

Abstract
Background
We evaluated whether next generation sequencing (NGS) of cfDNA could be used for patient selection and as a tumor clone response biomarker in patients with advanced cancers participating in early phase clinical trials of targeted drugs.
Methods
Plasma samples from patients with known tumor mutations who completed at least 2 courses of investigational targeted therapy were collected monthly, until disease progression. NGS was performed sequentially on the Ion Torrent PGM platform.
Results
cfDNA was extracted from 39 patients with various tumor types. Treatments administered targeted mailnly the PI3K-AKT-mTOR pathway (n=28) or MEK (n=7). Overall 159 plasma samples were sequenced with a mean sequencing coverage achieved of 1,685X across experiments. At trial initiation (C1D1), 23 of 39 (59%) patients had at least one mutation identified in cfDNA (mean 2, range 1-5). TP53, PIK3CA and KRAS were the top 3 mutated genes identified, with 16 (39%), 9 (22%) and 8 (17%) different mutations, respectively. Out of these 23 patients, 13 received a targeted drug matching their tumor profile. For the 23 patients with cfDNA mutation at C1D1, the monitoring of mutation allele frequency (AF) in consecutive plasma samples during treatment with targeted drugs demonstrated potential treatment associated clonal responses. Longitudinal monitoring of cfDNA samples with multiple mutations indicated the presence of separate clones behaving discordantly. Molecular changes at cfDNA mutation level were associated with time to disease progression by RECIST criteria.
Conclusion
Targeted NGS of cfDNA has potential clinical utility to monitor the delivery of targeted therapies.
Keywords: Next generation sequencing, plasma tumor DNA, early clinical trials
Introduction
Next generation sequencing (NGS) is becoming a key tool for molecular screening programs in anticancer drug development (1). The identification of patients with oncogenic driver mutations provides the opportunity to use the genomic information of individual tumors to guide the selection of rational therapeutics in an attempt to improve the outcome of patients with advanced cancers (2). The discovery of genomic aberrations like BRAF mutations and ALK rearrangements, and the subsequent accelerated development of vemurafenib or crizotinib for that targeted patient population, is an example of such a successful biomarker-driven drug development (3, 4).
However, the known spatial and temporal heterogeneity of tumors transforms such analyses into a much more complex challenge (5-7). Moreover, patients participating in early phase clinical trials form a highly heterogeneous cohort comprising a mixture of tumor types, prior treatment exposure, and the presence of multiple sites of disease that may each be molecularly heterogeneous. Thus, the validity of utilizing molecular characterization studies of archival tumor tissue acquired at diagnosis, often several years before, is questionable. In addition, the analysis of one tumor lesion may not provide sufficient information on this heterogeneity, but serial biopsies and sampling multiple lesions remain impractical.
Recent technological advances have delivered on the potential of circulating DNA (cfDNA) genotyping for the molecular characterization of tumors (8). Proof-of-concept studies have also shown that sequencing cfDNA can reveal important information on tumor-related genetic and epigenetic alterations relevant to oncogenesis and cancer progression (9, 10), tumor heterogeneity (11, 12) and mechanisms of response and resistance to therapy for a given patient (13, 14). In most of the reported studies, only a few mutations or genomic rearrangements have been interrogated and tracked (15), usually in selected tumor types, while published exome sequencing studies require high levels of cfDNA with high tumor DNA content and therefore may not be applicable to unselected populations (13).
Incorporating cfDNA next generation sequencing for patient selection can expedite patient molecular stratification and has the potential of circumventing the ethical and safety concerns associated with repeated fresh biopsies which may be very different to archival tumor samples acquired at diagnosis. In addition, similar to BCR-ABL monitoring in chronic myeloid leukemia (CML), the concept of tracking clonal or subclonal evolution can also be implemented in solid tumors, by serially tracking identified genomic alterations in cfDNA that are deemed relevant to the targeted treatment/s administered, while studying disease clonal evolution and potentially identifying emerging mechanisms of resistance. We therefore proceeded to evaluate the clinical utility of cfDNA by sequencing 159 serial plasma cfDNA samples obtained from 39 patients receiving targeted therapies on early phase clinical trials.
Material and Methods
Patients and sample collection
Tumor biopsies and plasma samples were prospectively collected from patients starting a Phase I clinical trial referred to the Drug Development Unit at the Royal Marsden hospital. These patients all had late stage metastatic solid tumors and had no available active anticancer treatment options. All provided informed consent for serial tumor and blood molecular characterization, utilizing a protocol that was approved by relevant regulatory and independent ethics committees. Twenty milliliters of blood was collected weekly during the first month, and monthly until disease progression in CPT tubes (BD, Oxford, UK); plasma was extracted and frozen within two hours. Formalin-fixed, paraffin embedded (FFPE) blocks or fresh tissue of diagnostic tumor tissue were obtained and reviewed by a pathologist to confirm diagnosis and tumor content. Macrodissection was performed on FFPE tumor tissue to enrich the tumor content to greater than 70%. Tumor DNA was first sequenced utilizing the MiSeq platform (Illumina); this sequencing impacted patient allocation to Phase I trials through tumor molecular stratification. Subsequently, patients with at least one mutation identified in their tumor by MiSeq analyses and who had completed at least 2 courses of investigational drug administration were selected. Matched patient tumor and multiple serial plasma samples from these selected patients were then sequenced on the PGM Ion Torrent platform (Life Technologies); we had previously shown that the MiSeq and PGM sequencing platforms provided highly concordant results in a range of solid tumors (1).
DNA Extraction and Quantification
FFPE or fresh tissue DNA was extracted using the FFPE Tissue DNA kit (Qiagen) and circulating plasma DNA was extracted from 2ml of plasma using the SnoMag™ Circulating DNA kit (Snova Biotechnologies) according to the manufacturer’s protocols. Buccal swab DNA was extracted, when necessary, using the QiaAmp DNA kit (Qiagen). The eluted DNA was quantified using the Quant-iT high sensitivity Picogreen double stranded DNA (dsDNA) Assay Kit (Invitrogen, CA, USA), according to the manufacturer’s recommendations. The concentration of cfDNA per milliliter of plasma at each timepoint analyzed was calculated.
Sequencing assay
Two commercialized and already validated panels, the Ion AmpliSeq™ Cancer Hotspot Panel v2 and Ion AmpliSeq™ Colon and Lung Cancer Panels were used. The Ion AmpliSeq™ Cancer Hotspot Panel v2 has been designed to amplify 207 amplicons covering approximately 2,800 COSMIC mutations from 50 oncogenes and tumor suppressor genes while the Ion AmpliSeq™ Colon and Lung Cancer Panels evaluated the regions of 22 genes implicated in colon and lung cancers. Three nanograms of circulating plasma DNA were used to generate these libraries using the Ion Ampliseq library preparation kit v2.0 (Life Technologies) according to the manufacturer’s protocol. The barcoded libraries were quantified using an Agilent 2100 Bioanalyser and Qubit 2.0 Fluorometer TM (Life Technologies) and then diluted to a final concentration of 10 pM for template preparation using the OneTouch 2 instrument and Ion One Touch Template kit v2 (Life Technologies). The resulting pooled libraries were quality control checked using the Ion Sphere quality control Kit on the Qubit 2.0 Fluorometer™. Libraries passing QC were then sequenced on the PGM Ion Torrent (Life Technologies) using a PGM 200 sequencing kit v2 and 318 Chip v2. A maximum of 16 libraries were pooled to achieve at least 500X coverage per target amplicon.
Inter-run and Intra-run Assay Reproducibility for cfDNA sequencing
Inter-run assay reproducibility was assessed by repeating the sequencing of 3 libraries prepared from 3 plasma DNA samples extracted from 3 different patients with 3 different tumor types (colon, bladder and breast cancers), across 2 independent multiplexed sequencing runs. To test intra-run assay reproducibility, libraries prepared from the same patient plasma DNA sample, indexed with 3 different barcodes, were multiplexed and sequenced on the same IonChip to compare the allele frequency.
Digital Polymerase Chain Reaction (ddPCR)
To validate cfDNA mutations ddPCR was performed on a QX100 droplet digital PCR system (Bio-Rad) with primers and probes (Supplementary Data 1) at a final concentration of 900nM primers and 250nM probes. Primers and probes for PIK3CA c.1624G>A (E542K), c.1633G>A (E545K) and TP53 c.743G>A (R248Q) were designed in-house using primer 3 (http://primer3.ut.ee/) and ordered as individual primers and probes from Life Technologies. Assays for KRAS mutant analysis were obtained from Bio-Rad: c.35G>T G12V: dHsaCP2000025 and WT: dHsaCP2000026; c.34G>T G12C: dHsaCP2000007 and WT: dHsaCP2000008 and c.35G>A G12D: dHsaCP2000001 and WT: dHsaCP2000002. PCR reactions were prepared with 3ng of DNA with 10ul of 2× ddPCR Supermix for probes (Bio-Rad 186-3010) in a total volume of 20ul, and partitioned into ~14,000 droplets per sample in a QX100 droplet generator according to the manufacturer’s instructions. Emulsified PCR reactions were run on a 96 well plate on a G-Storm GS4 thermal cycler as shown below:
-
A)
PIK3CA: 95 °C for 10 min followed by 45 cycles of 95 °C for 15 sec and 63.1 °C for 60 sec followed by 10 min incubation at 98 °C
-
B)
TP53: 95 °C for 10 min followed by 45 cycles of 95 °C for 15 sec and 60 °C for 60 sec followed by 10 min incubation at 98 °C
-
C)
KRAS G12V: 95 °C for 10 min followed by 50 cycles of 95 °C for 15 sec and 60.3 °C for 60 sec followed by 10 min incubation at 98 °C
-
D)
KRAS G12C: 95 °C for 10 min followed by 45 cycles of 95 °C for 30 sec and 54 °C for 60 sec followed by 10 min incubation at 98 °C
-
E)
KRAS G12D: 95 °C for 10 min followed by 50 cycles of 95 °C for 15 sec and 61.7 °C for 60 sec followed by 10 min incubation at 98 °C
The temperature ramp increment was 2.5 °C /sec for all steps. Plates were read on a Bio-Rad QX100 droplet reader using QuantaSoft v1.4.0.99 software from Bio-Rad to assess the number of droplets positive for mutant DNA, WT DNA, both or neither. At least two negative control wells with no DNA were included in every run. The concentration of mutant DNA (copies of mut DNA per droplet) is estimated from the Poisson distribution. Number of mutated copies per droplet Mmut = −ln (1−(nmut/n)), where nmut=number of droplets positive for mutant-FAM probe and n=total number of droplets. The DNA concentration in the reaction is estimated as follows MDNAconc = −ln (1−(nDNAcon/n)), where nDNAconc=number of droplets positive for mutant-FAM probe and WT-VIC probe and n=total number of droplets. The Fraction Mutant = Mmut/ MDNAconc; All other statistical analyses were two sided and performed with GraphPad Prism version 5.0 or Microsoft Excel.
Sequencing Data Analysis and variant calling
FASTQs were aligned to the human genome (hg19) and point mutations were identified with the Torrent Suite Software v3.0 and the Ion Torrent Variant Caller v4.0 Plugin using the somatic high stringency parameters and the targeted and hotspot pipelines. A 5% Allele Frequency (AF) threshold and 500X minimum coverage was set to report de novo mutations while 1% AF was selected to monitor previously identified and validated mutations during treatment (16). All the variants identified were established by visualizing the data through IGV 2.3 (Broad Institute).
Results
Patients and tumors analyzed
Between December 2012 and November 2013, 39 patients out of 84 screened fulfilled the study criteria. The main reasons for the exclusion of patients are shown in Supplementary Data 2. Patient demographics and treatment details are shown in Table 1. Briefly, the median age was 60 years (29-78) and the most common tumor types were colorectal, ovarian and breast cancers. ECOG performance status was 0-1 for all patients, with a median number of 2 [range: 0-8] prior lines of chemotherapy for metastatic disease. Investigational therapies administered included mainly small molecules targeting the PIK3CA-AKT-mTOR pathway (n=35/44), with 5 patients receiving 2 consecutive Phase 1 trial therapies (Supplementary Data 3). All tumors were sequenced with the Ion AmpliSeq™ Cancer Hotspot Panel v2 on the PGM Ion Torrent (Life Technologies), with 74 different mutations identified in 39 tumors, 30 of which were archival samples and 9 from fresh biopsies. The median number of detected molecular alterations per tumor was 2 (range 1-5). TP53, PIK3CA and KRAS were the most frequently mutated genes detected, with 29 (40%), 15 (21%) and 13 (18%) mutations identified, respectively (Tables 2 and 3).
Table 1.
Characteristics of the population (n=39) and targets of Phase I trial drug administered.
|
ECOG Performance Status
| ||
| 0 | 17 | |
| 1 | 22 | |
|
| ||
|
Gender
| ||
| Male | 18 | |
| Female | 21 | |
|
| ||
|
Histology
| ||
| Colorectal cancer | 12 | |
| Ovarian cancer | 9 | |
| Breast cancer | 7 | |
| Bladder cancer | 3 | |
| Glioblastoma | 2 | |
| Lung cancer adenocarcinoma | 2 | |
| Endometrial cancer | 1 | |
| Renal cell cancer | 1 | |
| Penile cancer | 1 | |
| Melanoma | 1 | |
|
| ||
|
Previous treatment
| ||
| Chemotherapy | 38 | |
| Median lines of treatment (Range) | 2 [0-8] | |
| Radiotherapy | 15 | |
| Hormonal therapy | 4 | |
|
| ||
|
Phase I trials targets
| ||
| PI3K-AKT-MTOR pathway | ||
| PI3K inhibitor | 10 | |
| PI3K inhibitor + MEK inhibitor ( | 4 | |
| TOR kinase inhibitor | 8 | |
| TOR kinase inhibitor + Taxane | 3 | |
| AKT inhibitor | 3 | |
| MEK pathway | ||
| MEK inhibitor + IGF-IR inhibitor | 5 | |
| MEK/RAF inhibitor | 2 | |
| Other | ||
| PARP inhibitor | 2 | |
| PIM kinase inhibitor | 2 | |
| Analog of Oleic Acid | 2 | |
| Androgen receptor inhibitor | 1 | |
| AGC kinase inhibitor | 1 | |
| Folate inhibitor | 1 | |
Table 2.
Comparison of mutations identified in archival tumor tissue (FFPE) and in plasma with the PGM platform
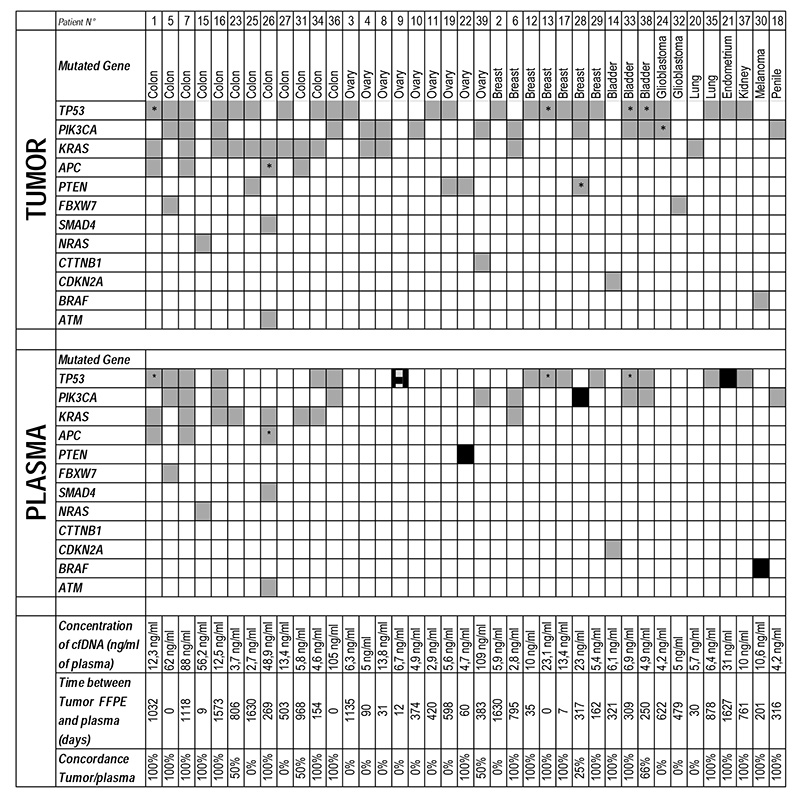
two different mutations for the same gene were identified in the sample.
 mutation identified at C1D1;
mutation identified at C1D1;
 mutation identified in plasma at C1D1 but absent in tumor tissue;
mutation identified in plasma at C1D1 but absent in tumor tissue;
 mutation identified only in plasma after C1D1.
mutation identified only in plasma after C1D1.
Table 3.
Details of mutations identified in tumor tissue and in plasma at Cycle 1 Day 1 on the PGM platform. ND: not detected.
| TUMOR | PLASMA | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PGM | PGM | ||||||||
| N° | Tumor Type | Gene | Mutation | % | Coverage | Gene | Mutation | % | Coverage |
| #1 | Colorectal cancer | KRAS | G12V | 46 | 820 | KRAS | G12V | 19 | 630 |
| TP53 | R248Q | 31 | 991 | TP53 | R248Q | 9 | 1997 | ||
| TP53 | R158fs*11 | 50 | 825 | TP53 | R158fs*11 | 16 | 1008 | ||
| APC | A1492fs*15 | 44 | 989 | APC | A1492fs*15 | 15 | 746 | ||
| #2 | Breast cancer | TP53 | H193Y | 37 | 1008 | TP53 | H193Y | ND | 3187 |
| #3 | Ovarian cancer | TP53 | S96fs*53 | 87 | 368 | TP53 | S96fs*53 | ND | 879 |
| #4 | Ovarian cancer | KRAS | G12D | 20 | 1555 | KRAS | G12D | ND | 473 |
| PIK3CA | E545K | 23 | 1717 | PIK3CA | E545K | ND | 678 | ||
| #5 | Colorectal cancer | FBXW7 | S582L | 31 | 1744 | FBXW7 | S582L | 46 | 1998 |
| PIK3CA | E542K | 31 | 1010 | PIK3CA | E542K | 58 | 1994 | ||
| TP53 | R282W | 31 | 1447 | TP53 | R282W | 43 | 904 | ||
| #6 | Breast cancer | KRAS | G12S | 43 | 533 | KRAS | G12S | 3 | 1519 |
| PIK3CA | N1044S | 39 | 516 | PIK3CA | N1044S | 9.1 | 986 | ||
| #7 | Colorectal cancer | PIK3CA | Q546K | 26 | 754 | PIK3CA | Q546K | 31 | 6509 |
| TP53 | Y234* | 28 | 640 | TP53 | Y234* | 57 | 8395 | ||
| KRAS | G12C | 19 | 772 | KRAS | G12C | 36 | 1723 | ||
| APC | E1374* | 30 | 712 | APC | E1374* | 33 | 2116 | ||
| #8 | Ovarian cancer | PIK3CA | E545K | 16 | 6676 | PIK3CA | E545K | ND | 2333 |
| KRAS | G12D | 29 | 6801 | KRAS | G12D | ND | 522 | ||
| #9 | Ovarian cancer | TP53 | Y220C | ND | 700 | TP53 | Y220C | 5.4 | 1162 |
| #10 | Ovarian cancer | PIK3CA | E542K | 40 | 7169 | PIK3CA | E542K | ND | 3895 |
| #11 | Ovarian cancer | TP53 | R175H | 67 | 255 | TP53 | R175H | ND | 858 |
| #12 | Breast cancer | TP53 | H179R | 90 | 353 | TP53 | H179R | 13.6 | 655 |
| #13 | Breast cancer | TP53 | R175H | 23 | 175 | TP53 | R175H | 7.6 | 1799 |
| TP53 | D281H | 17 | 281 | TP53 | D281H | 2 | 1756 | ||
| #14 | Bladder cancer | CDKN2A | H83Y | 76 | 212 | CDKN2A | H83Y | 4 | 1626 |
| #15 | Colorectal cancer | NRAS | G12S | 37 | 2662 | NRAS | G12S | 51 | 1088 |
| #16 | Colorectal cancer | KRAS | G12D | 25 | 907 | KRAS | G12D | 7.9 | 665 |
| PIK3CA | Q546R | 24 | 1130 | PIK3CA | Q546R | 13 | 1785 | ||
| TP53 | G244D | 33 | 930 | TP53 | G244D | 6,7 | 2100 | ||
| #17 | Breast cancer | TP53 | R342fs*2 | 30 | 238 | TP53 | R342fs*2 | 23 | 1214 |
| #18 | Penile cancer | PIK3CA | E545K | 9,1 | 1483 | PIK3CA | E545K | 4.9 | 1763 |
| #19 | Ovarian cancer | TP53 | A138P | 64 | 446 | TP53 | A138P | ND | 2413 |
| PTEN | EXON3-4S | 26 | 899 | PTEN | EXON3-4S | ND | 1237 | ||
| #20 | Lung adenocarcinoma | KRAS | G12C | 57 | 2084 | KRAS | G12C | ND | 849 |
| #21 | Endometrial cancer | TP53 | P190L | 89 | 1213 | TP53 | P190L | <1% | 7044 |
| #22 | Ovarian cancer | PTEN | L70fs*4 | 11,5 | 1795 | PTEN | L70fs*4 | ND | 546 |
| #23 | Colorectal cancer | KRAS | G12V | 40 | 1705 | KRAS | G12V | 4.4 | 1365 |
| TP53 | R175H | 58 | 151 | TP53 | R175H | ND | 686 | ||
| #24 | Glioblastoma | TP53 | K132N | 65 | 689 | TP53 | K132N | ND | 37094 |
| PIK3CA | E542K | 33 | 954 | PIK3CA | E542K | ND | 33631 | ||
| #25 | Colorectal cancer | KRAS | G12V | 43 | 2380 | KRAS | G12V | ND | 1119 |
| PTEN | V317fs* | 28 | 1938 | PTEN | V317fs* | ND | 1300 | ||
| TP53 | R273C | 65 | 2231 | TP53 | R273C | ND | 373 | ||
| #26 | Colorectal cancer | KRAS | Q61H | 75 | 5376 | KRAS | Q61H | 51 | 2960 |
| APC | S1501A | 57 | 937 | APC | S1501A | 28 | 1915 | ||
| APC | S1495fs*14 | 41 | 2213 | APC | S1495fs*14 | 26 | 1790 | ||
| SMAD4 | G386R | 49 | 1171 | SMAD4 | G386R | 26 | 280 | ||
| ATM | R337C | 42 | 1179 | ATM | R337C | 18 | 781 | ||
| #27 | Colorectal cancer | KRAS | G12V | 40 | 1897 | KRAS | G12V | ND | 4627 |
| TP53 | P151S | 45 | 247 | TP53 | P151S | ND | 2776 | ||
| #28 | Breast cancer | PIK3CA | E545K | 16 | 1183 | PIK3CA | E545K | ND | 2327 |
| PTEN | L325R | 22 | 851 | PTEN | L325R | ND | 1661 | ||
| PTEN | E235fs*2 | 11 | 922 | PTEN | E235fs*2 | ND | 1495 | ||
| TP53 | N210fs*54 | 39 | 524 | TP53 | N210fs*54 | ND | 3450 | ||
| #29 | Breast cancer | TP53 | E171* | 70 | 240 | TP53 | E171* | 6.5 | 740 |
| #30 | Melanoma | BRAF | V600E | 58 | 1395 | BRAF | V600E | ND | 2238 |
| #31 | Colorectal cancer | KRAS | G12D | 8 | 1845 | KRAS | G12D | 3.4 | 567 |
| APC | R1114* | 9 | 3491 | APC | R1114* | ND | 685 | ||
| #32 | Glioblastoma | FBXW7 | R484K | 6 | 439 | FBXW7 | R484K | ND | 5600 |
| #33 | Bladder cancer | PIK3CA | E545K | 28 | 3253 | PIK3CA | E545K | 8.5 | 778 |
| TP53 | E180K | 12 | 899 | TP53 | E180K | 6.1 | 836 | ||
| TP53 | R175H | 19 | 899 | TP53 | R175H | 8 | 840 | ||
| #34 | Colorectal cancer | KRAS | G12D | 33 | 769 | KRAS | G12D | 4.5 | 866 |
| TP53 | R175H | 29 | 879 | TP53 | R175H | 16 | 1119 | ||
| #35 | Lung adenocarcinoma | TP53 | G154V | 16 | 386 | TP53 | G154V | 4.6 | 1080 |
| #36 | Colorectal cancer | PIK3CA | H1074R | 56 | 819 | PIK3CA | H1074R | 40 | 1503 |
| TP53 | R175H | 57 | 205 | TP53 | R175H | 60 | 1287 | ||
| #37 | Renal cell cancer | TP53 | E285K | 51 | 226 | TP53 | E285K | 16.3 | 632 |
| #38 | Bladder cancer | PIK3CA | E545K | 56 | 2266 | PIK3CA | E545K | 9.4 | 1024 |
| TP53 | D281N | 44 | 950 | TP53 | D281N | 3.2 | 724 | ||
| TP53 | P278L | 44 | 951 | TP53 | P278L | ND | 732 | ||
| #39 | Ovarian cancer | CTNNB1 | S33C | 24 | 678 | CTNNB1 | S33C | ND | 1425 |
| PIK3CA | N345K | 41 | 946 | PIK3CA | N345K | 7.8 | 4139 | ||
Identification of cfDNA genomic alterations in plasma
A total of 159 plasma samples were sequenced from 39 patients. The mean sequencing coverage across the experiments was 1,685X. Plasma sequencing of baseline samples identified 44 different mutations in 23 patients (59%) (Table 3). By sequencing plasma samples at later time points, 4 additional patients were identified as having cfDNA mutations resulting in cfDNA mutations being detected in 27 of 39 (69%) patients. TP53, PIK3CA and KRAS were the commonest mutated genes identified in plasma, with 18 (42%), 9 (21%) and 8 (16%) different mutations, respectively. The median number of mutations detected in cfDNA was 2 (range 1-5) and the mean detected mutation allele frequency (AF) was 15% (range 1-60%). Overall, all cfDNA mutations had been previously identified in the matched tumor tissue except for a TP53 Y220C mutation (AF 5%) detected de novo in the plasma specimen of a patient with advanced ovarian cancer. The re-analysis of the MiSeq data on the archived FFPE tumor DNA and sequencing of germline DNA confirmed the absence of this mutation in tumor and its somatic origin.
The plasma cfDNA next generation sequencing results were cross validated with a different platform by performing digital PCR (ddPCR) assays on 19 random plasma samples with known TP53, KRAS or PIK3CA mutations in tumors (Supplementary Data 4). All 11 mutations identified in this set of plasma samples on the PGM platform (AF range 1-67.1%) were confirmed by ddPCR assay. Out of the 8 negative samples on the PGM platform, 5 were negative by ddPCR assay and 3 had a PIK3CA mutation identified (E545K or E542K) with an AF of less than 1% apart from 1 sample. AF on the two platforms showed excellent agreement correlation coefficient: 0.98; CI 95% [0.94-0.99]), p<0.0001.
In 4 patients, although not identified in cfDNA at C1D1 and despite good coverage of the genes of interest, 4 mutations detected in tumor tissue were identified in plasma cfDNA later during the course of their disease: one PIK3CA E545K mutation (AF, 5%) in a patient with breast cancer; one PTEN L70fs*4 mutation (AF, 10.6%) in a patient with ovarian cancer; and one TP53 P190L mutation (AF, 7%) in a patient with endometrial cancer. Interestingly, despite coverage of 2,238X, a BRAF V600E mutation was not identifed at the initial time point in a patient with BRAF V600E mutated melanoma who had discontinued vemurafenib for progression immediately before study entry. The patient received a RAF/MEK inhibitor in a Phase I trial, progressing after 2 cycles of treatment, with cfDNA sequenced at study progression confirming a BRAF V600E mutation at an AF of 5%.
Clinical factors associated with the detection of mutations in cfDNA
The mean cfDNA concentration at C1D1 was higher for patients with a cfDNA mutation identified compared to those without a mutation detected: 26.4ng/ml versus 9ng/ml (p<0.05). However, oncogenic mutations were identified in the plasma of 4 patients having a cfDNA concentration as low as 5ng/ml of plasma. Patients with colon/breast cancer or with lung or liver metastasis were more likely to have plasma cfDNA mutations detected.
Longitudinal monitoring of somatic genomic alterations in plasma cfDNA during targeted therapy
We performed serial monitoring of the detected genomic alterations in 159 different plasma samples from 39 patients. Critically, the intra and inter-run variability of the NGS assay in a sample was less than 12.5% (Supplementary 5). All the serial plasma samples of patients without cfDNA mutation identified at the beginning of treatment were also sequenced to track the potential emergence of mutations. For the 23 patients with identified cfDNA mutations at C1D1, the subsequent monitoring of mutation AF in consecutive plasma samples identified changes in AF in keeping the selective pressure generated by the targeted drug administration (Figure 1, 2, and Supplementary Data 6). Mutation analysis was utilized to match each patient to trial drugs targeting the identified genomic aberrations when possible; 13/23 patients with plasma cfDNA mutations had a targeted drug administered matching their detected profile. These were classified as “AF decrease” or “AF increase”.
Figure 1.


Monitoring of somatic genomic alterations in plasma during targeted therapy. Allele frequency (AF) of the identified mutations is represented on the left Y-axis while the sum of the target lesions on CT scan is represented on the right Y-axis. SD: stable disease according to RECIST criteria; PD: progressive disease. The colored box depicts the time on treatment. The second graph depicts the evolution of cfDNA concentration during this time frame in ng/ml of plasma. A. Patients with decreasing AF during therapy. B. Patients with increasing AF during therapy. C. Patients with mixed or discordant AF changes during therapy.
Figure 2.

Serial monitoring of cfDNA samples with multiple mutations. A. Samples with different mutations, and different AF, with similar trends during the course of disease. B. Patients with dominant mutations with AF with differing trends during treatment. These findings suggest the presence of heterogeneous clonal responses to drug administration.
Patients with “Allele Frequency Decrease” by plasma DNA NGS (Figure 1A)
Patient #1
A 76-year-old female patient with advanced colorectal adenocarcinoma had KRAS (G12V), TP53 (R248Q and R158fs*) and APC (A1492fs*) mutations in tumor tissue that were identified concurrently in cfDNA with an AF of 19.9, 9, 16, 15.5%respectively. She was allocated to a trial involving a combination of a PI3K inhibitor and MEK inhibitor. After 63 days of targeted drug administration, the AF of plasma cfDNA mutations fell to very low levels (4.1, 2, 1.3, 2% respectively) suggesting a clonal regression. Concurrent CT scan evaluation showed stable disease that was maintained for 119 days. At progression by CT scans, AF frequency in plasma increased to 18, 5.8, 9 and 10%respectively. After discontinuation of the trial drugs, the cfDNA mutation AF increased rapidly to 21.6, 55.7, 26.7 and 56.3%, respectively.
Patient #18
A 53-year-old patient with advanced penile cancer had a PIK3CA E545K mutation with an AF of 9.1% in tumor biopsy DNA. He was allocated to a trial of a PI3K inhibitor; after 40 days of treatment, this cfDNA PIK3CA E545K mutation decreased from 4.9% to 1% levels. Concurrently, his initial post-treatment CT scan showed stable disease by RECIST, but CT performed at day 75 showed cavitation of lung metastasis and a slightly less prominent lung metastasis, suggesting responding disease (Figure 2A). At day 68 however, the PIK3CA E545K mutation AF increased to 8.7% and the patient progressed radiologically at day 131. After PI3K inhibitor discontinuation, the plasma PIK3CA E545K AF rapidly increased to 18.6%, despite the administration of a TOR kinase inhibitor.
Patient #23
A 40-year-old female patient with KRAS (G12V) mutated colorectal cancer was treated with combined MEK and IGF1-R inhibition. The mutation was detectable in plasma at an AF level of 4.4% at initiation of treatment, but became undetectable after 28 days of therapy. She discontinued study after 139 days of treatment due to drug toxicity without any reappearance of the KRAS mutation in plasma.
Patient # 39
A 38-year-old female patient with clear cell ovarian cancer had PIK3CA (N345K) and CCTNB1 (S33C) mutations identified in tumor tissue. She received an AKT inhibitor; the PIK3CA mutation detected in plasma dropped from 7.8% to 3.1% after 45 days of treatment when CT scan revealed stable disease. After 66 days of AKT inhibition, the PIK3CA mutation became undetectable in plasma and the patient was still on trial 87 days after starting treatment with ongoing stable disease.
Patients with “AF increase” by plasma DNA NGS (Figure 1B, 2, Supplementary Data 6)
Patient #5 (Figure 2A)
A 67-year-old male patient with PIK3CA, FBXW7 and TP53 mutated colorectal cancer received a PI3K inhibitor within a phase I trial. The three mutations were identified in cfDNA and showed the same pattern of clonal evolution with an increasing AF from 58 to 72%, 46 to 60%, and 43 to 58% for the PIK3CA, FBXW7 and TP53 mutations, respectively, after 42 days of PI3K inhibition. The patient discontinued trial treatment shortly after for disease progression.
Patient #29(Figure 1B)
A 58-year-old patient with TP53 mutated breast cancer received a TORC1/2 inhibitor. At initiation of treatment, the mutation was at 6.5% and subsequently, the mutation increased to 37% at disease progression, leading to treatment discontinuation.
Patients with ‘discordant responses’ (Figure 1C)
Two patients had discordant responses detected radiologically. The first patient (Patient #6) had breast cancer with bone metastases, ascites, pleural and pericardial effusions. The tumor harbored PIK3CA N1044S and KRAS G12S mutations with PIK3CA and KRAS mutation being detected in plasma samples. She received a PI3K inhibitor and the cfDNA PIK3CA mutation decreased from 9.1% to undetectable levels after 76 days of therapy. Concurrently, the CT scan showed an increase in her right pleural effusion, and worsening pericardial effusion and ascites. The patient discontinued trial treatment and died shortly afterwards.
The second patient (Patient #16) had metastatic colorectal cancer with bone metastases and KRAS G12D, PIK3CA Q546R and TP53 G244D mutations were detected in tumor tissue; all were also identified in cfDNA. After administration of a RAF/MEK inhibitor for 29 days, an early MRI evaluation undertaken due to worsening symptoms of pain demonstrated an increase in the extent of epidural disease at T11 with cord impingement and progression of right femoral neck disease with suspicion of an incomplete pathological fracture. The patient thus discontinued trial therapy for disease progression. At this point, the monitoring of his plasma cfDNA mutations showed declining KRAS G12D AF, which decreased from 34.1% to 21.7 %; the PIK3CA Q546R also decreased from 41.3% to 19.5% and the TP53 G244D from 45.4 to 16.7%, with his CEA tumor marker also decreasing from 124 at C1D1 to 82 at C2D1 despite clinical disease progression.
Potential clonal evolution evidence by serial monitoring of multiple mutations within a sample
This assay permitted the multiplexed monitoring of several mutations concurrently in a single reaction. Multiple mutations were identified in 10 patients; these generally showed similar trends in serial plasma samples. However, in some cases, we observed discordant mutation AF changes during the course of targeted drug administration, suggesting disparate clonal evolution of more than one clone (Figure 2).
cfDNA concentration and AF dynamic changes quantification during targeted therapy and RECIST criteria comparison
The monitoring of total cfDNA concentration during therapies did not show a clear correlation of cfDNA dynamics and response to treatment (cfDNA concentrations are also depicted on the right side of AF frequency graphs).
For the 23 patients with identified cfDNA mutations at C1D1, we correlated the mutation AF changes after C2D1 with time to progression by RECIST assessment. These patients could be classified into two separate groups, as defined by cfDNA mutation AF changes: Group A with a decreasing mutation AF after C2D1 of more than 30% compared to baseline (n=9) and Group B for the remaining patients (n=14) (Supplementary Data 7). Time to progression for Group A versus Group B was significantly different: 111 versus 55 days respectively (p=0.0165). Of the 9 patients in Group A, 7 had stable disease (SD) by RECIST assessment, while 2 had progressing disease (PD). This was in contrast to patients in Group B, where 12 had PD and 2 had SD by RECIST assessment after 2 courses of treatment. Median time to progression (TTP) for patients in Group A with a declining cfDNA mutation AF, and that of patients with RECIST SD were similar (111 versus 111 days, p=0.728). Median TTP for patients in Group B with rising cfDNA mutation AF was also similar to that of patients with RECIST PD (55 versus 42 days, p=0.35).
Discussion
This study demonstrates the clinical utility of cfDNA for the monitoring of solid tumor responses to targeted drug administration. To our knowledge, this is the first study reporting serial monitoring of cfDNA in the context of Phase 1 trials of targeted drugs. We hypothesize that these findings may have important implications for the future of drug development, with regards to both predictive and response biomarker studies. Currently, molecular stratification relies on primary tumor or metastatic biopsy evaluation although several studies have demonstrated the utility of cfDNA for genotyping tumors (17-20). These cfDNA studies have primarily interrogated a few mutation hot spots or a larger DNA input (21). Using a much broader panel, we have identified mutations in 69% of unselected patients with only 3ng of cfDNA. This approach allows potentially a rapid and cost effective molecular characterization from contemporaneous disease. These cfDNA assays can therefore have major clinical utility, as supported by the recent positive opinion of the European Medicines Agency (EMA) on the use of cfDNA for the assessment of EGFR mutation status for gefitinib administration, a landmark decision for cfDNA studies (22). Further evaluation of these findings in this pilot study in larger, and preferably randomized trials, addressing the importance of these cfDNA allele frequency dynamics are now urgently required to determine the utility of these assays as predictive and response biomarkers. These studies should also attempt to address the issue of whether different mutations arise from geographically separate metastases, by acquiring multiple fresh biopsies at the same time as cfDNA and whether different mutations can be shed into blood at different rates.
Utilizing cfDNA as a multi-purpose biomarker for drug development requires analytic validation and clinical qualification. Our studies are a critically important part of this process; we have shown impressive sensitivity for cfDNA mutation detection that is platform dependent. The ddPCR assay detects mutations in cfDNA with frequencies as low as 0.01% (8). However, this platform mandates a priori knowledge of the exact genomic aberrations to be investigated. Utilizing next generation sequencing, we have identified tumor mutations in circulating cfDNA in 69% of unselected patients with advanced cancers. These results are consistent with a recent study using the same platform (23). The specificity of this assay is key and has been described for tumor tissue with this panel and platform (16); our data confirm the robustness of this assay.
Our studies pursued a pragmatic targeted approach that utilized a relatively inexpensive assay to generate rapid results for patients. Whole genome characterization can provide more data and is likely to become more feasible with improved bioinformatics pipelines, but for cfDNA analyses may be limited by the tumor:normal cfDNA ratio as recently demonstrated in selected patients with high cfDNA (13). Whole genome analyses may, however, support the detection of key rearrangements and copy-number changes (20, 24). This, however, currently demands protracted and complex bioinformatic analyses and no consensus on the optimal bioinformatic strategies for such analyses are currently available. Validated software suites are urgently needed to harmonize such studies since different methodological approaches can impact data interpretation. Conversely, we have utilized simple and ready-to-use algorithms for data analyses with results available within 7 days of cfDNA sample acquisition. We now postulate that the pursuit of this strategy can generate vital information on actionable genomic aberrations in early clinical trials.
The monitoring of tumor response to targeted drugs is critical to anticancer drug development; we demonstrate that cfDNA analyses can generate critically important quantitative and qualitative data that can supplement imaging data acquired by CT scan and also elucidate the impact of tumor heterogeneity on drug response. To date, few studies have investigated if cfDNA can be used to evaluate treatment responses (15, 25-28); almost all involve very small cohorts or chemotherapy-based treatment. We show in 23 patients, receiving various targeted drugs on our Phase I clinical trials program, that serial cfDNA sample sequencing may generate important data on disease clone response to therapy. For example, in patient #39, the disappearance in cfDNA of the PIK3CA mutation following AKT inhibition and of the PIK3CA mutation with a small molecule PI3K inhibitor (patient #18) suggest that these drugs are impacting sensitive clones in these patients. For patient #1, combined p110α and MEK inhibition led to the nearly disappearance of cfDNA KRAS, APC and TP53 mutations. For these last 2 patients, these cfDNA “responses” were associated with radiologically stable disease with increases in the AF of these mutations emerging at radiological progression. Our data also indicate that post-treatment changes in AF may be prognostic.
Drug development in oncology remains resource intensive, with a continuing high failure rate despite robust clinical data. Moreover, despite seemingly appropriate patient selection, most drugs result in very limited radiologically detectable antitumor activity when administered as single agents. Our studies suggest that imaging studies do not provide the whole story; we show that mutation AF decrease in plasma can be associated with disease progression by conventional imaging. These observations raise the possibility that the clearance of sensitive clone/s in patients without an imaged response may be deriving benefit from the drug undergoing evaluation. Such data may be vital for the optimal future development of such agents in drug combinations and understanding drug resistance. Conversely, the fact that most major mutations were concordant between archival tumor tissue and cfDNA, with a median time between acquisition of the two samples of 260 days [Range: 0-1627], indicates stable ‘truncal’ mutations during the course of these diseases.
Overall, therefore, cfDNA monitoring does allow the study of tumor clone dynamics as seen in patient #13 on our study; this breast cancer patient had 2 different TP53 mutations identified in tumor tissue and cfDNA. During the course of treatment, the TP53 R175H AF increased 10-fold, while the TP53 D281H remained unchanged in cfDNA. These data were associated with radiological findings suggesting a mixed response on CT scan with some progressing lesions, while others remained stable for more than 150 days of follow-up. These studies indicate that serial plasma ctDNA monitoring will provide critical important molecular information on such mixed radiological responses that can guide patient treatment and help us understand disease biology. Moreover, our studies have identified different scenarios. In some patients, after the disappearance of previously identified cfDNA mutations with treatment, mutations returned at radiological progression. In other patients, the ‘cleared’ mutation remained undetectable at disease progression. Critically, AF increased rapidly in some patients on targeted drug cessation, supporting the hypothesis that this therapeutic pressure was inhibiting specific clones despite disease progression, supporting the continued administration of such drugs (29). It is apparent that such cfDNA studies will provide us with greater insights into the study of tumor clone suppression.
Conclusion
Tumor cell genomic alterations can now be evaluated by sequencing cfDNA. This biomarker has important clinical utility for the delivery of more precise cancer therapy through the study of tumor cell clonal dynamics in early phase clinical trials. Our data support the further investigation of undertaking next generation sequencing in cfDNA as a prognostic, predictive and response biomarker.
Supplementary Material
Translational Relevance.
Drug development in oncology remains resource intensive with a continual high failure rate. An accurate prior stratification of patients is clearly needed, while monitoring tumor response to targeted drugs is critical. Tumor cell genomic alterations can now be evaluated by genotyping circulating cell free DNA (cfDNA). Incorporating cfDNA sequencing for patient selection and for subsequent monitoring of response to targeted drug administration can expedite patient molecular stratification and enhance our understanding of clonal changes after targeted therapy. This article demonstrates the feasibility of this approach in the context of developmental therapeutics in oncology. Furthermore, it appears that serial cfDNA sample sequencing can generate important data in populations treated on Phase I trial of targeted therapies. This biomarker has potential clinical utility to monitor the delivery of targeted therapies.
Acknowledgements
The authors were supported by a Cancer Research UK Centre grant, an Experimental Cancer Medical Centre (ECMC) grant from Cancer Research UK and the Department of Health (Ref: C51/A7401). JSF was funded by an ESMO Georges Mathé Translational Research Fellowship and is also supported by the Nuovo Soldati foundation and “La Fondation de France”. Raquel Perez-Lopez is a PhD candidate at the Department of Medicine, Universidad Autonoma de Barcelona (Spain). R.F was supported by The Wellcome Trust (Clinical PhD Programme). G.A. was supported by a Cancer Research UK Clinician Scientist Fellowship. D.R. and D.L. were the recipient of a grant from the Spanish Medical Oncology Society “BECA SEOM para la Investigación Traslacional en el Extranjero”. JM fellowship is funded by the Medical Research Council and Prostate Cancer UK / Movember Foundation. TAY is the recipient of grants from the Academy of Medical Sciences and the British Lung Foundation. The authors acknowledge NHS funding to the Royal Marsden NIHR Biomedical Research Centre.
References
- 1.Ong M, Carreira S, Goodall J, Mateo J, Figueiredo I, Rodrigues DN, et al. Validation and utilisation of high-coverage next-generation sequencing to deliver the pharmacological audit trail. British journal of cancer. 2014;111:828–36. doi: 10.1038/bjc.2014.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andre F, Bachelot T, Commo F, Campone M, Arnedos M, Dieras V, et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER) Lancet Oncol. 2014;15:267–74. doi: 10.1016/S1470-2045(13)70611-9. [DOI] [PubMed] [Google Scholar]
- 3.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. The New England journal of medicine. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. The New England journal of medicine. 2013;368:2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 5.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C. Intratumor heterogeneity: seeing the wood for the trees. Science translational medicine. 2012;4:127ps10. doi: 10.1126/scitranslmed.3003854. [DOI] [PubMed] [Google Scholar]
- 7.Zhao B, Hemann MT, Lauffenburger DA. Intratumor heterogeneity alters most effective drugs in designed combinations. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:10773–8. doi: 10.1073/pnas.1323934111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diaz LA, Jr., Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32:579–86. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Science translational medicine. 2012;4:136ra68. doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- 10.Perkins G, Yap TA, Pope L, Cassidy AM, Dukes JP, Riisnaes R, et al. Multi-purpose utility of circulating plasma DNA testing in patients with advanced cancers. PloS one. 2012;7:e47020. doi: 10.1371/journal.pone.0047020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, Miranda S, et al. Tumor clone dynamics in lethal prostate cancer. Science translational medicine. 2014;6:254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, Nuciforo P, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2014;25:1729–35. doi: 10.1093/annonc/mdu239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–12. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 14.Sakai K, Horiike A, Irwin DL, Kudo K, Fujita Y, Tanimoto A, et al. Detection of epidermal growth factor receptor T790M mutation in plasma DNA from patients refractory to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer science. 2013;104:1198–204. doi: 10.1111/cas.12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. The New England journal of medicine. 2013;368:1199–209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 16.Singh RR, Patel KP, Routbort MJ, Reddy NG, Barkoh BA, Handal B, et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. The Journal of molecular diagnostics: JMD. 2013;15:607–22. doi: 10.1016/j.jmoldx.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Diaz LA, Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–40. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, Powers P, et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:3462–9. doi: 10.1158/1078-0432.CCR-11-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Science translational medicine. 2012;4:162ra54. doi: 10.1126/scitranslmed.3004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lebofsky R, Decraene C, Bernard V, Kamal M, Blin A, Leroy Q, et al. Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Molecular oncology. 2015;9:783–90. doi: 10.1016/j.molonc.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. http://www.astrazeneca.com/Media/Press-releases/Article/20142609--iressa-receives-chmp-positive-opinion-to-include-blood-based-diagnostic-testing-in-european-label.
- 23.Couraud S, Vaca-Paniagua F, Villar S, Oliver J, Schuster T, Blanche H, et al. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: a proof-of-concept study from BioCAST/IFCT-1002. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:4613–24. doi: 10.1158/1078-0432.CCR-13-3063. [DOI] [PubMed] [Google Scholar]
- 24.Heitzer E, Ulz P, Belic J, Gutschi S, Quehenberger F, Fischereder K, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome medicine. 2013;5:30. doi: 10.1186/gm434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maier J, Lange T, Kerle I, Specht K, Bruegel M, Wickenhauser C, et al. Detection of mutant free circulating tumor DNA in the plasma of patients with gastrointestinal stromal tumor harboring activating mutations of CKIT or PDGFRA. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:4854–67. doi: 10.1158/1078-0432.CCR-13-0765. [DOI] [PubMed] [Google Scholar]
- 26.Oxnard GR, Paweletz CP, Kuang Y, Mach SL, O’Connell A, Messineo MM, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:1698–705. doi: 10.1158/1078-0432.CCR-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–90. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hyman DM, Diamond EL, Vibat CR, Hassaine L, Poole JC, Patel M, et al. Prospective Blinded Study of BRAFV600E Mutation Detection in Cell-Free DNA of Patients with Systemic Histiocytic Disorders. Cancer discovery. 2014 doi: 10.1158/2159-8290.CD-14-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campone M, Juin P, Andre F, Bachelot T. Resistance to HER2 inhibitors: is addition better than substitution? Rationale for the hypothetical concept of drug sedimentation. Critical reviews in oncology/hematology. 2011;78:195–205. doi: 10.1016/j.critrevonc.2010.04.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.