

Abstract
The factors influencing cancer susceptibility and why it varies across species are major open questions in the field of cancer biology. One underexplored source of variation in cancer susceptibility may arise from trade-offs between reproductive competitiveness (e.g. sexually selected traits, earlier reproduction and higher fertility) and cancer defence. We build a model that contrasts the probabilistic onset of cancer with other, extrinsic causes of mortality and use it to predict that intense reproductive competition will lower cancer defences and increase cancer incidence. We explore the trade-off between cancer defences and intraspecific competition across different extrinsic mortality conditions and different levels of trade-off intensity, and find the largest effect of competition on cancer in species where low extrinsic mortality combines with strong trade-offs. In such species, selection to delay cancer and selection to outcompete conspecifics are both strong, and the latter conflicts with the former. We discuss evidence for the assumed trade-off between reproductive competitiveness and cancer susceptibility. Sexually selected traits such as ornaments or large body size require high levels of cell proliferation and appear to be associated with greater cancer susceptibility. Similar associations exist for female traits such as continuous egg-laying in domestic hens and earlier reproductive maturity. Trade-offs between reproduction and cancer defences may be instantiated by a variety of mechanisms, including higher levels of growth factors and hormones, less efficient cell-cycle control and less DNA repair, or simply a larger number of cell divisions (relevant when reproductive success requires large body size or rapid reproductive cycles). These mechanisms can affect intra- and interspecific variation in cancer susceptibility arising from rapid cell proliferation during reproductive maturation, intrasexual competition and reproduction.
Keywords: cancer defences, life-history trade-offs, reproductive competition, sexual selection, comparative oncology
1. Introduction
Cancer incidence across species varies widely. These differences in cancer susceptibility are due to both environmental exposures [1], and evolutionary pressures shaping organism-level cancer susceptibility and cancer defences [2–4]. Beneficial traits such as defence against disease are often costly, and such trade-offs can maintain genes that contribute to disease susceptibility [5]. For example, trade-offs between reproduction and immune defence have been observed in a number of species [6,7]. Similar trade-offs have been proposed to underlie observed associations between reproductive competitiveness and cancer [8], though as of yet there has been no formal model describing these trade-offs and the factors affecting them.
Accumulating genetic and molecular evidence suggests that trade-offs between benefits early in life and disease susceptibility later in life may be widespread across disease (as reviewed in Carter & Nguyen [9] and Leroi et al. [10]). This phenomenon may also significantly shape the susceptibility to cancer [9]. Cancer suppressor genes such as P53 [11,12] and BRCA [13] play important roles in fertility and may be maintained owing to trade-offs between fertility enhancement and cancer risk.
Trade-offs between reproductive competitiveness and cancer risk can be expected for two main reasons: (i) selection for mechanisms that enable rapid cell proliferation could simultaneously enhance extreme trait expression as well as tumour formation and (ii) selection for increased allocation of energy to reproductive traits rather than to somatic maintenance could elevate cancer risk by increasing somatic mutation rates. The first type of trade-off may involve mutations or epigenetic silencing of cancer suppression genes. The second type of trade-off could be due to altered allocation of a finite energy pool towards reproduction at the expense of somatic maintenance, such as DNA repair or immune defences. Consequently, mutations that increase cancer risk arise more easily if energy must be diverted away from DNA repair to support development or expression of reproductive traits. These two pathways are distinct, but can potentially interact to produce trade-offs between reproductive competitiveness and cancer susceptibility. Even if somatic mutation rate per cell division did not change, reproductive success might require a larger number of cell divisions (e.g. when sexual selection favours large-bodied males), and this again increases the total risk.
In this paper, we explore the effect of reproductive trade-offs on the evolution of cancer defences, modelling reproductive competitiveness as the extent to which the most competitive individuals dominate reproduction. Reproductive competitiveness is an important force in the evolution of extreme morphologies and life histories, and is often impacted by sexual selection [14]. Traits such as large body size, extreme morphology (i.e. weapons or ornaments), larger and more frequent litter size, and aggressiveness can all have positive effects on fitness through preferential mating or differential fitness owing to higher reproductive output, but they can also be costly in terms of increasing mortality or morbidity. Large body size and extreme morphologies often require greater levels of cell proliferation via an increase in growth signalling mechanisms (including hormones and growth factors). Additionally, factors that accelerate organismal reproduction (e.g. growing fast, early maturation) or increase the frequency of reproduction might enhance cancer risk through the allocation of energy towards reproduction rather than through somatic maintenance, potentially increasing the likelihood of mutations in cancer suppressor genes.
Our model does not explicitly distinguish between these two types of trade-offs (cell-proliferation-related vulnerabilities and energy allocation trade-offs). For simplicity and generality, we base our model on the intensity of the trade-off between reproductive competitiveness and cancer defences. In addition to varying the intensity of the trade-offs, we also consider varying levels of extrinsic mortality. This allows us to explore the evolutionary viability of cancer defences in species under different reproductive, social and ecological conditions. Mortality (and the consequent expected lifespan) is important, because cancer defences can delay the onset of cancer, but delays bring about little selective benefit at an age where the organism is likely to have died of other causes. We use this model to make predictions about the patterns of cancer incidence across species and review a variety of mechanisms that might underlie trade-offs between reproductive competitiveness and cancer defence.
2. Model
(a). Stronger intraspecific competition is predicted to lead to higher cancer incidence
Can reproductive competition within a population lead to individuals adaptively ‘neglecting’, or never evolving, some of the possible defences against cancer? Here, we derive an optimality model to address this question, as well as whether populations with particularly strong intraspecific competitive effort should be expected to have an elevated risk of mortality owing to cancer (as opposed to some other cause). In this model, the population can refer to all individuals, regardless of their sex, but it is also possible to make statements about a subset of the biological population to the extent that competition occurs among ‘peers'. For example, the population may be split into males and females, which means that other males form the relevant peer group of males, whereas females compete with other females. Consequently, we also consider whether male defences against cancer are expected to differ from those of females.
(b). Model assumptions and derivations
(i). Cancer defence
First, we assume that variation in the strength of defences against cancer is present in the population. The model does not specify how the defence acts to delay cancer onset. Instead, we use an operational definition. Assuming that cancer takes n steps to form, with each occurring at a rate ki per cell, where i = 1, 2, … , n, if n steps have occurred in a particular cell lineage, then the whole organism has cancer. The organism possesses defence levels di against each step i, where 0 ≤ di ≤ 1. Each di denotes the reduction in the rate at which the corresponding step occurs: if, for example, di = 0.5, the ith step takes, on average, twice as long to happen than it would in the absence of any defence; if di = 0.75, it takes four times as long (only 25% of the rate then remains, and 1/0.25 = 4). One possible way to achieve di = 0.5 is to have DNA repair remove half of the critical mutations, but less direct ways include efficient immunocompetence that clears half of the infections before they can heighten the organism's cancer risk. Alternatively, the organism may simply eliminate half of the precancerous cells.
For simplicity, our examples are built assuming that all limiting steps occur at the same rate ki = k for all i, and that cancer defence likewise is the same across steps, di = d for all i. Without this assumption, the analysis would proceed as below but with specific multiplications for each i separately. The general model intends to make no statements about the relative efficiency of particular defence types against specific rate-limiting steps.
(ii). Reproductive competition within the peer group
Next, we specify how intraspecific competition impacts an individual's reproductive success. As described in §1, trade-offs between defence and investment in immediate reproductive success may occur for multiple reasons. For example, the latter may require fast growth, but this comes at a cost to the former. We assume competitiveness, c, is a function of two parameters α (the intensity of intraspecific competition) and β (the strength of trade-offs): the rate of fitness accumulation per time unit spent alive and cancer free is assumed proportional to cα = (1 – dβ)α.
This is specified as ‘proportional to’ rather than as ‘equal to’ to take into account the fact that fitness is relative to conspecifics. Thus, if surrounded by many other competitive individuals, fitness of the focal individual may remain low, but for the subsequent analysis contrasting alternative life histories of a focal individual the effect of others stays constant and does not have to be included [15]. Here, c = 1 – dβ describes the individual's competitiveness assuming it does not yet have cancer. Cancerous individuals are assumed to be non-competitive and thus gain zero reproductive success while alive with cancer.
Competitiveness, as defined above, is a declining function of cancer defences, which reflects the assumption that defences are costly. The parameter α describes the intensity of intraspecific competition (in many cases, limited to competition within a sex, i.e. intrasexual competition) such that resources obtained scale with competitiveness according to a power function cα (see parameter u in an analogous treatment in [15]). Thus, if α = 1, an individual whose competitiveness c exceeds that of its peer group by a certain percentage (say, 10%) will have an equivalently better (in this example 10%) access to resources. If α < 1, the benefits of increased competitiveness are lower: a 10% increase in c would, then, lead to lower than 10% improvement in reproductive gains. Conversely, α > 1 will make the resource–competitiveness relationship steeper, so that if α ≫ 1, competition is of a ‘winner-takes-all’ nature: a higher competitiveness, c, compared with the rest of the peer group will then lead to the focal individual reaping almost all the reproductive benefits.
The parameter β changes the shape of trade-offs: if β is small, then competitiveness declines in response to investing in other life-history components (here, cancer defence) even if the defence is not intensive; if β is large, then individuals can retain their competitiveness intact up to high (d ≈ 1) defence levels.
Individuals are assumed to gain reproductive payoffs (of magnitude cα) at a constant rate as long as they are cancer-free and have also avoided other sources of mortality.
(iii). Cancer-free lifespan
There are two ways in which an individual can end its reproductive career. The first is death owing to a source of mortality other than cancer, which we assume occurs at a constant rate, μ. A(t) is the probability that this type of death has happened by time t. The second way an individual can die is by cancer, that is, when one of the N cell lineages has passed through all the rate-limiting steps, denoted by B(t), the probability that this has happened by time t. The overall probability that the individual is alive is
| 2.1 |
where
| 2.2 |
and
| 2.3 |
B(t) is formed in the following way: 1 − e–(1 − d)kt is the probability that a single rate-limiting step has already happened by time t. When we assume that n steps are required before oncogenesis can end an individual's reproductively active life, (1 − e−(1−d)kt)n is the probability that all of them have happened in the same focal cell lineage. The complementary probability 1 − (1 − e−(1−d)kt)n consequently gives the probability that the focal lineage is healthy at time t. If there are N such lineages in an organism, (1 − (1 − e−(1−d)kt)n)N (which raises the above to the power N) is the probability that all cell lineages are healthy at time t. The complement of this quantity is the probability B(t) of cancer having already emerged in at least one cell lineage as indicated above.
In the absence of any defences against cancer, the probability that the individual has cancer by time t rises from zero to unity as time passes (figure 1). The effect of defence is to delay this increase in a very specific manner: for each value of d, the cumulative probability curves of cancer having ended the individual's cancer-free lifespan are shifted to the right such that any horizontal time line drawn from t = 0 to the probability curve has exactly the proportion d spent in the state of ‘cancer-free life prolonged due to defences'. Thus, if, for example, d = 0.75, one-quarter of the age that the organism managed to spend cancer-free was caused by ‘luck’, whereas three-quarters can be attributed to defences delaying the eventual inevitability of cancer.
Figure 1.
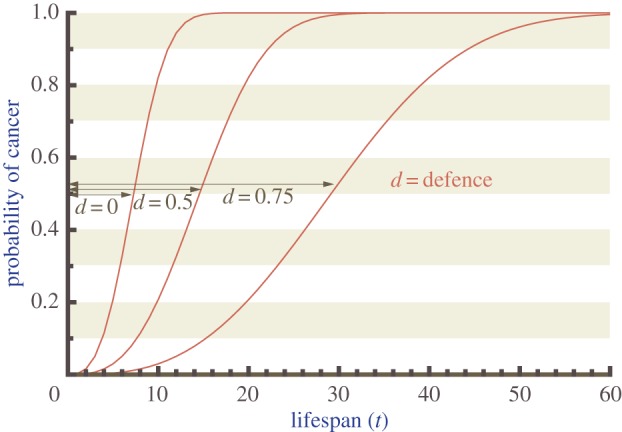
Higher cancer defence increases lifespan. The probability that cancer has arisen by age t for three different defences d = 0, d = 0.5 and d = 0.75. Note that for any probability level, d indicates the proportion of the arrow that extends beyond the d = 0 curve. Thus, e.g. with d = 0.75, three quarters of the delay from birth to cancer is due to the defence effort, one quarter owing to the time that mutations take to occur even if there is no cancer defence effort. Graphs are derived using parameters k = 0.01, n = 3 and N = 2000.
To sum up the effect of d on realized lifespan, however, it is not sufficient to stop at the information contained in figure 1, because other sources of mortality can make it unlikely that the organism is alive to reap the probabilistic benefits of the longest delays at the upper end of figure 1. In this context, it is insightful to use equation (2.3) to derive the time, denoted t(P, d), that it takes cancer incidence to reach any pre-specified value P, for a given level of defences d
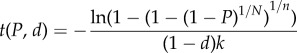 |
2.4 |
Expected lifespan then is an integral
| 2.5 |
Numerical integration of L(d) leads to a crucial insight. If extrinsic mortality (μ) is high (right side of figure 2), investing in defence, d, does not prolong lifespan. Even in the absence of defences, cancer remains an insignificant cause of death as it does not ‘have the time’ to develop before the organism has already been killed by predators or any other factors categorized as extrinsic mortality (μ). Only when these other sources of mortality are low (left side of figure 2), does investing in defence, d, have a substantial impact on lifespan. This highlights that in our aim to derive how optimal cancer defences depend on intraspecific competition (parameters α and β), we also must consider scenarios with a variety of extrinsic mortalities (μ).
Figure 2.
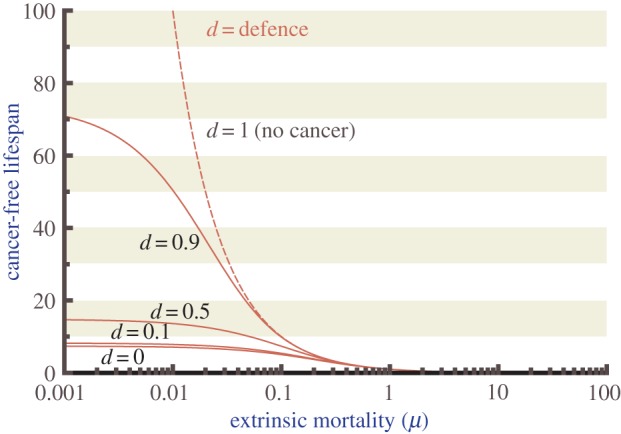
The effects of cancer defences on lifespan are strongest when extrinsic mortality is low. Model predictions for the expected duration of cancer-free life, which can be ended by either cancer or through other sources of mortality, as a function of extrinsic mortality μ, plotted for different values for defence effort d as indicated on the graph (lowest curve: d = 0, then d = 0.1, followed by d = 0.5 and d = 0.9). The dotted line gives the expected lifespan assuming no cancer ever occurs (d = 1). The impact of defence on lifespan varies from strong when extrinsic mortality is low, to negligible when lifespans are short owing to causes other than cancer. Parameters as in figure 1. Note that neither α nor β impact the graphs in figure 1 or in this figure.
To derive how optimal cancer defences depend on intraspecific competition, while considering scenarios with a variety of extrinsic mortalities (μ), we compute lifetime fitness, i.e. the quantity (1 – dβ)αL(d), for different extrinsic mortalities and intraspecific competition (α) and trade-off (β) values and find the maximum. The results (figure 3) show that in all the mortality scenarios (μ = 0.01, 0.1 and 1) and whether we assume weak or strong trade-offs (β) between cancer defences and reproductive success, the optimal defence always declines with increasing intraspecific competition (α), unless defence is already low to begin with (top right figure). The results remain similar across all values of n and N that we tested, though for simplicity only one set of parameters is shown. Lastly, we present alternative extrinsic mortality (μ) scenarios and illustrate how often life is actually ended by cancer, as opposed to all other causes. More intense intraspecific competition (α) increases mortality risk by cancer unless trade-offs (β) are low (figure 4).
Figure 3.

Intraspecific competitiveness has the strongest effect on cancer risk in environments with low extrinsic mortality and strong trade-offs. When extrinsic mortality is low (μ = 0.01) and trade-offs between cancer defence and reproductive competition is high (β = 1), as intraspecific competition increases (α), cancer defences remain high until a critical threshold is reached (when intraspecific competition is greater than 1) and then dramatically declines (upper left). Cancer defence declines in all cases with α, but is sometimes low to begin with (upper right corner where extrinsic mortality is high). Parameters as in figure 1 and figure 2, and additionally β = 1, 2 or 5 (top, middle and bottom row, respectively), and μ = 0.01, 0.1 or 1 (left, middle and right column, respectively). Colours indicate the fitness consequences of each possible level of defence effort (0 ≤ d ≤ 1 as indicated on the y axis) for value of intraspecific competition (α) ranging from 0.01 to 10. More yellow indicates higher fitness, more blue low fitness; the black line additionally indicates highest fitness (optimum).
Figure 4.

Stronger intraspecific competition increases the probability of cancer. The incidence of cancer, measured as the probability of cancer rather than extrinsic mortality ending an individual's reproductively active life, increases as a function of reproductive competition (α) under three different extrinsic mortalities (a) μ = 0.01, (b) μ = 0.1, (c) μ = 1, and three values of β (uppermost curve within each figure: β = 1, middle curve: β = 2, lowest curve: β = 5). Incidence always increases with intraspecific competition (α), but in (c) where extrinsic mortality is high, the increase does not yield high cancer incidence for any value of intraspecific competition (α).
3. Predictions from the model
(a). High intraspecific competition lowers cancer defences
Our model predicts that individuals may evolve traits that increase reproductive success even if this increases their cancer risk, but the extent to which this applies depends on the intensity of intraspecific (often intrasexual) competition as a determinant of reproductive success, and also other parameter values. In an environment with low extrinsic mortality (μ = 0.01), we expect more cancer in species where intraspecific competition (α) is very strong (at the extreme, of a winner-takes-all nature; figure 3 top left). Species in high extrinsic mortality environments (μ = 1), on the other hand, are expected to die from other causes than cancer, regardless of the intensity of intraspecific competition (α) (figure 4c). If extrinsic mortality is lower, then cancer incidence can depend strongly on the role that intraspecific competition (α) plays in shaping life histories (figure 4a,b). Intraspecific competition can vary between males and females, and thus we will discuss our predictions and review of the literature separately for each sex.
(i). Sexually selected traits may increase cancer risk in highly competitive males
Males often invest more energy in obtaining a mate than females and this can lead to high competition among individuals to reproduce. For example, males with high reproductive competitiveness participate in male–male competition and dominant males gain more opportunities to mate. This competition can favour the evolution of secondary sexual characteristics (e.g. extreme traits) [14,16], and it is well known that a shorter lifespan may be an ‘acceptable’ cost of such traits in the sense that net selection favours their exaggeration, whereas male lifespan is shortened. According to our model, we predict sexually dimorphic species, such as individuals with extreme ornaments and weapons or larger body size, to be subject to increased cancer risk (figure 5); similarly, we expect more cancer in species with strong sperm competition (particularly in the relevant tissues such as the testes). These trade-offs may be a result of vulnerabilities arising from increased cell proliferation (e.g. growth promoting signals to produce large body size or weapons; more of the continually dividing cells in larger testes) or as a result of allocation of energy towards reproductive competition at the expense of somatic maintenance (e.g. DNA repair, immune defences). However, it is clearly difficult to obtain cancer incidence for species in the wild, especially when male–male competition is intense: poor performance of an individual after the onset of cancer can lead to a death (e.g. taken by a predator) that is not easy to trace back to cancer.
Figure 5.
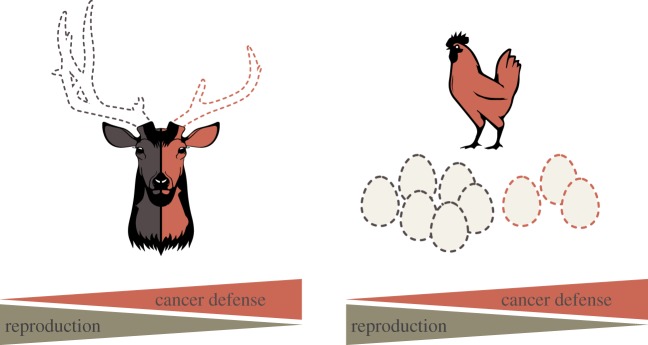
Increased reproductive effort through mating effort or fertility may lower cancer defences. An illustration of our hypothesis–selection to enhance reproductive competition decreases cancer defences. Traits selected to increase reproductive effort include body size, ornaments or weapons in males, or fertility frequency and productivity in females. Hypothetical trade-offs are illustrated above depicting examples of reproductive competition. For example, the smaller antler on the deer illustrates less reproductive competitiveness, but better protection against cancer. Similarly, increased reproductive output in domesticated chickens may trade off with lower cancer defences.
There is some evidence to suggest that selection for large body size in males can lead to higher cancer susceptibility. For example, Xiphophorus maculatus, a freshwater fish called the Southern platyfish, carries a dominant oncogene (Xiphophorus melanoma receptor kinase, Xmrk) that is normally suppressed but can result in the formation of male-biased malignant melanomas in hybrids [17–20]. Body size can strongly predict reproductive success in this species, and the Xmrk genotype is positively correlated with a larger body size in both males and females. Females prefer to mate with males carrying a larger black spot [17,18], and males with melanoma and Xmrk were significantly longer than both males with Xmrk and no melanoma and males without Xmrk [17]. Xmrk is derived from a gene duplication event of an epidermal growth factor receptor that controls cell proliferation [21], and it may be involved in binding to growth factor ligands that increase both body size and melanoma formation in several Xiphophorus species [22].
Another intriguing example of a trait that can enhance reproductive competitiveness of males, but also may increase cancer risk is the production of sexually selected weapons, such as antlers. In most deer species, only males grow antlers, and growth is initiated at puberty [23,24]. New antler growth and casting is seasonal, and thought to be controlled by levels of circulating testosterone [25,26]. It has been hypothesized that there is also a period of androgen-independent growth during antler formation [23], where circulating levels of insulin-like growth factor-1 (IGF-1) were significantly increased in male deer growing their antlers [27]. Male–male competition can lead to strong selection for growth to produce such elaborate traits, but this comes with a risk: disregulation in the system can lead to uncontrolled cellular growth. Species with antlers are accordingly susceptible to antleromas, which are massive growths found on the antlers of free ranging deer [28–35]. Antleromas can be artificially produced when androgen production is disrupted (e.g. castration) and circulating testosterone does not cycle [32], suggesting that interactions between hormone levels and growth factors underlie the mechanisms creating susceptibility to antleromas.
(ii). Factors that enhance female fertility may lead to elevated cancer risk
In many species, females invest typically more heavily in other components of reproductive effort rather than mating effort. Determinants of reproductive success for females often include components such as fertility timing and frequency, as well as size of the litter and investment in individual offspring. Our model is not sex-specific per se: although intraspecific competition (α) is probably lower for most females compared with males (reproduction is not of a ‘winner-takes-all’ nature), it still predicts that investment in traits that elevate reproductive competitiveness can elevate susceptibility to cancer, especially if very high reproductive output requires rapid cell proliferation (figure 4). Given that early reproductive maturity and rapid offspring production both require rapid cell proliferation, and may also involve the allocation of energy towards reproduction at the expense of DNA repair, we consider it possible that selection for improved reproductive success can lead to higher cancer risk in females, too.
Once again, detecting cases in the wild remains challenging: natural selection can yield life histories where most deaths occur before the expected onset of cancer (figure 4). Artificial breeding, however, offers instructive examples for revealing underlying trade-offs: intense selection for reproductive traits can heighten cancer risk considerably (figure 5). The domesticated Jungle fowl hen (Gallus gallus domesticus) has been artificially selected for daily ovulation and continuous egg-laying. This hen is the only known non-human animal with a high incidence of spontaneous ovarian cancer [36]. Ovarian cancer is observed in hens as young as 2 years of age, and 30–35% develop the highly malignant disease by 3.5 years of age [36,37].
4. Discussion
(a). Life-history trade-offs between cancer defences and reproduction
One way reproductive competition may be lowering cancer defences is through energy allocation trade-offs [38]. Organisms have limited energy to divide between soma maintenance and reproduction, with allocations dependent on life history (see ‘disposable soma theory’ [39]). Short-lived organisms allocate more resources towards reproduction in order to reproduce successfully within their lifespan, whereas investment in somatic maintenance (among them cancer defences) has allowed species to extend their lifespans [40,41]. When all else is equal, short-lived rodents are more prone to cancer [42,43] than are long-lived species, such as elephants and whales [3]. Indicative of lower defences, cells of shorter-lived organisms overcome intrinsic growth limitations more easily (i.e. transformation in cell culture) [44], allocate less time to DNA repair (short-lived species have more unresolved DNA damage in the form of micronuclei) [45] and proliferate rapidly in vitro [46]. Our results suggest that large-bodied animals must invest more in somatic maintenance, including tumour suppression, to build and maintain a large body for a sufficiently long time for reproduction to occur [3,47]. It has also been demonstrated that DNA break recognition is better in mammals with long lifespans [48]. Increasing lifespan may thus select for new tumour suppressor mechanisms or a shift in energy allocation towards more prolonged somatic tissue maintenance [49].
Some of the genetic and molecular mechanisms that mediate trade-offs between cancer defences and reproduction are becoming better understood. For example, the cancer suppressor genes P53 [11,12] and BRCA [13] are likely candidates given that mutations in these genes appear to be associated with enhanced fertility in some cases. Current evidence suggests that the P53 family of tumour suppressors (which includes also p63 and p73) may be involved in fertility and reproduction as well as cell cycle regulation [50]. Mouse models have demonstrated that p63 may have effects on the quality and survival of the oocyte pool [51], p73 plays a role in early blastocyst division [52], and p53 may help regulate the implantation of the fertilized egg as well as litter size in knockout mice [53].
DNA repair is a critical mechanism for cancer suppression and somatic maintenance [54,55]. BRCA1 and BRCA2 (breast cancer susceptibility genes 1 and 2) genes encode for tumour suppressor proteins important in DNA repair pathways [56]. Mutations in these genes increase the risk for breast and ovarian cancers [57]. Interestingly, one study demonstrated that women with BRCA1 and BRCA2 mutations have greater susceptibility to breast cancer, but also higher fertility. Women with BRCA mutations had significantly more children, shorter birth intervals and end childbearing later than the controls [13]. However, the mechanism for increased fertility among women with BRCA mutations is unknown and murine models of BRCA mutations do not support these findings [58].
Another potential example of an association between fertility and cancer risk involves the Kisspeptin (KISS1) gene in humans. Kisspeptin is a G protein coupled receptor ligand that is required for normal maturation and puberty. Defects in KISS1 lead to the absence of sexual maturation, indicating a critical role in the timing of puberty [59,60]. Kisspeptin also induces production of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) and is required for menstruation. Interestingly, KISS1 was first discovered as a metastasis suppressor gene and thought to play a role in suppression of metastasis of breast cancers and melanomas [61]. Recently, KISS1 has been also implicated in the inhibition of trophoblast invasion and angiogenesis in the placenta [62,63]. Although the exact mechanisms are yet to be determined, it may be that the capacity to suppress trophoblast invasion might also confer capacities to suppress cell invasion more generally and thus reduce the risk of invasive/metastatic cancer.
Similarities in the underlying biological mechanisms of cancer invasion and deep placentation, such as degradation of the extracellular matrix and angiogenesis, suggest an evolutionary link between these processes [64]. Effective placental invasion may increase fertility at the cost of higher cancer risk. Given that eutherian mammals vary in the degree of placental invasiveness, cancer risk would be expected to covary, and some evidence supports this prediction. Equines and bovines have the least invasive placenta type (epitheliochorial placentation) and were shown to have lower rates of metastatic cancer than felines and canines, who have characteristics of deeper placentation (endotheliochorial placentation) [65]. Shallower placentation has evolved numerous times across eutherian mammals [66], suggesting that females of those species have also evolved mechanisms to suppress trophoblast invasion, and these may be the same underlying mechanisms that suppress metastatic disease. Additional studies, linking metastatic cancer risk and placentation penetration using species with all placental types, including those with the most invasive haemochorial placentas, are needed to confirm this relationship.
(b). Environmental and ecological factors affect the strength of trade-offs
The strength of a trade-off between cancer defence and reproduction may be dramatically affected by ecological conditions, especially available resources (when the trade-off is due to energy allocation). Low resource levels have been found to increase the intensity of trade-offs between reproduction and immune function in a number of species [6], including the immunosuppressive effect of testosterone [67]. Differences in the intensity of trade-offs between reproduction and survival have been found in human populations with varying levels of socio-economic status, with more intense trade-offs between reproduction and longevity occurring in lower socio-economic status groups [68–70]. This suggests that environmental and ecological conditions are likely to play important roles in modulating the intensity of trade-offs between reproduction and cancer defence, especially when this trade-off is mediated by energy allocations.
In our model, we explored the implications of varying the intensity of the trade-off (β; figure 3). We found that in conditions with higher trade-offs owing to decreased resource availability (upper panels), elevated levels of cancer defence result in very low fitness, especially when extrinsic mortality is high. In contrast, in conditions with less intense trade-offs, corresponding to more plentiful resources, high levels of cancer defence result in extended lifespan and higher fitness. The importance of both resource levels and extrinsic mortality (which for non-humans may be caused by predation or other ecological factors) in determining the fitness of varying levels of cancer defence suggests that future work should consider these trade-offs in their ecological context.
(c). Selection for rapid growth may mediate sexually selected traits and cancer risk
Rapid growth may not only enhance within-species reproductive competitiveness, but also lead to greater cancer risk because of vulnerabilities associated with rapid cell proliferation. The development and expression of many key aspects of morphology and physiology influenced by sexual selection and reproduction are mediated by growth factors, including steroid hormones. Growth factors stimulate cell proliferation and play a pivotal role in increasing growth during development [71,72]. Growth factors are synthesized in most tissues in the body and their action is modulated by a network of molecules that promote cell cycle progression and inhibition of apoptosis [73]. Insulin signalling has been proposed as the mechanism by which a variety of organisms, from beetles to mammals, developmentally regulate expression of exaggerated structures, such as horns and antlers [74]. IGF has also been implicated in numerous cancer phenotypes [73,75,76]. Low levels of IGF-1 may extend the time to malignant proliferation [77]. IGF-1 mutant mouse models live longer and are resistant to cancer [78]. Increased levels of IGF may exacerbate cancer phenotypes through a reduction in apoptosis, an increase in cell turnover or an amplification of effects owing to DNA damage. In humans, mutations in growth hormone receptor genes have been found to be associated with lower cancer risk [79]. Serum from these individuals was found to reduce DNA breaks and increase apoptosis of human epithelial mammary cells in culture [79].
Hormones control many of the physiological processes involved in reproduction from development of sex organs to the timing and frequency of reproduction. Growth factors, and specifically steroid hormones, have been implicated in the growth and progression of many cancers [80–84]. Steroid hormones bind to nuclear receptors within cells and stimulate a complex signalling cascade involved in many cellular actions, including proliferation. Larger body size and larger ornaments or weapons can increase reproductive success; however, this may lead to sexual antagonism and favour reproductive competition over long-term survival [85,86]. One important component of tumour suppression is likely the suppression and slowing of somatic evolution. However, organismal-level selection for growth may relax constraints on somatic evolution within the body. Rapid proliferation may be instantiated through a shorter generation time, which could lead to a faster rate of somatic evolution. A consequence of this may be greater susceptibility to cancer in organisms selected for rapid growth.
(d). Does reproductive competition influence cancer risk in humans?
Although cancer in the wild is poorly studied, predictions from our model as well as a literature review find examples where sexually selected traits—e.g. body size in fish and antlers in deer—appear to influence cancer susceptibility in these species. Do we see similar trends in humans, a much better studied species with respect to cancer? Our model suggests that differences in overall cancer risk between men and women [87,88] might be partially explained by higher reproductive competition in men than women. Within sexes, reproductive competitiveness appears to be associated with cancer risk for a number of traits. Taller men are reproductively more successful than shorter men, indicating that there is active selection for stature in some human populations [89–91]. In general, taller humans have an increased risk for cancer susceptibility, including melanoma [92] and testicular cancer [93]. Rapid growth prior to reproductive maturity (i.e. becoming bigger faster) is also associated with cancer in humans. Early growth in adolescents and the rate of this growth can influence the risk of prostate cancer in men [94] and breast cancer in women [95]. Outcome of height in an individual depends on many growth factors, including IGF [96]. As noted in §4c, low levels of IGF-1 have been associated with a decreased incidence to several forms of cancer in animals and humans [76,97].
High testosterone levels are associated with increased aggressiveness, high reproductive effort and mating success [85,98–100]. Lifetime exposure to androgens, such as testosterone, appears to be associated with risk of prostate cancer in human males [101]. However, this relationship has been difficult to confirm, as prostate cancer typically has a late age of onset, and testosterone diminishes with age [102]. Additionally, expression levels of androgen receptor, which binds testosterone, may influence fertility in males [85,103] and are associated with an increased risk of prostate cancer [104]. There are open questions whether humans and other species can facultatively trade off between cancer suppression and reproductive competitiveness. Future work on adaptive calibration of physiology based on physical and social environmental inputs during development [105] could provide insights into these open questions.
Within humans, energy budgets can affect the strength of trade-offs between reproduction and somatic maintenance, as seen in figure 3. Forager–horticulturalists, Tsimane, of the Bolivian Amazon live in a resource-limited environment compared with those of industrialized societies. Tsimane men are therefore likely to have stronger trade-offs between reproduction (i.e. testosterone levels) and somatic maintenance (i.e. immune function). Tsimane men, who live in caloric restricted environments with high parasite load, have significantly lower baseline levels of salivary testosterone when compared with age-matched US males [106]. Further, infections, through exposures to pathogens and parasites dramatically decrease testosterone levels [107]. Together, these results suggest that with lower energy budgets there may be a stronger trade-off between somatic maintenance (in the form of immune function) and reproductive competitiveness (as measured by circulating testosterone).
Reproductive competitiveness, including fertility timing, frequency and attractiveness, may influence cancer susceptibility in human females. High oestrogen levels are associated with attractiveness in human females [108–110], and oestrogen is an important component of follicle maturation and oocyte quality [111]. However, the relationship between reproduction and cancer risk in human females is complex and differs for different types of cancer. Even within breast cancer, a recent meta-analysis showed hormone positive breast cancer has been associated with greater exposure to cyclical hormones through earlier menarche, later reproduction and lower parity, whereas hormone negative breast cancer is associated only with early menarche [112]. These findings suggest that hormone positive breast cancer risk may be explained by a mismatch between ancestral reproductive conditions and modern ones, with the result that present-day women experience higher exposure to hormones and suboptimally high rates of cell proliferation, resulting in increased vulnerability to cancer. Hormone negative breast cancer risk, on the other hand, might be mediated by other factors, including perhaps energetic trade-offs favouring reproduction over somatic maintenance.
Other factors that may influence a women's risk for cancer include effects on fertility and pregnancy. As stated earlier, women with BRCA1/2 mutations were shown to have significantly different reproductive profiles than age-matched controls. While BRCA1/2 mutations increase the risk for breast cancer, these women had significantly more children and shorter interbirth intervals [13], suggesting that there may be a fertility advantage for BRCA mutations, possibly accounting for the frequency of BRCA mutations in certain human populations [113]. Additionally, physiological processes involved in gestation, including placentation (as noted in §4a) could influence disease risk. Humans have the most invasive placental type, haemochorial. According to the positive pleiotropy hypothesis, which claims that placental invasiveness should be correlated with susceptibility to metastatic disease [65], a women's cancer risk may be higher with greater depth of placentation. Interestingly, a negative correlation has been found between pre-eclampsia, characterized by abnormally shallow placentation during pregnancy, and breast cancer risk [114,115]. However, this does not hold true for all populations studied [116], and there are likely additional factors influencing cancer susceptibility.
5. Conclusion
The role of reproductive investment as a determinant of cancer susceptibility is only beginning to be understood. Here, we contribute to this emerging understanding by presenting a model in which investment in reproductive competitiveness occurs in the presence of a trade-off with investment in cancer defence. We use this model to make predictions about cancer incidence within and across species in various ecological circumstances. Review of relevant literature reveals several examples that are consistent with the model assumptions and predictions. Future comparative oncology and genomic studies will be needed from species with diverse life histories to tease apart the molecular underpinnings that may influence both reproduction and cancer.
Acknowledgements
The authors gratefully acknowledge the support of the Wissenschaftskolleg zu Berlin (Institute for Advanced Study, Berlin) and the members of both the Cancer Evolution and Extreme Traits working groups for providing an intellectual environment that made this paper possible during the 2013–2014 academic year.
Funding statement
This work was supported in part by grants from the US National Institutes of Health (NIH) R01 CA170595 and the National Science Foundation (NSF) DEB 0952260 to G.S.W.
Authors' contributions
All of the authors contributed to the conception of the paper as well as the discovery and interpretation of relevant literature. H.K. developed and ran the model. A.M.B., C.A.A. and H.K. drafted the manuscript. G.S.W. and F.B. revised the manuscript for intellectual content. All authors have given final approval for the manuscript.
Competing interests
We have no competing interests.
References
- 1.Mix MC. 1986. Cancerous diseases in aquatic animals and their association with environmental pollutants: a critical literature review. Mar. Environ. Res. 20, 1–141. ( 10.1016/0141-1136(86)90013-9) [DOI] [Google Scholar]
- 2.Aktipis CA, Boddy AM, Jansen G, Hibner U, Hochberg ME, Maley CC, Wilkinson GS. 2015. Cancer across the tree of life: cooperation and cheating in multicellularity. Phil. Trans. R. Soc. B 370, 20140219 ( 10.1098/rstb.2014.0219) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caulin AF, Maley CC. 2011. Peto's paradox: evolution's prescription for cancer prevention. Trends Ecol. Evol. 26, 175–182. ( 10.1016/j.tree.2011.01.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leroi AM, Koufopanou V, Burt A. 2003. Cancer selection. Nat. Rev. Cancer 3, 226–231. ( 10.1038/nrc1016) [DOI] [PubMed] [Google Scholar]
- 5.Williams GC. 1957. Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411. ( 10.2307/2406060) [DOI] [Google Scholar]
- 6.Sandland GJ, Minchella DJ. 2003. Costs of immune defense: an enigma wrapped in an environmental cloak? Trends Parasitol. 19, 571–574. ( 10.1016/j.pt.2003.10.006) [DOI] [PubMed] [Google Scholar]
- 7.Sheldon BC, Verhulst S. 1996. Ecological immunology: costly parasite defences and trade-offs in evolutionary ecology. Trends Ecol. Evol. 11, 317–321. ( 10.1016/0169-5347(96)10039-2) [DOI] [PubMed] [Google Scholar]
- 8.Crespi BJ, Summers K. 2006. Positive selection in the evolution of cancer. Biol. Rev. 81, 407–424. ( 10.1017/S1464793106007056) [DOI] [PubMed] [Google Scholar]
- 9.Carter AJ, Nguyen AQ. 2011. Antagonistic pleiotropy as a widespread mechanism for the maintenance of polymorphic disease alleles. BMC Med. Genet. 12, 160 ( 10.1186/1471-2350-12-160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leroi AM, et al. 2005. What evidence is there for the existence of individual genes with antagonistic pleiotropic effects? Mech. Ageing Dev. 126, 421–429. ( 10.1016/j.mad.2004.07.012) [DOI] [PubMed] [Google Scholar]
- 11.Kang H-J, Feng Z, Sun Y, Atwal G, Murphy ME, Rebbeck TR, Rosenwaks Z, Levine AJ, Hu W. 2009. Single-nucleotide polymorphisms in the p53 pathway regulate fertility in humans. Proc. Natl Acad. Sci. USA 106, 9761–9766. ( 10.1073/pnas.0904280106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ungewitter E, Scrable H. 2009. Antagonistic pleiotropy and p53. Mech. Ageing Dev. 130, 10–17. ( 10.1016/j.mad.2008.06.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith KR, Hanson HA, Mineau GP, Buys SS. 2012. Effects of BRCA1 and BRCA2 mutations on female fertility. Proc. R. Soc. B 279, 1389–1395. ( 10.1098/rspb.2011.1697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersson MB. 1994. Sexual selection. Princeton, NJ: Princeton University Press. [Google Scholar]
- 15.Rankin DJ, Dieckmann U, Kokko H. 2011. Sexual conflict and the tragedy of the commons. Am. Nat. 177, 780–791. ( 10.1086/659947) [DOI] [PubMed] [Google Scholar]
- 16.Darwin C. 1871. The descent of man and selection in relation to sex. New York, NY: Appleton. [Google Scholar]
- 17.Fernandez AA, Bowser PR. 2010. Selection for a dominant oncogene and large male size as a risk factor for melanoma in the Xiphophorus animal model. Mol. Ecol. 19, 3114–3123. ( 10.1111/j.1365-294X.2010.04738.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernandez AA, Morris MR. 2008. Mate choice for more melanin as a mechanism to maintain a functional oncogene. Proc. Natl Acad. Sci. USA 105, 13 503–13 507. ( 10.1073/pnas.0803851105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon M. 1927. The genetics of a viviparous top-minnow Platypoecilus: the inheritance of two kinds of melanophores. Genetics 12, 253–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schartl M. 2008. Evolution of Xmrk: an oncogene, but also a speciation gene? Bioessays 30, 822–832. ( 10.1002/bies.20807) [DOI] [PubMed] [Google Scholar]
- 21.Summers K, Crespi BJ. 2010. Xmrks the spot: life history tradeoffs, sexual selection and the evolutionary ecology of oncogenesis. Mol. Ecol. 19, 3022–3024. ( 10.1111/j.1365-294X.2010.04739.x) [DOI] [PubMed] [Google Scholar]
- 22.Patton EE, Mathers ME, Schartl M. 2011. Generating and analyzing fish models of melanoma. Methods Cell Biol. 105, 339–366. ( 10.1016/B978-0-12-381320-6.00014-X) [DOI] [PubMed] [Google Scholar]
- 23.Li C, Zhao H, Liu Z, McMahon C. 2014. Deer antler–a novel model for studying organ regeneration in mammals. Int. J. Biochem. Cell Biol. 56, 111–122. ( 10.1016/j.biocel.2014.07.007) [DOI] [PubMed] [Google Scholar]
- 24.Suttie J, Fennessy P, Lapwood K, Corson I. 1995. Role of steroids in antler growth of red deer stags. J. Exp. Zool. 271, 120–130. ( 10.1002/jez.1402710207) [DOI] [PubMed] [Google Scholar]
- 25.Bubenik G, Schams D, Coenen G. 1987. The effect of artificial photoperiodicity and antiandrogen treatment on the antler growth and plasma levels of LH, FSH, testosterone, prolactin and alkaline phosphatase in the male white-tailed deer. Comp. Biochem. Physiol. A, Physiol. 87, 551–559. ( 10.1016/0300-9629(87)90359-8) [DOI] [PubMed] [Google Scholar]
- 26.Goss R. 1990. Tumor-like growth of antlers in castrated fallow deer: an electron microscopic study. Scann. Microsc. 4, 715–720; discussion 720–711. [PubMed] [Google Scholar]
- 27.Suttie JM, Gluckman PD, Butler JH, Fennessy PF, Corson ID, Laas FJ. 1985. Insulin-like growth factor 1 (IGF-1) antler-stimulating hormone? Endocrinology 116, 846–848. ( 10.1210/endo-116-2-846) [DOI] [PubMed] [Google Scholar]
- 28.Carrasco L, Fierro Y, Sánchez-Castillejo J, Hervas J, Perez J, Gómez-Villamandos J. 1997. Abnormal antler growth associated with testicular hypogonadism in red deer. J. Wildl. Dis. 33, 670–672. ( 10.7589/0090-3558-33.3.670) [DOI] [PubMed] [Google Scholar]
- 29.Demartini JC, Connolly GE. 1975. Testicular atrophy in Columbian black-tailed deer in California. J. Wildl. Dis. 11, 101–106. ( 10.7589/0090-3558-11.1.101) [DOI] [PubMed] [Google Scholar]
- 30.Goss RJ. 2012. Deer antlers: regeneration, function and evolution. San Diego, CA: Academic Press. [Google Scholar]
- 31.Kierdorf U, Li C, Price JS. 2009. Improbable appendages: deer antler renewal as a unique case of mammalian regeneration. In Seminars in cell and developmental biology, pp. 535–542. Elsevier. [DOI] [PubMed] [Google Scholar]
- 32.Munk B, Garrison E, Clemons B, Keel MK. 2014. Antleroma in a free-ranging white-tailed deer (Odocoileus virginianus). Vet. Pathol. 52, 213–216. ( 10.1177/0300985814528216) [DOI] [PubMed] [Google Scholar]
- 33.Taylor D, Thomas JW, Marburger R. 1964. Abnormal antler growth associated with hypogonadism in white-tailed deer in Texas. Am. J. Vet. Res. 25, 179–185. [PubMed] [Google Scholar]
- 34.Tiller BL, Dagle GE, Cadwell LL. 1997. Testicular atrophy in a mule deer population. J. Wildl. Dis. 33, 420–429. ( 10.7589/0090-3558-33.3.420) [DOI] [PubMed] [Google Scholar]
- 35.Veeramachaneni DR, Amann RP, Jacobson JP. 2006. Testis and antler dysgenesis in Sitka black-tailed deer on Kodiak Island, Alaska: sequela of environmental endocrine disruption? Environ. Health Perspect. 114, 51–59. ( 10.1289/ehp.8052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson PA, Giles JR. 2013. The hen as a model of ovarian cancer. Nat. Rev. Cancer 13, 432–436. ( 10.1038/nrc3535) [DOI] [PubMed] [Google Scholar]
- 37.Fredrickson T. 1987. Ovarian tumors of the hen. Environ. Health Perspect. 73, 35–51. ( 10.1289/ehp.877335) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stearns SC. 1989. Trade-offs in life-history evolution. Funct. Ecol. 3, 259–268. ( 10.2307/2389364) [DOI] [Google Scholar]
- 39.Kirkwood TB. 1977. Evolution of ageing. Nature 270, 301–304. ( 10.1038/270301a0) [DOI] [PubMed] [Google Scholar]
- 40.Campisi J. 2005. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120, 513–522. ( 10.1016/j.cell.2005.02.003) [DOI] [PubMed] [Google Scholar]
- 41.Lorenzini A, Stamato T, Sell C. 2011. The disposable soma theory revisited: time as a resource in the theories of aging. Cell Cycle 10, 3853–3856. ( 10.4161/cc.10.22.18302) [DOI] [PubMed] [Google Scholar]
- 42.Lipman R, Galecki A, Burke DT, Miller RA. 2004. Genetic loci that influence cause of death in a heterogeneous mouse stock. J. Gerontol. A, Biol. Sci. Med. Sci. 59, B977–B983. ( 10.1093/gerona/59.10.B977) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burek J, Hollander C. 1977. Incidence patterns of spontaneous tumors in BN/Bi rats. J. Natl Cancer Inst. 58, 99–105. [DOI] [PubMed] [Google Scholar]
- 44.Todaro GJ, Green H. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17, 299–313. ( 10.1083/jcb.17.2.299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fink LS, Roell M, Caiazza E, Lerner C, Stamato T, Hrelia S, Lorenzini A, Sell C. 2011. 53BP1 contributes to a robust genomic stability in human fibroblasts. Aging (Albany NY) 3, 836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seluanov A, Hine C, Bozzella M, Hall A, Sasahara TH, Ribeiro AA, Catania KC, Presgraves DC, Gorbunova V. 2008. Distinct tumor suppressor mechanisms evolve in rodent species that differ in size and lifespan. Aging Cell 7, 813–823. ( 10.1111/j.1474-9726.2008.00431.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roche B, Sprouffske K, Hbid H, Missé D, Thomas F. 2013. Peto's paradox revisited: theoretical evolutionary dynamics of cancer in wild populations. Evol. Appl. 6, 109–116. ( 10.1111/eva.12025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lorenzini A, Johnson FB, Oliver A, Tresini M, Smith JS, Hdeib M, Sell C, Cristofalo VJ, Stamato TD. 2009. Significant correlation of species longevity with DNA double strand break recognition but not with telomere length. Mech. Ageing Dev. 130, 784–792. ( 10.1016/j.mad.2009.10.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeGregori J. 2011. Evolved tumor suppression: why are we so good at not getting cancer? Cancer Res. 71, 3739–3744. ( 10.1158/0008-5472.CAN-11-0342) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. 2011. The p53 family: guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 12, 259–265. ( 10.1038/nrm3086) [DOI] [PubMed] [Google Scholar]
- 51.Suh E-K, et al. 2006. p63 protects the female germ line during meiotic arrest. Nature 444, 624–628. ( 10.1038/nature05337) [DOI] [PubMed] [Google Scholar]
- 52.Tomasini R, et al. 2008. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 22, 2677–2691. ( 10.1101/gad.1695308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu W. 2009. The role of p53 gene family in reproduction. Cold Spring Harb. Perspect. Biol. 1, a001073 ( 10.1101/cshperspect.a001073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hanahan D, Weinberg RA. 2000. The hallmarks of cancer. Cell 100, 57–70. ( 10.1016/S0092-8674(00)81683-9) [DOI] [PubMed] [Google Scholar]
- 55.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144, 646–674. ( 10.1016/j.cell.2011.02.013) [DOI] [PubMed] [Google Scholar]
- 56.Tutt A, Ashworth A. 2002. The relationship between the roles of BRCA genes in DNA repair and cancer predisposition. Trends Mol. Med. 8, 571–576. ( 10.1016/S1471-4914(02)02434-6) [DOI] [PubMed] [Google Scholar]
- 57.Chen S, Parmigiani G. 2007. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 25, 1329–1333. ( 10.1200/JCO.2006.09.1066) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu C-Y, Flesken-Nikitin A, Li S, Zeng Y, Lee W-H. 1996. Inactivation of the mouse Brca1 gene leads to failure in the morphogenesis of the egg cylinder in early postimplantation development. Genes Dev. 10, 1835–1843. ( 10.1101/gad.10.14.1835) [DOI] [PubMed] [Google Scholar]
- 59.Messager S, et al. 2005. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc. Natl Acad. Sci. USA 102, 1761–1766. ( 10.1073/pnas.0409330102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Skorupskaite K, George JT, Anderson RA. 2014. The kisspeptin-GnRH pathway in human reproductive health and disease. Hum. Reprod. Update 20, 485–500. ( 10.1093/humupd/dmu009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee J-H, Miele ME, Hicks DJ, Phillips KK, Trent JM, Weissman BE, Welch DR. 1996. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J. Natl Cancer Inst. 88, 1731–1737. ( 10.1093/jnci/88.23.1731) [DOI] [PubMed] [Google Scholar]
- 62.Hiden U, Bilban M, Knöfler M, Desoye G. 2007. Kisspeptins and the placenta: regulation of trophoblast invasion. Rev. Endocr. Metab. Disord. 8, 31–39. ( 10.1007/s11154-007-9030-8) [DOI] [PubMed] [Google Scholar]
- 63.Cartwright JE, Williams PJ. 2012. Altered placental expression of kisspeptin and its receptor in pre-eclampsia. J. Endocrinol. 214, 79–85. ( 10.1530/JOE-12-0091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stearns SC. 2012. Evolutionary medicine: its scope, interest and potential. Proc. R. Soc. B 279, 4305–4321. ( 10.1098/rspb.2012.1326) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.D'Souza AW, Wagner GP. 2014. Malignant cancer and invasive placentation A case for positive pleiotropy between endometrial and malignancy phenotypes. Evol. Med. Public Health 2014, 136–145. ( 10.1093/emph/eou022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wildman DE, Chen C, Erez O, Grossman LI, Goodman M, Romero R. 2006. Evolution of the mammalian placenta revealed by phylogenetic analysis. Proc. Natl Acad. Sci. USA 103, 3203–3208. ( 10.1073/pnas.0511344103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Braude S, Tang-Martinez Z, Taylor GT. 1999. Stress, testosterone, and the immunoredistribution hypothesis. Behav. Ecol. 10, 345–350. ( 10.1093/beheco/10.3.345) [DOI] [Google Scholar]
- 68.Dribe M. 2004. Long-term effects of childbearing on mortality: evidence from pre-industrial Sweden. Popul. Stud. 58, 297–310. ( 10.1080/0032472042000272357) [DOI] [PubMed] [Google Scholar]
- 69.Lycett JE, Dunbar R, Voland E. 2000. Longevity and the costs of reproduction in a historical human population. Proc. R. Soc. Lond. B 267, 31–35. ( 10.1098/rspb.2000.0962) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jasienska G. 2009. Reproduction and lifespan: trade-offs, overall energy budgets, intergenerational costs, and costs neglected by research. Am. J. Hum. Biol. 21, 524–532. ( 10.1002/ajhb.20931) [DOI] [PubMed] [Google Scholar]
- 71.Juul A, Dalgaard P, Blum WF, Bang P, Hall K, Michaelsen KF, Müller J, Skakkebæk NE. 1995. Serum levels of insulin-like growth factor (IGF)-binding protein-3 (IGFBP-3) in healthy infants, children, and adolescents: the relation to IGF-I, IGF-II, IGFBP-1, IGFBP-2, age, sex, body mass index, and pubertal maturation. J. Clin. Endocrinol. Metab. 80, 2534–2542. ( 10.1210/jc.80.8.2534) [DOI] [PubMed] [Google Scholar]
- 72.Butler AA, Roith DL. 2001. Control of growth by the somatropic axis: growth hormone and the insulin-like growth factors have related and independent roles. Annu. Rev. Physiol. 63, 141–164. ( 10.1146/annurev.physiol.63.1.141) [DOI] [PubMed] [Google Scholar]
- 73.Moschos SJ, Mantzoros CS. 2001. The role of the IGF system in cancer: from basic to clinical studies and clinical applications. Oncology 63, 317–332. ( 10.1159/000066230) [DOI] [PubMed] [Google Scholar]
- 74.Emlen DJ, Warren IA, Johns A, Dworkin I, Lavine LC. 2012. A mechanism of extreme growth and reliable signaling in sexually selected ornaments and weapons. Science 337, 860–864. ( 10.1126/science.1224286) [DOI] [PubMed] [Google Scholar]
- 75.Chaves J, Saif MW. 2011. IGF system in cancer: from bench to clinic. Anti-cancer Drugs 22, 206–212. ( 10.1097/CAD.0b013e32834258a1) [DOI] [PubMed] [Google Scholar]
- 76.Renehan AG, Zwahlen M, Minder C, O'Dwyer S, Shalet SM, Egger M. 2004. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet 363, 1346–1353. ( 10.1016/S0140-6736(04)16044-3) [DOI] [PubMed] [Google Scholar]
- 77.Milman S, Atzmon G, Huffman DM, Wan J, Crandall JP, Cohen P, Barzilai N. 2014. Low insulin-like growth factor-1 level predicts survival in humans with exceptional longevity. Aging Cell 13, 769–771. ( 10.1111/acel.12213) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Anisimov VN, Bartke A. 2013. The key role of growth hormone–insulin–IGF-1 signaling in aging and cancer. Crit. Rev. Oncol. Hematol. 87, 201–223. ( 10.1016/j.critrevonc.2013.01.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guevara-Aguirre J, et al. 2011. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 3, 70ra13 ( 10.1126/scitranslmed.3001845) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Group EH.BC.C. 2003. Body mass index, serum sex hormones, and breast cancer risk in postmenopausal women. J. Natl Cancer Inst. 95, 1218–1226. ( 10.1093/jnci/djg022) [DOI] [PubMed] [Google Scholar]
- 81.Holly J, Gunnell D, Smith GD. 1999. Growth hormone, IGF-I and cancer. Less intervention to avoid cancer? More intervention to prevent cancer? J. Endocrinol. 162, 321–330. ( 10.1677/joe.0.1620321) [DOI] [PubMed] [Google Scholar]
- 82.Hyde Z, Flicker L, McCaul KA, Almeida OP, Hankey GJ, Chubb SP, Yeap BB. 2012. Associations between testosterone levels and incident prostate, lung, and colorectal cancer. A population-based study. Cancer Epidemiol. Biomark. Prev. 21, 1319–1329. ( 10.1158/1055-9965.EPI-12-0129) [DOI] [PubMed] [Google Scholar]
- 83.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. 2008. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 371, 569–578. ( 10.1016/S0140-6736(08)60269-X) [DOI] [PubMed] [Google Scholar]
- 84.Stingl J. 2011. Estrogen and progesterone in normal mammary gland development and in cancer. Horm. Cancer 2, 85–90. ( 10.1007/s12672-010-0055-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Summers K, Crespi B. 2008. The androgen receptor and prostate cancer: a role for sexual selection and sexual conflict? Med. Hypotheses 70, 435–443. ( 10.1016/j.mehy.2007.04.044) [DOI] [PubMed] [Google Scholar]
- 86.Trivers R. 1972 Parental investment and sexual selection. In Sexual selection and the descent of man 1871–1971 (ed. BG Campbell), pp. 136–179. Chicago, IL: Aldine. [Google Scholar]
- 87.Dorak MT, Karpuzoglu E. 2012. Gender differences in cancer susceptibility: an inadequately addressed issue. Front. Genet. 3, 268 ( 10.3389/fgene.2012.00268) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Syed DN, Mukhtar H. 2012. Gender bias in skin cancer: role of catalase revealed. J. Invest. Dermatol. 132, 512–514. ( 10.1038/jid.2011.423) [DOI] [PubMed] [Google Scholar]
- 89.Pawlowski B, Dunbar R, Lipowicz A. 2000. Evolutionary fitness: tall men have more reproductive success. Nature 403, 156 ( 10.1038/35003107) [DOI] [PubMed] [Google Scholar]
- 90.Nettle D. 2002. Height and reproductive success in a cohort of British men. Hum. Nat. 13, 473–491. ( 10.1007/s12110-002-1004-7) [DOI] [PubMed] [Google Scholar]
- 91.Mueller U, Mazur A. 2001. Evidence of unconstrained directional selection for male tallness. Behav. Ecol. Sociobiol. 50, 302–311. ( 10.1007/s002650100370) [DOI] [Google Scholar]
- 92.Shors AR, Solomon C, McTiernan A, White E. 2001. Melanoma risk in relation to height, weight, and exercise (United States). Cancer Causes Control 12, 599–606. ( 10.1023/A:1011211615524) [DOI] [PubMed] [Google Scholar]
- 93.Rasmussen F, Gunnell D, Ekbom A, Hallqvist J, Tynelius P. 2003. Birth weight, adult height, and testicular cancer: cohort study of 337,249 Swedish young men. Cancer Causes Control 14, 595–598. ( 10.1023/A:1024860826830) [DOI] [PubMed] [Google Scholar]
- 94.Giles GG, Severi G, English DR, McCredie MR, MacInnis R, Boyle P, Hopper JL. 2003. Early growth, adult body size and prostate cancer risk. Int. J. Cancer 103, 241–245. ( 10.1002/ijc.10810) [DOI] [PubMed] [Google Scholar]
- 95.Ahlgren M, Melbye M, Wohlfahrt J, Sørensen TI. 2004. Growth patterns and the risk of breast cancer in women. N. Engl. J. Med. 351, 1619–1626. ( 10.1056/NEJMoa040576) [DOI] [PubMed] [Google Scholar]
- 96.Robson H, Siebler T, Shalet SM, Williams GR. 2002. Interactions between GH, IGF-I, glucocorticoids, and thyroid hormones during skeletal growth. Pediatr. Res. 52, 137–147. ( 10.1203/00006450-200208000-00003) [DOI] [PubMed] [Google Scholar]
- 97.Ikeno Y, Bronson RT, Hubbard GB, Lee S, Bartke A. 2003. Delayed occurrence of fatal neoplastic diseases in Ames dwarf mice: correlation to extended longevity. J. Gerontol. A, Biol. Sci. Med. Sci. 58, B291–B296. ( 10.1093/gerona/58.4.B291) [DOI] [PubMed] [Google Scholar]
- 98.Archer J. 2006. Testosterone and human aggression: an evaluation of the challenge hypothesis. Neurosci. Biobehav. Rev. 30, 319–345. ( 10.1016/j.neubiorev.2004.12.007) [DOI] [PubMed] [Google Scholar]
- 99.Gettler LT, McDade TW, Feranil AB, Kuzawa CW. 2011. Longitudinal evidence that fatherhood decreases testosterone in human males. Proc. Natl Acad. Sci. USA 108, 16 194–16 199. ( 10.1073/pnas.1105403108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Peters M, Simmons LW, Rhodes G. 2008. Testosterone is associated with mating success but not attractiveness or masculinity in human males. Anim. Behav. 76, 297–303. ( 10.1016/j.anbehav.2008.02.008) [DOI] [Google Scholar]
- 101.Alvarado LC. 2013. Do evolutionary life-history trade-offs influence prostate cancer risk? A review of population variation in testosterone levels and prostate cancer disparities. Evol. Appl. 6, 117–133. ( 10.1111/eva.12036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vermeulen A, Rubens R, Verdonck L. 1972. Testosterone secretion and metabolism in male senescence. J. Clin. Endocrinol. Metab. 34, 730–735. ( 10.1210/jcem-34-4-730) [DOI] [PubMed] [Google Scholar]
- 103.Wang R-S, Yeh S, Tzeng C-R, Chang C. 2009. Androgen receptor roles in spermatogenesis and fertility: lessons from testicular cell-specific androgen receptor knockout mice. Endocr. Rev. 30, 119–132. ( 10.1210/er.2008-0025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Heinlein CA, Chang C. 2004. Androgen receptor in prostate cancer. Endocr. Rev. 25, 276–308. ( 10.1210/er.2002-0032) [DOI] [PubMed] [Google Scholar]
- 105.Del Giudice M, Ellis BJ, Shirtcliff EA. 2011. The adaptive calibration model of stress responsivity. Neurosci. Biobehav. Rev. 35, 1562–1592. ( 10.1016/j.neubiorev.2010.11.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Trumble BC, Cummings D, von Rueden C, O'Connor KA, Smith EA, Gurven M, Kaplan H. 2012. Physical competition increases testosterone among Amazonian forager-horticulturalists: a test of the ‘challenge hypothesis’. Proc. R. Soc. B 279, 2907–2912. ( 10.1098/rspb.2012.0455) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Trumble BC, Brindle E, Kupsik M, O'Connor KA. 2010. Responsiveness of the reproductive axis to a single missed evening meal in young adult males. Am. J. Hum. Biol. 22, 775–781. ( 10.1002/ajhb.21079) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Henss R. 2000. Waist-to-hip ratio and female attractiveness. Evidence from photographic stimuli and methodological considerations. Pers. Ind. Differences 28, 501–513. ( 10.1016/S0191-8869(99)00115-4) [DOI] [Google Scholar]
- 109.Jasienska G, Lipson SF, Ellison PT, Thune I, Ziomkiewicz A. 2006. Symmetrical women have higher potential fertility. Evol. Hum. Behav. 27, 390–400. ( 10.1016/j.evolhumbehav.2006.01.001) [DOI] [Google Scholar]
- 110.Singh D. 1993. Body shape and women's attractiveness. Hum. Nat. 4, 297–321. ( 10.1007/BF02692203) [DOI] [PubMed] [Google Scholar]
- 111.Revelli A, Delle Piane L, Casano S, Molinari E, Massobrio M, Rinaudo P. 2009. Follicular fluid content and oocyte quality: from single biochemical markers to metabolomics. Reprod. Biol. Endocrinol. 7, 4330–4337. ( 10.1186/1477-7827-7-40) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Aktipis CA, Ellis BJ, Nishimura KK, Hiatt RA. 2014. Modern reproductive patterns associated with estrogen receptor positive but not negative breast cancer susceptibility. Evol. Med. Public Health B 1, 52–74. ( 10.1093/emph/eou028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Whittemore AS, Gong G, John EM, McGuire V, Li FP, Ostrow KL, DiCioccio R, Felberg A, West DW. 2004. Prevalence of BRCA1 mutation carriers among US non-Hispanic Whites. Cancer Epidemiol. Biomark. Prev. 13, 2078–2083. [PubMed] [Google Scholar]
- 114.Kim JS, et al. 2013. The relationship between preeclampsia, pregnancy-induced hypertension and maternal risk of breast cancer: a meta-analysis. Acta Oncologica (Stockholm, Sweden) 52, 1643–1648. ( 10.3109/0284186x.2012.750033) [DOI] [PubMed] [Google Scholar]
- 115.Vatten L, Romundstad P, Trichopoulos D, Skjaerven R. 2002. Pre-eclampsia in pregnancy and subsequent risk for breast cancer. Br. J. Cancer 87, 971–973. ( 10.1038/sj.bjc.6600581) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Calderon-Margalit R, Friedlander Y, Yanetz R, Deutsch L, Perrin MC, Kleinhaus K, Tiram E, Harlap S, Paltiel O. 2009. Preeclampsia and subsequent risk of cancer: update from the Jerusalem Perinatal Study. Am. J. Obstet. Gynecol. 200, e61–e68. ( 10.1016/j.ajog.2008.06.057) [DOI] [PMC free article] [PubMed] [Google Scholar]