

Abstract
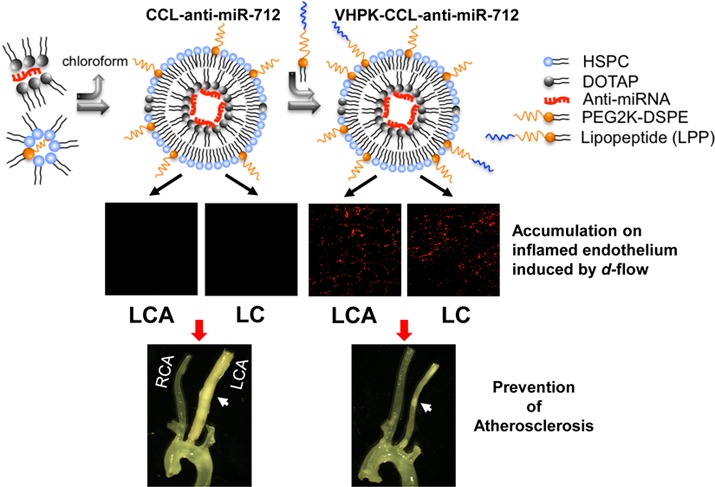
The current study presents an effective and selective multifunctional nanoparticle used to deliver antiatherogenic therapeutics to inflamed pro-atherogenic regions without off-target changes in gene expression or particle-induced toxicities. MicroRNAs (miRNAs) regulate gene expression, playing a critical role in biology and disease including atherosclerosis. While anti-miRNA are emerging as therapeutics, numerous challenges remain due to their potential off-target effects, and therefore the development of carriers for selective delivery to diseased sites is important. Yet, co-optimization of multifunctional nanoparticles with high loading efficiency, a hidden cationic domain to facilitate lysosomal escape and a dense, stable incorporation of targeting moieties is challenging. Here, we create coated, cationic lipoparticles (CCLs), containing anti-miR-712 (∼1400 molecules, >95% loading efficiency) within the core and with a neutral coating, decorated with 5 mol % of peptide (VHPK) to target vascular cell adhesion molecule 1 (VCAM1). Optical imaging validated disease-specific accumulation as anti-miR-712 was efficiently delivered to inflamed mouse aortic endothelial cells in vitro and in vivo. As with the naked anti-miR-712, the delivery of VHPK-CCL-anti-miR-712 effectively downregulated the d-flow induced expression of miR-712 and also rescued the expression of its target genes tissue inhibitor of metalloproteinase 3 (TIMP3) and reversion-inducing-cysteine-rich protein with kazal motifs (RECK) in the endothelium, resulting in inhibition of metalloproteinase activity. Moreover, an 80% lower dose of VHPK-CCL-anti-miR-712 (1 mg/kg dose given twice a week), as compared with naked anti-miR-712, prevented atheroma formation in a mouse model of atherosclerosis. While delivery of naked anti-miR-712 alters expression in multiple organs, miR-712 expression in nontargeted organs was unchanged following VHPK-CCL-anti-miR-712 delivery.
Keywords: microRNA, anti-miRNA, multifunctional particles, targeted delivery, atherosclerosis, endothelial inflammation
Atherosclerosis is an inflammatory disease of the arterial system that leads to myocardial infarction, ischemic stroke and peripheral arterial disease.1 Atherosclerotic plaques preferentially develop in regions exposed to disturbed flow (d-flow) while arterial regions exposed to stable flow (s-flow) are resistant to plaque formation.2−6 Vascular endothelial cells respond to blood flow through mechanosensors, which transduce the shear stress associated with flow into cell signaling events and ultimately result in gene expression.4,5,7 Oligonucleotide-based inhibitors such as anti-miRs or miR-mimic activators have emerged as promising therapeutics to treat various diseases.8 Using the partial carotid ligation mouse model, we have recently shown that miR-712 is a pro-atherogenic, mechanosensitive miRNA that is upregulated by d-flow in endothelial cells both in vitro and in vivo.7,9 Further, we showed that treatment of ApoE–/– mice with naked anti-miR-712 dissolved in saline (delivered via subcutaneous injections at 5 mg/kg dose, twice a week) effectively silenced miR-712 level while rescuing its target gene tissue inhibitor of metalloproteinase-3 (TIMP3) and repressing metalloproteinase activities of matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinases (ADAMs) in mouse arterial endothelium exposed to d-flow.9 Moreover, the systemic anti-miR-712 treatment prevented atherosclerosis development as well as abdominal aortic aneurysm (AAA) formation in Angiotensin-II infused ApoE–/– mice.10
However, several challenges remain to be addressed in using anti-miRs and miR-mimics as therapeutics. One challenge is off-target effects; for example, systemic delivery of naked anti-miR-712 silenced miR-712 levels not only in the intended target cells (inflamed endothelial cells in d-flow regions) but also in other tissues and cells, raising concerns for unexpected and undesired consequences.11 This could be addressed by a targeted delivery of anti-miRs specifically to the inflamed endothelial cells in disease-prone areas. Therefore, a critical challenge in successful gene or RNA therapy is the ability to selectively deliver the therapeutics only to the target cells.
Lipid nanoparticles are an attractive platform for the creation of gene delivery systems.12 Such particles have previously shown efficient uptake and transfection of the liver, lung and in tumors with leaky endothelium where the particles can accumulate via the enhanced permeability and retention effect;13−15 however, therapeutically effective endothelial transfection in a vessel site-specific manner has been elusive. Cationic lipid particles have long been shown to be effective in enhancing transfection both by increasing the rate of internalization and by enhancing the escape of nucleic acids from lysosomal or endosomal compartments. However, particles with exposed cationic membranes induce aggregation and increase cellular and systemic toxicity.
In order to selectively deliver anti-miR-712, we developed a novel strategy to deliver anti-miR-712 (a prototypic therapeutic) encapsulated using lipid particles that contain an aqueous core of nucleic acids complexed with cationic lipid constituting an inner leaflet. The outer leaflet is composed of neutral lipid, which thus creates an asymmetrical bilayer. Such coated cationic lipoparticles (CCLs) have been applied previously to deliver siRNA to the liver and tumors.16−24 In order to target this particle, we have developed a methodology to postinsert a conjugate combining a targeting peptide, polymer and lipid (lipo-PEG-peptide) within the outer leaflet. With this combined strategy, the toxicity of the particle is minimized, the stability of the anti-miR is preserved and loading efficiency and efficacy are maximized.
In order to target VCAM1, a VCAM1-internalizing sequence, VHPKQHR (VHPK) is incorporated within the lipo-PEG-peptide and exposed above the outer leaflet to yield a specific and high-affinity particle that binds to VCAM1 on endothelial surface in atherosclerotic lesions.25−28 The VHPK peptide has previously been conjugated with imaging agents to identify surface VCAM1 in atherosclerotic plaques in vivo.27 Importantly, the VHPK peptide not only binds to VCAM1 but when conjugated to nanoparticles is avidly internalized in endothelial cells,25,28 which could be exploited for targeted delivery of therapeutics to treat atherosclerosis.
Here, we evaluate the use of these targeted CCLs to deliver anti-miR-712 to the inflamed vasculature, the specificity of delivery, and the efficacy and toxicity of the resulting therapy. We demonstrate that VHPK-conjugated to CCLs can serve as an effective anti-miR-712 delivery system specifically to the inflamed endothelium expressing VCAM1 in d-flow regions to inhibit atherosclerosis without significant off-target tissue effects.
Results
Synthesis and Characterization of VHPK-CCL-anti-miR-712 and CCL-anti-miR-712
The CCL synthesis and characterization are described in detail in the Material and Methods and Supporting Information. Briefly, anti-miR-712 was encapsulated into a formulation of DOTAP:DSPE-PEG2k:HSPC:chol (9.3:3.1:52.6:35, molar ratio) in a multistep process (Figure 1A). First, an electrostatic interaction was formed between the negatively charged oligonucleic acid and the positively charged lipid, DOTAP. By employing the concept of Bligh and Dyer monophase,29 the hydrophilic oligonucleic acids were extracted into the organic phase and formed hydrophobic anti-miR-712-DOTAP particles. Anti-miR-712 is a 15-mer oligonucleic acid and stoichiometrically can bind to up to 15 mol of DOTAP. To optimize the various steps involved in the loading procedure and maximize the loading of anti-miR-712, anti-miR-712 was first labeled with carboxyfluorescein (anti-miR-712-FAM). In a series of pilot studies, the amount of DOTAP was kept constant at 0.72 mg (1.03 μmol) and the amount of FAM-labeled anti-miR-712 (anti-miR-712-FAM) was gradually increased to fully react with DOTAP. We found that with 480 μg of anti-miR-712-FAM, 95% of the total was extracted into the organic phase and thus loaded into the particles.
Figure 1.
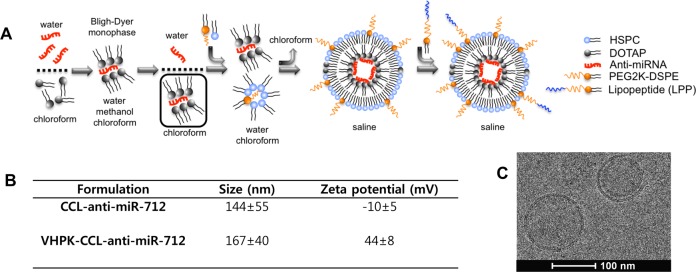
Preparation and characterization of coated-cationic lipoparticles (CCLs) and VHPK-CCLs encapsulating anti-miR-712. (A) The hydrophilic anti-miR-712 in water and hydrophobic DOTAP in chloroform are brought to interaction by addition of methanol to induce the Bligh–Dyer monophase and create the hydrophobic DOTAP-anti-miR-712 complex. Following the addition of water and chloroform to produce a biphasic solution, the hydrophobic complex is extracted in the organic phase and added to a mixture of neutral and PEG lipids. A stable water-in-oil microemulsion was induced upon addition of water. Gradual removal of chloroform resulted in formation of CCLs with asymmetric lipid bilayers entrapping anti-miR-712 within the core. The VCAM1 targeting was achieved by postinsertion of VHPK-LPP into the outer leaflet of CCL-anti-miR-712. (B) Diameter and zeta potential of nontargeted-CCL-anti-miR-712 and VHPK-CCL-anti-miR-712. (C) Electron microscopy image of CCL-anti-miR-712.
The hydrophobic particles in chloroform were then coated with neutral and PEG lipids to enhance the stability and reduce the nonspecific binding. Following extraction of anti-miR-712-FAM with DOTAP in chloroform, the isolated organic phase was added to the coating lipids composed of HSPC:DSPE-PEG2k:chol. Increasing the HSPC-to-DOTAP wt. ratio from 4.5 to 5.0, and 5.7 increased the loading, with the maximum achieved at 5.7 mg of HSPC to 1 mg of DOTAP. Deionized distilled water was then added to induce the water-in-oil emulsion, a reverse phase evaporation30 was carried out and multilamellar liposomes were formed without aggregation or precipitation. After extrusion, CCLs encapsulating anti-miR-712-FAM (CCL-anti-miR-712-FAM) had an average diameter of 144 ± 55 nm and zeta potential of −10 mV, confirming the successful coating of the cationic lipid, where the DSPE-PEG2k adds a slightly negative charge (Figure 1B and C). Upon elution, the CCL-anti-miR-712 was mostly round in shape with either a single or bilayer membrane, as analyzed by electron microscopy (Figure 1C).
Following the purification of CCL-anti-miR-712-FAM, the loading was assessed based on the quenched FAM fluorescence. The fluorescence of CCL-anti-miR-712-FAM eluted in early fractions (fraction #2 and #3) was quenched, and free oligonucleic acid could not be detected (SI Figure S1A). Following particle lysis with Triton X-100, a 7-fold increase in the fluorescence of CCL-anti-miR-712-FAM was observed, indicating that anti-miR-712-FAM is sequestered in the particle interior with a high loading efficiency (SI Figure S1A). While FAM fluorescence is quenched within the CCLs, Alexa555 fluorescence is not quenched within the CCLs and therefore was used in the following in vitro and in vivo studies. Alexa555-labeled anti-miR-712 showed an identical elution pattern with the fluorescent anti-miR-712 eluted only with liposomal fractions (SI Figure S1B).
Peptide VHPK (Boc-VHPKQHRGGSK(ivDde)GC) was prepared using standard Fmoc mediated solid-phase peptide synthesis. For targeting CCLs with VHPK, we compared coupling of VHPK to the surface of CCL-anti-miR712 via maleimide–thiol coupling and via postinsertion of targeting moieties. When 5 mol % DSPE-PEG5000-Maleimide was added to the coating lipids, the anti-miR712 loading efficiency within the final particle was reduced by 50% (SI Figure S1C). Therefore, to maintain the 95% loading of anti-miR-712 in CCLs, the LPP ligand was postinserted into the preformed CCL-anti-miR-712.31
The VHPK-LPP was prepared by conjugation of Fmoc-NH-(PEG)27-COOH (x3) to the antepenultimate lysine on the C-terminus, followed by conjugation of Fmoc-Lys(Fmoc)-OH and two stearic acid chains, as in the literature.32 The purified LPP was confirmed by MALDI (SI Figure S2A). Incubation of VHPK-LPP at 9 mol percent with CCL-anti-miR-712 did not release the encapsulated anti-miR-712, as revealed in the elution profile of FAM-loaded CCLs (SI Figure S2B).
Increasing the molar ratio of VHPK-LPP from 3 to 9% linearly improved the binding potential of VHPK-CCL-anti-miR-712-FAM to HUVECs (SI Figure S3A), and incubation with 9 mol % VHPK-LPP was used to produce VHPK-CCL-anti-miR-712 in all further studies. Following the purification of VHPK-CCL-anti-miR-712-FAM, the amount of VHPK on the surface of CCLs was measured as 4.9 mol % by HPLC (SI Figure S3B). Binding of the VHPK-CCL-anti-miR-712 to HUVECs was confirmed (SI Figure S4A) and a positive surface charge of approximately 44 mV was observed after purification (SI Figure S4B). Assuming the extruded CCLs to be unilamellar vesicles of ∼140 nm with an encapsulated 5.8% anti-miR-712-to-lipid mass ratio and a surface area of approximately 65 × 10–2 nm2 per HSPC molecule,33,34 we encapsulated ∼1400 molecules of anti-miR-712 in each CCL or VHPK-CCL.35,36 Particles with or without the VHPK peptide preserved their size, the surface charge, and the encapsulated anti-miR-712 over 2 months after storage at 4–6 °C; longer periods were not evaluated.
As noted above, in the absence of the targeting moiety, the zeta potential of CCL-anti-miR-712-FAM was −10 mV; however, a positive charge resulted from the addition of the VHPK peptide. Therefore, we assessed the biodistribution and blood circulation of VHPK-CCL-anti-miR-712 by radiolabeling the liposomal outer shell with 64Cu (SI Figure S5 and SI Table S1). No significant difference in organ/tissue accumulation of VHPK-CCL-anti-miR-712 was observed when compared to VHPK-liposomes with a lipid formulation similar to that of the outer membrane of VHPK-CCL, but lacking the cationic lipid and anti-miRNA. The circulation half-life of VHPK-CCL-anti-miR-712 and VHPK-liposomes was 190.8 and 167.7 s, respectively, with no significant difference detected. In each case, the circulation of these targeted particles is far shorter than that (>12 h) reported for nontargeted liposomes.37,38 The results clearly indicate that cationic lipids and anti-miRNA do not alter the biodistribution and blood circulation of VHPK-CCL.
Intracellular Delivery and Silencing Efficacy of VHPK-CCL-anti-miR-712 in Endothelial Cells In Vitro
Since the VHPK-conjugated nanoparticles are known to be internalized via endocytosis,25,39,40 we first determined whether VHPK-CCL-anti-miR-712 can be internalized into endothelial cells. For this study, anti-miR-712 labeled with Alexa555 (easier to detect by fluorescence microscopy than FAM against autofluorescence background in arteries) was encapsulated into either VHPK-CCLs (VHPK-CCL-anti-miR-712-Alexa555) or CCLs (CCL-anti-miR-712-Alexa555) as a nontargeting control. Mouse endothelial cells (iMAECs) were pretreated with TNFα (3 ng/mL), a well-known inducer of VCAM1 expression on the endothelial surface, and incubated with VHPK-CCL-anti-miR-712-Alexa555 or CCL-anti-miR-712-Alexa555 for 30 min and imaged using fluorescence microscopy. When compared to CCL-anti-miR-712, however, VHPK-CCL-anti-miR-712 was abundantly observed in the cytosol of TNFα-treated endothelial cells but not in untreated cells, suggesting its VCAM1-dependent intracellular delivery (Figure 2A).
Figure 2.

VCAM1-dependent delivery of VHPK-CCL-anti-miR-712 and its silencing efficiency in iMAECs. (A) Anti-miR-712 labeled with Alexa555 (200 nM) was delivered to iMAECs in 2 different ways, VHPK-CCL-anti-miR-712 or CCL-anti-miR-712 for 30 min. iMAECs were pretreated with or without TNFα (3 ng/mL) to induce VCAM1. Fluorescence microscopy imaging of iMAECs shows delivery of anti-miR-712 (red) in the TNFα-treated VHPK-CCL group (iv). (B–F) To test the silencing efficiency of 3 different nanoparticles, iMAECs pretreated without or with TNFα were incubated with VHPK-CCL-anti-miR-712, VHPK-CCL-mismatched, and CCL-anti-miR-712 for 24 h. (B) VCAM1 induction in TNFα-treated cells by qPCR. (C) Basal miR-712 expression that was reduced in VHPK-CCL-anti-miR-712 treated cells. (D–F) iMAECs transfected with pre-miR-712 (20 nM) plus TNFα were further incubated with the lipid nanoparticles for 24 h and expression of miR-712 (D), TIMP3 (E) and RECK (F) was determined by qPCR and normalized to 18S. n = 5, data shown as mean ± s.e.m; *p < 0.05 as determined by Student’s t-test.
Next, we compared the miR-712 silencing efficiency of VHPK-CCL-anti-miR-712 to two controls: VHPK-CCL containing a mismatched anti-miR-712 (VHPK-CCL-mismatched) and nontargeted CCLs containing anti-miR-712 (CCL-anti-miR-712). For this, we determined the miR-712 expression, and expression of its target genes TIMP3 and RECK.9,10 To induce VCAM1 expression on endothelial cells, iMAECs were treated with TNFα (3 ng/mL for 30 min). As expected, TNFα significantly increased VCAM1 expression compared to the vehicle control in iMAECs (Figure 2B). In these cells, the basal expression of miR-712 was significantly reduced in iMAECs only when treated with VHPK-CCL-anti-miR-712 in TNFα-treated cells, but not in any other control groups (Figure 2C). The efficacy of VHPK-CCL-anti-miR-712 in silencing miR-712 expression in iMAECs in the VCAM1-targeted manner was further demonstrated in cells transfected with pre-miR-712 to overexpress it. As shown in Figure 2D, pre-miR-712 increased miR-712 levels in iMAECs by ∼30-fold over controls and it was significantly reduced only in cells treated with TNFα and VHPK-CCL-anti-miR-712, but not by other control CCLs or in TNFα-untreated cells. These results demonstrate that VHPK-CCL-anti-miR-712 effectively silenced miR-712 expression only in TNFα-treated iMAECs suggesting its VCAM1-targeted delivery. Consistent with this result, VHPK-CCL-anti-miR-712 was able to rescue expression of the well-known miR-712 target genes, TIMP3 and RECK (Figure 2E and F). However, the effect of anti-miR-712 on the target gene rescue was not observed in non-TNFα-treated cells, VHPK-CCL-mismatched or CCL-anti-miR-712 (Figure 2E and F). These results further support that VHPK-CCL-anti-miR-712 effectively silenced miR-712 expression and rescued expression of its downregulated target genes in a VCAM1-targeted manner.
VCAM1-Targeted Delivery of VHPK-CCL-anti-miR-712 to Inflamed Endothelium in d-flow Regions In Vivo
We first confirmed expression of VCAM1 as a marker of inflammation in various regions of mouse arterial endothelium exposed to d-flow or s-flow by en face confocal imaging. As we showed previously,2 the mouse partial carotid ligation surgery induces d-flow in the left carotid artery (LCA) while the contralateral right carotid artery (RCA) continues to experience s-flow (Figure 3A). VCAM1 protein expression (shown in red) significantly increased in the LCA endothelium compared to the RCA at 4-days post partial carotid ligation surgery (Figure 3B). Further, expression of VCAM1 was significantly increased in the lesser curvature (LC) region of the aortic arch (naturally occurring pro-atherogenic d-flow region) compared to the greater curvature (GC) region (naturally occurring athero-resistant s-flow region) in control C57BL/6 mice as well (Figure 3A and B). These results confirmed that VCAM1 expression was significantly higher in endothelial cells exposed to d-flow conditions induced either by surgery or naturally in curved arteries.
Figure 3.

Targeted delivery of VHPK-CCLs to inflamed endothelium expressing VCAM1 in d-flow regions. (A) The partial carotid ligation surgery in the left common carotid artery (LCA) and contralateral RCA along with the naturally occurring d-flow (lesser curvature, LC) and s-flow (greater curvature, GC) regions in the aortic arch are indicated. (B) VCAM1 antibody staining (red) in arterial endothelium was carried out using the LCA and RCA as well as the aortic arches (GC and LC) obtained from C57BL/6 mice at 4 days post partial carotid ligation surgery. Shown are representative (n = 3) en face confocal images of VCAM1 (red), DAPI (blue) and internal elastic lamina (IEL, green autofluorescence). (C) At 4 days postpartial ligation, anti-miR-712 was conjugated with Alexa555 and mice were tail-vein injected with VHPK-CCL-anti-miR-712 or CCL-anti-miR-712 (1 mg/kg). One hour later, the carotids and aortic arch were dissected out and en face confocal imaged. Representative images (n = 3) showed that Alexa555 (red) was visible in mouse arteries injected with VHPK-CCL-Alexa only in d-flow regions (LCA and LC). DAPI (blue) and IEL (green autofluorescence).
We next tested whether VHPK-CCLs or nontargeted CCLs carrying anti-miR-712 can be delivered to VCAM1-expressing endothelial cells in the surgically induced and naturally occurring d-flow regions in vivo. For identification, anti-miR-712 conjugated with Alexa555 was loaded within CCLs. On day 4 post partial ligation, mice were injected with either VHPK-CCL-anti-miR-712-Alexa555 or nontargeted-CCL-anti-miR-712-Alexa555 via the tail vein. One hour after the injection, en face confocal imaging of arterial endothelium showed significant Alexa555 signals in d-flow regions (both LCA and LC) but not in the s-flow regions (RCA and GC), corresponding to the VCAM1 expression in these regions (Figure 3C). This result demonstrated that VHPK-CCLs can effectively and selectively deliver anti-miR-712 to the pro-atherogenic, inflamed endothelium expressing VCAM1 in d-flow regions in vivo. Further confocal en face imaging studies showed that VHPK-CCL-anti-miR-712-Alexa555 were found only in the intimal layer, especially within the endothelial cells (SI Figure S6), consistent with the intracellular uptake of the VHPK peptide upon VCAM1 targeting.25,28 VHPK-CCL-encapsulating Alexa555 without anti-miR-712 also showed similar endothelial accumulation (SI Figure S7), further suggesting that preferential accumulation of the targeted CCLs is determined by VHPK targeting but not influenced by the therapeutic effect of anti-miR-712.
Selective and Efficient Silencing Effect of VHPK-CCL-anti-miR-712 in Inflamed Endothelium In Vivo
Next, we determined if anti-miR-712 delivery using VHPK-CCLs can effectively silence increased miR-712 expression in inflamed LCA endothelium of partially ligated mice. For this study, VHPK-CCL-anti-miR-712, VHPK-CCL-mismatched control, or nontargeted-CCL-anti-miR-712 (1 mg/kg each) were injected into C57BL/6 mice via the tail vein 4 and 5 days post partial carotid ligation. At 7 days post partial ligation, endothelial-enriched RNA was isolated from the RCA and LCA, respectively. The expression of miR-712 was increased in the LCA endothelium (EC-enriched) compared to the RCA (Figure 4A) which was significantly silenced in endothelium only in the mice treated with VHPK-CCL-anti-miR-712 but not in VHPK-CCL-mismatched or nontargeting CCL-anti-miR-712 controls. Interestingly, however, this silencing effect of VHPK-CCL-anti-miR-712 was observed only in endothelial layer but not in the media and adventitia (Figure 4A and B). The mechanism for the lack of anti-miR-712 effect in the media and adventitia is currently unclear, but correlates with the lack of anti-miR-712-Alexa555 in the media (SI Figure S6).
Figure 4.

Selective and efficient silencing effect of VHPK-CCL-anti-miR-712 in inflamed endothelium in d-flow regions of mice. At 2 days following partial carotid ligation, ApoE–/– mice were tail-vein injected with VHPK-CCL-anti-miR-712, VHPK-CCL-mismatched control (VHPK-CCL-anti-m.m) or CCL-anti-miR-712 (1 mg/kg). Mice were sacrificed 2 days later and endothelial-enriched RNA was extracted from the RCA and LCA. Expression of miR-712 (A) and its target genes TIMP3 and RECK (C, E) was determined by qPCR. RNA from the left over (LO) samples (containing media and adventitia) were also tested for expression of miR-712 (B) and its target genes TIMP3 and RECK (D, F) for comparison. Expression of miRNA and mRNAs was normalized to RNU6b and 18S, respectively as internal controls. n = 5, data shown as mean ± s.e.m; *p < 0.05 as determined by Student’s t-test. In some studies, frozen sections of the RCA and LCA (n = 5 each) were prepared and immunostained with TIMP3 (red) (G); and in situ gelatinase activity assay using DQ-gelatin (green) (H) was performed as shown by the representative images (n = 5). Inset shows zoomed sections of endothelial regions showing expression of TIMP3 under different treatment conditions. L = lumen. Internal elastic lamina (green autofluorescence), nuclei (blue).
Next, we examined the expression of two well-known miR-712 target genes, TIMP3 and RECK, in the same samples used above. As expected, TIMP3 and RECK expression was significantly reduced in LCA endothelium (Figure 4C and E) and was restored to its RCA control levels in mice treated with VHPK-CCL-anti-miR-712 but not in VHPK-CCL-mismatched or nontargeting CCL-anti-miR-712 controls. Similar to the above miR-712 result, TIMP3 and RECK expression was restored by VHPK-CCL-anti-miR-712 only in the endothelium, but not in the medial and adventitial samples (Figure 4D and F).
We next tested whether VHPK-CCL-anti-miR-712 treatment can rescue the loss of TIMP3 at the protein level as well by d-flow in vivo. Confocal immunostaining shows that d-flow induced a significant loss of TIMP3 protein expression (shown in red) in LCA and was rescued by VHPK-CCL-anti-miR-712 treatment but not with VHPK-CCL-mismatched or CCL-anti-miR-712 (Figure 4G). Since we previously showed that TIMP3 is a potent regulator of metalloproteinase activity,9 we further tested the functional consequence of rescuing TIMP3 by VHPK-CCL-anti-miR-712 by in situ zymography. D-flow increased gelatinase activity as shown by increased green fluorescence from cleaved DQ-Gelatin which was prevented by VHPK-CCL-anti-miR-712 treatment but not by VHPK-CCL-mismatched or CCL-anti-miR-712 (Figure 4H). These results clearly demonstrate that anti-miR-712 delivered via VHPK-CCL specifically and efficiently reduced miR-712 expression in inflamed endothelium in d-flow regions. Moreover, VHPK-CCL-anti-miR-712 treatment effectively restored the loss of its targets (especially TIMP3) at the gene, protein and functional activity levels.
Antiatherogenic Effect of VHPK-CCL-anti-miR-712
We tested whether VHPK-CCL-anti-miR-712 treatment can inhibit atherosclerosis development using the partial carotid ligation model using ApoE–/– mice. As shown previously,2,9 partial carotid ligation surgery plus a high-fat diet rapidly induced robust atherosclerosis (indicated with white arrow) in the LCA within 2 weeks, as shown by bright field imaging (top) and cross-sectional Oil-Red-O stained images of LCA and RCA (mid and bottom panels) (Figure 5A). Intravenous injection of VHPK-CCL-anti-miR-712 (1 mg/kg, twice a week for 2 weeks) significantly reduced atherosclerotic lesion development (Figure 5A and B), compared to the mismatched or nontargeting controls. Further, lipid profiling showed no significant difference between the three CCL groups, suggesting that the antiatherogenic effect of VHPK-CCL-anti-miR-712 was not due to changes in the lipid profile (SI Table S2).
Figure 5.

Treatment with VHPK-CCL-anti-miR-712 inhibits atherosclerosis in ApoE–/– mice. ApoE–/– mice received the partial carotid ligation surgery plus high-fat diet for 2 weeks to rapidly develop atherosclerosis in the LCA. At 4, 5, 8, and 11 days post partial ligation surgery, mice were injected with VHPK-CCL-anti-miR-712, VHPK-CCL-anti-miR-mismatched control, or CCL-anti-miR-712 (1 mg/kg anti-miR-712 dose each injected in 150 μL via tail-vein). Mice were sacrificed at 2 weeks postsurgery, arterial trees dissected out for bright field imaging and histological evaluation (A) and plaque size quantification by image analysis (B). n = 7 mice each, data shown as mean ± s.e.m; *p < 0.05 as determined by 1-way ANOVA. Following bright field imaging, LCA and RCA were frozen sectioned (from the mid regions indicated by the arrows) and stained with Oil-Red-O for further immunohistochemical examination as shown by representative images (A). (C) VHPK-CCL-anti-miR-712 did not affect miR-712 expression in other tissues and cells. Following extraction of arteries, various tissues and whole blood were also obtained from these mice, total RNA extracted, and expression of miR-712 in liver, kidneys, lungs, spleen, thymus, bone marrow and PBMCs was determined by qPCR. n = 7 mice each; data shown as mean ± s.e.m.
Off-Target Effects and Toxicity
We determined whether anti-miR-712 delivered via VHPK-CCLs showed any silencing effects in nontargeted tissues other than the inflamed arterial endothelial cells. For this study, we extracted the RNAs from tissue samples, liver, kidney, lung, spleen, thymus, bone marrow and peripheral blood mononuclear cells (PBMCs), harvested from the same animals used for the atherosclerosis studies above. We found no significant difference in expression of miR-712 across all 3 treatment groups in all tissues tested (Figure 5C). Taken together, these findings clearly demonstrate that delivery of anti-miR-712 using the VHPK-CCLs as delivery vehicles effectively inhibited atherosclerosis development without discernible off-target effects.
In an initial in vitro assessment of toxicity, we found that increased C3a activation was induced by CCL and VHPK-CCL; however, the levels of C3a induced by VHPK-CCL and CCL injection were 5.7- and 10-fold significantly lower than the level of activation produced by the injection of the positive control, Zymosan, (p < 0.001), respectively (SI Figure S8). Further, we assessed the acute toxicity of CCL-anti-miR-712, VHPK-CCL-anti-miR-712, VHPK-CCL-mismatched and VHPK-vacant liposomes without an encapsulated oligonucleotide. After a single injection and 24 h circulation period, analysis of blood samples did not reveal a significant difference in circulating proteins, blood chemicals, and counts of various blood cell types as compared with control (saline-treated) animals (SI Figure S9). Further, no change in the animal body weight or the weight of the dissected heart, liver, and spleen was observed across the types of CCLs and liposomes studied (SI Figure S9).
In order to evaluate the toxicity of the multiple treatments used within this study, we examined the mouse and organ weight, blood chemistry, complete blood count and tissue histology after 5 injections over 2 weeks of VHPK-CCL-anti-miR-712 or control (saline injection). The animal weight, the weight of the major organs and the blood chemistry and complete blood count were unchanged by treatment (SI Figure S10A–D). Histological sections of skeletal muscle, heart, liver and spleen did not show any significant differences between the corresponding tissue in VHPK-CCL- and saline-treated animals (SI Figure S11). Circulating cytokines (IL-1β, IL-10, IL-6, IL-12, GM-CSF, IL-5, IFN-γ, TNF-α, IL-2, IL-4) were below the detection limit for both VHPK-CCL- and saline-treated animals.
Discussion
We have developed a new VCAM1-targeted anti-miR delivery strategy by packaging anti-miR-712 in CCLs conjugated to VHPK peptides. We have demonstrated for the first time, to our knowledge, that this VCAM1-targeted strategy effectively delivers anti-miR-712 to inflamed endothelial cells in the pro-atherogenic d-flow regions. Further, our data showed that anti-miR-712 was internalized into the inflamed endothelium and effectively silenced miR-712 and rescued its downstream gene targets (TIMP3 and RECK) both in vitro and in vivo. Importantly, VHPK-CCL-anti-miR-712 was able to inhibit atherosclerosis development in ApoE–/– mice demonstrating the proof-of-concept that this VCAM1-targeted anti-miR delivery strategy could be used as a novel, effective antiatherogenic therapy without apparent off-tissue target effects and at a much lower dose compared to systemically delivered naked anti-miR-712. While, naked anti-miR-712 delivered via subcutaneous injection required a 5 mg/kg dose (twice a week),9 the VCAM1-targeted lipid nanoparticle approach showed a similar antiatherogenic effect at a 1 mg/kg dose (twice a week via tail vein injection).
In our previous study based on subcutaneous injection of naked anti-miR-712, we observed significant miR-712 silencing in nonendothelial cells including whole blood cells and the spleen with silencing trends shown in the liver, thymus and lymph nodes.9 In fact, we previously reported that administration of the naked anti-miR-712 resulted in miR-712 expression (relative to saline injection) of 0.11 ± 0.03, 0.30 ± 0.14, 0.24 ± 0.27 and 0.20 ± 0.03 in the liver, spleen, thymus and PBMCs, respectively.9 With the targeted, encapsulated delivery, mean miR-712 expression remained above 0.91 at each site and no significant change was observed. Thus, miR-712 silencing was not observed in those organs and in fact was limited to the inflamed endothelial cells of the vascular wall. These two studies were carried out in an overlapping time period by the same investigators in the same lab using the same batches of reagents including anti-miR-712 and same ApoE–/– mice (all purchased from Jackson Lab), and therefore can be directly compared in terms of the specificity and efficacy of anti-miR-712 in endothelial and nonendothelial tissues. The subcutaneous injection of naked anti-miRs is a widely accepted current method of choice. While this method has been successfully used in many animal studies and even an ongoing Phase II clinical trial, anti-miRs or miR-mimics delivered in this fashion have access to all tissues, making it difficult to deliver them specifically to the target tissues. Indiscriminate tissue-wide delivery of anti-miRs or miR-mimics may provoke undesirable effects in some tissues and cells raising safety concerns. Therefore, better strategies such as our VHPK-CCLs that can deliver these anti-miRs specifically to the intended cells and tissues at the site of diseased areas are urgently needed.
Lipid-based nanoparticles are an attractive platform for the creation of gene delivery systems because they are bioinert, nontoxic and carry and protect a large payload.12−15 Cationic lipid structures have long been shown to be particularly effective in enhancing transfection both by increasing the rate of internalization and by enhancing the escape of nucleic acids from lysosomal or endosomal compartments. However, particles with exposed cationic membranes can increase systemic toxicity, thereby limiting dosing and delivery. In order to minimize toxicity, methods to coat cationic structures have been developed and typically result in asymmetrical bilayers.16−24 One such structure is the CCL, in which nucleic acids such as anti-miRs are complexed with cationic lipids and the resulting nanoparticle is coated with neutral lipid. The zeta potential of the resulting nontargeted CCL was −10 mV with a positive zeta potential detected only after the addition of the VHPK cationic peptide. We confirmed that the blood half-life and biodistribution of the empty liposome and CCL were similar. Our resulting strategy for particle assembly resulted in efficient internalization without any loss of functional activity of anti-miR-712, as indicated by its silencing efficiency toward miR-712, both in vitro and in vivo. As expected, a low level of complement activity was observed with the targeted CCL, where the level is 5.7 fold less than the positive control and is below that previously observed by our group for targeted liposomes and microbubbles.41,42 Therefore, we do not anticipate a significant issue with complement activation. Through a toxicity study with a dosing protocol that mimicked the therapeutic study (5 intravenous administrations of VHPK-CCLs encapsulating anti-miR-712 at 1 mg anti-miR/kg body weight) we demonstrated that systemic toxicity was not detected by weight, blood chemistry, complete blood counts, circulating cytokines or histology. Taken together the results suggest that our CCL strategy enhanced transfection efficiency while reducing off-target effects and toxicity.
Another critical challenge in successful gene or RNA therapy is the ability to selectively deliver the nucleic acids only to the target cells, such as inflamed endothelial cells expressing VCAM1 on their surface. Recently, lipid-based nanoparticles have been developed to effectively deliver siRNAs to endothelial cells in various vascular beds including lung, heart and tumor,13−15 but they were not specifically delivered to a specific site, such as inflamed endothelium in pro-atherogenic regions. To overcome this challenge, we used VHPK, the VCAM1-internalizing peptide, which was discovered by Kelly et al. as a peptide homologous to the integrin Very Late Antigen-4 that plays a role in leukocyte adhesion to endothelium.25 Two important characteristics of the VHPK peptide are that it binds to VCAM1 on the endothelial cell surface and it is subsequently internalized, potentially via the clathrin-mediated endocytosis pathway.39,40 The VHPK peptide has been used successfully for imaging of atherosclerotic plaques in vivo,26,27 but to our knowledge it has not been reported as a targeted therapeutic delivery system.
One of the greatest challenges in the creation of multifunctional particles is to incorporate the targeting moiety without reducing the loading or stability. Our previous work with peptide-conjugated particles targeted to the heart demonstrated that targeted accumulation on the endothelium of arterioles, venules and larger vessels increases with peptide concentrations up to 6 mol %.32 With the postinsertion strategy employed here, VHPK peptide incorporation, co-optimized with 95% anti-miR loading peaked at ∼5 mol %. The postinsertion strategy used here was superior to other postconjugation strategies, as the incorporation of lipids terminated in functional groups (such as maleimide) reduced the loading and stability.
Intracellular delivery of VHPK-CCL-anti-miR-712 was supported by our confocal imaging studies both in vitro and in vivo (Figures 2A and 3C; SI Figures S6 and S7), and more importantly by the efficient silencing of miR-712 in inflamed endothelial cells both in vitro and in vivo (Figures 2C and D, 4A), but not in the medial smooth muscle cells (Figure 4B). We have previously demonstrated that a small molecule encapsulated within heart-targeted liposomes travels from endothelial cells to fibroblasts and to a lesser degree to myocytes.43 Here, while the anti-miR was internalized by the endothelial cells, it was not transported further into the vessel wall. Still, silencing of miR-712 with the downstream effect on TIMP3 and therefore presumably on MMPs was sufficient to prevent the development of atherosclerosis in this model of vascular inflammation and disturbed flow.
Conclusion
Here, we developed and tested a strategy to accomplish targeted delivery of anti-miR-712 using VHPK-CCLs to inflamed endothelium in the pro-atherogenic d-flow regions. Our approach can easily be applied to other therapeutics such as miRNA inhibitors, miRNA mimics, siRNAs, plasmids and drugs. Like VHPK, other targeting ligands that are found in disease specific areas such as cancers may be used for targeted delivery of the CCL-carrying therapeutics to their site of action. Our treatment method may benefit human atherosclerosis prevention along with other lifestyle changes such as diet modification and exercise. In summary, we showed that the VCAM1-targeted cationic nanoparticles carrying anti-miRs can serve as a new gene delivery vehicle to perform targeted antiatherogenic therapy while minimizing potential off-target tissue effects.
Materials and Methods
Study Design and Animal Numbers
The main goal of this study was first to develop lipid nanoparticles that (1) are coated with cationic lipids (CCLs) to enhance the nucleotide-based anti-miR transfection to endothelial cells in vitro and in vivo, (2) carry VHPK peptide postinserted after the CCL preparation to ensure proper placement of the peptide on the CCL surface, and (3) encapsulate anti-miR-712 within CCL for its efficient escape from the lysosomal compartment in the cell. The targeting specificity and efficiency of VHPK-CCL-anti-miR-712 to inflamed endothelial cells was determined by comparing to proper controls (targeted vs nontargeting peptide as well as mismatched anti-miR control) in vitro using iMAECs and in vivo using C57BL/6 and ApoE–/– mice with or without partial carotid ligation surgery to induced d-flow in the LCA in comparison to the contralateral RCA. All in vitro studies were carried out using at least three biologically independent replicates while in vivo therapeutic and targeting studies were carried out with n = 5 mice and toxicity studies with n = 4 mice based on our previous experience for these types of studies. For atherosclerosis studies using the partial ligation, n = 7 was chosen based on a Power Calculation analysis. For all mouse studies, only male mice were used for reproducibility within a gender.
Materials
Hydrogenated soy phosphatidylcholine (HSPC), cholesterol (chol), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000]. (DSPE-PEG2k), and 1,2-dioleoyl-3-trimethylammonium propane (DOTAP) were from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). 5′-NH2-modified anti-miRNA composed of locked nucleic acid (LNA) and phosphorothioate backbone was purchased from Exiqon (Woburn, MA). (Expanded material is found in the Supporting Information).
Optical Imaging
Two fluorescent probes were used to label anti-miR-712. FAM is a quenchable fluorophore (Ex: 495 nm, Em: 516 nm) and was tagged to anti-miR-712 for in vitro optimization of the loading of anti-miR-712 in the interior. FAM-labeled anti-miR-712 was quenched when loaded in the interior aqueous compartment and fluorescence was restored upon disintegration of particle. In later studies of tracking encapsulated anti-miR-712 (in which quenching was not desirable and a very bright fluorophore was required), Alexa555 (Ex: 556 nm, Em: 573 nm) was used for labeling anti-miR-712 when encapsulated in CCLs. For these studies, the dye conjugation was performed in house. We verified that the conjugation of these dyes does not affect the functionality of the anti-miR using the methods below.
Labeling of Anti-miRNA with Alexa555- and FAM-NHS Ester
5′-C6-NH2-modified anti-miRNA (1 mg) was attached to either Alexa555- or FAM-NHS ester by using amine-reactive cross-linker reaction (See Supporting Information on line). The prepared product was purified and desalted by liquid chromatography and desalting column, respectively.
Preparation of VHPK-PEG-peptide
Peptide VHPK (Boc VHPKQHRGGSK(ivDde)GC) was conjugated to Fmoc-NH-(PEG)27-COOH using solid phase peptide conjugation chemistry, as described in32 and in the expanded methods online. Crude LPP (∼11 mg/run) was purified using semipreparative high performance liquid chromatography (HPLC) and the purified LPP was confirmed by mass spectroscopy (SI Figure S2A).
Preparation of Coated Cationic Lipoparticles (CCLs) Encapsulating Anti-miR-712
For preparation of 1 mL of CCLs with 7.75 mg of total lipids, a 480-mg portion of anti-miR-712-FAM (equivalent to 85.3 nanomole of anti-miR-712) was dissolved in 500 μL of distilled deionized water. DOTAP, 1.04 μmol, dissolved in 500 μL of chloroform and 1040 μL of methanol was added, and the mixture was gently mixed to form a monophase, as schematically illustrated in Figure 1A. After a 30 min incubation at room temperature, 500 μL of distilled deionized water and 500 μL of chloroform were added to form two separate phases. Upon mixing and centrifugation at 800g for 8 min at 5 °C, the organic phase containing the DOTAP-anti-miR-712-FAM complex was extracted. Under these conditions, 95% of anti-miR-712-FAM was recovered in the organic phase. The resulting anti-miR-712-to-DOTAP molar ratio was 1:12, and a positive-to-negative charge ratio of ∼1:1 was obtained.
The extracted organic phase was added to dried lipid composed of HSPC:DSPE-PEG2k:Chol (5.8:0.34:3.8 μmol) to give a PC-to-DOTAP ratio of 5.6:1. Next, 500 μL of distilled deionized water was added and the mixture was vortexed for 20 s and then emulsified by sonication for 2 min. The organic phase was evaporated under a vacuum on a rotary evaporator. After 30 min, 500 μL of distilled deionized water was added and the mixture was kept under a vacuum in the rotary evaporator for another 30 min. Lipo-particles (1 mL) which form as the organic phase evaporates, were extruded 21 passages through a 100 nm polycarbonate membrane at 55 °C. The size of the CCLs encapsulating anti-miR-712-FAM (CCL-anti-miR-712-FAM) was determined using a Malvern Zeta-Sizer Instrument (UK). Lipid concentration was measured using the Phospholipids C assay kit (Wako Chemicals USA, Richmond, VA) according to the manufacturer’s instructions.
Postinsertion of VHPK-lipo-PEG-peptide into CCL-anti-miR-712
Lipo-PEG-peptide (LPP) containing VHPK (VHPK-LPP) was dissolved in chloroform and subsequently dried under a stream of nitrogen. CCL-anti-miR-712-FAM was then added to the dried LPP and stirred slowly. The resulting VHPK-CCL-anti-miR-712-FAM and nontargeted CCL-anti-miR-712-FAM were purified using Sepharose CL-4B columns. LPP incorporation was analyzed by HPLC. The HPLC column used for the analysis was Phenomenex Jupiter C4, 5 μm, 250 × 4.6 mm). A standard curve was obtained from a known concentration of LPP solution by area integration from HPLC chromatogram.
Preparation of VHPK-Vacant Liposomes or VHPK-Liposomes-Alexa555
Lipid composition of VHPK-liposomes was matched with that of the lipid coating of CCLs. Lipids composed of HSPC:DSPE-PEG2k:chol (57:3:35, mol %) were mixed with 5 mol % VHPK-LPP in chloroform. Chloroform was evaporated under stream of nitrogen gas and the remaining traces of solvent were removed with lyophilization overnight. The dried lipid and VHPK-LPP were hydrated with PBS (−/−) (with or without Alexa) at 62–64 °C for 30 min. The resulting multilamellar VHPK-liposomes were extruded 21 passages through a 100 nm polycarbonate membrane at 62–64 °C. The extruded liposomes were eluted through a Sephadex G-75 column equilibrated with PBS (−/−) and rich fractions of purified liposomes were collected and combined. The liposomes had an average diameter of 159 ± 52 nm with a zeta potential of 26 mV.
In Vitro miRNA Knockdown and Targeted Gene Effects of CCLs
Immortalized mouse aortic endothelial cells (iMAECs) were used for this study. Cells were pretreated with 3 ng/mL of mouse TNFα to induce expression of surface VCAM1. To induce the expression of miR-712, cells were transfected with 20 nM pre-miR-712 or control pre-miR. To study the effect of CCLs on miR-712 and its target genes, cells were incubated with either Alexa555-labeled-anti-miR-712 encapsulated in VHPK-CCLs or in nontargeting CCLs for various time points.
In Vitro Imaging of Cell-CCL Interactions
For imaging iMAECs, cells were washed with ice cold DPBS including 0.9 mM calcium and 0.5 mM magnesium (DPBS+/+) supplemented with 1% w/v bovine serum albumin (BSA) X3 and images were acquired using a fluorescence microscope. Post imaging, the regular culture media was replaced and cells were kept in a CO2 incubator for 24 h and total RNA was extracted to study expression of miR-712 and its target gene TIMP3 and RECK by qPCR. All in vitro experiments included non-CCL exposed controls as well as a naked Alexa555-labeled anti-miR-712 control.
Evaluation of CCL Toxicity
First, complement activation was assessed in complement-preserved human serum. Second, the acute toxicity of three types of CCLs: CCL-anti-miR-712, VHPK-CCL-anti-miR-712 and VHPK-CCL-mismatched, and VHPK-vacant liposomes (not containing anti-miR-712) was evaluated after single dose intravenous administration to C57BL/6 mice at 1 mg anti-miR-712/kg-body weight (n = 5 per group). The mice were weighed and euthanized after 24 h; blood was analyzed for complete blood count (CBC) and circulating proteins as a measure of liver/kidney function. Third, multidose toxicity was assessed in ApoE–/– C57BL/6 mice on a high fat diet as described in the SI. After 5 injections over 14 days, the mice were euthanized, blood was analyzed for CBC and circulating proteins and skeletal muscle, heart, liver, and spleen were weighed and processed for hematoxylin and eosin. In addition, cytokine levels were assessed treatment termination on day 18.
Mouse Partial Carotid Ligation Surgery
All animal procedures carried out for this study were approved by the Animal Care and Use Committees at Emory University and University of California at Davis. Partial ligation surgery was performed as described previously.2,44 Following surgery, animals were fed a Paigen’s high-fat diet (HFD; Science Diets) containing 1.25% cholesterol, 15% fat, and 0.5% cholic acid45 for 2 weeks.
Isolation of Endothelial-Enriched RNA
Following partial carotid ligation in C57BL/6 mice, endothelial enriched RNA from the carotids and aortic arch regions was extracted as described previously.2,44,9 For each experiment, the LC and GC regions from two to four mice were pooled. qPCR for endothelium (PECAM1), smooth muscle (αSMA) and immune cell (CD45) marker genes was used to determine the enrichment of endothelial RNA in each prep, and confirmed that the endothelial-enriched fraction did not contain a discernible contamination of RNAs from smooth muscle cells and immune cells (SI Figure S12).
Plaque Lesion Analysis
Aorta and carotid arteries were isolated en bloc as described above from ligated ApoE–/– mice fed a high-fat diet for 2 weeks. RCA and LCA were photographed using a CCD camera attached to a dissection microscope and the opaque area covered by plaque and total artery area of LCA were quantified using NIH ImageJ software.
Quantitative Real-Time PCR (qPCR)
Intimal RNAs from three LCAs or RCAs were pooled to obtain ∼30 ng total RNA.2 RNA samples were reverse-transcribed for cDNA synthesis and qPCR was performed to determine mRNA expression as described previously.2,44,9 Fold changes between LCA and RCA were determined for all targets using the ΔΔCt method. Sequences for primers used for mRNA expression studies were previously published by us.9,10
Mature and precursor miRNA assays were performed using miRNA quantification via SYBR green qPCR with a miScript reverse transcription kit (Qiagen) according to the manufacturer’s instructions. qPCR was performed using miScript SYBR Green PCR kit (Qiagen) with miScript universal primer (U6B) and the miRNA-specific forward primers and relative fold change was calculated. The specific mature and pre-miR primers were purchased from Qiagen.
En Face Staining
C57BL/6 mice were sacrificed by CO2 inhalation and perfused first with saline containing heparin and then with formalin. Aortas were excised, fixed in 4% paraformaldehyde, and tissue samples from LCA and RCA as well as the greater and lesser curvature of the aortic arch taken. Samples were counterstained using DAPI (Sigma) and mounted on glass slides using fluorescence mounting medium (Dako). Samples were imaged using a Zeiss LSM 510 META confocal microscope (Carl Zeiss).
Immunohistochemistry
Following treatment, ApoE–/– mice were euthanized and perfused with saline containing heparin. LCA and RCA were collected en bloc along with the heart, aortic arch, trachea, esophagus, and surrounding fat tissue. Tissue was embedded in optimal cutting temperature (OCT) compound (Tissue-Tek), frozen on liquid nitrogen and stored at −80 °C until used. To visualize atherosclerosis development, Oil red O staining was carried out as described previously.9 Sections were fixed in methanol/acetone and then blocked (1 h, at RT) using 10% (v/v) donkey serum in PBS. Immunohistochemical staining was carried out using following antibodies: TIMP3 (Abcam) (1:100) and RECK (cell signaling) (1:100) overnight at 4 °C. Samples were imaged using a Zeiss LSM 510 META confocal microscope (Carl Zeiss).
In Situ Zymography
Gelatinolytic activity was analyzed in unfixed cryostat sections (8 μm thick) using DQ-gelatin as a substrate (Molecular Probes) as we recently described.10
Transient Transfections of Naked Anti-miRs
Cells were transiently transfected with naked anti-miR-712 (400 nM; Exiqon), mismatched anti-miR control (400 nM; Exiqon), pre-miR-712 (20 nM; Ambion), or control-pre-miR (20 nM; Ambion) using Oligofectamine (Invitrogen), following manufacturer’s protocol as described previously.9,46
Statistical Analyses
Statistical analyses were carried out with Graph-Pad Prism (GraphPad Software). All error bars reported are SEM unless otherwise indicated. Pairwise comparisons were performed using one-way Student t-tests. Multiple comparisons of means were performed using 1-way ANOVA followed by Tukey’s multiple comparison tests. Differences between groups were considered significant at P values below 0.05. All miRNA array data were analyzed with GenomeStudio software (Illumina).
Acknowledgments
This work was supported by funding from National Institutes of Health grants HL119798, HL113451, HL095070 and HL124879 to HJ and HL124879 and the Research Investments in Science and Engineering program to KF. This work was also supported by the National Heart Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology award HHSN268201000043C to HJ and KF. SK is an American Heart Association Postdoctoral fellow.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.5b02611.
Additional information for the Materials and Methods, Figures S1–S12, and Tables S1–S2. (PDF)
Author Contributions
⊥ AK and CWK contributed equally to this work. KWF and HJ also contributed equally to this paper and are co-corresponding authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Libby P.; Theroux P. Pathophysiology of Coronary Artery Disease. Circulation 2005, 111, 3481–3488 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- Nam D.; Ni C. W.; Rezvan A.; Suo J.; Budzyn K.; Llanos A.; Harrison D.; Giddens D.; Jo H. Partial Carotid Ligation Is a Model of Acutely Induced Disturbed Flow, Leading to Rapid Endothelial Dysfunction and Atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1535–43 10.1152/ajpheart.00510.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarbell J. M.; Shi Z.-D.; Dunn J.; Jo H. Fluid Mechanics, Arterial Disease, and Gene Expression. Annu. Rev. Fluid Mech. 2014, 46, 591–614 10.1146/annurev-fluid-010313-141309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak B. R.; Back M.; Bochaton-Piallat M. L.; Caligiuri G.; Daemen M. J.; Davies P. F.; Hoefer I. E.; Holvoet P.; Jo H.; Krams R.; et al. Biomechanical Factors in Atherosclerosis: Mechanisms and Clinical Implications. Eur. Heart J. 2014, 3530133020a–3020d 10.1093/eurheartj/ehu353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies P. F. Endothelial Mechanisms of Flow-Mediated Athero-Protection and Susceptibility. Circ. Res. 2007, 101, 10–2 10.1161/CIRCRESAHA.107.156539. [DOI] [PubMed] [Google Scholar]
- Davies P. F. Hemodynamic Shear Stress and the Endothelium in Cardiovascular Pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26 10.1038/ncpcardio1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Kim C. W.; Simmons R. D.; Jo H. Role of Flow-Sensitive MicroRNAs in Endothelial Dysfunction and Atherosclerosis: Mechanosensitive Athero-MiRs. Arterioscler., Thromb., Vasc. Biol. 2014, 34, 2206–16 10.1161/ATVBAHA.114.303425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E.; Purcell A. L.; Levin A. A. Developing MicroRNA Therapeutics. Circ. Res. 2012, 110, 496–507 10.1161/CIRCRESAHA.111.247916. [DOI] [PubMed] [Google Scholar]
- Son D. J.; Kumar S.; Takabe W.; Kim C. W.; Ni C. W.; Alberts-Grill N.; Jang I. H.; Kim S.; Kim W.; Won Kang S.; et al. The Atypical Mechanosensitive MicroRNA-712 Derived from Pre-Ribosomal RNA Induces Endothelial Inflammation and Atherosclerosis. Nat. Commun. 2013, 4, 3000. 10.1038/ncomms4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C. W.; Kumar S.; Son D. J.; Jang I. H.; Griendling K. K.; Jo H. Prevention of Abdominal Aortic Aneurysm by Anti-MicroRNA-712 or Anti-MicroRNA-205 in Angiotensin Ii-Infused Mice. Arterioscler., Thromb., Vasc. Biol. 2014, 34, 1412–21 10.1161/ATVBAHA.113.303134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. A.; Betel D.; Miller M. L.; Sander C.; Leslie C. S.; Marks D. S. Transfection of Small RNAs Globally Perturbs Gene Regulation by Endogenous MicroRNAs. Nat. Biotechnol. 2009, 27, 549–55 10.1038/nbt.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrecilla J.; Rodriguez-Gascon A.; Solinis M. A.; del Pozo-Rodriguez A. Lipid Nanoparticles as Carriers for RNAi against Viral Infections: Current Status and Future Perspectives. Biomed. Res. Int. 2014, 2014, 17. 10.1155/2014/161794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santel A.; Aleku M.; Keil O.; Endruschat J.; Esche V.; Fisch G.; Dames S.; Loffler K.; Fechtner M.; Arnold W.; et al. A Novel SiRNA-Lipoplex Technology for RNA Interference in the Mouse Vascular Endothelium. Gene Ther. 2006, 13, 1222–34 10.1038/sj.gt.3302777. [DOI] [PubMed] [Google Scholar]
- Schultheis B.; Strumberg D.; Santel A.; Vank C.; Gebhardt F.; Keil O.; Lange C.; Giese K.; Kaufmann J.; Khan M.; et al. First-in-Human Phase I Study of the Liposomal RNA Interference Therapeutic Atu027 in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2014, 32, 4141–8 10.1200/JCO.2013.55.0376. [DOI] [PubMed] [Google Scholar]
- Dahlman J. E.; Barnes C.; Khan O. F.; Thiriot A.; Jhunjunwala S.; Shaw T. E.; Xing Y.; Sager H. B.; Sahay G.; Speciner L.; et al. In Vivo Endothelial SiRNA Delivery Using Polymeric Nanoparticles with Low Molecular Weight. Nat. Nanotechnol. 2014, 9, 648–55 10.1038/nnano.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignole C.; Pagnan G.; Marimpietri D.; Cosimo E.; Allen T. M.; Ponzoni M.; Pastorino F. Targeted Delivery System for Antisense Oligonucleotides: A Novel Experimental Strategy for Neuroblastoma Treatment. Cancer Lett. 2003, 197, 231–235 10.1016/S0304-3835(03)00107-1. [DOI] [PubMed] [Google Scholar]
- Hussain S.; Plueckthun A.; Allen T. M.; Zangemeister-Wittke U. Chemosensitization of Carcinoma Cells Using Epithelial Cell Adhesion Molecule-Targeted Liposomal Antisense against Bcl-2/Bcl-Xl. Mol. Cancer Ther. 2006, 5, 3170–3180 10.1158/1535-7163.MCT-06-0412. [DOI] [PubMed] [Google Scholar]
- Pagnan G.; Stuart D. D.; Pastorino F.; Raffaghello L.; Montaldo P. G.; Allen T. M.; Calabretta B.; Ponzoni M. Delivery of C-Myb Antisense Oligodeoxynucleotides to Human Neuroblastoma Cells Via Disialoganglioside Gd(2)-Targeted Immunoliposomes: Antitumor Effects. J. Natl. Cancer Inst. 2000, 92, 253–261 10.1093/jnci/92.3.253. [DOI] [PubMed] [Google Scholar]
- Pastorino F.; Brignole C.; Marimpietri D.; Pagnan G.; Morando A.; Ribatti D.; Semple S. C.; Gambini C.; Allen T. M.; Ponzoni M. Targeted Liposomal C-Myc Antisense Oligodeoxynucleotides Induce Apoptosis and Inhibit Tumor Growth and Metastases in Human Melanoma Models. Clin. Cancer Res. 2003, 9, 4595–4605. [PubMed] [Google Scholar]
- Pastorino F.; Mumbengegwi D. R.; Ribatti D.; Ponzoni M.; Allen T. M. Increase of Therapeutic Effects by Treating Melanoma with Targeted Combinations of C-Myc Antisense and Doxorubicin. J. Controlled Release 2008, 126, 85–94 10.1016/j.jconrel.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Stuart D. D.; Allen T. M. A New Liposomal Formulation for Antisense Oligodeoxynucleotides with Small Size, High Incorporation Efficiency and Good Stability. Biochim. Biophys. Acta Biomembr. 2000, 1463, 219–229 10.1016/S0005-2736(99)00209-6. [DOI] [PubMed] [Google Scholar]
- Stuart D. D.; Kao G. Y.; Allen T. M. A Novel, Long-Circulating, and Functional Liposomal Formulation of Antisense Oligodeoxynucleotides Targeted against Mdr1. Cancer Gene Ther. 2000, 7, 466–475 10.1038/sj.cgt.7700145. [DOI] [PubMed] [Google Scholar]
- Li J.; Chen Y.-C.; Tseng Y.-C.; Mozumdar S.; Huang L. Biodegradable Calcium Phosphate Nanoparticle with Lipid Coating for Systemic SiRNA Delivery. J. Controlled Release 2010, 142, 416–421 10.1016/j.jconrel.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Yang Y.; Huang L. Calcium Phosphate Nanoparticles with an Asymmetric Lipid Bilayer Coating for SiRNA Delivery to the Tumor. J. Controlled Release 2012, 158, 108–114 10.1016/j.jconrel.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly K. A.; Nahrendorf M.; Yu A. M.; Reynolds F.; Weissleder R. In Vivo Phage Display Selection Yields Atherosclerotic Plaque Targeted Peptides for Imaging. Mol. Imaging Biol. 2006, 8, 201–7 10.1007/s11307-006-0043-6. [DOI] [PubMed] [Google Scholar]
- Nahrendorf M.; Keliher E.; Panizzi P.; Zhang H.; Hembrador S.; Figueiredo J. L.; Aikawa E.; Kelly K.; Libby P.; Weissleder R. 18f-4v for Pet-Ct Imaging of VCAM-1 Expression in Atherosclerosis. JACC Cardiovasc. Imaging 2009, 2, 1213–22 10.1016/j.jcmg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M.; Jaffer F. A.; Kelly K. A.; Sosnovik D. E.; Aikawa E.; Libby P.; Weissleder R. Noninvasive Vascular Cell Adhesion Molecule-1 Imaging Identifies Inflammatory Activation of Cells in Atherosclerosis. Circulation 2006, 114, 1504–11 10.1161/CIRCULATIONAHA.106.646380. [DOI] [PubMed] [Google Scholar]
- Kelly K. A.; Allport J. R.; Tsourkas A.; Shinde-Patil V. R.; Josephson L.; Weissleder R. Detection of Vascular Adhesion Molecule-1 Expression Using a Novel Multimodal Nanoparticle. Circ. Res. 2005, 96, 327–336 10.1161/01.RES.0000155722.17881.dd. [DOI] [PubMed] [Google Scholar]
- Bligh E. G.; Dyer W. J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Biochem. Physiol. 1959, 37, 911–917 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Szoka F.; Papahadjopoulos D. Procedure for Preparation of Liposomes with Large Internal Aqueous Space and High Capture by Reverse-Phase Evaporation. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 4194–4198 10.1073/pnas.75.9.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira J. N.; Ishida T.; Gaspar R.; Allen T. M. Use of the Post-Insertion Technique to Insert Peptide Ligands into Pre-Formed Stealth Liposomes with Retention of Binding Activity and Cytotoxicity. Pharm. Res. 2002, 19, 265–9 10.1023/A:1014434732752. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Kusunose J.; Kheirolomoom A.; Seo J. W.; Qi J.; Watson K. D.; Lindfors H. A.; Ruoslahti E.; Sutcliffe J. L.; Ferrara K. W. Dynamic Imaging of Arginine-Rich Heart-Targeted Vehicles in a Mouse Model. Biomaterials 2008, 29, 1976–88 10.1016/j.biomaterials.2007.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Mason J. T. Geometric Packing Constraints in Egg Phosphatidylcholine Vesicles. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 308–310 10.1073/pnas.75.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucerka N.; Kiselev M. A.; Balgavy P. Determination of Bilayer Thickness and Lipid Surface Area in Unilamellar Dimyristoylphosphatidylcholine Vesicles from Small-Angle Neutron Scattering Curves: A Comparison of Evaluation Methods. Eur. Biophys. J. 2004, 33, 328–34 10.1007/s00249-003-0349-0. [DOI] [PubMed] [Google Scholar]
- Kheirolomoom A.; Ferrara K. W. Cholesterol Transport from Liposomal Delivery Vehicles. Biomaterials 2007, 28, 4311–20 10.1016/j.biomaterials.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheirolomoom A.; Dayton P. A.; Lum A. F.; Little E.; Paoli E. E.; Zheng H.; Ferrara K. W. Acoustically-Active Microbubbles Conjugated to Liposomes: Characterization of a Proposed Drug Delivery Vehicle. J. Controlled Release 2007, 118, 275–84 10.1016/j.jconrel.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J. W.; Mahakian L. M.; Tam S.; Qin S.; Ingham E. S.; Meares C. F.; Ferrara K. W. The Pharmacokinetics of Zr-89 Labeled Liposomes over Extended Periods in a Murine Tumor Model. Nucl. Med. Biol. 2015, 42, 155–163 10.1016/j.nucmedbio.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J. W.; Zhang H.; Kukis D. L.; Meares C. F.; Ferrara K. W. A Novel Method to Label Preformed Liposomes with 64cu for Positron Emission Tomography (Pet) Imaging. Bioconjugate Chem. 2008, 19, 2577–2584 10.1021/bc8002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinea M.; Manduteanu I.; Dragomir E.; Capraru M.; Simionescu M. Immunoliposomes Directed toward Vcam-1 Interact Specifically with Activated Endothelial Cells--a Potential Tool for Specific Drug Delivery. Pharm. Res. 2005, 22, 1906–17 10.1007/s11095-005-7247-3. [DOI] [PubMed] [Google Scholar]
- Ricard I.; Payet M. D.; Dupuis G. Vcam-1 Is Internalized by a Clathrin-Related Pathway in Human Endothelial Cells but Its Alpha 4 Beta 1 Integrin Counter-Receptor Remains Associated with the Plasma Membrane in Human T Lymphocytes. Eur. J. Immunol. 1998, 28, 1708–18. [DOI] [PubMed] [Google Scholar]
- Borden M. A.; Zhang H.; Gillies R. J.; Dayton P. A.; Ferrara K. W. A Stimulus-Responsive Contrast Agent for Ultrasound Molecular Imaging. Biomaterials 2008, 29, 597–606 10.1016/j.biomaterials.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Tam S.; Ingham E. S.; Mahakian L. M.; Lai C. Y.; Tumbale S. K.; Teesalu T.; Hubbard N. E.; Borowsky A. D.; Ferrara K. W. Ultrasound Molecular Imaging of Tumor Angiogenesis with a Neuropilin-1-Targeted Microbubble. Biomaterials 2015, 56, 104–113 10.1016/j.biomaterials.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Li N.; Sirish P.; Mahakian L.; Ingham E.; Curry F. R.; Yamada S.; Chiamvimonvat N.; Ferrara K. W. The Cargo of Crppr-Conjugated Liposomes Crosses the Intact Murine Cardiac Endothelium. J. Controlled Release 2012, 163, 10–17 10.1016/j.jconrel.2012.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam D.; Ni C. W.; Rezvan A.; Suo J.; Budzyn K.; Llanos A.; Harrison D. G.; Giddens D. P.; Jo H. A Model of Disturbed Flow-Induced Atherosclerosis in Mouse Carotid Artery by Partial Ligation and a Simple Method of RNA Isolation from Carotid Endothelium. J. Visualized Exp. 2010, 10.3791/1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni C. W.; Qiu H.; Rezvan A.; Kwon K.; Nam D.; Son D. J.; Visvader J. E.; Jo H. Discovery of Novel Mechanosensitive Genes In Vivo Using Mouse Carotid Artery Endothelium Exposed to Disturbed Flow. Blood 2010, 116, e66–73 10.1182/blood-2010-04-278192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni C. W.; Qiu H.; Jo H. MicroRNA-663 Upregulated by Oscillatory Shear Stress Plays a Role in Inflammatory Response of Endothelial Cells. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1762–9 10.1152/ajpheart.00829.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.