

Abstract
The past several decades have seen a paradigm shift with the integration of evolutionary thinking into studying cancer. The evolutionary lens is most commonly employed in understanding cancer emergence, tumour growth and metastasis, but there is an increasing realization that cancer defences both between tissues within the individual and between species have been influenced by natural selection. This special issue focuses on discoveries of these deeper evolutionary phenomena in the emerging area of ‘comparative oncology’. Comparing cancer dynamics in different tissues or species can lead to insights into how biology and ecology have led to differences in carcinogenesis, and the diversity, incidence and lethality of cancers. In this introduction to the special issue, we review the history of the field and outline how the contributions use empirical, comparative and theoretical approaches to address the processes and patterns associated with ‘Peto's paradox’, the lack of a statistical relationship of cancer incidence with body size and longevity. This burgeoning area of research can help us understand that cancer is not only a disease but is also a driving force in biological systems and species life histories. Comparative oncology will be key to understanding globally important health issues, including cancer epidemiology, prevention and improved therapies.
Keywords: Peto's paradox, comparative oncology, evolution, life-history theory, cancer, modelling
1. Introduction
Cancer is a phenomenon whereby cells in multicellular organisms fail to contribute to normal tissue or organ functions, and instead divide selfishly, resulting in local tissue invasion, metastasis and often death of the individual. The vast majority of cancer research focuses on the development of a mechanistic understanding of the molecular and cellular processes that characterize particular cancers. This research has yielded two important unifying concepts. First, cancer emerges and progresses in a multistage process, typically requiring both multiple mutations (and/or epigenetic changes) and altered microenvironmental conditions. Second, all cancers have in common a set of perhaps 10 ‘hallmarks’, six originally proposed by Hanahan & Weinberg [1], plus two enabling and two emerging hallmarks that were added later [1]. These hallmarks characterize the finite set of cellular states and behaviours corresponding to emergence, progression and metastasis.
The hallmarks of cancer are defined largely from the perspective of the cancer cell: they are the features necessary for the ‘success’ of a cancer. Alternatively, we can view them from the perspective of the multicellular individual, where each hallmark reflects the failure of a set of defences that generally protect it from cancer [2,3]. In this conflict between the individual and its constituent cells, we can see two distinct adaptive processes, both driven by natural selection but showing a critically important difference: selection on cells operates at very much shorter time scale than selection on individuals. Natural selection is generally strongest at the level with the shortest generation time, and as we know, the evolution of a population of cancer cells is indeed a very effective process: once cancer is initiated it is very difficult to arrest tissue invasion and metastasis due to extremely large cancer cell population sizes and corresponding phenotypic diversity. However, selection on host genomes against cancer acting at the longer generation time of host individuals [4,5] can be effective in evolving increased defences that suppress cancer and/or promote surveillance and destruction of cancer cells if they arise, e.g. enhanced policing [6] such as in immune surveillance [7].
The evolutionary process occurring within a tumour is a critical component of cancer biology and selects cancer cells that express the hallmarks described by Hanahan & Weinberg [1,8]. Nowell [9] was the first to outline some details and consequences of this evolutionary process [9], and his perspective has been extended to incorporate recent developments [10]. With recent advances in sequencing technology, we can now study the genetics of clonal expansion in exquisite detail through genomic sequencing of tumour cells (e.g. [11,12]). There is no question that such studies of the evolution of tumour cells are very important for developing anti-tumour therapies; however, as noted above, natural selection acting at the time scale of the cell population is a very powerful process, and the evolution of resistance to therapy always remains a clinical concern.
Most cancer research focuses on the mechanisms of tumour growth, with a significant part of this research attempting to identify methods of limiting the growth of a tumour once it can be diagnosed. A parallel avenue of research for addressing the problem of cancer as a health risk is to gain enough understanding of the mechanisms of cancer suppression to stop tumours forming in the first place (e.g. [13,14]). This perspective shifts the focus on the principles that govern how these defence mechanisms evolve, and the comparative interspecific study of variation in cancer defences. Comparative studies are in their infancy, but work comparing multiple species of rodent has already proved productive [15], and there is growing interest in investigating the origins of naturally occurring cancer in dogs [16,17]. Progress towards developing the study of ‘comparative oncology’ will require a multidisciplinary approach, integrating mathematical and computational theory, empirical and comparative analysis, and controlled experimental investigation. This area is still in its youth, and below we present current knowledge and progress to date, and future research avenues.
2. Multistage carcinogenesis
Insights into the mechanism of cancer suppression began with Nordling [18], who noted that the age-specific incidence of cancer in males was consistent with a simple model of the stepwise accumulation of about seven somatic mutations [18]. He also made the prescient observations that, based on the analysis of Iversen & Arley [19], the number of mutations needed to induce cancer in mice seemed to be less than in humans, and that data on the largely non-epithelial cancers of childhood were not consistent with a model requiring seven mutations. Armitage & Doll [20] tested Nordling's [18] proposal by examining the age-specific incidence of a range of cancers in both sexes [20]. They found that the data for colon, rectum, stomach, oesophagus and pancreas were consistent with a model of 6–7 hits; however, they noted that a second group of cancers (lung, plus bladder and prostate in men, and breast, ovary, cervix and uterus in women) did not. Specifically, data for the second group tended to flatten at the older ages relative to the seven-hit model. Armitage & Doll [20] suggested that environmental and hormonal factors were playing a confounding role in this second group [20]. The seven-hit model of multistage carcinogenesis was further challenged as a general explanation for the development of cancer by Knudson's [21] insight that the onset of retinoblastoma could be fully explained by a two-hit model [21]. However, the earlier data have stood the test of time, and overall it still appears that tumours require 2–8 ‘driver’ mutations, depending upon the tissue [22]. The reasons for these tissue-specific and cancer subtype differences have never been fully resolved by the cancer research community, but such differences fit very naturally within an evolutionary paradigm. As outlined in §5a, tissues differ in their intrinsic susceptibility to cancer. As a result, once it is recognized that the suppression of cancer is an evolving trait, it is expected that tissues will differ in their anti-cancer defences [4].
3. Peto's paradox
The strongest support for the evolution of cancer suppression originates with a prediction of the multistage model that was succinctly expressed by Peto [23] when he observed that because humans are around 1000 times larger than mice and live about 30 times longer, the risk of cancer should be many orders of magnitude greater in humans [23]. However, the overall incidence of cancer in mice and humans (scaled for lifespan) is very similar [24]. This disconnect between prediction and observation has become known as Peto's paradox [4].
Is it reasonable to assume that large size and long life increase the risk of cancer? There is no question that age is the biggest risk factor for cancer in humans; one need only look at the age-specific incidence data used by Nordling [18] and Armitage & Doll [20]. But what about size? Nunney [25] noted that until recently it was uncertain if body size was actually a risk factor for cancer, but that this is no longer the case [25]. In particular, the large-scale analysis of Green et al. [26] showed conclusively that the incidence of most human cancers increases significantly with height [26]. The same appears to be true in canines with larger dogs displaying increased cancer risks compared with miniature breeds [27].
Peto assumed that increased cancer resistance of humans was a by-product of evolving to be larger and longer lived; however, given that ‘human epithelia could be described as being a billion times more cancer proof than mouse epithelia’ [23, p. 1414], we can be more specific and propose that such a huge change in the ability of cells to suppress cancer would require adaptive evolution. It is currently unknown, however, to what extent the coevolution between life-history traits (such as body size and longevity) with cancer defences occurs in single, matched alternative steps and/or large changes in defence followed by one or more sequential increases in body size/longevity [28].
Cairns [29] previously recognized the same problem in the context of the rapidly dividing epithelial cells of long-lived animals such as humans [29]. He proposed that the patterns of cell renewal in these tissues must have evolved to minimize the risk of somatic mutation and discussed several possible mechanisms. In doing so, he recognized that different tissues are subject to different evolutionary pressures, especially in comparing those typically vulnerable to higher versus lower levels of somatic mutations, e.g. those giving rise to carcinomas versus sarcomas and leukaemias.
Nunney used the multistage model to examine the evolutionary resolution of Peto's paradox across species and among tissues within a species [4,30]. This approach quantified the expected patterns. First, species that evolve to be large and/or long-lived experience an increase in the overall cancer risk that drives natural selection for additional cancer suppression (either general or tissue-specific). As a result, almost every paper discussing the evolution of cancer suppression notes that we should all be studying whales, the largest and longest lived mammalian species of all. However, the most effective way of studying this problem is to control for common ancestry through taking into account phylogeny, for example, through comparisons of longevity and body size among rodents [15]. Second, tissues within an individual that are large and/or rapidly dividing will have more robust defences (requiring more ‘hits’ to be overcome), and cancers in these tissues will tend to predominate in old age. However, such modelling does not predict how natural selection will increase cancer suppression, although an increase in the number of tumours suppressor genes regulating cell growth is one obvious possibility. Telomerase suppression is another possibility and has been found to correlate with body size in rodents [31]. Caulin & Maley [32] provided an in-depth review of the range of additional hypotheses that could potentially resolve Peto's paradox. One filter through which to view these hypotheses is that, given the magnitude of the differences in cancer susceptibility originally outlined by Peto [23], any hypotheses proposed must be consistent with an evolutionary model.
4. Evolutionary trade-offs
Recognizing that cancer suppression is an evolving trait facilitates a comparative paradigm and allows us to apply basic evolutionary principles. One of the most important evolutionary principles is the idea of a trade-off among traits, so that change often comes at a cost, i.e. a beneficial change in one trait results in a detrimental change in one or more others [33]. For example, we have already noted that as animals evolve to become larger and/or longer lived, they will become more vulnerable to cancer. However, such trade-offs can often be resolved. In this example, the increased cancer incidence will, in turn, drive natural selection for a compensatory response of enhanced cancer suppression.
Along these lines, Leroi et al. [34] described the interesting example of the platyfish, Xiphophorus maculatus [34]. This species of platyfish has evolved black spots that developed using the same mechanism that causes melanoma in a close relative (X. helleri), suggesting compensatory evolution of suppression in X. maculatus. The same authors also suggested that, in humans, some evolutionary changes in our growth may have been too recent to have been successfully compensated, resulting in the relatively high incidence of some pediatric cancers. Crespi & Summers [35] further emphasized how recent changes in the human environment could lead to a temporary evolutionary disequilibrium, but they also noted that some evolutionary conflicts do not resolve and may affect cancer rates, notably parent–offspring conflict and sexual antagonism [35]. More recently, Aktipis et al. [33] focused specifically on the important evolutionary trade-offs involved in life-history evolution, applying these ideas to the progression of a cancer [33]. However, one of the themes ripe for development is the way in which this same approach can be applied at the level of the individual to define how cancer suppression and life-history evolution interact.
5. The objectives of this issue
Animals have been evolving mechanisms to suppress cancer ever since the origin of multicellularity, making cancer a pervasive, fundamental constraint on the emergence, maintenance and diversification of multicellularity, both in terms of biological complexity and emergent life-history characteristics. As noted in §2, life-history traits such as lifespan and body size are expected to covary with the intrinsic risk of developing cancer. Recognizing that Peto's paradox is resolved through adaptive evolution allows us to apply the power of evolutionary and population genetic theory to investigate differences among taxa in their cancer biology and differences among specific cancers. It is then the hope of comparative oncology to apply and clinically translate this knowledge to effective prevention and treatment strategies in humans and veterinary care.
In this issue, we begin with a review of the ubiquitous nature of cancer [2], followed by a set of papers that illustrate how evolutionary models can be applied to understand the patterns seen in the incidence of cancer. One of the unavoidable conclusions in any discussion of Peto's paradox is that humans and mice must differ to some degree in their cancer suppression, and yet the vast majority of cancer research has focused on just these two species, often with the implicit assumption that mice and humans do not differ in their mechanisms of cancer suppression. The next set of papers reflect the growing efforts to broaden our horizons beyond these two species. This research will ultimately allow us to begin to understand the diversity of ways in which different taxa have evolved to minimize the threat of cancer, an approach that leads logically to the last set of papers that represent the first steps along the path of comparative oncology.
(a). Theoretical considerations of Peto's paradox and comparative oncology
Peto's paradox rests on the observation that every cell has some chance of generating a cancer, and that chance increases with time and numbers. In comparative oncology studies, we typically use body mass and lifespan as proxies for the underlying cell numbers and processes of mutation accumulation. In fact, age is the strongest known risk factor for most cancer. There are two reasons for this: first, DNA damage and mutations accumulate with time as a result of physical processes and exposures to carcinogens such as radiation and chemicals. Second, cell divisions are the targets of mutations. Errors occur during DNA synthesis and mitosis. In organisms with ongoing proliferation in adult tissues, such as vertebrates, age correlates with number of cell divisions and thus number of uncorrected mutations.
Inspired by a recent controversial analysis of the relationship between cancer rates, the number of stem cells and proliferation rates across organs [36], Noble et al. [37] as well as Nunney & Muir [3] point out that Peto's paradox applies to the comparison of organs within a body. Organs with more stem cells and more cell turnover ought to have a higher incidence of cancer, and in general, they do [36]. Noble et al. [37] demonstrate that grouping cancers according to anatomical site explains significantly more variation in incidence, suggesting common evolutionary paths of different tissue and organ types in achieving some degree of cancer prevention and defence. However, interesting exceptions have been described, including specific organs with both surprisingly high (e.g. gallbladder non-papillary adenocarcinoma) or low (e.g. small intestine adenocarcinoma) incidences of cancer [37]. Study of the mechanisms of cancer suppression and vulnerability in those organs should lead into important discoveries about the biology of cancer susceptibility.
Data are consistent with the interpretation that evolution explains Peto's paradox: larger, longer lived organisms have had to evolve better cancer suppression mechanisms than their smaller, shorter-lived cousins, increasing the likelihood of survival and reproduction. What remain unclear are the mechanisms that evolution ‘discovered’ for achieving this. It is also unclear how much the biology of an organism would have to change in order to compensate for the 1000× difference in mass between a human and a blue whale. Models can help to give approximate (and qualified) answers to that question. Caulin et al. [32] show that a mere 3× decrease in mutation rate, a 3× reduction in the stem cell division rate, or the addition of one to two copies of a tumour suppressor gene would be sufficient to compensate for a 1000× increase in the number of cells in an organism.
However, these estimates are based on models of carcinogenesis that do not include natural selection (i.e. clonal expansion). Natural selection makes many realistic mathematical models intractable, because cell lineages can no longer be treated as independent of each other. The models used by Caulin et al. [32], the algebraic models of Nunney [4] and Calabrese & Shibata [38] and a stochastic Fisher–Wright model [39] all assume neutral somatic evolution. So do the models of [3,37,40].
Peto's paradox implies that the life-history strategy of a species has led to selection for more or less cancer suppression. Life-history theory provides a powerful tool for developing the theory of Peto's paradox. Three chapters in this issue have capitalized on this opportunity. Brown et al. [28] use the Euler–Lotka population model to formalize the trade-offs between fecundity and cancer suppression. Cancer suppression should be strongest in organisms where the chance of non-cancer death decreases with age (e.g. alligators), fecundity increases with age (e.g. male elephants), maturation is delayed and fecundity rates are low. Their model can explain the unusually high rates of cancer experienced by modern humans in comparison to other species. Brown & Aktipis [41] extend this to point out that, contrary to a common misconception, cancer can indeed impose a selective cost post-reproduction. This is because many organisms, especially humans, demonstrate significant levels of parental (and grandparental) investment in their offspring. In addition, species with cooperative breeding, in which some individuals sacrifice some of their own reproductive potential to help other parents, have potentially been under selective pressure to suppress cancer past their own reproductive years. Boddy et al. [42] further develop the application of life-history theory to comparative oncology by modelling the trade-offs between cancer suppression and intraspecific competition, particularly with respect to mate competition. Larger body size in males often results in successful competition for mates, and thus there may be selection for large body sizes even at the expense of greater cancer susceptibility in males. This can been seen in the association between sexually selected extreme traits like antlers, and related cancers such as antleromas, and may explain the greater cancer incidence in men compared to women.
Haig [43], working in the same vein as Boddy et al. [42], argues that intra-genomic conflict has led to genes with carcinogenic functions, which may explain why animals, and particularly mammals, seem to be more cancer-prone than other forms of multicellularity [2,44]. Genes for placental development have evolved to invade maternal tissues and evade the maternal immune system. Paternally derived genes have been selected to stimulate cellular growth and evade growth control from maternally derived genes. The weapons that have evolved in these arms races are then readily available to wreak havoc later in mammalian lifespan. In these cases, evolution is essentially playing with fire, and the likely result is cancer susceptibility in mammals.
Peto's paradox is an important question within the larger field of comparative oncology. What can we learn from comparing cancer, and cancer suppression, across species? The lead article in this issue from Aktipis et al. [2] contributes both a survey of cancer-like phenomena across the tree of life as well as an important theoretical framework for understanding cancer that can be universally applied across species, even in organisms such as plants and algae that have very different tissue structures from animals. They argue that all forms of complex multicellularity involve five forms of cellular cooperation in order to (i) inhibit somatic cell proliferation, (ii) enforce controlled cell death, (iii) transport and allocate resources throughout the body, (iv) specialize on tasks for the good of the organisms with a division of labour, and (v) maintain the extracellular environment. Cancer, in any organism, can be understood as cheating within each of these forms of cooperation.
Comparative oncology can also help to reveal the mechanisms of carcinogenesis. The Ewalds [45] show that one regularity across species is the fact that infectious agents often cause neoplasms. This can be seen in the galls and boils in plants as well as many of the cancers reported in zoos and domesticated animals. The dynamics of infectious agents adds a further level of selection to the already multilevel selection involved in cancer. Most carcinogenic agents are viruses, probably because the virus gains a selective advantage from causing their host cells to replicate.
Comparative oncology also highlights the importance of metabolism in cancer. There is a strong negative correlation between organism size and metabolic rate, and this may provide a general mechanism to explain Peto's paradox, regardless of cancer type [32]. Dang [46] reviews the growing evidence that metabolism is closely linked to somatic mutation rates. But the link between cancer and metabolism goes beyond mutation rates. Mutations in metabolic genes are often carcinogenic, and mutations in many cancer genes alter the neoplastic cell's metabolism.
(b). Cancer in different mammalian species
Very few data exist on cancer rates in non-human mammalian species in the wild. Furthermore, despite the common practice of necropsies on animals in captivity (a regular practice performed in zoos), we still have limited data compiled about the true prevalence of cancer in nature in different animal species [47]. This is compounded by the issue of not knowing how much the captive environment, with its decreased stress and artificial diet compared with the wild, will increase or decrease the development of cancer. As encouraged by several of the articles in this special issue, a comprehensive compendium is required of cancers occurring in wild, captive and domesticated mammalian species. As has been true in human cancer studies through a variety of databases and cohorts, these types of epidemiological investigations will shed great light on the development of cancer across varying species.
In this issue, Varki & Varki [48] discuss the common occurrence of carcinomas originating from epithelial tissue in humans (including lung, breast, prostate, colon, ovary and pancreas). Surprisingly, captive chimpanzees have not been found to have the same high rates of carcinoma. As discussed by Varki & Varki [48], ascertainment bias is a concern, although many zoo veterinarians perform necropsies as described above and the observed carcinoma rate in chimpanzees remains relatively low. Varki & Varki offer several explanations for the different carcinoma rate between such genetically similar primates as chimpanzees and humans including inadequacy of numbers surveyed, differences in life expectancy, diet (chimpanzees do not eat highly processed foods like humans), genetic susceptibility, immune responses or their microbiomes, and other potential environmental factors (chimpanzees do not smoke like humans). Given the data currently available, this difference in carcinoma incidence appears to be real, although a thorough epidemiological survey is still required to support this supposition in both captive and wild chimpanzees. Intriguingly, it has been reported by Arora et al. [49] that human cells display reduced apoptotic function relative to chimpanzee cells; although apoptosis was investigated in relationship to neuronal development, the same authors hypothesized in an earlier publication [50] that the selection of decreased programmed cell death in human brains may have led to increased cognition over chimpanzees with a trade-off of increased cancer risk due to decreased apoptosis of mutated cells.
More insight into human cancer development can be found in the California sea lion (Zalophus californianus), as described by Browning et al. [51]. Naturally occurring cancers in mammals in the wild, such as the California sea lion, more closely resemble human tumours than laboratory-induced tumours owing to a more similar overlap of genetic and environmental interactions. Browning et al. [51] report on the very high prevalence of urogenital carcinoma in the California sea lion population, including aetiology related to genotype, persistent organic pollutants, and even a cancer-associated herpesvirus. As discussed, the California sea lions are a useful organism for understanding the origins of similar anatomical human cancers that result from very similar risk factors. However, similar to cancer in chimpanzees and other non-human primates, challenges exist to documenting and studying cancer in a wild population. Nevertheless, Browning et al. [51] make the same argument as Varki & Varki [48] that a comprehensive survey is required better to understand cancer in our mammalian relatives.
Picking up on this point, Guy et al. [17] report on the very exciting Golden Retriever Lifetime Study (GRLS) that has successfully established an observational cohort study of a single dog breed at high risk for several cancers with direct translational relevance for human health. Their article describes the GRLS as the first prospective longitudinal study in veterinary medicine comprehensively to detail the major dietary, genetic and environmental risk factors for cancer in dogs. The goal of GRLS is to enroll and follow 3000 purebred golden retrievers throughout their lives with an extensive battery of questionnaires, physical examinations and annual collections of biological samples for comparative oncology analysis (including genomics of tumours as they develop). Taking advantage of the high rate of cancer in a single purebred dog that lives in the same environment as humans, the GRLS will be an unparalleled resource for investigating the complex associations between genes and environmental influences leading to cancer.
(c). The promise of comparative oncology
The exploration of cancer development in different animal species holds great potential in its translation to humans, including shedding light on our own cancer susceptibility and treatment. As Kokko & Hochberg [40] caution, we need to be careful to include the influence of cancer on reproductive lifespan in our interpretation of interspecies body size evolution and life-history theory. As they explain, even in the wild, if an animal has developed cancer, this will ultimately affect its fitness and ability to survive (regardless of whether that particular individual does or does not directly die from its cancer). The observation of life-history patterns cannot be separated from cancer risk and development, and tumour patterns across species can differ from within-species predictions. Kokko & Hochberg also provide modelling to predict that males will be more cancer-prone than females in sexually dimorphic species, an observation certainly seen in humans on both the population [52] and molecular level [53]. As seen in other articles in this issue, mathematical modelling around the evolutionary concepts of Peto's paradox is important to consider in the field of comparative oncology when devising hypotheses to test in the laboratory.
Perhaps one of the best examples of the promise of comparative oncology for improving health is described by Schiffman & Breen [16] in their comprehensive review of canine cancer. On average, and driven by purebred dogs, the rate of cancer development is estimated to be over 10 times higher in dogs than in humans. As Schiffman & Breen discuss, much of this increase in cancer susceptibility is due to the numerous genetic bottlenecks created during the phenotypic selection of purebred traits. In fact, specific breeds remain at extraordinarily high rates for development of specific cancer subtypes (e.g. Bernese mountain dogs and histiocytic sarcoma, golden retrievers and lymphomas, Scottish terriers and urothelial carcinoma, and boxers and glioblastomas). Owing to the overlap between regions in the canine and human genomes, the study of germline cancer risk and subsequent somatic tumour genomics in dogs proves to be an unparalleled resource for understanding universal concepts of cancer development. Schiffman & Breen [16] review the current genome-wide association studies (GWAS) in canine cancer and their comparison to the equivalent GWAS results in human cancer, as well as describe many of the comparative genomic investigations into actual tumours including sarcoma, haematological malignancies, bladder cancer, intracranial neoplasms and melanoma. As highlighted in their review, the study of genomes of different dog breeds and the cancers that they develop accelerates cancer gene discovery in the field of comparative oncology.
Finally, as a fitting conclusion to our special issue, Lawrence et al. [54] offer a tangible description of how different species respond to cancer chemotherapy. They detail the development of chemotherapy in humans and its relatively recent adoption into veterinary medicine. As emphasized in their paper, Lawrence et al. [54] describe the poor predictability of tumour responses to cancer chemotherapy drugs in rodent models leading to the subsequent failure of drugs that might otherwise benefit different species such as dogs and humans. In fact, pet dogs may actually be the ideal ‘pre-clinical’ models for Phase I, II and III human chemotherapeutic testing due to similar drug metabolism (both functionally and genetically), similar cancers that occur naturally, and overall large number of affected individuals to test [16,54]. This advantage already has been recognized in the field of pediatric oncology where trials are ongoing for novel drug therapies for osteosarcoma, a rare bone tumour in humans but quite common in dogs [55–57]. As Lawrence et al. [54] highlight, to capture fully the benefits of comparative oncology for therapeutic advances in cancer treatment for humans and dogs, we still need to understand better the differences and similarities in pharmacodynamics, pharmacokinetics and pharmacogenomics between humans and dogs.
6. Concluding thoughts
This collection of papers reflects a sea change in the integration of evolutionary ideas into understanding all aspects of cancer biology. The number of papers published with the word ‘evolution (or evolutionary)’ combined with the word ‘cancer’ in the title has increased from an average of less than 20 in the early 1980s, to about 40 in the late 1990s, to more than 150 in each of the past two years, and perhaps even more importantly, the number of citations to such papers has increased 30-fold over the same period (figure 1).
Figure 1.
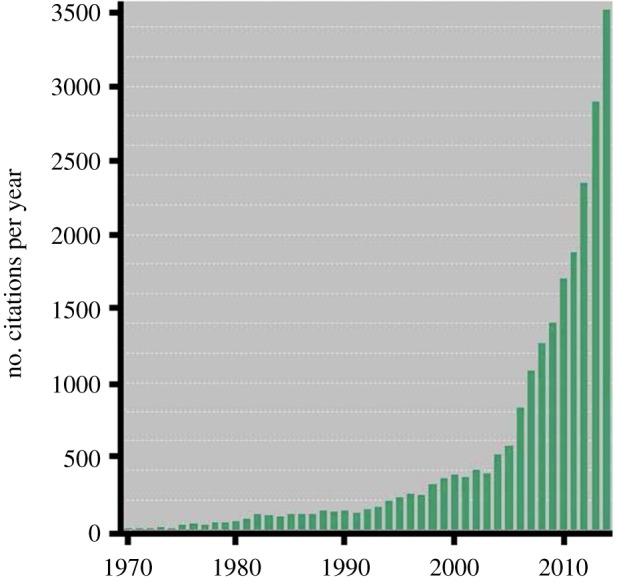
The increasing number of citations to articles with ‘cancer’ and ‘evolution’ (or ‘evolutionary’) in the title from 1970 to 2014. Data from Web of Science (webofknowledge.com). (Online version in colour.)
Being a relatively new field, the study of cancer from an evolutionary perspective offers enormous research potential with many major unexplored questions. In this special issue, we have focused primarily on the evolution of host defences, a topic that has at its core the simple question so clearly expressed by Peto [23]: why don't big, long-lived animals get more cancer? From this starting point, this special issue highlights two important areas of research. First, it illustrates that a rich theoretical literature has been developing that can be used to test hypotheses about the resolution of Peto's paradox and more generally to understand the variation in cancer incidence seen in nature. Second, it highlights new empirical approaches that become possible as data on cancer incidence in non-model organisms begins to accumulate. The frequencies of the types of cancer observed in animals often differ substantially from what we see in humans (e.g. [16,24]), again driving testable hypotheses, and in some cases allowing us to initiate large-scale studies of cancers that are too rare in humans to study at the population level.
In the past, many of these questions that can be included in the broad paradigm of an evolutionary approach to cancer could be posed, but we did not have the tools to address them. With the increasing availability of genome sequences of non-model organisms and RNAseq expression data from their tissues, as well as the continued development of other new genomic technologies, we can expect the interplay between theory and empirical data to be increasingly productive. As this research effort expands, there is great promise for advances in cancer prevention in both medical and veterinary science.
Funding
We gratefully acknowledge the support of the Wissenschaftskolleg zu Berlin (Institute for Advanced Study, Berlin) for making the organization and contribution to this special issue possible. M.E.H. acknowledges the Agence National de la Recherche (ANR-13-BSV7–0003-01) and ITMO (PC201306) for funding. J.D.S. and M.B. gratefully acknowledge support of the Skippy Frank Fund for Life Sciences and Translational Research/Rockefeller Philanthropy Advisors. J.D.S. holds the Edward B. Clark, MD Chair in Pediatric Research and receives support through the Primary Children's Hospital (PCH) Pediatric Cancer Program funded by the Intermountain Healthcare Foundation and the PCH Foundation. M.B. holds the Oscar J. Fletcher Distinguished Professorship of Comparative Oncology Genetics at North Carolina State University.
References
- 1.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144, 646–674. ( 10.1016/j.cell.2011.02.013) [DOI] [PubMed] [Google Scholar]
- 2.Aktipis CA, Boddy AM, Jansen G, Hibner U, Hochberg ME, Maley CC, Wilkinson GS. 2015. Cancer across the tree of life: cooperation and cheating in multicellularity. Phil. Trans. R. Soc. B 370, 20140219 ( 10.1098/rstb.2014.0219) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nunney L, Muir B. 2015. Peto's paradox and the hallmarks of cancer: constructing an evolutionary framework for understanding the incidence of cancer. Phil. Trans. R. Soc. B 370, 20150161 ( 10.1098/rstb.2015.0161) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nunney L. 1999. Lineage selection and the evolution of multistage carcinogenesis. Proc. R. Soc. Lond. B 266, 493–498. ( 10.1098/rspb.1999.0664) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nunney L. 1999. Lineage selection: natural selection for long-term benefit. In. Levels of selection in evolution (ed. L Keller), pp. 238–258. [Google Scholar]
- 6.Frank SA. 1995. Mutual policing and repression of competition in the evolution of cooperative groups. Nature 377, 520–522. ( 10.1038/377520a0) [DOI] [PubMed] [Google Scholar]
- 7.de Visser KE, Eichten A, Coussens LM. 2006. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 6, 24–37. ( 10.1038/nrc1782) [DOI] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. 2000. The hallmarks of cancer. Cell 100, 57–70. ( 10.1016/S0092-8674(00)81683-9) [DOI] [PubMed] [Google Scholar]
- 9.Nowell PC. 1976. The clonal evolution of tumor cell populations. Science 194, 23–28. ( 10.1126/science.959840) [DOI] [PubMed] [Google Scholar]
- 10.Greaves M, Maley CC. 2012. Clonal evolution in cancer. Nature 481, 306–313. ( 10.1038/nature10762) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navin N, et al. 2011. Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94. ( 10.1038/nature09807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang XC, et al. 2013. Tumor evolution and intratumor heterogeneity of an oropharyngeal squamous cell carcinoma revealed by whole-genome sequencing. Neoplasia 15, 1371–1378. ( 10.1593/neo.131400) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hochberg ME, Thomas F, Assenat E, Hibner U. 2013. Preventive evolutionary medicine of cancers. Evol. Appl. 6, 134–143. ( 10.1111/eva.12033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maley CC, Szabo E, Reid BJ. 2011. Somatic evolution in neoplastic progression and cancer prevention. In Pre-invasive disease: pathogenesis and clinical management (ed. RC Fitzgerald), pp. 111–117. New York, NY: Springer Science+Business Media. [Google Scholar]
- 15.Gorbunova V, Seluanov A, Zhang Z, Gladyshev VN, Vijg J. 2014. Comparative genetics of longevity and cancer: insights from long-lived rodents. Nat. Rev. Genet. 15, 531–540. ( 10.1038/nrg3728) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schiffman JD, Breen M. 2015. Comparative oncology: what dogs and other species can teach us about humans with cancer. Phil. Trans. R. Soc. B 370, 20140231 ( 10.1098/rstb.2014.0231) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guy MK, Page RL, Jensen WA, Olson PN, Haworth JD, Searfoss EE, Brown DE. 2015. The Golden Retriever Lifetime Study: establishing an observational cohort study with translational relevance for human health. Phil. Trans. R. Soc. B 370, 20140230 ( 10.1098/rstb.2014.0230) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nordling CO. 1953. A new theory on cancer-inducing mechanism. Br. J. Cancer 7, 68–72. ( 10.1038/bjc.1953.8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iversen S, Arley N. 1950. On the mechanism of experimental carcinogenesis. Acta Path. Microbiol. Scand. 27, 773–803. ( 10.1111/j.1699-0463.1950.tb00081.x) [DOI] [PubMed] [Google Scholar]
- 20.Armitage P, Doll R. 2004. The age distribution of cancer and a multi-stage theory of carcinogenesis. Intl J. Epidemiol. 33, 1174–1179. ( 10.1093/ije/dyh216) [DOI] [PubMed] [Google Scholar]
- 21.Knudson AG., Jr 1971. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl Acad. Sci. USA 68, 820–823. ( 10.1073/pnas.68.4.820) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. 2013. Cancer genome landscapes. Science 339, 1546–1558. ( 10.1126/science.1235122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peto R. 1977. Epidemiology, multistage models, and short-term mutagenicity tests. In The origins of human cancer (eds HH Hiatt, JD Watson, JA Winsten). pp. 1403–1428. Cold Spring Harbor Conferences on Cell Proliferation, vol. 4 NY: Cold Spring Harbor Laboratory. [Google Scholar]
- 24.Rangarajan A, Weinberg RA. 2003. Opinion: comparative biology of mouse versus human cells: modelling human cancer in mice. Nat. Rev. Cancer 3, 952–959. ( 10.1038/nrc1235) [DOI] [PubMed] [Google Scholar]
- 25.Nunney L. 2013. The real war on cancer: the evolutionary dynamics of cancer suppression. Evol. Appl. 6, 11–19. ( 10.1111/eva.12018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green J, Cairns BJ, Casabonne D, Wright FL, Reeves G, Beral V. 2011. Height and cancer incidence in the Million Women Study: prospective cohort, and meta-analysis of prospective studies of height and total cancer risk. Lancet Oncol. 12, 785–794. ( 10.1016/S1470-2045(11)70154-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fleming JM, Creevy KE, Promislow DE. 2011. Mortality in North American dogs from 1984 to 2004: an investigation into age-, size-, and breed-related causes of death. J. Vet. Intern. Med. 25, 187–198. ( 10.1111/j.1939-1676.2011.0695.x) [DOI] [PubMed] [Google Scholar]
- 28.Brown JS, Cunningham JJ, Gatenby RA. 2015. The multiple facets of Peto's paradox: a life-history model for the evolution of cancer suppression. Phil. Trans. R. Soc. B 370, 20140221 ( 10.1098/rstb.2014.0221) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cairns J. 1975. Mutation selection and the natural history of cancer. Nature 255, 197–200. ( 10.1038/255197a0) [DOI] [PubMed] [Google Scholar]
- 30.Nunney L. 2003. The population genetics of multistage carcinogenesis. Proc. R. Soc. Lond. B 270, 1183–1191. ( 10.1098/rspb.2003.2351) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seluanov A, Chen Z, Hine C, Sasahara TH, Ribeiro AA, Catania KC, Presgraves DC, Gorbunova V. 2007. Telomerase activity coevolves with body mass not lifespan. Aging Cell 6, 45–52. ( 10.1111/j.1474-9726.2006.00262.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caulin AF, Graham TA, Wang L-S, Maley CC. 2015. Solutions to Peto's paradox revealed by mathematical modelling and cross-species cancer gene analysis. Phil. Trans. R. Soc. B 370, 20140222 ( 10.1098/rstb.2014.0222) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aktipis CA, Boddy AM, Gatenby RA, Brown JS, Maley CC. 2013. Life history trade-offs in cancer evolution. Nat. Rev. Cancer 13, 883–892. ( 10.1038/nrc3606) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leroi AM, Koufopanou V, Burt A. 2003. Cancer selection. Nat. Rev. Cancer 3, 226–231. ( 10.1038/nrc1016) [DOI] [PubMed] [Google Scholar]
- 35.Crespi B, Summers K. 2005. Evolutionary biology of cancer. Trends Ecol. Evol. 20, 545–552. ( 10.1016/j.tree.2005.07.007) [DOI] [PubMed] [Google Scholar]
- 36.Tomasetti C, Vogelstein B. 2015. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81. ( 10.1126/science.1260825) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noble R, Kaltz O, Hochberg ME. 2015. Peto's paradox and human cancers. Phil. Trans. R. Soc. B 370, 20150104 ( 10.1098/rstb.2015.0104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calabrese P, Shibata D. 2010. A simple algebraic cancer equation: calculating how cancers may arise with normal mutation rates. BMC Cancer 10, 3 ( 10.1186/1471-2407-10-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beerenwinkel N, Antal T, Dingli D, Traulsen A, Kinzler KW, Velculescu VE, Vogelstein B, Nowak MA. 2007. Genetic progression and the waiting time to cancer. PLoS Comput. Biol. 3, e225 ( 10.1371/journal.pcbi.0030225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kokko H, Hochberg ME. 2015. Towards cancer-aware life-history modelling. Phil. Trans. R. Soc. B 370, 20140234 ( 10.1098/rstb.2014.0234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown JS, Aktipis A. 2015. Inclusive fitness effects can select for cancer suppression into old age. Phil. Trans. R. Soc. B 370, 20150160 ( 10.1098/rstb.2015.0160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boddy AM, Kokko H, Breden F, Wilkinson GS, Aktipis CA. 2015. Cancer susceptibility and reproductive trade-offs: a model of the evolution of cancer defences. Phil. Trans. R. Soc. B 370, 20140220 ( 10.1098/rstb.2014.0220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haig D. 2015. Maternal–fetal conflict, genomic imprinting and mammalian vulnerabilities to cancer. Phil. Trans. R. Soc. B 370, 20140178 ( 10.1098/rstb.2014.0178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Effron M, Griner L, Benirschke K. 1977. Nature and rate of neoplasia found in captive wild mammals, birds, and reptiles at necropsy. J. Natl Cancer Inst. 59, 185–198. [DOI] [PubMed] [Google Scholar]
- 45.Ewald PW, Swain Ewald HA. 2015. Infection and cancer in multicellular organisms. Phil. Trans. R. Soc. B 370, 20140224 ( 10.1098/rstb.2014.0224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dang CV. 2015. A metabolic perspective of Peto's paradox and cancer. Phil. Trans. R. Soc. B 370, 20140223 ( 10.1098/rstb.2014.0223) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vittecoq M, et al. 2013. Cancer: a missing link in ecosystem functioning? Trends Ecol. Evol. 28, 628–635. ( 10.1016/j.tree.2013.07.005) [DOI] [PubMed] [Google Scholar]
- 48.Varki NM, Varki A. 2015. On the apparent rarity of epithelial cancers in captive chimpanzees. Phil. Trans. R. Soc. B 370, 20140225 ( 10.1098/rstb.2014.0225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arora G, Mezencev R, McDonald JF. 2012. Human cells display reduced apoptotic function relative to chimpanzee cells. PLoS ONE 7, e46182 ( 10.1371/journal.pone.0046182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arora G, Polavarapu N, McDonald JF. 2009. Did natural selection for increased cognitive ability in humans lead to an elevated risk of cancer? Med. Hypotheses 73, 453–456. ( 10.1016/j.mehy.2009.03.035) [DOI] [PubMed] [Google Scholar]
- 51.Browning HM, Gulland FMD, Hammond JA, Colegrove KM, Hall AJ. 2015. Common cancer in a wild animal: the California sea lion (Zalophus californianus) as an emerging model for carcinogenesis. Phil. Trans. R. Soc. B 370, 20140228 ( 10.1098/rstb.2014.0228) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siegel RL, Miller KD, Jemal A. 2015. Cancer statistics, 2015. Cancer J. Clin. 65, 5–29. ( 10.3322/caac.21254) [DOI] [PubMed] [Google Scholar]
- 53.Sun T, Warrington NM, Luo J, Brooks MD, Dahiya S, Snyder SC, Sengupta R, Rubin JB. 2014. Sexually dimorphic RB inactivation underlies mesenchymal glioblastoma prevalence in males. J. Clin. Invest. 124, 4123–4133. ( 10.1172/JCI71048) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lawrence J, Cameron D, Argyle D. 2015. Species differences in tumour responses to cancer chemotherapy. Phil. Trans. R. Soc. B 370, 20140233 ( 10.1098/rstb.2014.0233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fenger JM, London CA, Kisseberth WC. 2014. Canine osteosarcoma: a naturally occurring disease to inform pediatric oncology. ILAR J. 55, 69–85. ( 10.1093/ilar/ilu009) [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez CO., Jr 2014. Using canine osteosarcoma as a model to assess efficacy of novel therapies: can old dogs teach us new tricks? Adv. Exp. Med. Biol. 804, 237–256. ( 10.1007/978-3-319-04843-7_13) [DOI] [PubMed] [Google Scholar]
- 57.Rankin KS, Starkey M, Lunec J, Gerrand CH, Murphy S, Biswas S. 2012. Of dogs and men: comparative biology as a tool for the discovery of novel biomarkers and drug development targets in osteosarcoma. Pediatric Blood Cancer 58, 327–333. ( 10.1002/pbc.23341) [DOI] [PubMed] [Google Scholar]