, and the daily fluctuations in plasma concentrations were low (mean peak-to-trough ratio, 1.25). As a result of the low daily fluctuations, plasma profiles at steady state resembled a constant infusion. The enzalutamide Ctrough values in individual patients remained constant (i.e. did not show an upward or downward trend over time) beyond 1 month of long-term therapy, demonstrating time-linear pharmacokinetics once steady state was achieved. The coefficient of variation (standard deviation/mean) for AUCτ, Ctrough, and Cmax was ≤30 %, demonstrating low intersubject variability. With daily oral administration, enzalutamide accumulated approximately 8.3-fold relative to the single dose. Cmax increased with increasing dose, with the exception of 480 and 600 mg, which were given as a divided dose (240 and 300 mg twice daily, respectively), with approximately 12 h between the two doses. To assess dose-proportionality of AUC, a power model was applied to AUCτ values for doses ranging from 30 to 360 mg/day (insufficient data were available at 480 and 600 mg/day). The estimated slope was 0.872 with a 90 % CI of 0.799–0.944, and the p-value for goodness of fit was 0.0043 (Fig. 2). As the 90 % CI of the slope was mostly contained within the interval of 0.800–1.25, and the p value for the goodness of fit was ≤0.05, the analysis indicated no major deviations from dose proportionality.
Table 4.
Pharmacokinetic parameters of enzalutamide in castration-resistant prostate cancer patients after multiple oral dose administration
| Study | Dose (mg/day)a | Subjects (n) | C max (μg/mL)b | AUCτ (μg·h/mL)b | t max (h)c | CL/F (L/h)b |
|---|---|---|---|---|---|---|
| Dose escalationd | 30 | 3 | 2.8 ± 0.6 | 61 ± 12 | 2.1 [1.0–3.9] | 0.51 ± 0.11 |
| 60 | 21 | 5.7 ± 1.5 | 115 ± 34 | 1.1 [0.5–23.7] | 0.58 ± 0.25 | |
| 150 | 23 | 14.5 ± 3.3 | 300 ± 68 | 1.0 [0.0–25.8] | 0.53 ± 0.15 | |
| 240 | 29 | 19.5 ± 5.0 | 410 ± 112 | 1.1 [0.0–26.2] | 0.63 ± 0.18 | |
| 360 | 16 | 25.1 ± 5.2 | 502 ± 119 | 1.6 [0.5–24.1] | 0.76 ± 0.18 | |
| 480 | 1 | 27.9 | 463 | 0.0e | 1.1 | |
| All doses combined | 93 | NC | NC | 1.1 [0.0–26.2] | 0.61 ± 0.20 | |
AUC τ area under the plasma concentration–time curve for one 24-h dosing interval at steady state, CL/F apparent oral clearance, C max maximum plasma concentration, NC not calculated, t max time to reach C max
aEnzalutamide was administered by once-daily dosing in all dose groups except for 480 mg/day, in which it was administered as a divided dose (240 mg twice daily), with approximately 12 h between the two doses
bValues are expressed as mean ± standard deviation
cValues are expressed as median [range]
dDose-escalation study, multiple-dose period [5]; pharmacokinetics were assessed after approximately 3 months of dosing
e t max was observed in the predose sample
Fig. 2.

Dose-proportionality assessment of enzalutamide for doses ranging from 30 to 360 mg/day. A linear regression (power model) analysis was applied to the mean values for the AUCτ after approximately 3 months of treatment. Circles denote individual patients and the gray area depicts 90 % CIs. AUC τ area under the plasma concentration–time curve during one 24-h dose interval at steady state, CIs confidence intervals
Mass Balance and Biotransformation Study
Enzalutamide was absorbed rapidly after oral administration, with a median tmax of 1.75 h (range 1–3 h; Table 5). The mean t½ of enzalutamide in plasma was 69.8 h (2.9 days). N-desmethyl enzalutamide and the carboxylic acid metabolite appeared to be formed slowly, with median tmax in plasma of 132 and 96 h (5.5 and 4.0 days) postdose, respectively. The mean t½ of N-desmethyl enzalutamide and the carboxylic acid metabolite in plasma were 186 and 223 h (7.8 and 9.3 days), respectively.
Table 5.
Pharmacokinetic parameters of enzalutamide after a single oral dose in healthy male volunteers and male subjects with impaired hepatic function
| Study | Dose (mg) | Subjects (n) | C max (μg/mL)a | t max (h)b | AUC∞ (μg·h/mL)a | t ½ (days)a | CL/F (L/h)a | V z/F (L)a |
|---|---|---|---|---|---|---|---|---|
| Mass balancec | 160 (fasted) | 6 | 4.5 ± 0.9 | 1.8 [1.0–3.0] | 237 ± 50 | 2.9 ± 0.3 | 0.71 ± 0.17 | 72 ± 21 |
| Food-effectd | 160 (fasted) | 30 | 5.3 ± 1.1 | 1.0 [0.8–3.1] | 292 ± 88 | 3.9 ± 1.3 | 0.60 ± 0.19 | 76 ± 22 |
| 160 (fed) | 30 | 3.7 ± 1.2 | 2.0 [0.5–6.0] | 285 ± 73 | 3.6 ± 1.0 | 0.60 ± 0.16 | 72 ± 17 | |
| Hepatice | 160 (fasted)f | 6 | 3.7 ± 1.3 | 1.3[0.5–2.0] | 246 ± 68 | 4.8 ± 1.8 | 0.69 ± 0.18 | 117 ± 58 |
| 160 (fasted)g | 8 | 3.8 ± 0.8 | 1.0 [0.8–2.0] | 225 ± 51 | 4.5 ± 2.2 | 0.75 ± 0.21 | 109 ± 41 | |
| 160 (fasted)h | 6 | 4.4 ± 0.8 | 0.8 [0.5–2.0] | 253 ± 42 | 3.5 ± 1.0 | 0.65 ± 0.12 | 77 ± 23 | |
| 160 (fasted)i | 8 | 3.7 ± 2.1 | 1.0 [0.5–2.0] | 303 ± 126 | 8.2 ± 7.7 | 0.60 ± 0.23 | 142 ± 105 |
AUC ∞ area under the plasma concentration–time curve from time zero to infinity, CL/F apparent oral clearance, C max maximum plasma concentration, t ½ half-life, t max time to reach C max, V z /F apparent volume of distribution during the terminal phase
aValues are expressed as mean ± standard deviation
bValues are expressed as median [range]
cMass balance and biotransformation study
dFood-effect study
eHepatic impairment study
fSubjects with normal hepatic function who served as controls for the mild hepatic impairment arm
gSubjects with normal hepatic function who served as controls for the moderate hepatic impairment arm
hSubjects with mild hepatic impairment
iSubjects with moderate hepatic impairment
The mean concentration-time profiles of 14C-concentrations in plasma and whole blood were essentially parallel (Fig. 3). The overall whole blood to plasma ratio was 0.55, indicating that the radioactivity was preferentially retained in the plasma component of blood. Enzalutamide, N-desmethyl enzalutamide, and the carboxylic acid metabolite accounted for 88 % of the 14C-radioactivity in plasma, representing 30, 49, and 10 % of the total AUC, respectively. In consideration of these percentages, N-desmethyl enzalutamide and the carboxylic acid metabolite are major human metabolites.
Fig. 3.

Mean concentration–time profiles after a single oral dose of 14C-enzalutamide (160 mg, 100 µCi) in the mass balance and biotransformation study (n = 6 healthy adult males)
The overall mean total recovery of 14C-radioactivity in urine and feces was 84.6 % of the administered dose (Fig. 4). The major route of excretion of 14C-radioactivity was urine (71.0 % of dose), primarily as metabolites. The remainder of the radioactivity was excreted in feces (13.6 % of dose). Based on LC-MS/MS analysis, the main component in urine was the carboxylic acid metabolite, which accounted for 62.7 % of the dose. A trace amount of unchanged parent enzalutamide was excreted in urine. N-desmethyl enzalutamide was too low to quantitate in urine by LC–MS/MS. Based on this information, renal excretion is a minor elimination pathway for unchanged parent enzalutamide and N-desmethyl enzalutamide.
Fig. 4.

Mean cumulative 14C-radioactivity recovery–time profiles after a single oral dose of 14C-enzalutamide (160 mg, 100 µCi) in the mass balance and biotransformation study(n = 6 healthy adult males). The majority of excretion of drug or drug-related product in urine was in the form of carboxylic acid metabolite
In feces, 0.39 % of the dose was recovered as unchanged parent enzalutamide. Given that overall recovery in excreta was 84.6 % of the dose, at least 84.2 % of the dose was absorbed.
Food-Effect Study
Enzalutamide was absorbed rapidly after oral administration, with a median tmax of 1 and 2 h postdose for the fasted and fed conditions, respectively (Table 6).
Table 6.
Statistical summary of food-effect comparisons
| Molecule and pharmacokinetic parameter | Adjusted geometric means | Ratio (test/reference) | 90 % CI for ratio | |
|---|---|---|---|---|
| Fed (test) | Fasted (reference) | |||
| Enzalutamide | ||||
| N, n | 30, 30 | 27, 27 | – | – |
| AUClast, µg·h/mL | 269 | 270 | 1.00 | 0.88–1.13 |
| AUC∞, µg·h/mL | 276 | 279 | 0.99 | 0.87–1.12 |
| C max, µg/mL | 3.61 | 5.13 | 0.70 | 0.63–0.79 |
| N-desmethyl enzalutamide | ||||
| N, n | 30, 30 | 27, 25 | – | – |
| AUClast, µg·h/mL | 386 | 355 | 1.09 | 0.98–1.21 |
| AUC∞, µg·h/mL | 410 | 379 | 1.08 | 0.96–1.21 |
| C max, µg/mL | 0.81 | 0.76 | 1.06 | 0.95–1.18 |
AUC ∞ area under the plasma concentration–time curve from time zero to infinity, AUC last area under the plasma concentration–time curve from time zero to the last quantifiable concentration, CI confidence interval, C max maximum plasma concentration, N number of subjects for whom AUClast and C max were calculated, n number of subjects for whom AUC∞ was determined
Based on AUC values for enzalutamide (Table 6), the extent of absorption was similar after a high-fat meal and under fasting conditions. The geometric mean AUC values were approximately 1 % lower in fed than in fasted subjects, and the 90 % CI for the ratio of treatment mean AUC values were within the 0.80–1.25 range, establishing the absence of a food effect. Based on Cmax and tmax values, the rate of absorption after a high-fat meal was slower than under fasting conditions. The geometric mean Cmax was approximately 30 % lower and median tmax was approximately 1 h later with food. The 90 % CI for the ratio of treatment mean Cmax values was 0.63–0.78, which is below the 0.80–1.25 range to establish absence of a food effect. Taken together, the results show that there was a negligible change in AUC and a slightly lower Cmax when enzalutamide was taken after a high-fat meal. These Cmax changes are not considered clinically relevant.
Summary statistics for N-desmethyl enzalutamide (Table 6) show that food does not have a clinically meaningful effect on exposure to the active metabolite.
Hepatic Impairment Study
The mean plasma concentration–time profiles for the sum of enzalutamide plus N-desmethyl enzalutamide after single-dose oral administration of enzalutamide 160 mg in subjects with mild or moderate hepatic impairment and in matched control subjects are presented in Fig. 5. Exposure parameters (AUC∞, Cmax) for enzalutamide, N-desmethyl enzalutamide, and the sum of enzalutamide plus N-desmethyl enzalutamide were no more than 1.30-fold higher in subjects with mild or moderate hepatic impairment than in matched control subjects with normal hepatic function (Table 7). Exposure parameters (AUClast, Cmax) for the carboxylic acid metabolite were unchanged with mild hepatic impairment and tended to be approximately 40 % lower in subjects with moderate hepatic impairment.
Fig. 5.
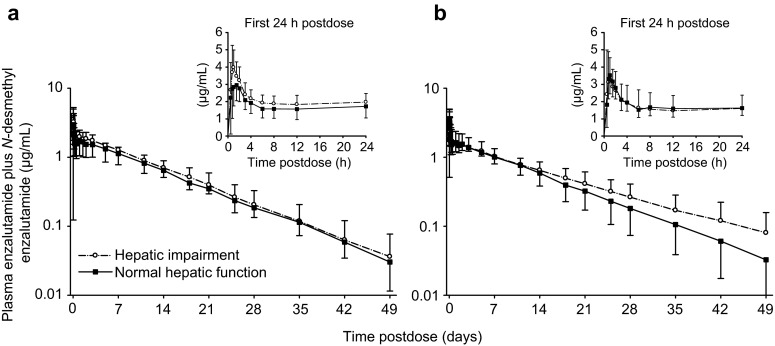
Concentration–time profiles (mean ± standard deviation) for the sum of enzalutamide plus N-desmethyl enzalutamide after a single oral dose of enzalutamide 160 mg in male subjects: a subjects with mild hepatic impairment (Child–Pugh Class A; n = 8) and the age- and BMI-matched control subjects with normal hepatic function (n = 8); b subjects with moderate hepatic impairment (Child–Pugh Class B; n = 6) and the age- and BMI-matched control subjects with normal hepatic function (n = 6). BMI body mass index
Table 7.
Statistical summary of hepatic impairment comparisons
| Molecule and parameter | Geometric mean ratio (90 % CI) | |
|---|---|---|
| Mild hepatic impairmenta | Moderate hepatic impairmentb | |
| Enzalutamide | ||
| AUC∞ | 1.05 (0.79–1.39) | 1.29 (1.01–1.65) |
| C max | 1.24 (0.92–1.66) | 0.89 (0.69–1.16) |
| N-desmethyl enzalutamide | ||
| AUC∞ | 1.18 (0.90–1.54) | 1.07 (0.85–1.35) |
| C max | 1.26 (0.92–1.72) | 0.85 (0.65–1.11) |
| Enzalutamide + N-desmethyl enzalutamide | ||
| AUC∞ | 1.13 (0.89–1.43) | 1.18 (0.96–1.45) |
| C max | 1.23 (0.92–1.66) | 0.89 (0.69–1.15) |
| Carboxylic acid metabolite | ||
| AUClast c | 1.14 (0.71–1.81) | 0.57 (0.38–0.86) |
| C max | 1.30 (0.85–1.99) | 0.63 (0.44–0.91) |
AUC ∞ area under the plasma concentration–time curve from time zero to infinity, AUC last area under the plasma concentration–time curve from time zero to the last quantifiable concentration, CI confidence interval, C max maximum plasma concentration
aBased on a comparison of n = 6 subjects with mild hepatic function and n = 6 subjects with normal hepatic function after a single oral dose of enzalutamide 160 mg
bBased on a comparison of n = 8 subjects with moderate hepatic function and n = 8 subjects with normal hepatic function after a single oral dose of enzalutamide 160 mg
cComparisons were based on AUClast because the sampling period was of insufficient duration to calculate AUC∞
The mean values for CL/F were similar in subjects with normal hepatic function, mild hepatic impairment, and moderate hepatic impairment (Table 5). The mean values for Vz/F and t½ were similar in subjects with normal hepatic function and mild hepatic impairment, while in subjects with moderate hepatic impairment, the mean values for Vz/F and t½ were approximately twofold higher. The mean unbound fraction for enzalutamide ranged from 1.49 to 2.39 %, from 2.62 to 3.63 % for N-desmethyl enzalutamide, and from 1.43 to 2.28 % for the carboxylic acid metabolite. The mean values for the unbound fractions were similar in subjects with normal hepatic function and subjects with impaired hepatic function, and there were no trends in the data to suggest alterations in the unbound fraction with mild or moderate hepatic impairment for any of the three molecules. Comparisons of AUC and Cmax based on the unbound concentrations of enzalutamide and its metabolites produced essentially the same ratios as AUC and Cmax based on total concentrations.
Phase III Efficacy and Safety Study
Pharmacokinetic assessments in patients taking enzalutamide 160 mg/day were based on predose Ctrough samples. Beyond week 5 (for enzalutamide) and week 9 (for the metabolites), the steady-state Ctrough values in individual patients remained constant (i.e. no general upward or downward trends with time) during more than 1 year of long-term therapy (Fig. 6), demonstrating time-linear pharmacokinetics once steady state was achieved. At week 13, the mean ± standard deviation Ctrough for enzalutamide was 11.4 ± 2.95 μg/mL (n = 679), 13.0 ± 3.78 μg/mL (n = 680) for N-desmethyl enzalutamide, and 8.44 ± 6.77 μg/mL (n = 680) for the carboxylic acid metabolite. Intersubject variability in Ctrough values for enzalutamide and N-desmethyl enzalutamide were ≤29 percent coefficient of variation (%CV), and 80 %CV for the carboxylic acid metabolite.
Fig. 6.

C trough versus time profiles for enzalutamide, N-desmethyl enzalutamide, and the carboxylic acid metabolite in an mCRPC patient taking enzalutamide 160 mg/day in the phase III trial (AFFIRM) [2]. C trough trough plasma concentration (measured concentration in a predose sample taken directly before the next administration), mCRPC metastatic castration-resistant prostate cancer
The exposure-response analysis examined the relationships between Ctrough quartiles for the sum of enzalutamide plus N-desmethyl enzalutamide and the efficacy endpoint of overall survival (ESM 3). The Kaplan–Meier exposure–response curves of overall survival for the composite sum of enzalutamide plus N-desmethyl enzalutamide are shown in Fig. 7. In all pairwise comparisons versus placebo, the effects of the active treatment Ctrough quartile groups were statistically significant (p ≤ 0.0001) in favor of active treatment, supporting the conclusion that the enzalutamide treatment was statistically superior to placebo for overall survival (Fig. 8). In addition, all assessments of overall survival versus the sum of enzalutamide plus N-desmethyl enzalutamide Ctrough values as a continuous variable resulted in statistically significant slopes (p < 0.0001). Although this suggests an association between higher levels of exposure and improved prognosis for patients, pairwise tests showed no difference in the risk of events among active treatment Ctrough quartiles (p ≥ 0.5499) (Fig. 7). Thus, the active treatment Ctrough quartile groups were uniformly beneficial relative to placebo, and there was no specific threshold of plasma concentrations in patients receiving enzalutamide that was associated with achieving a statistically significant better response. Similar results were obtained for exposure–response analyses based on Ctrough values for enzalutamide alone and N-desmethyl enzalutamide alone. Overall, the exposure‐response analysis showed similar efficacy in patients across the concentration/exposure range associated with a fixed oral dose of enzalutamide 160 mg/day.
Fig. 7.

Kaplan–Meier exposure–response analysis for exposure to enzalutamide plus N-desmethyl enzalutamide versus overall survival in the intent-to-treat population in the phase III clinical trial (AFFIRM) [2]. Exposure was based on time‐averaged steady-state predose (C trough) plasma concentrations that were classified into quartiles. The analysis involved 1103 patients (n = 176 patients in each of Q1, Q2, Q3, and Q4, and n = 399 placebo patients). C trough trough plasma concentration, Q exposure quartile
Fig. 8.

Cox proportional hazard model exposure–response analysis for overall survival in the enzalutamide plus N-desmethyl enzalutamide intent-to-treat population. Q exposure quartile
Discussion
Combined pharmacokinetic data from patients and healthy subjects provided insights into the absorption, distribution, and elimination properties of enzalutamide. With regard to absorption, after single- and multiple-dose oral administration, the tmax generally occurred around 1 h postdose, showing that enzalutamide is rapidly absorbed. Based on excretion of metabolites in urine and feces in the mass balance and biotransformation study, the extent of absorption of enzalutamide after oral administration is at least 84.2 %. With regard to distribution, the mean Vz/F of enzalutamide in patients (110 L) was approximately 2.6 times greater than the volume of total body water (42 L [10]), and approximately 37 times greater than the plasma volume (3 L [10]), suggesting extensive extravascular distribution of the drug. With regard to elimination, urinary and fecal recovery data after oral administration of 14C-enzalutamide showed that enzalutamide is primarily eliminated by hepatic metabolism, and renal excretion is an important pathway for the carboxylic acid metabolite and an insignificant elimination pathway for unchanged parent enzalutamide and N-desmethyl enzalutamide. The mean CL/F of enzalutamide was 0.56 L/h in patients, corresponding to approximately 1 % of liver plasma flow (48.7 L/h [10]); thus, enzalutamide is a low extraction ratio drug.
With daily oral administration, enzalutamide achieved steady state in patients by day 28, and accumulated approximately 8.3-fold relative to a single dose, which is consistent with the long t½ (5.8 days). Due to the long t½, daily fluctuations in plasma concentrations were low (mean peak-to-trough ratio, 1.25), and enzalutamide therefore had a relatively flat concentration profile over the dose interval with once-daily dosing. The steady-state Ctrough values for enzalutamide and N-desmethyl enzalutamide in individual patients remained constant during more than 1 year of long-term therapy, demonstrating time-linear pharmacokinetics once steady state was achieved. At steady state, enzalutamide showed no major deviations from dose proportionality over the daily dose range of 30–360 mg. In patients, %CV for AUCτ, Ctrough, and Cmax was ≤30 %, demonstrating low intersubject variability.
Consumption of a high-fat meal did not affect the extent of enzalutamide absorption but did result in a slower rate of absorption, as evidenced by a geometric mean Cmax that was approximately 30 % lower and a median tmax that was approximately 1 h later. Given the long t½ (5.8 days) and low peak-to-trough ratio (1.25) with once-daily dosing, these changes in the rate of absorption have essentially no effect on the pharmacokinetic profile of enzalutamide under conditions of clinical use; therefore, enzalutamide can be taken with or without food.
Mild hepatic impairment had no impact on the pharmacokinetics of enzalutamide or its major metabolites. Moderate hepatic impairment had no impact on exposure parameters (AUC∞ and Cmax) for enzalutamide active moieties (enzalutamide and N-desmethyl enzalutamide) but was associated with a decrease of approximately 40 % in exposure parameters for the carboxylic acid, which is of no clinical importance because this metabolite is inactive. Subjects with moderate hepatic impairment showed no apparent change in CL/F for enzalutamide but did demonstrate an approximately twofold increase in Vz/F and t½. The reason for the increase in Vz/F is unknown but may be attributed to a combination of factors typically associated with hepatic impairment, namely decreased concentrations of serum albumin, increased extracellular fluid (due to ascites and edema), and decreased muscle mass. As t½ is determined by the ratio of Vz/F to CL/F, the increase in t½ is attributed to the increase in Vz/F. Importantly, as the composite AUC∞ and Cmax of enzalutamide plus N-desmethyl enzalutamide were similar in subjects with mild or moderate hepatic impairment relative to subjects with normal hepatic function, no starting dose adjustment is needed when treating patients with mild or moderate hepatic impairment with enzalutamide. The effects of baseline severe hepatic impairment (Child–Pugh Class C) on enzalutamide pharmacokinetics have not yet been assessed.
Based on the results of the phase III trial in patients with mCRPC, there is no apparent exposure–response relationship for the efficacy endpoint of overall survival within a single fixed oral dose of 160 mg/day. As there was no specific threshold of plasma concentration in patients receiving enzalutamide that was associated with achieving a statistically significant better response, the analysis supports the suitability of a fixed oral dose of enzalutamide 160 mg/day for the treatment of patients with mCRPC.
Conclusions
Data from the studies summarized in this paper show that orally administered enzalutamide has a predictable pharmacokinetic profile with low intersubject variability, and similar efficacy is observed in patients across the concentration/exposure range associated with a fixed oral dose of enzalutamide 160 mg/day.
Electronic supplementary material
Acknowledgments
Jacqueline Gibbons and Joyce Mordenti are employees of Medivation, Inc, and Vanessa Beddo is an employee of ICON Clinical Research. Yoshiaki Ohtsu is an employee of Astellas Pharma, Inc. All other authors are employees of Astellas Pharma Europe B.V. The research described in this study was funded by Medivation, Inc. and Astellas Pharma, Inc., and individuals from these organizations were involved in the design and conduct of the study, collection of the data, and analysis and interpretation of the data. Copy editing assistance was provided by Shannon Davis of Infusion Communications and funded by Medivation, Inc. The authors take full responsibility for the content of the paper.
Footnotes
J. A. Gibbons and T. Ouatas contributed equally to this work.
References
- 1.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, AFFIRM Investigators et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 3.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate before chemotherapy. N Engl J Med. 2014;371(5):424–433. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.US Department of Health and Human Services. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. 2000. Available at: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070246. Accessed 13 Feb 2015.
- 5.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, et al. Prostate Cancer Foundation/Department of Defense Prostate Cancer Clinical Trials Consortium. Antitumour activity of enzalutamide in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;24;375(9724):1437–46. [DOI] [PMC free article] [PubMed]
- 6.US Department of Health and Human Services. Bioanalytical method validation. 2001. Available at: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107. Accessed 13 Feb 2015.
- 7.Bennett D, Gibbons JA, Mol R, Ohtsu Y, Williard C. Validation of a method for quantifying enzalutamide and its major metabolites in human plasma by LC–MS/MS. Bioanalysis. 2014;6(6):737–744. doi: 10.4155/bio.13.325. [DOI] [PubMed] [Google Scholar]
- 8.US Department of Health and Human Services. Food-effect bioavailability and fed bioequivalence studies. 2003. Available at: http://www.fda.gov/RegulatoryInformation/Guidances/UCM126833. Accessed 13 Feb 2015.
- 9.US Department of Health and Human Services. Pharmacokinetics in patients with impaired hepatic function: study design, data analysis, and impact on dosing and labeling. 2005. Available at: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072123. Accessed 13 Feb 2015.
- 10.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093–1095. doi: 10.1023/A:1018943613122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.