

Abstract
Background
Recent advances in molecular genetics have enabled to determine the genetic causes of non-syndromic hearing loss, and more than 100 genes have been related to the phenotype. Due to this extraordinary genetic heterogeneity, a large percentage of patients remain without any molecular diagnosis. This condition imply the need for new methodological strategies in order to detect a greater number of mutations in multiple genes. In this work, we optimized and tested a panel of 86 mutations in 17 different genes screened using a high-throughput genotyping technology to determine the molecular etiology of hearing loss.
Methods
The technology used in this work was the MassARRAY iPLEX® platform. This technology uses silicon chips and DNA amplification products for accurate genotyping by mass spectrometry of previous reported mutations. The generated results were validated using conventional techniques, as direct sequencing, multiplex PCR and RFLP-PCR.
Results
An initial genotyping of control subjects, showed failures in 20 % of the selected alterations. To optimize these results, the failed tests were re-designed and new primers were synthesized. Then, the specificity and sensitivity of the panel demonstrated values above 97 %. Additionally, a group of 180 individuals with NSHL without a molecular diagnosis was screened to test the diagnostic value of our panel, and mutations were identified in 30 % of the cases. In 20 % of the individuals, it was possible to explain the etiology of the HL. Mutations in GJB2 gene were the most prevalent, followed by other mutations in in SLC26A4, CDH23, MT-RNR1, MYO15A, and OTOF genes.
Conclusions
The MassARRAY technology has the potential for high-throughput identification of genetic variations. However, we demonstrated that optimization is required to increase the genotyping success and accuracy. The developed panel proved to be efficient and cost-effective, being suitable for applications involving the molecular diagnosis of hearing loss.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-015-0232-8) contains supplementary material, which is available to authorized users.
Keywords: Hearing loss, Genetic testing, MassARRAY
Background
Hearing loss (HL) is one of the most common sensory disorders, affecting around one in a thousand individuals, and can be caused by a variety of environmental and genetic factors [1]. In recent years, there have been significant advances in understanding the genetic causes of HL, which has assisted in disease diagnosis and other clinical practices.
Hereditary HL can be classified as syndromic or non-syndromic. The syndromic type is associated with distinctive clinical features and accounts for 30 % of congenital hereditary HL. Hearing loss that occurs in the absence of any other abnormal physical findings is known as non-syndromic hearing loss (NSHL) and accounts for the other 70 % [2–4].
More than 80 genes have been associated with NSHL [5–8]. Due to this high genetic heterogeneity, there is still great difficulty in determining the molecular etiology of HL. The most frequent genes associated with autosomal recessive inheritance that have been identified in NSHL are (in order of frequency) GJB2, SLC26A4, MYO15A, OTOF, CDH23, and TMC1 [9, 10]. For each of these genes, at least 20 mutations have been reported. One specific mutation, c.35delG in the GJB2 gene, is the most frequent in Caucasians, found in from 10 to 63 % of homozygous NSHL cases [11, 12]. None of the genes associated with non-syndromic autosomal dominant HL are very frequent, although the most common are WFS1, KCNQ4, COCH, and GJB2 [9].
Identification of hearing impairment is extremely important because it allows positive cases to be properly referred for medical intervention and/or rehabilitation programs, and enables genetic counseling of the families affected. The extreme genetic heterogeneity of NSHL makes genetic diagnosis based on Sanger sequencing impractical, because this is a very expensive and time-consuming technique, besides not being feasible for genes with several exons. Thus, only a small number of genes are currently screened for determining the cause of HL, while a large percentage of patients remain without any genetic diagnosis. This indicates the need for new methodological strategies for detection of a greater number of mutations in multiple genes.
With the emergence of high-throughput technologies, this gap is being filled due the possibility to perform multiple simultaneous analyses, using small volumes of samples and reagents in the reactions, combining high accuracy with simplicity [13]. One of this promising technologies is the MassARRAY® platform (Sequenom Inc., San Diego, USA), that provides rapid measurement of DNA products, with modest multiplexing and minimal assay setup costs due to the use of unmodified oligonucleotide primers.
In order to address the genetic heterogeneity of HL and the labor and expense of conventional techniques, we developed a panel for diagnosis of non-syndromic hearing loss based on genotyping of 86 of the most frequent mutations that have already been described, using the MassARRAY® iPLEX mass spectrometry system. The power of the panel to detect mutations was demonstrated by applying it to 180 patients with presumed NSHL.
Methods
Patient recruitment and assessment
The study was carried out at the Human Molecular Genetics Laboratory of the Molecular Biology and Genetic Engineering Center (CBMEG) of the University of Campinas, in collaboration with the ENT department of the University of Campinas Teaching Hospital (São Paulo, Brazil). Blood samples were collected for genetic testing after obtaining written informed consent. The project was previously approved by the Research Ethics Committee of the Faculty of Medical Sciences of the University of Campinas (Report number 396/2006).
The technique was standardized using negative and positive control samples from 25 affected individuals for whom previous sequencing results were available for the GJB2 gene and/or the SLC26A4 gene, and for the m.1555A > G mitochondrial mutation in the MT-RNR1 gene. Additionally, analysis was made of 180 unrelated Brazilian individuals with NSHL and without a molecular diagnosis. Inclusion criteria were: (1) bilateral HL, (2) no apparent syndromic features, (3) existence of audiometric and physical examinations, (4) a complete history to rule out obvious environmental causes of HL, and (5) onset of HL at age <50 years.
Obvious environmental causes of HL identified by the case history and physical examination included: prenatal factors (measles and CMV infections, and exposure to teratogenic agents during pregnancy), perinatal factors (anoxia and prematurity/care in a neonatal intensive care unit), postnatal factors (meningitis, otitis and head trauma), and other clinical features (otosclerosis, osteogenesis imperfecta, Usher syndrome, and Ménière’s disease).
For those patients with postlingual HL, hearing loss severity was defined according to pure-tone threshold hearing levels: mild (26–40 dB); moderate (41–70 dB); severe (71–90 dB); and profound (>90 dB) [14]. OAE, BERA, and infant audiometric testing were used to diagnose children with prelingual HL. All subjects were considered to have moderate to profound bilateral sensorioneural HL.
Sample preparation
Genomic DNA was extracted from the leukocytes present in 4–8 mL of peripheral blood. The extraction was performed according to standard phenol-chloroform protocols. The purity and concentration of the samples were checked using a NanoDrop® 8000 spectrophotometer (Thermo Scientific) and a Qubit® 2.0 fluorometer (Invitrogen), respectively. Genomic DNA samples were diluted to obtain a final concentration of 10 ng/μL.
The Sequenom MassARRAY® iPLEX platform
This technology uses silicon chips and DNA amplification products for accurate genotyping by mass spectrometry. Genomic DNAs are submitted to iPLEX Gold reaction and the product are transferred to chip wells by a robot. The genotypes are detected in situ by using matrix-assisted laser desorption ionization mass spectrometry (MALDI-TOF). This miniaturized method has the potential for accurate, high-throughput, low-cost identification of genetic variations [15].
The iPLEX Gold technology consists of an initial locus-specific PCR reaction, followed by single base extension (SBE) using mass-modified dideoxynucleotide terminators of an oligonucleotide primer, which anneals immediately upstream of the polymorphic site of interest (Fig. 1) [15]. The product of these reactions are directly applied in a silicon chip. The mass of the extended primer is determined by means of MALDI-TOF mass spectrometry. The mass of the primer indicates the mutation of interest and the mass of added bases indicate the alleles present at the polymorphic site. Sequenom supplies a software (SpectroTYPER) that automatically translates the mass of the observed primers into a genotype for each reaction [16].
Fig. 1.
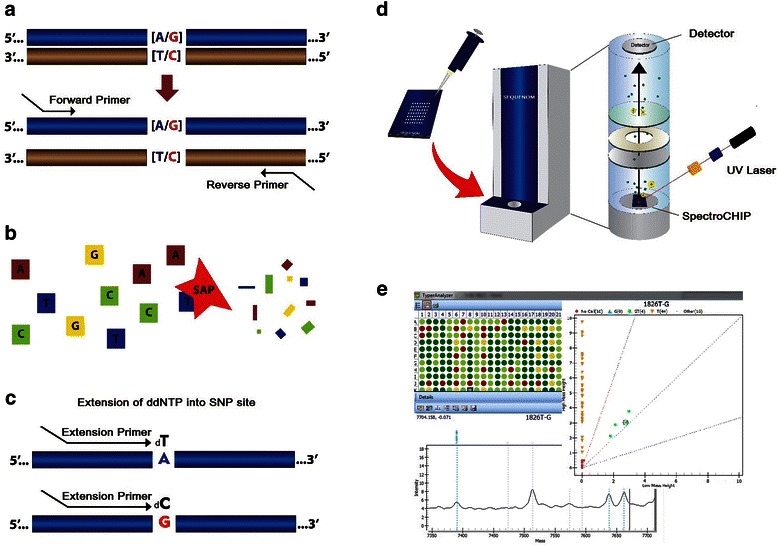
Steps in genotyping using MassARRAY® iPLEX System. A schematic of the genotype reaction of an A-to-G SNP. a. Locus-specific amplification reaction b. Treatment with SAP enzyme to neutralize unincorporated dNTPs. c. Locus-specific primer extension reaction (iPLEX assay). In this reaction, an oligonucleotide primer anneals immediately upstream of the polymorphic site being genotyped, the primer and amplified target DNA are incubated with mass-modified dideoxynucleotide terminators. The primer extension is made according to the sequence of the variant site. d. The products of the reactions are spotted on a spectroCHIP. The CHIP is placed into the mass spectrometer and each spot is then shot with a laser under vacuum by the matrix-assisted laser desorption ionization–time-of-flight (MALDI-TOF) method. A laser beam serves as desorption and ionization source in MALDI mass spectrometry. Once the sample molecules are vaporized and ionized, they are transferred electrostatically into a time-of-flight mass spectrometer (TOF-MS), where they individually detected. The mass of the extended primer is determined. e. Sequenom supplies software that automatically translates the mass of the observed primers into a genotype for each reaction
Mutation selection and assay design
After a bibliographic search for genetic alterations related to HL in different populations, 94 alterations in 17 genes were selected for development of the panel (Table 1). The choice of genes to be studied was based on the most important genes involved in HL [9]. All these genes were described as affecting protein structures related to hearing, and were selected based on the frequency of reported mutations.
Table 1.
Patterns of inheritance, genes and number of alterations selected for the panel
| Inheritance | Gene | Locus | No. of selected alterations | OMIM | Transcripts IDs |
|---|---|---|---|---|---|
| Autosomal recessive | GJB2 | DFNB1 | 22 | 121011 | NM_004004 |
| SLC26A4 | DFNB4 | 14 | 605646 | NM_000441 | |
| MYO15A | DFNB3 | 5 | 602666 | NM_016239 | |
| OTOF | DFNB9 | 16 | 603681 | NM_194248 | |
| CDH23 | DFNB12 | 11 | 605516 | NM_022124 | |
| TMC1 | DFNB7/11 | 4 | 606706 | NM_138691 | |
| TMPRSS3 | DFNB8/10 | 5 | 605511 | NM_024022 | |
| TRIOBP | DFNB28 | 1 | 609761 | NM_001039141 | |
| TMIE | DFNB6 | 2 | 607237 | NM_147196 | |
| DFNB59 | DFNB59 | 1 | 610219 | NM_001042702 | |
| Autosomal dominant | GJB2 | DFNA3 | 1 | 121011 | NM_004004 |
| WFS1 | DFNA6/14 | 1 | 606201 | NM_006005.3 | |
| KCNQ4 | DFNA2 | 1 | 603537 | NM_004700 | |
| COCH | DFNA9 | 1 | 603196 | NM_004086 | |
| TECTA | DFNA8/12 | 4 | 602574 | NM_005422 | |
| MIR-96 | DFNA50 | 2 | 613074 | NR_029512 | |
| Mitochondrial | MT-RNR1 | 12S rRNA | 2 | 561000 | - |
| MT-TS1 | tRNASer(UCN) | 1 | 590080 | - |
The alterations in the GJB2 gene were selected based on files for about 2500 individuals with HL, who had been previously screened at Human Molecular genetics Laboratory at CBMEG (Campinas/Brazil). For other genes, mutations were selected based on how often they were reported in the literature, and only mutations that had been found in two or more families were included. Regarding the OTOF, SLC26A4, and MYO15A genes, all mutations that were identified in the Brazilian population were selected [17–19]. To check all selected alterations, see Additional file 1.
It was not possible to elaborate assays for the del(GJB6-D13S1830) and del(GJB6-D13S1854) deletions in the GJB6 gene, because the MassARRRAY spectrometry technique presents a limitation to detect large delections/insertions.
The sequences covering the selected alterations were taken from the database of the National Center for Biotechnology Information [http://www.ncbi.nlm.nih.gov/] and Ensembl [http://www.ensembl.org/index.html]. Amplification and extension primers were designed using MassARRAY Assay Design v. 4.0 software. To avoid interaction among the primers, the software divided the PCR amplification and the Single Base Extension (SBE) into multiplex reactions. At the end of the standardization, 86 assays functioned properly, and the mutations were divided into 8 groups, containing 20, 18, 14, 9, 9, 8, 5 and 3 assays, respectively.
Genotyping using the Sequenom MassARRAY® iPLEX platform
The whole process was performed according to the manufacturer’s instructions for the multiplex reactions, including the PCR amplification, the Shrimp Alkaline Phosphatase (SAP) treatment, and the primer extension reactions using the iPLEX Gold assay (Sequenom Inc., San Diego, USA). The reaction products were then dispensed onto a 384-SpectroCHIP using the MassARRAY nanodispenser and analyzed using the MassARRAY platform. Mass signals for the different alleles were captured with high accuracy by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). Typer v. 4.0 (Sequenom Inc., San Diego, USA) was used to process the raw data obtained from the assays.
Specificity and sensitivity calculations
The calculations were made individually for 38 alterations that had been previously detected in the control group. Specificity is considered the proportion of true negatives that are correctly identified by the test [20]. The calculation was made by the fraction of the genotypes who do not have mutations (True Negatives) to those who truly do not have mutations (True Negatives + False Positives).
Sensitivity is the proportion of true positives that are correctly identified by the test [20]. Sensitivity is a fraction of genotypes with mutations detected by the test (True Positives) among those who actually have mutations (True Positives + False Negatives)
Validation of the results
All samples were tested for the mutations c.35delG and c.-23 + 1G > A (commonly known as IVS1 + 1G > A) in the GJB2 gene, the mutations del(GJB6-D13S1830) and del(GJB6-D13S1854) in the GJB6 gene, and the m.1555A > G mutation in the MTRNR1 gene.
Allele-specific PCR was used to detect the c.35delG mutation, as described elsewhere [21]. GJB2 mutations were screened by direct sequencing of the coding region of the gene [22, 23]. A multiplex PCR methodology was used to detect del(GJB6-D13S1830) and del(GJB6-D13S1854) mutations, according to the procedures reported previously [24, 25]. Analysis of m.1555A > G was performed by PCR amplification followed by digestion with the BsmAI restriction endonuclease [26]. To validate mutations found in other genes, only the corresponding exon containing the mutation was sequenced.
Results
Optimization
In the first genotyping using mass spectrometry of 15 control patients in triplicate, the genotyping call was calculated to be 74 %. Additionally, in all the control samples, 19 out of 94 mutations presented a failure.
In an attempt to improve the results, and minimize the occurrence of primer interactions and incorrect primer annealing, we diminished the number of mutations to be screened in each multiplex reaction, and the maximum number of mutations was limited to 20 for each well. In addition, the extension primers of all the 19 reactions that failed were redesigned in reverse sequences using the MassARRAY Assay Design software. This resulted in a greater number of groups of multiplex reactions (eight rather than six) used to analyze all the 94 mutations in each patient.
After optimization, new screening of 25 individuals of the control group was performed indicating that the genotyping call rate of the technique increased to 91 % and the sensitivity and specificity of the panel exceeded 97 %. These calculations were made comparing results of 38 mutations previously analyzed by Sanger sequencing with those results obtained by MassARRAY (Table 2). Following primer redesign, of 19 assays that were not working, 11 assays started to function properly, but 8 assays still failed (Table 3). In seven of these assays, no allele was detected, and one assay (p.V153I in the GJB2 gene) continued to show false negative results, even after redesigning the extension primer. These eight mutations were excluded from the panel and were not considered in subsequent analyses.
Table 2.
Comparison of genotyping results of 38 alterations obtained by MassARRAY® iPLEX system before and after optimization of the panel
| TP | FP | TN | FN | GC | SP | SN | |
|---|---|---|---|---|---|---|---|
| Before optimization (%) | 3.2 | 3.1 | 93.23 | 0.47 | 74 | 97 | 87 |
| After optimization (%) | 4.04 | 1.96 | 93.89 | 0.12 | 91 | 98 | 97 |
TP True positives, FP False positives, TN True negatives, FN False negatives, GC Genotyping Call, SP Specificity, SN Sensitivity
Table 3.
Mutations that failed and had the assays excluded of the panel
| Gene | Alteration | Protein change |
|---|---|---|
| GJB2 | c.283G > A | p.V95M |
| GJB2 | c.457G > A | p.V153I |
| MYO15A | c.6796G > A | p.V2266M |
| OTOF | c.2122C > T | p.R708* |
| TMPRSS3 | c.1221C > T | p.P404L |
| TECTA | c.3107G > A | p.C1036Y |
| TMIE | c.241C > T | p.R81C |
| CDH23 | c.6133G > A | p.D2045N |
*translation termination (stop) codon.
Mutation screening
After standardizing the panel with 86 mutations, this set of variants was screened using the mass spectrometry system in a group of 180 individuals with moderate to profound non-syndromic HL. The technique identified genetic alterations in seven genes in a total of 54 individuals (Table 4). Mutations in the GJB2 gene were found in 43 cases, with c.35delG being the most prevalent, identified in at least one allele in 28 individuals. The gene with the second largest number of mutations detected by mass spectrometry in our sample was SLC26A4, with p.V609G being the most frequent variant (detected in four cases). Mutations in the MT-RNR1, MT-TS1, OTOF, MYO15A, and CDH23 genes were also identified.
Table 4.
Molecular alterations detected in 180 individuals
| No. of cases | Alterations detected | Gene(s) | Onset | Severity |
|---|---|---|---|---|
| 20 | c.35delG/c.35delG | GJB2 | Prelingual | Severe/profound |
| 3 | p.V27I/wt | GJB2 | Prelingual/postlingual | Moderade/profound |
| 2 | c.35delG/wt | GJB2 | Postlingual | Moderade/profound |
| 2 | p.M34T/wt | GJB2 | Prelingual/postlingual | Moderade/profound |
| 2 | p.V609G/wt | SLC26A4 | Prelingual/postlingual | Severe/profound |
| 2 | m.1555A > G | MT-RNR1 | Postlingual | Moderade/profound |
| 1 | c.35delG/wt; ΔGJB6-D13S1830/wt | GJB2; GJB6 | Prelingual | Profound |
| 1 | c.35deG/p.L90P | GJB2 | Postlingual | Moderade |
| 1 | c.35delG/c.-23 + 1G > A | GJB2 | Prelingual | Profound |
| 1 | c.35delG/c.167delT | GJB2 | Prelingual | Moderade-severe |
| 1 | c.35delG/p.W172* | GJB2 | Prelingual | Profound |
| 1 | p.35delG/p.M34T | GJB2 | Postlingual | Moderade |
| 1 | p.E47*/wt; ΔGJB6-D13S1830/wt | GJB; GJB6 | Prelingual | Profound |
| 1 | p.K168R/wt | GJB2 | Postlingual | Profound |
| 1 | p.M34T/p.V37I | GJB2 | Postlingual | Moderade |
| 1 | p.L90P/wt | GJB2 | Postlingual | Moderade |
| 1 | p.V37I/p.V95M | GJB2 | Postlingual | Profound |
| 1 | p.R184P/c.35delG | GJB2 | Prelingual | Profound |
| 1 | p.V27I/p.E114G | GJB2 | Postlingual | Moderade |
| 1 | p.W24*/p.W24* | GJB2 | Postlingual | Profound |
| 1 | p.R75Q/wt | GJB2 | Postlingual | Moderade |
| 1 | p.V27I/wt; p.V609G/wt | GJB2; SLC26A4 | Postlingual | Profound |
| 1 | p.V138F/wt | SLC26A4 | Postlingual | Profound |
| 1 | p.V609G/wt; c.5800_5801dupC/wt | SLC26A4; OTOF | Postlingual | Profound |
| 1 | p.R776C/wt | SLC26A4 | Postlingual | Profound |
| 1 | p.R1746Q/wt | CDH23 | Postlingual | Moderade |
| 1 | m.7445A > G | MT-TS1 | Prelingual | Profound |
| 1 | p.S3525G/wt | MYO15A | Prelingual | Profound |
| 1 | ΔGJB6-D13S1854/wt | GJB6 | Postlingual | Moderade |
wt wild type. *translation termination (stop) codon. Alterations in bold were detected by conventional techniques
A comparison of the results obtained by mass spectrometry and conventional techniques is provided in Table 4, where the alterations shown in bold type were only detected by the conventional techniques because they were not included in the panel.
The c.10573delA mutation in MYO15A gene was identified in one patient by mass spectrometry. However, the Sanger sequencing revealed a different alteration at the same site, c.10573A > G (p.S3525G). This occurred because iPLEX MassARRAY® technique detects only one base after the extension primer binding region, and for both alterations the base was a guanine instead of an adenine. The c.10573delA mutation corresponds to the deletion of an adenine, and the next base in the sequence is a guanine. In the case of the p.S3525G mutation, a substitution of the adenine for a guanine occurs at the same site. The p.S3525G alteration has been reported as a single nucleotide variation (SNV), according to the National Center for Biotechnology Information [http://www.ncbi.nlm.nih.gov/], and no data of clinical significance was available. To assess the possible damaging effect of amino acid substitution, four different in silico software analyses were used: SIFT [http://sift.jcvi.org/]; PolyPhen [http://www.polyphen.com]; Align-GVGD [http://agvgd.iarc.fr/]; and Mutation Taster [http://www.mutationtaster.org]. In all analyses, the effect was damaging for the myosin XV protein.
The genotyping cost using the developed panel to screen 4128 reactions is US$ 950 per SpectroCHIP (Table 5). Then, the cost per reaction (SNP/mutation) is approximately US$ 0.23. Taking into consideration that our panel uses eight wells per patient, it is possible to genotype 48 patients in a single 384-SpectroCHIP. Therefore, the costs per patient, including materials and reagents, is approximately US$ 20. These values do not include the labor, equipment costs, maintenances, and operational expenditure.
Table 5.
Costs in dollars to genotype a set of 86 alterations related to NSHL using the MassARRAY® iPLEX System
| Materials/reagents | Cost per chip | Cost per patient |
|---|---|---|
| Chips and kits sequenom | US$ 800.00 | US$ 16.7 |
| Primers | US$ 100.00 | US$ 2.08 |
| Consumables | US$ 50.00 | US$ 1.04 |
| Total | US$ 950.00 | US$ 19.8 |
Discussion
The MassARRAY iPLEX technology has the potential for high-throughput identification of genetic variations; however, our results demonstrated that to increase the genotyping success and accuracy, the optimization is required. After redesigning the assays that failed in the initial tests and genotyping control samples, sensitivity of the technique raised from 87 % to 97 %, the specificity increases from 97 % to 98 % and genotyping call rate increases from 74 % to 91 %.
Studies that have screened multiple mutations simultaneously in a substantial number of patients are scarce in the literature. In this study, the etiology of NSHL was investigated in a sample of 180 Brazilian individuals. These patients were selected carefully, and those cases with possible environmental etiology or with clinical features not compatible with sensorineural NSHL, were excluded.
After screening by mass spectrometry, we identified mutations in 32 patients with prelingual HL, and 22 patients with postlingual HL, totaling 54 patients. 32 out of 54 patients, (59.3 %) had family history of HL. Among the 17 genes analyzed, mutations were identified in seven of them, five autosomal (GJB2, SLC26A4, MYO15A, OTOF, and CDH23) and two mitochondrial (MT-RNR1 and MT-TS1). The results indicated a predominance of mutations in connexin 26 (GJB2), in concordance with previous works [9, 23–25, 27].
The c.35delG mutation in the GJB2 gene was identified in 15.6 % of the patients, and in 11.1 % of cases, this mutation was found in homozygosis, explaining the etiology of deafness. All of the individuals that were homozygous for this mutation had profound prelingual HL. The molecular identification of the c.35delG is usually made by allele-specific PCR or PCR followed by enzymatic restriction. These reactions are relatively inexpensive and are recommended as the first genetic test performed.
Other mutations in the GJB2 gene were identified by mass spectrometry in 20 of the analyzed patients. To detect GJB2 mutations, the coding exon of this gene is usually sequenced. As Sanger sequencing is expensive and time consuming, mass spectrometry offers an useful alternative procedure.
Mutations in the SLC26A4 gene are the second most frequent cause of NSHL [9]. We identified six individuals with three different mutations in this gene. The p.V609G variant was the most prevalent, found in four individuals (2.2 % of the sample population). Functional studies have demonstrated that there is a partial loss of protein activity when this alteration is present, indicating a potential pathogenic effect, especially if associated with other genetic or environmental factors [28]. In all the four individuals, the p.V609G was identified in only one allele of the gene, so the cause of HL was not elucidated. The SLC26A4 gene has 21 exons, so it would be expensive to perform a routine sequencing of all of them. Therefore, genotyping the selected set of mutations with mass spectrometry could be a useful option for initial analysis of this gene.
The del(GJB6-D13S1830) and del(GJB6-D13S1854) deletions in the GJB6 gene [24, 25], could not be included in our panel. However, these mutations were screened in all patients, as was previously described. The del(GJB6-D13S1854) deletion was found in one heterozygous individual, and compound heterozygous genotypes involving del(GJB6-D13S1830) and GJB2 mutations were identified in two individuals.
Three other mutations: p.R1746Q, c.5800_5801dupC, and p.S3525G, were identified in heterozygous genotypes, in the genes CDH23, OTOF, and MYO15A, respectively. These mutations do not explain the etiology of HL because they segregate with autosomal recessive inheritance. In subsequent studies, it would be necessary to sequence these genes in an attempt to find mutations in the second allele that could explain the cause of HL in these individuals.
The p.S3525G (c.10573A > G) mutation in the MYO15A gene was identified here by Sanger sequencing. No significant clinical data were available for this alteration, so in silico analyses were performed using four different software packages in order to assess the possible damaging effect of amino acid substitution. All these analyses indicated protein damage as an outcome. It is possible that this alteration contributes to hearing loss if it is present in both alleles or associated with other mutation in MYO15A.
Two mutations in mitochondrial genes were identified: m.1555A > G in MT-RNR1, detected in two individuals, and m.7445A > G in MT-TS1, detected in one case. According to previous studies, m.1555A > G is a common cause of genetic HL in Brazil. It was found in approximately 2 % of unselected cases of HL, and it was recommended for the inclusion in molecular diagnostic testing for HL [29, 30]. Early identification of patients with HL due to mutations in mitochondrial DNA can influence genetic counseling regarding maternal inheritance, enable avoidance of known risk factors, and assist pharmacological strategies for the prevention or reduction of HL progression [31].
Taking into account our data and the need to minimize costs and time, a pipeline trial is recommended before proceeding with mutations analysis using high-throughput methods. The first step should be to evaluate the clinical history of the patient in order to exclude obvious environmental etiologies. The second one is to assess the age of onset and the degree of HL. In cases of prelingual HL, it is recommend screening for the c.35delG mutation and GJB6 deletions by conventional techniques, due to the high frequency of these mutations as a cause of prelingual HL. For the remaining cases where molecular diagnosis is unclear, as well as cases of postlingual HL, the screening for the main mutations involved in HL by high throughput or conventional methods is recommended.
The genetic evaluation identified mutations in 30 % of the analyzed patients, and emphasized the importance of molecular testing, however in only 20 % of the cases the etiology of HL was concluded. The high number of cases with unknown etiology was possibly due to the broad genetic heterogeneity involved in NSHL, and also to the selection of mutations made to this study, since this selection was based on studies of other populations, including countries with high rates of consanguineous marriages, and might not be representative of the Brazilian population.
The continuous advances in DNA sequencing technologies allowed us to identify genetic alterations in a growing number of individuals. Therefore, they enabled the identification of the most frequent mutations in different populations. With this knowledge, the screening of specific mutations by high-throughput technologies could be really interesting to use in routine tests, since panels with specific assays could be customized for each population, reducing costs and enabling the rapid screening of a large number of patients.
There are several reliable, relatively simple and inexpensive methods currently in use to detecting mutations. Recently, array-based tests have been developed for NSHL, using different genotyping technologies: APEX microarray [32], designed Affymetrix resequencing microarrays [33] and TaqMan® OpenArray™ [34]. All these technologies have in common the use of oligonucleotides applied on a solid surface and fluorescent compounds. The OpenArray and APEX array focus specifically on the detection of previously reported mutations, whereas the Affymetrix resequencing array allows for the discovery of new mutations. The APEX microarray allows some versatility, since it is possible add SNPs after ordering the custom test. In contrast, OpenArray and resequencing arrays do not allow for modifications after achievement of the plates/chips. With reference to costs, the screening of mutations with Affymetrix resequencing arrays are quite high in comparison with the others, making it unfeasible the use of this technique in a routine laboratory setting [35, 36].
In contrast to these technologies, the MassARRAY iPLEX does not uses fluorescent compounds, and analyses direct DNA products. As the OpenArray and APEX, this technique has the limitation of detect only previously reported mutations. However, the great advantage of MassARRAY iPLEX is the versatility and flexibility, since the assays are not pre-spotted into the chip by the manufacturer. Therefore, you can easily add or remove tests and genotyping different sets of mutations on the same chip. Moreover, the number of wells analyzed at a time can be variable, since the same chip can be used in different experiments.
All the techniques mentioned above, including MassARRAY, need be confirmed by other techniques when mutations were found. Our results demonstrated that even after optimization, false positive results were observed in 1.96 % of the genotypes. Therefore, the validation of positive cases must be carried out to ensure the reliability of the results.
Sanger sequencing is considered the golden standard for mutation identification due to its accuracy. However, Sanger sequencing is labor intensive and has high per-base sequencing costs. It can be effectively only if used to screen genes with a limited number of exons, or to confirm mutations found with other technologies.
Next generation sequencing is the most efficient method to identify mutations, especially in heterogeneous disorders such as hereditary hearing loss. Recent developments in NGS and DNA capture technologies provide a potential approach for accurate molecular diagnosis of HL [37–39]. The main advantage of NGS over current diagnostic methods is that a significantly higher rate of successful diagnosis can be obtained by screening both known and novel mutations in all HL genes simultaneously. However, its high cost and delay in data analysis make it impractical for application in the diagnosis of many individuals.
The high-throughput techniques such as IPLEX MassARRAY® system is a good option for initial screening of the most frequent mutations, since it enables genotyping a substantial set of known alterations simultaneously, in shorter periods of time, and at lower cost, compared to the screening of the main mutations performed with conventional techniques like PCR, Sanger sequencing and enzymatic restriction. The cost of conventional routine screening of mutations in the GJB2, GJB6, and MT-RNR1 genes for diagnosis of hearing loss is approximately US$ 30 per patient. In contrast, the cost of mass spectrometry screening of 86 genetic mutations in nine genes is approximately US$ 20 per patient, evidencing, this way, a better cost-benefit. This comparison only include the costs of reagents and materials used in our research laboratory.
Conclusions
We have developed and tested a panel based on MassARRAY technology for screening 86 mutations related to NSHL. This technology has the potential for high-throughput identification of genetic variations; however, we demonstrated that to increase the genotyping success and accuracy, optimization is required. The panel proved to be cost-effective and efficient, due to the high values obtained in sensitivity and specificity. We tested the panel in affected Brazilian individuals with NSHL without a molecular diagnosis. Genetic alterations were identified in 30 % of them, and the etiology of NSHL was concluded in 20 % of the cases. These results demonstrate that the developed panel is suitable and helpful to be applied in the diagnosis of hearing loss.
Funding
The authors thank the patients who participated and everyone involved in this study. The work was supported by São Paulo Research Foundation (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Abbreviations
- BERA
Brainstem evoked response audiometry
- CMV
Cytomegalovirus
- HL
Hearing loss
- NSHL
Non-syndromic hearing loss
- OAE
Otoacoustic emissions
- PCR
Polymerase chain reaction
- RFLP
Restriction fragment length polymorphism
- SBE
Single base extension
- SNP
Single nucleotide polymorphism
- SNV
Single nucleotide variation
- US$
United States dollar
Additional file
Patterns of inheritance, mutations selected for the study, and their respective genes and references. Table with all alterations selected for the panel and references. [42-92] (DOCX 20 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MCCMS carried out all experiments, including samples preparation, genotyping, and validation. SMSC, PZR, NZPS and FTAM helped with sample preparation, validation and with the manuscript review. AMC and ELS contributed with comments and written of the manuscript. All the authors read and accepted the final version of the manuscript.
Contributor Information
Maria Carolina CCosta CMelo Svidnicki, Email: mcarolinasvid@gmail.com.
Sueli Matilde Silva-Costa, Email: silvacosta.sm@gmail.com.
Priscila Zonzini Ramos, Email: priscila.zonzini@yahoo.com.br.
Nathalia Zocal Pereira dos Santos, Email: nathalia.zps@gmail.com.
Fábio Tadeu Arrojo Martins, Email: fabio.arrojo@gmail.com.
Arthur Menino Castilho, Email: arthurcastilho@gmail.com.
Edi Lúcia Sartorato, Email: sartor@unicamp.br.
References
- 1.Dror AA, Avraham KB. Hearing loss: Mechanisms revealed by genetics and cell biology. Annu Rev Gen. 2009;43:411–437. doi: 10.1146/annurev-genet-102108-134135. [DOI] [PubMed] [Google Scholar]
- 2.Willems PJ. Genetic causes of hearing loss. N Engl J Med. 2000;342:1101–1109. doi: 10.1056/NEJM200004133421506. [DOI] [PubMed] [Google Scholar]
- 3.Smith RJ, Bale JF, Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365:879–890. doi: 10.1016/S0140-6736(05)71047-3. [DOI] [PubMed] [Google Scholar]
- 4.Morton CC, Nance WE. Newborn hearing screening - a silent revolution. N Engl J Med. 2006;354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 5.Petit C, Richardson GP. Linking genes underlying deafness to hair-bundle development and function. Nat Neurosci. 2009;12:703–710. doi: 10.1038/nn.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dror AA, Avraham KB. Hearing impairment: a panoply of genes and functions. Neuron. 2010;68:293–308. doi: 10.1016/j.neuron.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 7.Duman D, Tekin M. Autosomal recessive nonsyndromic deafness genes: a review. Front Biosci. 2012;17:2213–2236. doi: 10.2741/4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shearer AE, Hildebrand MS, Sloan CM, Smith RJ. Deafness in the genomics era. Hear Res. 2011;282:1–9. doi: 10.1016/j.heares.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hilgert N, Smith RJH, Van Camp G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutation Res. 2009;681:189–196. doi: 10.1016/j.mrrev.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hutchin T, Coy NN, Conlon H, Telford E, Bromelow K, Blaydon D, Taylor G, Coghill E, Brown S, Trembath R, Liu XZ, Bitner-Glindzicz M, Mueller R. Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK - implications for genetic testing. Clin Genet. 2005;68(6):506–12. doi: 10.1111/j.1399-0004.2005.00539.x. [DOI] [PubMed] [Google Scholar]
- 11.Gasparini P, Rabionet R, Barbujani G, Melchionda S, Petersen M, Brøndum-Nielsen K, Metspalu A, Oitmaa E, Pisano M, Fortina P, Zelante L, Estivill X. High carrier frequency of the 35delG deafness mutation in European populations. Genetic analysis consortium of GJB2 35delG. Eur J Hum Genet. 2000;8:19–23. doi: 10.1038/sj.ejhg.5200406. [DOI] [PubMed] [Google Scholar]
- 12.Petersen M, Willems P. Non-syndromic, autosomal-recessive deafness. Clin Genet. 2006;69:371–392. doi: 10.1111/j.1399-0004.2006.00613.x. [DOI] [PubMed] [Google Scholar]
- 13.Morrison T, Hurley J, Garcia J, Yoder K, Katz A, Roberts D, Cho J, Kanigan T, Ilyin SE, Horowitz D, Dixon JM, Brenan CJ. Nanoliter high throughput quantitative PCR. Nucleic Acids Res. 2006;34(18):123. doi: 10.1093/nar/gkl639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis H, Silverman SR. Auditory Test Hearing Aids. In: Davis H, Silvermann SR, editors. Hearing and deafness. New York: Holt: Rinehart and Winston; 1970. pp. 83–139. [Google Scholar]
- 15.Tang K, Fu DJ, Julien D, Braun A, Cantor CR, Köster H. Chip-based genotyping by mass spectrometry. Proc Natl Acad Sci. 1999;96(18):10016–20. doi: 10.1073/pnas.96.18.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gabriel S, Ziaugra L, Tabbaa D. SNP Genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc Hum Genet. 2009;2(12):1–18. doi: 10.1002/0471142905.hg0212s60. [DOI] [PubMed] [Google Scholar]
- 17.Romanos J, Kimura L, Fávero ML, Izarra FA, de Mello Auricchio MT, Batissoco AC, Lezirovitz K, Abreu-Silva RS, Mingroni-Netto RC. Novel OTOF mutations in Brazilian patients with auditory neuropathy. J Hum Genet. 2009;54(7):382–5. doi: 10.1038/jhg.2009.45. [DOI] [PubMed] [Google Scholar]
- 18.de Moraes VC, Dos Santos NZ, Ramos PZ, Svidnicki MC, Castilho AM, Sartorato EL. Molecular analysis of SLC26A4 gene in patients with nonsyndromic hearing loss and EVA: Identification of two novel mutations in Brazilian patients. Int J Pediatr Otorhinolaryngol. 2013;77(3):410–413. doi: 10.1016/j.ijporl.2012.11.042. [DOI] [PubMed] [Google Scholar]
- 19.Lezirovitz K, Pardono E, de Mello Auricchio MT, de Carvalho E, Silva FL, Lopes JJ, Abreu-Silva RS, Romanos J, Batissoco AC, Mingroni-Netto RC. Unexpected genetic heterogeneity in a large consanguineous Brazilian pedigree presenting deafness. Eur J Hum Genet. 2008;16(1):89–96. doi: 10.1038/sj.ejhg.5201917. [DOI] [PubMed] [Google Scholar]
- 20.Altman DG, Bland JM. Diagnostic test 1: sensitivity and specificity. BMJ. 1994;308:1552. doi: 10.1136/bmj.308.6943.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott DA, Kraft ML, Carmi R, Ramesh A, Elbedour K, Yairi Y, et al. Identification of mutations in the connexin 26 gene that cause autosomal recessive nonsyndromic hearing loss. Hum Mutat. 1998;11:387–394. doi: 10.1002/(SICI)1098-1004(1998)11:5<387::AID-HUMU6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 22.Denoyelle F, Weil D, Maw MA, Wilcox SA, Lench NJ, Allen-Powell DR, et al. Prelingual deafness: High prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet. 1997;6:2173–2177. doi: 10.1093/hmg/6.12.2173. [DOI] [PubMed] [Google Scholar]
- 23.Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 24.Del Castillo I, Moreno-Pelayo MA, Del Castillo FJ, Brownstein Z, Marlin S, Adina Q, et al. Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. Am J Hum Genet. 2003;73:1452–1458. doi: 10.1086/380205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Del Castillo FJ, Rodríguez-Ballesteros M, Alvarez A, Hutchin T, Leonardi E, de Oliveira CA, et al. A novel deletion involving the connexin-30 gene, del(GJB6-D13S1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet. 2005;42:588–594. doi: 10.1136/jmg.2004.028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prezant TR, Agapian JV, Bohlman MC, Bu X, Öztas S. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. 1993;4:289–294. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- 27.Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, Waligora J, et al. GJB2 mutations and degree of hearing loss: A multicenter study. Am J Hum Genet. 2005;77:945–957. doi: 10.1086/497996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dossena S, Bizhanova A, Nofziger C, Bernardinelli E, Ramsauer J, Kopp P, Paulmichl M. Identification of allelic variants of pendrin (SLC26A4) with loss and gain of function. Cell Physiol Biochem. 2011;28(3):467–76. doi: 10.1159/000335108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mingroni-Netto RC, Abreu-Silva RS, Braga MCC, Lezirovitz K, Della-Rosa VA, Pirana S, et al. Mitochondrial mutation A1555G (12S rRNA) and connexin 26 35delG mutation are frequent causes of deafness in Brazil. Am J Hum Genet. 2001;69(suppl):A2124. [Google Scholar]
- 30.Pupo AC, Pirana S, Spinelli M, Lezirovitz K, Mingroni-Netto RC, et al. Study of a Brazilian family presenting non-syndromic hearing loss with mitochondrial inheritance. Braz J Otorhinolaryngol. 2008;74:786–789. doi: 10.1016/S1808-8694(15)31392-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.King PJ, Ouyang X, Du L, Yan D, Angeli SI, Liu XZ. Etiologic diagnosis of nonsyndromic genetic hearing loss in adult vs. pediatric populations. Otolaryngol Head Neck Surg. 2012;147:932–936. doi: 10.1177/0194599812453553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez-Paris J, Pique L, Colen T, Roberson J, Gardner P, Schrijver I. Genotyping with a 198 mutation arrayed primer extension array for hereditary hearing loss: assessment of its diagnostic value for medical practice. PLoS One. 2010;5(7):e11804. doi: 10.1371/journal.pone.0011804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kothiyal P, Cox S, Ebert J, Husami A, Kenna MA, Greinwald JH, Aronow BJ, Rehm HL. High-throughput detection of mutations responsible for childhood hearing loss using resequencing microarrays. BMC Biotechnol. 2010;10:10. doi: 10.1186/1472-6750-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martins FT, Ramos PZ, Svidnicki MC, Castilho AM, Sartorato EL. Optimization of simultaneous screening of the main mutations involved in non-syndromic deafness using the TaqMan® OpenArray™ Genotyping platform. BMC Med Genet. 2013;14:112. doi: 10.1186/1471-2350-14-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cremers FP, Kimberling WJ, Külm M, de Brouwer AP, van Wijk E, te Brinke H, Cremers CW, Hoefsloot LH, Banfi S, Simonelli F, Fleischhauer JC, Berger W, Kelley PM, Haralambous E, Bitner-Glindzicz M, Webster AR, Saihan Z, De Baere E, Leroy BP, Silvestri G, McKay GJ, Koenekoop RK, Millan JM, Rosenberg T, Joensuu T, Sankila EM, Weil D, Weston MD, Wissinger B, Kremer H. Development of a genotyping microarray for Usher syndrome. J Med Genet. 2007;44(2):153–60. doi: 10.1136/jmg.2006.044784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cremonesi L, Ferrari M, Giordano PC, Harteveld CL, Kleanthous M, Papasavva T, Patrinos GP, Traeger-Synodinos J. An overview of current microarray-based human globin gene mutation detection methods. Hemoglobin. 2007;31(3):289–311. doi: 10.1080/03630260701459366. [DOI] [PubMed] [Google Scholar]
- 37.Shahzad M, Sivakumaran TA, Qaiser TA, Schultz JM, Hussain Z, Flanagan M, Bhinder MA, Kissell D, Greinwald JH, Jr, Khan SN, Friedman TB, Zhang K, Riazuddin S, Riazuddin S, Ahmed ZM. Genetic analysis through OtoSeq of Pakistani families segregating prelingual hearing loss. Otolaryngol Head Neck Surg. 2013;149(3):478–87. doi: 10.1177/0194599813493075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shearer AE, Black-Ziegelbein EA, Hildebrand MS, Eppsteiner RW, Ravi H, Joshi S, Guiffre AC, Sloan CM, Happe S, Howard SD, Novak B, Deluca AP, Taylor KR, Scheetz TE, Braun TA, Casavant TL, Kimberling WJ, Leproust EM, Smith RJ. Advancing genetic testing for deafness with genomic technology. J Med Genet. 2013;50(9):627–34. doi: 10.1136/jmedgenet-2013-101749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang W, Qian D, Ahmad S, Mattox D, Todd NW, Han H, Huang S, Li Y, Wang Y, Li H, Lin X. A low-cost exon capture method suitable for large-scale screening of genetic deafness by the massively-parallel sequencing approach. Genet Test Mol Biomarkers. 2012;16(6):536–42. doi: 10.1089/gtmb.2011.0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJ. Carrier rates in the Midwestern United States for GJB2 mutations causing inherited deafness. JAMA. 1999;281(23):2211–6. doi: 10.1001/jama.281.23.2211. [DOI] [PubMed] [Google Scholar]
- 41.Zelante L, Gasparini P, Estivill X, Melchionda S, D’Agruma L, Govea N, Milá M, Monica MD, Lutfi J, Shohat M, Mansfield E, Delgrosso K, Rappaport E, Surrey S, Fortina P. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum Mol Genet. 1997;6(9):1605–1609. doi: 10.1093/hmg/6.9.1605. [DOI] [PubMed] [Google Scholar]
- 42.Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, Kimberling WJ. Novel Mutations in the Connexin 26 Gene (GJB2) That Cause Autosomal Recessive (DFNB1) Hearing Loss. Am J Hum Genet. 1998;62:792–799. doi: 10.1086/301807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuse Y, Doi K, Hasegawa T, Sugii A, Hibino H, Kubo T. Three novel connexin26 gene mutations in autosomal recessive non-syndromic deafness. Neuroreport. 1999;10(9):1853–7. doi: 10.1097/00001756-199906230-00010. [DOI] [PubMed] [Google Scholar]
- 44.Murgia A, Orzan E, Polli R, Martella M, Vinanzi C, Leonardi E, Arslan E, Zacchello F. Cx26 deafness: mutation analysis and clinical variability. J Med Genet. 1999;36(11):829–32. [PMC free article] [PubMed] [Google Scholar]
- 45.Wu BL, Lindeman N, Lip V, Adams A, Amato RS, Cox G, Irons M, Kenna M, Korf B, Raisen J, Platt O. Effectiveness of sequencing connexin 26 (GJB2) in cases of familial or sporadic childhood deafness referred for molecular diagnostic testing. Genet Med. 2002;4(4):279–88. doi: 10.1097/00125817-200207000-00006. [DOI] [PubMed] [Google Scholar]
- 46.Kenna MA, Wu BL, Cotanche DA, Korf BR, Rehm HL. Connexin 26 studies in patients with sensorineural hearing loss. Arch Otolaryngol Head Neck Surg. 2001;127(9):1037–42. doi: 10.1001/archotol.127.9.1037. [DOI] [PubMed] [Google Scholar]
- 47.Brobby GW, Müller-Myhsok B, Horstmann RD. Connexin 26 R143W mutation associated with recessive nonsyndromic sensorineural deafness in Africa. N Engl J Med. 1998;338(8):548–50. doi: 10.1056/NEJM199802193380813. [DOI] [PubMed] [Google Scholar]
- 48.Frei K, Lucas T, Ramsebner R, Schöfer C, Baumgartner WD, Weipoltshammer K, Erginel-Unaltuna N, Wachtler FJ, Kirschhofer K. A novel connexin 26 mutation associated with autosomal recessive sensorineural deafness. Audiol Neurootol. 2004;9(1):47–50. doi: 10.1159/000074186. [DOI] [PubMed] [Google Scholar]
- 49.Marlin S, Garabédian EN, Roger G, Moatti L, Matha N, Lewin P, Petit C, Denoyelle F. Connexin 26 gene mutations in congenitally deaf children: pitfalls for genetic counseling. Arch Otolaryngol Head Neck Surg. 2002;127(8):927–33. doi: 10.1001/archotol.127.8.927. [DOI] [PubMed] [Google Scholar]
- 50.Samanich J, Lowes C, Burk R, Shanske S, Lu J, Shanske A, Morrow BE. Mutations in GJB2, GJB6, and Mitochondrial DNA Are Rare in African American and Caribbean Hispanic Individuals With Hearing Impairment. Am J of Med Genet. 2007;143A(8):830–8. doi: 10.1002/ajmg.a.31668. [DOI] [PubMed] [Google Scholar]
- 51.Posukh O, Pallares-Ruiz N, Tadinova V, Osipova L, Claustres M, Roux AF. First molecular screening of deafness in the Altai Republic population. BMC Med Genet. 2005;24(6):2. doi: 10.1186/1471-2350-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilcox SA, Saunders K, Osborn AH, et al. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet. 2000;106:399–405. doi: 10.1007/s004390000273. [DOI] [PubMed] [Google Scholar]
- 53.Kopp P, Arseven OK, Sabacan L, Kotlar T, Dupuis J, Cavaliere H, Santos CL, Jameson JL, Medeiros-Neto G. Phenocopies for deafness and goiter development in a large inbred Brazilian kindred with Pendred’s syndrome associated with a novel mutation in the PDS gene. J Clin Endocrinol Metab. 1999;84(1):336–41. doi: 10.1210/jcem.84.1.5398. [DOI] [PubMed] [Google Scholar]
- 54.Van Hauwe P, Everett LA, Coucke P, Scott DA, Kraft ML, Ris-Stalpers C, Bolder C, Otten B, de Vijlder JJ, Dietrich NL, Ramesh A, Srisailapathy SC, Parving A, Cremers CW, Willems PJ, Smith RJ, Green ED, Van Camp G. Two frequent missense mutations in Pendred syndrome. Hum Mol Genet. 1998;7(7):1099–104. doi: 10.1093/hmg/7.7.1099. [DOI] [PubMed] [Google Scholar]
- 55.Park HJ, Lee SJ, Jin HS, Lee JO, Go SH, Jang HS, Moon SK, Lee SC, Chun YM, Lee HK, Choi JY, Jung SC, Griffith AJ, Koo SK. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin Genet. 2005;67(2):160–5. doi: 10.1111/j.1399-0004.2004.00386.x. [DOI] [PubMed] [Google Scholar]
- 56.Adato A, Raskin L, Petit C, Bonne-Tamir B. Deafness heterogeneity in a Druze isolate from the Middle East: novel OTOF and PDS mutations, low prevalence of GJB2 35delG mutation and indication for a new DFNB locus. Eur J Hum Genet. 2000;8(6):437–42. doi: 10.1038/sj.ejhg.5200489. [DOI] [PubMed] [Google Scholar]
- 57.Coyle B, Reardon W, Herbrick JA, Tsui LC, Gausden E, Lee J, et al. Molecular analysis of the PDS gene in Pendred syndrome. Hum Mol Gene. 1998;7(7):1105–12. doi: 10.1093/hmg/7.7.1105. [DOI] [PubMed] [Google Scholar]
- 58.Ji YB, Han DY, Wang DY, Zhou Y, Zhao C, Wang H, Lan L, Wang QJ. Evaluation of deaf-mute patients with sensitive deafness gene screening in Shandong province. Zhonghua Yi Xue Za Zhi. 2009;89(36):2531–5. [PubMed] [Google Scholar]
- 59.Pryor SP, Madeo AC, Reynolds JC, Sarlis NJ, Arnos KS, Nance WE, Yang Y, Zalewski CK, Brewer CC, Butman JA, Griffith AJ. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet. 2005;42(2):159–65. doi: 10.1136/jmg.2004.024208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nal N, Ahmed ZM, Erkal E, Alper OM, Lüleci G, Dinç O, Waryah AM, Ain Q, Tasneem S, Husnain T, Chattaraj P, Riazuddin S, Boger E, Ghosh M, Kabra M, Riazuddin S, Morell RJ, Friedman TB. Mutational spectrum of MYO15A: the large N-terminal extension of myosin XVA is required for hearing. Hum Mutat. 2007;28(10):1014–9. doi: 10.1002/humu.20556. [DOI] [PubMed] [Google Scholar]
- 61.Rodriguez-Ballesteros M, Reynoso R, Olarte M, Villamar M, Morera C, Santarelli R, Arslan E, Meda C, Curet C, Volter C. A multicenter study on the prevalence and spectrum of mutations in the OTOFerlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum Mutat. 2008;29(6):823–831. doi: 10.1002/humu.20708. [DOI] [PubMed] [Google Scholar]
- 62.Rodríguez-Ballesteros M, del Castillo FJ, Martín Y, Moreno-Pelayo MA, Morera C, Prieto F, Marco J, Morant A, Gallo-Terán J, Morales-Angulo C, Navas C, Trinidad G, Tapia MC, Moreno F, del Castillo I. Auditory neuropathy in patients carrying mutations in the OTOFerlin gene (OTOF) Hum Mutat. 2003;22(6):451–6. doi: 10.1002/humu.10274. [DOI] [PubMed] [Google Scholar]
- 63.Varga R, Avenarius MR, Kelley PM, Keats BJ, Berlin CI, Hood LJ, Morlet TG, Brashears SM, Starr A, Cohn ES, Smith RJ, Kimberling WJ. OTOF mutations revealed by genetic analysis of hearing loss families including a potential temperature sensitive auditory neuropathy allele. J Med Genet. 2006;43(7):576–81. doi: 10.1136/jmg.2005.038612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Migliosi V, Modamio-Høybjør S, Moreno-Pelayo MA, Rodríguez-Ballesteros M, Villamar M, Tellería D, Menéndez I, Moreno F, Del Castillo I. Q829X, a novel mutation in the gene encoding OTOFerlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss. J Med Genet. 2002;39(7):502–6. doi: 10.1136/jmg.39.7.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C. A mutation in OTOF, encoding OTOFerlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999;21(4):363–9. doi: 10.1038/7693. [DOI] [PubMed] [Google Scholar]
- 66.Wagatsuma M, Kitoh R, Suzuki H, Fukuoka H, Takumi Y, Usami S. Distribution and frequencies of CDH23 mutations in Japanese patients with non-syndromic hearing loss. Clin Genet. 2007;72(4):339–44. doi: 10.1111/j.1399-0004.2007.00833.x. [DOI] [PubMed] [Google Scholar]
- 67.Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, Wayne S, Bellman S, Desmukh D, Ahmed Z, Khan SN, Kaloustian VM, Li XC, Lalwani A, Riazuddin S, Bitner-Glindzicz M, Nance WE, Liu XZ, Wistow G, Smith RJ, Griffith AJ, Wilcox ER, Friedman TB, Morell RJ. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001;68(1):26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Astuto LM, Bork JM, Weston MD, Askew JW, Fields RR, Orten DJ, Ohliger SJ, Riazuddin S, Morell RJ, Khan S, Riazuddin S, Kremer H, van Hauwe P, Moller CG, Cremers CW, Ayuso C, Heckenlively JR, Rohrschneider K, Spandau U, Greenberg J, Ramesar R, Reardon W, Bitoun P, Millan J, Legge R, Friedman TB, Kimberling WJ. CDH23 mutation and phenotype heterogeneity: a profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am J Hum Genet. 2002;71(2):262–75. doi: 10.1086/341558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bolz H, von Brederlow B, Ramírez A, Bryda EC, Kutsche K, Nothwang HG, Seeliger M, del C-Salcedó CM, Vila MC, Molina OP, Gal A, Kubisch C. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet. 2001;27(1):108–12. doi: 10.1038/83667. [DOI] [PubMed] [Google Scholar]
- 70.von Brederlow B, Bolz H, Janecke A, La O, Cabrera A, Rudolph G, Lorenz B, Schwinger E, Gal A. Identification and in vitro expression of novel CDH23 mutations of patients with Usher syndrome type 1D. Hum Mutat. 2002;19(3):268–73. doi: 10.1002/humu.10049. [DOI] [PubMed] [Google Scholar]
- 71.Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, Arnaud D, Drury S, Mo J, Makishima T, Ghosh M, Menon PS, Deshmukh D, Oddoux C, Ostrer H, Khan S, Riazuddin S, Deininger PL, Hampton LL, Sullivan SL, Battey JF, Jr, Keats BJ, Wilcox ER, Friedman TB, Griffith AJ. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet. 2002;30(3):277–84. doi: 10.1038/ng842. [DOI] [PubMed] [Google Scholar]
- 72.Meyer CG, Gasmelseed NM, Mergani A, Magzoub MM, Muntau B, Thye T, Horstmann RD. Novel TMC1 structural and splice variants associated with congenital nonsyndromic deafness in a Sudanese pedigree. Hum Mutat. 2005;25(1):100. doi: 10.1002/humu.9302. [DOI] [PubMed] [Google Scholar]
- 73.Santos RL, Wajid M, Khan MN, McArthur N, Pham TL, Bhatti A, Lee K, Irshad S, Mir A, Yan K, Chahrour MH, Ansar M, Ahmad W, Leal SM. Novel sequence variants in the TMC1 gene in Pakistani families with autosomal recessive hearing impairment. Hum Mutat. 2005;26(4):396. doi: 10.1002/humu.9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brownstein Z, Friedman LM, Shahin H, Oron-Karni V, Kol N, Abu Rayyan A, Parzefall T, Lev D, Shalev S, Frydman M, Davidov B, Shohat M, Rahile M, Lieberman S, Levy-Lahad E, Lee MK, Shomron N, King MC, Walsh T, Kanaan M, Avraham KB. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in Middle Eastern families. Genome Biol. 2011;12(9):R89. doi: 10.1186/gb-2011-12-9-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wattenhofer M, Di Iorio MV, Rabionet R, Dougherty L, Pampanos A, Schwede T, Montserrat-Sentis B, Arbones ML, Iliades T, Pasquadibisceglie A, D’Amelio M, Alwan S, Rossier C, Dahl HH, Petersen MB, Estivill X, Gasparini P, Scott HS, Antonarakis SE. Mutations in the TMPRSS3 gene are a rare cause of childhood nonsyndromic deafness in Caucasian patients. J Mol Med. 2002;80(2):124–31. doi: 10.1007/s00109-001-0310-6. [DOI] [PubMed] [Google Scholar]
- 76.Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry A, Chrast R, Guipponi M, Wang J, Kawasaki K, Asakawa S, Minoshima S, Younus F, Mehdi SQ, Radhakrishna U, Papasavvas MP, Gehrig C, Rossier C, Korostishevsky M, Gal A, Shimizu N, Bonne-Tamir B, Antonarakis SE. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet. 2001;27(1):59–63. doi: 10.1038/83768. [DOI] [PubMed] [Google Scholar]
- 77.Elbracht M, Senderek J, Eggermann T, Thürmer C, Park J, Westhofen M, Zerres K. Autosomal recessive postlingual hearing loss (DFNB8): compound heterozygosity for two novel TMPRSS3 mutations in German siblings. J Med Genet. 2007;44(6):e81. doi: 10.1136/jmg.2007.049122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Masmoudi S, Antonarakis SE, Schwede T, Ghorbel AM, Gratri M, Pappasavas MP, Drira M, Elgaied-Boulila A, Wattenhofer M, Rossier C, Scott HS, Ayadi H, Guipponi M. Novel missense mutations of TMPRSS3 in two consanguineous Tunisian families with non-syndromic autosomal recessive deafness. Hum Mutat. 2001;18(2):101–8. doi: 10.1002/humu.1159. [DOI] [PubMed] [Google Scholar]
- 79.Shahin H, Walsh T, Sobe T, Abu Sa’ed J, Abu Rayan A, Lynch ED, Lee MK, Avraham KB, King MC, Kanaan M. Mutations in a novel isoform of TRIOBP that encodes a filamentous-actin binding protein are responsible for DFNB28 recessive nonsyndromic hearing loss. Am J Hum Genet. 2006;78(1):144–52. doi: 10.1086/499495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Naz S, Giguere CM, Kohrman DC, Mitchem KL, Riazuddin S, Morell RJ, Ramesh A, Srisailpathy S, Deshmukh D, Riazuddin S, Griffith AJ, Friedman TB, Smith RJ, Wilcox ER. Mutations in a novel gene, TMIE, are associated with hearing loss linked to the DFNB6 locus. Am J Hum Genet. 2002;71(3):632–6. doi: 10.1086/342193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Delmaghani S, del Castillo FJ, Michel V, Leibovici M, Aghaie A, Ron U, Van Laer L, Ben-Tal N, Van Camp G, Weil D, Langa F, Lathrop M, Avan P, Petit C. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat Genet. 2006;38(7):770–8. doi: 10.1038/ng1829. [DOI] [PubMed] [Google Scholar]
- 82.Uyguner O, Tukel T, Baykal C, Eris H, Emiroglu M, Hafiz G, Ghanbari A, Baserer N, Yuksel-Apak M, Wollnik B. The novel R75Q mutation in the GJB2 gene causes autosomal dominant hearing loss and palmoplantar keratoderma in a Turkish family. Clin Genet. 2002;62(4):306–9. doi: 10.1034/j.1399-0004.2002.620409.x. [DOI] [PubMed] [Google Scholar]
- 83.Bespalova IN, Van Camp G, Bom SJ, Brown DJ, Cryns K, DeWan AT, Erson AE, Flothmann K, Kunst HP, Kurnool P, Sivakumaran TA, Cremers CW, Leal SM, Burmeister M, Lesperance MM. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet. 2001;10(22):2501–8. doi: 10.1093/hmg/10.22.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Coucke PJ, Van Hauwe P, Kelley PM, Kunst H, Schatteman I, Van Velzen D, Meyers J, Ensink RJ, Verstreken M, Declau F, Marres H, Kastury K, Bhasin S, McGuirt WT, Smith RJ, Cremers CW, Van de Heyning P, Willems PJ, Smith SD, Van Camp G. Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum Mol Genet. 1999;8(7):1321–8. doi: 10.1093/hmg/8.7.1321. [DOI] [PubMed] [Google Scholar]
- 85.de Kok YJ, Bom SJ, Brunt TM, Kemperman MH, van Beusekom E, van der Velde-Visser SD, Robertson NG, Morton CC, Huygen PL, Verhagen WI, Brunner HG, Cremers CW, Cremers FP. A Pro51Ser mutation in the COCH gene is associated with late onset autosomal dominant progressive sensorineural hearing loss with vestibular defects. Hum Mol Genet. 1999;8(2):361–6. doi: 10.1093/hmg/8.2.361. [DOI] [PubMed] [Google Scholar]
- 86.Hildebrand MS, Morín M, Meyer NC, Mayo F, Modamio-Hoybjor S, Mencía A, Olavarrieta L, Morales-Angulo C, Nishimura CJ, Workman H, DeLuca AP, del Castillo I, Taylor KR, Tompkins B, Goodman CW, Schrauwen I, Wesemael MV, Lachlan K, Shearer AE, Braun TA, Huygen PL, Kremer H, Van Camp G, Moreno F, Casavant TL, Smith RJ, Moreno-Pelayo MA. DFNA8/12 caused by TECTA mutations is the most identified subtype of nonsyndromic autosomal dominant hearing loss. Hum Mutat. 2011;32(7):825–34. doi: 10.1002/humu.21512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moreno-Pelayo MA, del Castillo I, Villamar M, Romero L, Hernández-Calvín FJ, Herraiz C, Barberá R, Navas C, Moreno F. A cysteine substitution in the zona pellucida domain of alpha-tectorin results in autosomal dominant, postlingual, progressive, mid frequency hearing loss in a Spanish family. J Med Genet. 2001;38(5):E13. doi: 10.1136/jmg.38.5.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sagong B, Park R, Kim YH, Lee KY, Baek JI, Cho HJ, Cho IJ, Kim UK, Lee SH. Two novel missense mutations in the TECTA gene in Korean families with autosomal dominant nonsyndromic hearing loss. Ann Clin Lab Sci. 2010;40(4):380–5. [PubMed] [Google Scholar]
- 89.Plantinga RF, de Brouwer AP, Huygen PL, Kunst HP, Kremer H, Cremers CW. A novel TECTA mutation in a Dutch DFNA8/12 family confirms genotype-phenotype correlation. J Assoc Res Otolaryngol. 2006;7(2):173–81. doi: 10.1007/s10162-006-0033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mencía A, Modamio-Høybjør S, Redshaw N, Morín M, Mayo-Merino F, Olavarrieta L, Aguirre LA, del Castillo I, Steel KP, Dalmay T, Moreno F, Moreno-Pelayo MA. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet. 2009;41(5):609–13. doi: 10.1038/ng.355. [DOI] [PubMed] [Google Scholar]
- 91.Hyslop SJ, James AM, Maw M, Fischel-Ghodsian N, Murphy MP. The effect on mitochondrial function of the tRNA Ser(UCN)/COI A7445G mtDNA point mutation associated with maternally-inherited sensorineural deafness. Biochem Mol Biol Int. 1997;42(3):567–75. doi: 10.1080/15216549700202971. [DOI] [PubMed] [Google Scholar]
- 92.Fischel-Ghodsian N, Prezant TR, Bu X, Oztas S. Mitochondrial ribosomal RNA gene mutation in a patient with sporadic aminoglycoside ototoxicity. Am J Otolaryngo. 1993;14(6):399–403. doi: 10.1016/0196-0709(93)90113-L. [DOI] [PubMed] [Google Scholar]