

Summary
Replication origins are under tight regulation to ensure activation occurs only once per cell cycle [1, 2]. Origin re-firing in a single S-phase leads to the generation of DNA double-strand breaks (DSBs) and activation of the DNA damage checkpoint [2–7]. If the checkpoint is blocked, cells enter mitosis with partially re-replicated DNA that generates chromosome breaks and fusions [5]. These types of chromosomal aberrations are common in numerous human cancers, suggesting re-replication events contribute to cancer progression. It was proposed that fork instability and DSBs formed during re-replication are the result of head-to-tail collisions and collapse of adjacent replication forks [3]. However, previously studied systems lack the resolution to determine whether the observed DSBs are generated at sites of fork collisions. Here we utilize the Drosophila ovarian follicle cells, which exhibit re-replication under precise developmental control [8–10], to model the consequences of re-replication at actively elongating forks. Re-replication occurs from specific replication origins at six genomic loci, termed Drosophila Amplicons in Follicle Cells (DAFCs) [10–12]. Precise developmental timing of DAFC origin firing permits identification of forks at defined points after origin initiation [13, 14]. Here we show that DAFC re-replication causes fork instability and generates DSBs at sites of potential fork collisions. Immunofluorescence and ChIP-seq demonstrate the DSB marker γH2Av is enriched at elongating forks. Fork progression is reduced in the absence of DNA damage checkpoint components and nonhomologous end-joining (NHEJ), but not homologous recombination. NHEJ appears to continually repair forks during re-replication to maintain elongation.
Graphical abstract
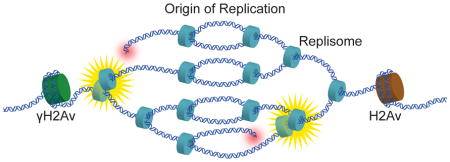
Results & Discussion
Fork instability and double-strand breaks occur during amplification
Drosophila marks DSBs by phosphorylation the H2Av histone tail, forming γH2Av [15], which can therefore be used to monitor DSB generation. The nuclear localization of γH2Av was visualized by immunofluorescence in amplifying follicle cells using a phospho-specific antibody. Follicle cells were co-labeled with the thymidine analog ethynyl deoxyuridine (EdU), which specifically marks the DAFCs due to the absence of genome-wide replication [9, 13]. Drosophila egg chambers are divided into developmental stages based on their distinct morphologies, each of which lasts for a defined period of time. This enables isolation of the follicle cells at specific times in development by ovary dissection. Origin firing at the DAFCs begins at stage 10B across all follicle cells of a given egg chamber [9]. At this stage EdU is visible in single foci corresponding to each DAFC origin and the surrounding forks (Fig. 1A, C) [9, 13]. By stages 12 and 13, the origin of the most highly amplified site, DAFC-66D, no longer fires, but existing replication forks continue to travel; this results in the resolution of two adjacent EdU foci around the DAFC-66D origin, called the double-bar structure [13]. (Fig. 1A, F).
Figure 1. Markers of DNA damage and replication fork stress co-localize with sites of re-replication.

(A) The onion skin model of amplification. EdU is drawn in red overlaying sites of actively replicating DNA. EdU labeling during origin initiation and fork progression in stage 10B results in incorporation throughout the amplicons (left). In stage 13 when forks continue to progress without further origin firing events, EdU incorporation gives rise to the double-bar structure (right).
(B–G) Immunofluorescence images of stage 10B (B–D) and 13 (E–G) follicle cell nuclei reveal the double-strand break marker γH2Av (D, G) co-localizes with EdU (C, F). As forks progress in stage 13 and EdU incorporation forms the double-bar structure (F), the γH2Av signal also resolves into to double-bars (G). This co-localization pattern was present in every follicle cell nucleus of every egg chamber observed (53 stage 10Bs and 49 stage 13s). (B, E) Merged image with EdU is shown in red, γH2Av in green, DAPI in blue. Each image is a single plane of nucleus. The prominent EdU focus corresponds to DAFC-66D (arrows). Scale bars, 1μm.
(H–M) RPA immunofluorescence reveals direct overlap with EdU in stage 10B (H–J) and 13 (K–M) follicle cells. RPA follows the pattern of fork progression highlighted by EdU, resolving into a double-bar structure in stage 13 (M). This co-localization pattern was present in every follicle cell nucleus of every egg chamber observed (51 stage 10Bs and 60 stage 13s). (H, K) Merged image with EdU is shown in red, RPA in green, DAPI in blue. Each image is a single plane of a follicle cell nucleus. The prominent EdU focus corresponds to DAFC-66D (arrows). Scale bars, 1 μm.
We found that intense γH2Av staining directly overlaps with sites of EdU incorporation in all amplifying follicle cells observed (Fig. 1B–G). In stage 10B when replication forks have just begun to progress away from the origin, γH2Av was already visible at each EdU focus (Fig. 1B, D). Strikingly, in stage 13 γH2Av resolved into a double-bar pattern overlapping EdU (Fig. 1E, G). These results demonstrate that DSBs are generated during amplification. Additionally, the resolution of γH2Av into double-bars in stage 13 strongly suggests that DSBs are occurring at the active replication forks and that these breaks are repaired as the forks progress.
The γH2Av localization pattern was confirmed using a second antibody (Fig. S1A) [16]. The antibody specificity was confirmed in H2AvΔCT mutant follicle cells, in which the only form of H2Av expressed lacks the phosphorylation site [17]. No γH2Av signal was detected during any stage of amplification in H2AvΔCT follicle cells (Fig. S1B). To confirm the observed DNA damage was not generated by EdU incorporation, follicle cells were co-labelled for γH2Av and the DAFC fork marker DUP (Fig. S1E) [13]. Here γH2Av signal overlapped with DUP as single foci in stage 10B and double-bars in stage 13, as was seen with EdU.
To determine if the γH2Av signal at the DAFCs is generated by DSBs or single-stranded DNA (ssDNA), staining was done in follicle cells lacking ATR and ATM activity (Fig. S1F–H). Both activated kinases phosphorylate H2Av; ATR is activated in response to extended RPA tracks on ssDNA, whereas ATM is specifically activated by DSBs [18]. In the absence of either single kinase, γH2Av localization was the same as in wild-type follicle cells (Fig. S1G–H). However, when neither kinase was active, γH2Av was completely absent (Fig. S1F). This demonstrates that both ssDNA and DSBs generate γH2Av during re-replication.
To confirm our results with γH2Av staining, we sought to localize RPA as a second marker of fork stalling and damage. RPA forms long tracks on single-stranded DNA caused by fork stalling, as well as after resection of DSBs [19, 20]. RPA staining therefore marks both fork stress and sites of DSB repair. Similar to γH2Av, we found that strong RPA staining directly overlapped with sites of EdU incorporation during all amplification stages observed (Fig. 1H–M). Additionally, RPA resolved into a double-bar structure in stage 13, following the pattern of EdU (Fig. 1M). Together the RPA and γH2Av results indicate replication forks stall and collapse during re-replication at the DAFCs.
To confirm that the RPA and γH2Av signals observed were not general markers of DNA replication, we examined staining in earlier follicle cells undergoing S-phase. Prior to the onset of amplification, the follicle cells undergo three endocycles [9]. The endocycle is an alternative cell cycle that undergoes consecutive G- and S-phases without an intervening mitosis. S-phase of the endocycle resembles that of a canonical S-phase in that origins fire only once per cell cycle, and therefore do not exhibit re-replication [21]. Although diffuse nuclear staining was detected for both RPA and γH2Av, neither signal was specific to EdU positive cells (Fig. S1C–D). This shows that neither RPA nor γH2Av can be detected at replication forks during S-phase in the absence of fork stress. There was γH2Av at genomic regions outside the DAFCs during amplification stages, which was absent in staining controls (S1B, F) indicating it is specific and generated in response to DNA damage. The appearance of γH2Av throughout the nucleus during the endocycles is consistent with previous observations that DSBs occur in the heterochromatin in follicle cells [22, 23]. The γH2Av staining that is not coincident with the amplicons also is at heterochromatin as evidenced by intense DAPI staining.
To evaluate γH2Av localization at the DAFCs and across the genome at the molecular level, we analyzed γH2Av enrichment by ChIP-seq. Enrichment was assessed in both stage 10B and 13 follicle cell nuclei to observe changes in γH2Av accumulation at the initial and final points in amplification. The same ChIP-seq experiment was done from H2AvΔCT follicle cells to control for non-specific antibody binding (Fig. S2). To determine where γH2Av is enriched along each DAFC, the position of γH2Av peaks was compared to Comparative Genomic Hybridization (CGH) analysis from wild-type egg chambers. CGH analysis measures the DNA copy number over chromosomal position, and demonstrates that fork progression expands the amplification gradient of each DAFC between stages 10B to 13 (Fig. 2, first and third lines). Comparison of ChIP-seq with the CGH gradients enabled us to analyze the γH2Av enrichment profile relative to the active replication forks.
Figure 2. γH2Av enrichment at the DAFCs during re-replication stages.

CGH and γH2Av ChIP-seq from OrR stage 10B and 13 follicle cells at each of the six DAFCs. Chromosomal position is indicated above each panel. CGH profiles are the log2 ratio (0–5) of egg chamber to embryonic DNA (first and third lines). ChIP-seq enrichment is the RPM of ChIP/input (0–26) for 1kb windows sliding every 100bp, and is the geometric mean of two biological replicates (second and fourth lines). Genomic coordinates are displayed above.
We found that γH2Av was significantly enriched at all six DAFCs in both stages compared to enrichment across the genome and in the H2AvΔCT control (Table S1). The ChIP-seq enrichment profiles shifted from a single broad region of enrichment in stage 10B to two adjacent peaks on either side of the origin in stage 13, reflecting the double-bar structure seen by γH2Av immunofluorescence at DAFC-66D (Fig. 2, second and fourth lines). Previous analysis of DAFC-66D measured a 70kb gap between the double-bars by stage 13 [13], much larger than the gaps between γH2Av ChIP-seq peaks. However, previous measurements were made at individual follicle cells, whereas the ChIP-seq data is averaged across 3×106 cells. Co-localization of γH2Av staining and EdU indicate γH2Av is at active replication forks (Fig. 1E–G). Therefore the reduced double-bar distance measured by ChIP-seq is likely the result of the large population average.
Interestingly, the resolution provided by ChIP-seq revealed enrichment at DAFC-66D is resolved into double-bars by stage 10B. In stage 13, the positions are maintained with increased levels of enrichment. We propose that fork stress and accumulation of DSBs in the double-bar structure early at DAFC-66D increases the frequency of fork collisions in those same positions. Therefore γH2Av enrichment is enhanced over the same sequences, rather than spreading away from the origin between stages 10B and 13. Together our ChIP-seq and cytological results demonstrate extensive fork stalling and DSBs occur at the active replication forks during re-replication.
The DNA damage response is essential for fork progression after re-replication
DSBs are generated from the earliest point of amplification in stage 10B, yet replication fork progression is still continues until the end of follicle cell development in stage 13. This suggests that the DNA damage response (DDR) and DSB repair may be essential for continued fork movement during re-replication. To test the requirement for repair at active replication forks, we measured fork progression at the DAFCs in several DDR mutants by Comparative Genomic Hybridization (CGH) analysis (Fig. 3). The shape of the amplification gradients generated by CGH is reflective of replication fork progression. A gradual decrease in copy number indicates uninhibited fork movement, whereas a rapid decrease indicates fork movement is impeded (Fig. 3A). CGH analysis is therefore a powerful tool to compare fork progression between different mutant lines. CGH analysis was performed at each site of re-replication except DAFC-22B; this site is a strain-specific amplicon [12], and therefore could not be compared across different genetic backgrounds. The number and timing of replication initiation events was first measured for each mutant by quantitative PCR. None of the mutants analyzed significantly affected replication initiation (Fig. 3D), confirming that any changes in the amplification gradients are not due to altered initiation kinetics. Additionally, fork progression was measured in appropriate controls to rule out changes in fork progression due to differences in genetic backgrounds (Fig. S3A and S4A).
Figure 3. Fork progression is reduced in the absence of DDR components.

(A) Blocked fork progression causes adjacent forks to pile up, resulting in close spacing as demonstrated by the replication forks highlighted in red (top). This is reflected in the CGH gradient by a sharp decrease in copy number. An example of one such region is highlighted in red on the wild-type DAFC-66D gradient (bottom).
(B) CGH of DAFC-66D from DDR mutants reveals impaired replication fork progression. DNA from stage 13 egg chambers was competitively hybridized with diploid embryonic DNA to microarrays with approximately one probe every 125bp. Chromosomal position is plotted on the x-axis, the log2 ratio of stage 13 DNA to embryonic DNA is plotted on the y-axis. In all mutants shown, the amplification gradient exhibits a rapid decrease in copy number compared to the wild type (top).
(C) The half-maximum distance was calculated in the wild-type and mutant backgrounds for each DAFC. Each half-maximum value is the average of three biological replicates. Significance measured by the Dunnett test for multiple comparisons, asterisks indicate p<0.05 and n.s. indicates not significant.
(D) The level of amplification was measured at the DAFC-66D origin of replication in each DSB signaling and repair mutant by quantitative real-time PCR. The copy number in stages 10B and 13 egg chambers is relative to the nonamplified rosy locus. Error bars are standard error of three replicates. None of the mutants were significantly different from the wild type as measured by the Dunnett test for multiple comparisons.
CGH analysis was done for a collection of mutants previously shown to be involved in various stages of the DDR: H2AvΔCT, mus101D1, chk11, chk2P6 and [15, 18, 24]. To measure fork progression quantitatively, we calculated the half-maximum distance for each DAFC from the wild type and DDR mutant CGH data. The half-maximum distance is the number of basepairs between the left and right position of half-maximal copy number. Since inhibited fork movement causes a more rapid decrease in copy number, a reduced half-maximum distance indicates fork progression is impeded. The half-maximum distance was significantly reduced at nearly all DAFCs in the H2AvΔCT, mus101D1, grp1 (chk1)1, chk2P6 mutant follicle cells (Fig. 3B–C, S3B). Together these results show that impairing the DNA checkpoint prevents complete fork progression, suggesting that checkpoint-mediated fork stabilization and repair are utilized during re-replication.
One site, DAFC-30B, does not exhibit a significant decrease in the half-maximum distance in H2AvΔCT or chk11 (Fig. 3C). This site only undergoes two origin firings before the completion of stage 10B [11]. It is likely that because this site completes re-replication at the earliest stages, these forks have enough time to repair and progress close to the wild-type distance by stage 13 even when DDR signaling is dampened.
It is well established that activation of Chk1 during S-phase prevents late origin firing [2]. However, the number of origin firings at each DAFC was unaffected by loss of Chk1 (Fig. 3D). It has been shown that Chk1 does not globally block origin firing, but rather limits new initiations to origins nearby stressed replication forks [25]. Therefore, it is not surprising that activation of the DNA damage checkpoint does not influence origin activation at the DAFCs. It is more likely that amplification results from the ability of these origins to escape re-initiation controls, rather than inactivity of the DNA damage checkpoint.
Double-strand break repair is required for continued fork progression during re-replication
To elucidate the mechanism of repair, fork progression was measured in mutants known to be defective in specific repair pathways. The half-maximum distance was measured in the null mutants spnA093 (Rad51 homolog) [26] and brca2KO [27] to test the role of homologous recombination (HR), and ligIV169 [28] to examine nonhomologous end joining (NHEJ) in repair after re-replication. We found that the half-maximum distance at each DAFC was significantly decreased in ligIV169, but not spnA093 or brca2KO follicle cells (Fig. 4, S4B). We demonstrated the effect was specific to loss of ligIV by testing the parental strain in which the excision was generated (Fig. S4). These results indicate HR is dispensable, whereas NHEJ is utilized for DSB repair during re-replication. The dependence of fork progression on DSB repair machinery further demonstrates that re-replication generates DSBs at the active replication forks, and these breaks must be repaired for subsequent forks to continue elongating.
Figure 4. LigIV is utilized for DSB repair during re-replication.

(A) CGH of DAFC-66D reveals impaired replication fork progression in the ligIV169, but not the spnA093 or brca2KO mutants. DNA from stage 13 egg chambers was competitively hybridized with diploid embryonic DNA to microarrays with approximately one probe every 125bp. Chromosomal position is plotted on the x-axis, the log2 ratio of stage 13 DNA to embryonic DNA is plotted on the y-axis.
(B) The half-maximum distance was calculated in the wild-type and mutant backgrounds for each DAFC. Each half-maximum value is the average of three biological replicates. Significance measured by the Dunnett test for multiple comparisons, asterisks indicate p<0.05. The spnA093 and brca2KO mutants are not significantly different from wild type.
The half-maximum measurements from DDR and ligIV mutants show only a 25–30% decrease in fork progression at each re-replicated site, rather than a complete replication block. There are two possible explanations for this effect: 1) each signaling and repair component is required to repair 30% of breaks that form on every copy of re-replicated DNA; or 2) DSBs are generated on 30% of the amplified strands. The former explanation seems unlikely for this collection of mutants, which represent diverse functions at different stages of DNA damage detection and repair. We therefore prefer the latter argument, which can be explained by replication fork collisions. Such collision events are expected to be stochastic, and will not occur at the same position on each copy of DNA in every cell. Additionally, this variation in break position is averaged across all the copies of amplified DNA in each 16C cell and the approximately 100,000 cells per CGH experiment, explaining why copy number decreases as a gradient rather than a sharp drop at sites of damage. In addition to replication fork collision, it is possible the DAFC replication forks are inherently unstable.
Our results indicate that the NHEJ repair pathway is utilized to maintain fork progression at the DAFCs, whereas inhibition of HR has no significant effect. These results are supported by a recent study that found deletions within DAFC-66D from amplification stage follicles, consistent with end-joining repair [29]. HR is often the preferred repair mechanism when homologous sequences are available to copy [30]. The follicle cells undergo endocycles prior to amplification, increasing the genome ploidy to 16C [8]. This increase in genome content, coupled with amplification, provides many identical copies of the DAFCs available for HR repair. It was thus initially surprising that the follicle cells instead utilize the mutagenic NHEJ pathway. It is possible that the presence of too many templates is problematic for HR repair, and generates DNA structures that could actually slow repair and fork progression. Repair by NHEJ is also much faster than HR [30], allowing the cells to repair the damage as soon as possible so that replication forks can continue. The presence of multiple broken DNA ends within the DAFCs would also provide many substrates for NHEJ repair. Additionally, because the follicle cells are sloughed off the oocyte soon after amplification ends, potential mutations produced by NHEJ will not have deleterious effects for the organism. We propose that fast kinetics, coupled to the terminal differentiation of the follicle cells, make NHEJ the ideal mechanism to repair damage generated during re-replication.
Conclusions
The gene amplification system is a well-established model of DNA replication. We establish for the first time that the gene amplification also is ideal to study how DNA damage is generated and repaired during re-replication. The resolution of the DAFC system enabled us to visualize DSBs directly at active forks, providing more direct evidence for the cause-and-effect relationship between re-replication and DSB generation. We show that loss of various checkpoint and repair components impairs fork progression, illustrating that checkpoint signaling is essential for repair of forks that are damaged during re-replication. Additionally, we propose that the DAFCs are a model of general fork instability that can be used to elucidate the pathways responsible for maintenance of fork progression under replication stress.
Supplementary Material
Highlights.
DNA breaks are generated at active replication forks during re-replication
The DNA damage checkpoint is required for complete fork elongation
Loss of nonhomologus end-joining repair inhibits fork progression
Acknowledgments
We thank Brian Hua for sharing unpublished data and for affinity purified DUP antibody. Bioinformatics support was provided by George Bell for CGH half-maximum analysis. Paul Fisher provided the RPA antibody, Scott Hawley and Jeff Sekelsky the γH2Av monoclonal antibody, and Kim McKim, Trudi Schupbach, Mitch McVey, and the Bloomington Stock Center were the sources for Drosophila stocks. We thank Tom DiCesare for graphics help. We are grateful for helpful comments on the manuscript from Mitch McVey, Steve Bell and Adam Martin. This work was supported by NIH grant GM57940 to Terry Orr-Weaver and the MIT School of Science Fellowship in Cancer Research. Terry Orr-Weaver is an American Cancer Society Research Professor.
Footnotes
Accession Numbers
The CGH and ChIP-seq data sets have been deposited in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). CGH data sets for wild-type controls are under accession numbers GSM432742 and GSM1354444. All mutant CGH and all ChIP-seq data sets are under accession number GSE66691.
Supplemental information includes Supplemental Experimental Procedures, four figures and one table.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- 2.Abbas T, Keaton MA, Dutta A. Genomic instability in cancer. Cold Spring Harb Perspect Biol. 2013;5:a012914. doi: 10.1101/cshperspect.a012914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davidson IF, Anatoily L, Blow JJ. Deregulated replication licensing causes DNA fragmentation consistent with head-to-tail fork collision. Mol Cell. 2006;24:433–443. doi: 10.1016/j.molcel.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green BM, Li JJ. Loss of rereplication control in Saccharomyces cerevisiae results in extensive DNA damage. Mol Biol Cell. 2005;16:421–432. doi: 10.1091/mbc.E04-09-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melixetian M, Ballabeni A, Masiero L, Gasparini P, Zamponi R, Bartek J, Lukas J, Helin K. Loss of Geminin induces rereplication in the presence of functional p53. J Cell Biol. 2004;165:473–482. doi: 10.1083/jcb.200403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mihaylov IS, Kondo T, Jones L, Ryzhikov S, Tanaka J, Zheng J, Higa LA, Minamino N, Cooley L, Zhang H. Control of DNA replication and chromosome ploidy by Geminin and Cyclin A. Mol Cell Biol. 2002;22:1868–1880. doi: 10.1128/MCB.22.6.1868-1880.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu W, Chen Y, Dutta A. Rereplication by depletion of Geminin is seen regardless of p53 status and activates a G2/M checkpoint. Mol Cell Biol. 2004;16:7140–7150. doi: 10.1128/MCB.24.16.7140-7150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spradling AC, Mahowald AP. Amplification of genes for chorion proteins during oogenesis in Drosophila melanogaster. Proc Natl Acad Sci USA. 1980;77:1096–1100. doi: 10.1073/pnas.77.2.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calvi BR, Lilly MA, Spradling AC. Cell cycle control of chorion gene amplification. Genes Dev. 1998;12:734–744. doi: 10.1101/gad.12.5.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Claycomb JM, Orr-Weaver TL. Developmental gene amplification: insights into DNA replication and gene expression. Trends Genet. 2005;21:149–62. doi: 10.1016/j.tig.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Claycomb JM, Benasutti M, Bosco G, Fenger DD, Orr-Weaver TL. Gene amplification as a developmental strategy: isolation of two developmental amplicons in Drosophila. Dev Cell. 2004;6:145–155. doi: 10.1016/s1534-5807(03)00398-8. [DOI] [PubMed] [Google Scholar]
- 12.Kim JC, Nordman J, Xie F, Kashevsky H, Eng T, Li S, MacAlpine DM, Orr-Weaver TL. Integrative analysis of gene amplification in Drosophila follicle cells: parameters of origin activation and repression. Genes Dev. 2011;25:1384–1398. doi: 10.1101/gad.2043111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Claycomb JM, MacAlpine DM, Evans JG, Bell SP, Orr-Weaver TL. Visualization of replication initiation and elongation in Drosophila. J Cell Biol. 2002;159:225–236. doi: 10.1083/jcb.200207046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park EA, MacAlpine DM, Orr-Weaver TL. Drosophila follicle cell amplicons as models for metazoan DNA replication: a cyclinE mutant exhibits increased replication fork elongation. Proc Natl Acad Sci USA. 2007;104:16739–16746. doi: 10.1073/pnas.0707804104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madigan AP, Chotkowski HL, Glaser RL. DNA double-strand break-induced phosphorylation of Drosophila histone variant H2Av helps prevent radiation-induced apoptosis. Nucleic Acids Res. 2002;30:3698–3705. doi: 10.1093/nar/gkf496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lake CM, Holsclaw JK, Bellendir SP, Sekelsky J, Hawley RS. The Development of a monoclonal antibody recognizing the Drosophila melanogaster phosphorylated histone H2A variant (γ-H2AV) G3. 2013;3:1539–1543. doi: 10.1534/g3.113.006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarkson MJ, Wells JRE, Gibson F, Saint R, Tremethick DJ. Regions of variant histone His2AvD required for Drosophila development. Nature. 1999;399:694–697. doi: 10.1038/21436. [DOI] [PubMed] [Google Scholar]
- 18.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 19.Byun TS, Pacek M, Yee M, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GCM, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 21.Nordman J, Li S, Eng T, MacAlpine D, Orr-Weaver TL. Developmental control of the DNA replication and transcription programs. Genome Res. 2011;21:175–181. doi: 10.1101/gr.114611.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong A, Narbonne-Reveau K, Riesgo-Escovar J, Fu H, Aladjem MI, Lilly MA. The cyclin-dependent kinase inhibitor Dacapo promotes replication licensing during Drosophila endocycles. EMBO J. 2007;26:2071–2082. doi: 10.1038/sj.emboj.7601648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehrotra S, Maqbool SB, Kolpakas A, Murnen K, Calvi BR. Endocycling cells do not apoptose in response to DNA rereplication genotoxic stress. Genes Dev. 2008;22:3158–3171. doi: 10.1101/gad.1710208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kondo S, Perrimon N. A genome-wide RNAi screen identifies core components of the G2-M DNA damage checkpoint. Sci Signal. 2011;4:rs1. doi: 10.1126/scisignal.2001350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ge XQ, Blow JJ. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol. 2010;191:1285–1297. doi: 10.1083/jcb.201007074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staeva-Vieira E, Yoo S, Lehmann R. An essential role of DmRad51/SpnA in DNA repair and meiotic checkpoint control. EMBO. 2003;22:5863–5874. doi: 10.1093/emboj/cdg564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klovstad M, Abdu U, Schüpbach T. Drosophila brca2 is required for mitotic and meiotic DNA repair and efficient activation of the meiotic recombination checkpoint. PLoS Genet. 2008;4:e31. doi: 10.1371/journal.pgen.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McVey M, Radut D, Sekelsky JJ. End-joining repair of double-strand breaks in Drosophila melanogaster is largely DNA ligase IV independent. Genetics. 2004;168:2067–2076. doi: 10.1534/genetics.104.033902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yarosh W, Spradling AC. Incomplete replication generates somatic DNA alterations within Drosophila polytene salivary gland cells. Genes Dev. 2014;28:1840–1855. doi: 10.1101/gad.245811.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.