

Abstract
Triatoma dimidiata (Latreille, 1811) is the most abundant and significant insect vector of the parasite Trypanosoma cruzi in Central America, and particularly in Guatemala. Tr. cruzi is the causative agent of Chagas disease, and successful disease control requires understanding the geographic distribution and degree of migration of vectors such as T. dimidiata that frequently re-infest houses within months following insecticide application. The population genetic structure of T. dimidiata collected from six villages in southern Guatemala was studied to gain insight into the migration patterns of the insects in this region where populations are largely domestic. This study provided insight into the likelihood of eliminating T. dimidiata by pesticide application as has been observed in some areas for other domestic triatomines such as Triatoma infestans. Genotypes of microsatellite loci for 178 insects from six villages were found to represent five genetic clusters using a Bayesian Markov Chain Monte Carlo method. Individual clusters were found in multiple villages, with multiple clusters in the same house. Although migration occurred, there was statistically significant genetic differentiation among villages (FRT = 0.05) and high genetic differentiation among houses within villages (FSR = 0.11). Relatedness of insects within houses varied from 0 to 0.25, i.e., from unrelated to half-sibs. The results suggest that T. dimidiata in southern Guatemala moves between houses and villages often enough that recolonization is likely, implying the use of insecticides alone is not sufficient for effective control of Chagas disease in this region and more sustainable solutions are required.
Keywords: Chagas disease, Triatoma dimidiata, dispersal, vector control, genetic diversity
Understanding migration and gene flow of insect vectors is critical to effective control and elimination of vector-borne diseases. Of more than 140 species of triatomines, less than a couple dozen are responsible for the bulk of the transmission to humans of Tr. cruzi, the parasite that causes Chagas disease (Stevens et al. 2011). Chagas disease is the most serious parasitic disease in Latin America, with an estimated seven million people infected with Tr. cruzi, 60 million people living at risk of contracting the disease, and 23,000 deaths annually (Hotez et al. 2008). The triatomine species that transmit Tr. cruzi to people differ in their epidemiological importance, and this difference is strongly influenced by vector movement (Lent and Wygodzinsky 1979). Immature nymphs can migrate, actively walking tens of meters, and adults of some species are known to fly up to 200 m, or even 2 km (Schweigmann et al. 1988). Triatomines can also migrate passively by carriage in wood, personal effects, agricultural products, or by birds (Lent and Wygodzinsky 1979). The field of population genetics can be used to understand the genetic structure of triatomine species and to quantify migration and gene flow, important parameters for designing control strategies effective against local species and populations.
Triatoma dimidiata (Latreille, 1811) is the major Chagas disease vector in Central America. Its range extends from southern Mexico throughout Central America, and into South America (Venezuela, Colombia, Ecuador, and northern Peru; Ramirez et al. 2005). Triatoma dimidiata varies morphologically, genetically, and biochemically over this geographic range with an unnamed sibling species identified in northern Guatemala (Petén); Yucatán, Mexico; and Belize (Panzera et al. 2006, Dorn et al. 2007, Bargues et al. 2008, Dorn et al. 2009). Triatoma dimidiata shows variability in inhabiting domestic, peri-domestic, and sylvatic habitats (Zeledon et al. 1973, Nakagawa et al. 2003, Zeledon et al. 2005, Dorn et al. 2007, Blandon-Naranjo et al. 2010), and the tendency to colonize houses is of particular importance for transmission to people. In the hyperendemic area of southern Guatemala, as well as in Ecuador, only a single lineage of T. dimidiata occurs and it is found exclusively in domestic and peri-domestic habitats, often with high house infestation (Monroy et al. 2003b, Bargues et al. 2008). Within southern Guatemala, the department of Jutiapa has had some of the highest house infestation rates in Guatemala (35% infestation), and bugs are not found in the surrounding remnant forest (Tabaru et al. 1999, Monroy et al. 2003b, Zeledon and Rojas 2006). In contrast, in northern Guatemala, Costa Rica, and Colombia, T. dimidiata remains in the sylvan environment, only rarely visiting houses (Monroy et al. 2003c; Zeledon and Rojas 2006). In the Yucatán, Mexico, T. dimidiata enters houses seasonally from April to June (Dumonteil et al. 2002, Monroy et al. 2003a).
Although information is available regarding the habitat preference of T. dimidiata in different localities (Zeledon et al. 1973, Monroy et al. 2003b, Zeledon et al. 2005, Dorn et al. 2007, Bargues et al. 2008, Blandon-Naranjo et al. 2010), little is known if migration and gene flow differ among localities and habitats. Preliminary studies examining genetic differentiation among sylvatic locations 80–200 km apart, using Random Amplified Polymorphic DNA by the Polymerase Chain Reaction (RAPD-PCR), showed significant genetic differentiation (D = 0.121–0.189, Nei’s genetic distance), indicating little gene flow (Calderon et al. 2004). However, the same study found domestic populations 40–100 km apart showed more gene flow, indicated by lower genetic differentiation (D = 0.05–0.085). On a smaller spatial scale of meters rather than kilometers, domestic populations showed migration among three households 80–160 m apart within the rural community of Aguazarca, Santa Rosa, Guatemala (D = 0.013–0.022) and between nearby villages Aguazarca and El Cuje (∼25 km distant, D = 0.0199) by RAPD-PCR (Dorn et al. 2003). The low genetic differentiation indicates that gene flow, i.e., migration or passive transport, occurs among households as well as among villages. These results used RAPD markers, which can be inaccurate, and were based on only three houses and two villages (Dorn et al. 2003) or among three villages (Calderon et al. 2004). To gain a better picture of the ecology of T. dimidiata, we conducted further population genetic studies to determine if these observations apply elsewhere, in particular in an endemic area of Guatemala.
Microsatellite loci are valuable markers for understanding the genetic structure of vector populations (e.g., Anderson et al. 2002, Dumonteil et al. 2007, Pizarro et al. 2008). These loci are highly variable, differing in the number of repeats due to polymerase slippage during replication (Brinkmann et al. 1998). The large number of alleles allows fine-scale discrimination of closely related individuals, providing the statistical power to understand gene flow and the genetic structure of populations (Hardy et al. 2003). In addition, microsatellite loci are more reproducible than RAPD-PCR markers and most microsatellite loci (except perhaps tri-nucleotide repeats) are noncoding, hence neutral, markers.
To understand migration and gene flow of T. dimidiata, seven microsatellite loci were examined in 178 individuals to analyze the population genetic structure and gene flow, within and among six villages, in southern Guatemala where Chagas disease is hyperendemic.
Materials and Methods
Specimen Collection
The 178 T. dimidiata specimens examined were collected by trained personnel from the Monroy laboratory and the Guatemalan Ministry of Health using the person/hour method (Monroy et al. 2003 c) from houses in six villages in the Department (State) of Jutiapa, Guatemala, during August–September 2001 and January 2002 before fumigation (Fig. 1; Table 1). The study was approved for informed oral consent by two review boards: the World Health Organization Tropical Disease Research (WHO-TDR) and the Dirección General de Investigación de la Universidad de San Carlos. All residents provided informed oral consent, which was documented on a survey sheet prior to bug collection. Five villages, Calderas, El Carpintero, El Copante, El Tablón, and Tunillas, are located within the Municipality of San José Acatempa and one village, La Brea, is located within the Municipality of Quezada. All are rural communities with houses at least 50 m apart. The region is experiencing rapid deforestation, and the villages are nestled among forest remnants and agricultural crops on rolling hills. There are no major topographical barriers, such as mountains or large rivers, in the area.
Fig. 1.
Map showing the location of the six villages in Jutiapa, Guatemala.
Table 1.
T. dimidiata collection information for six villages in Jutiapa, Guatemala
| Feature | Calderas | El Tablón | El Carpintero | La Brea | Tunillas | El Copante |
|---|---|---|---|---|---|---|
| No. insects (n) | 30 | 30 | 30 | 30 | 30 | 28 |
| n/housea average | 2.7 | 3.0 | 2.2 | 2.1 | 2.5 | 2.6 |
| n/house range | 1–6 | 1–6 | 1–5 | 1–7 | 1–5 | 1–6 |
| No. of houses | 6 | 9 | 13 | 13 | 6 | 7 |
| House unknown | 14 | 3 | 2 | 0 | 15 | 12 |
| Latitude (N) | 14° 13′16′′ | 14° 14′35′′ | 14° 14′12′′ | 14° 13′44′′ | 14° 19′43 | 14° 15′00′′ |
| Longitude (W) | 90° 04′52′′ | 90° 07′50′′ | 90° 06′30 | 90° 03′40′′ | 90° 03′50′′ | 90° 06′40′′ |
| Meters above sea level | 1240 | 1310 | 1245′′ | 1140 | 1250 | 1350 |
a Only for insects where the house of collection is known.
The live insects were placed in vials with folded paper, and in most cases the house where the insect was collected was recorded. Insects were transported to the Universidad de San Carlos, Guatemala City, Guatemala, where the life cycle stage (first- through fifth-instar nymphs) or sex (for adults) and infection status with Tr. cruzi (assessed by microscopic examination of the contents of the intestines and rectum; [Pizarro et al. 2007]) were recorded. The legs of each bug were removed and placed in 95% alcohol and 5% glycerol and stored at −20°C. For each village, the number of insects analyzed, the average number of insects per house (for insects where the house of collection is known), and the number of houses examined are shown in Table 1.
Molecular Genetic Analysis
DNA Isolation
Insect legs were transported to Loyola University New Orleans and stored at −80°C until DNA was isolated using published methods (Dorn et al. 2003).
Microsatellite Amplification
PCR amplification of seven microsatellite loci (Table 2 ) was performed in 10 µl reactions: 5 µl Multiplex PCR Master Mix 2x (Qiagen,Valencia, CA), 2 µl H2O, 1 µl Q-solution, 1 µl primer mix, and 200 ng of DNA. The reactions were multiplexed in two groups. The loci in group 1 (and their fluorescent dye) included—TDMS09 (HEX), TDAK01 (FAM), TDAK17 (FAM), and TDAK20 (NED). Group 2 included—TDMS22 (HEX), TDAK04 (NED), and TDAK49 (FAM). TDMS22 is a tetra-nucleotide repeat; the other six loci are di-nucleotide repeats. The amplification consisted of 95°C for 15 min, followed by 35 cycles of 94°C for 30 s, 52°C for 90 s, 72°C for 60 s, and a final extension at 72°C for 10 min, using a Techne Thermocycler (TC-512, Burlington, NJ). The PCR product was diluted 1/20 with water, and 0.5 µl of diluted product from each insect was mixed with 14.6 µl of formamide and 0.40 µl of LIZ 500 size standard (Applied Biosystems, Foster City, CA). The sizes of the resulting fluorescently labeled DNA fragments were determined using either an Applied BioSystems 3100-Avant Genetic Analyzer (University of Vermont Cancer Center, DNA Analysis Facility) or Applied BioSystems 3730xl DNA Analyzer (Cornell University, Life Sciences Core Laboratories Center) and data recorded in .fsa format data files.
Table 2.
Genetic information for the seven microsatellite loci analyzed in 28–30 T. dimidiata from each of six villages in Jutiapa, Guatemala
| Locusa | Accession number | Forward primer (5′-3′) | Reverse primer (5′-3′) | Size (bp) | Na | null alleles (%) |
|---|---|---|---|---|---|---|
| TDMS09 | AF487985 | cat tgc aat cgt gtc gaa at | tgc cca aaa ttt tcg tgt ct | 157–205 | 10.2 | 1.7 |
| TDMS22 | AF487989 | tat tat ggt tgc cgg tat taa gg | cca ccc aag ttt tac tat cca | 98–160 | 5.8 | 0 |
| TDAK01 | AY695416 | gaa att agc tca aga aca aca ctc c | acc aaa gca tgc caa gta gg | 169–215 | 11.0 | 11.2 |
| TDAK04 | AY695418 | agc gat tgt tct cct ttg c | ttc ttt tcg gtc ccg tag tg | 130–208 | 13.5 | 2.2 |
| TDAK17 | AY695419 | tct tcc aac cat ttt tcg ttt tg | ctg cta cta aag gat cta tca aat tt | 120–142 | 8.5 | 0 |
| TDAK20 | AY695420 | tgt cgt cca agt tta ttt gct c | tga ttt ctt cta ttt cgt ttg gaa | 119–188 | 11.8 | 6.2 |
| TDAK49 | AY695423 | tat cat aga ccc aca cgg t | taa atg gca ggt tgg att cg | 144–178 | 9.8 | 1.1 |
The locus name, based on previous publication or information in GenBank for unpublished loci, GenBank accession number, nucleotide sequences for forward and reverse primers, size of the amplified fragment in base pairs (bp), the average number of alleles per village (Na), and percent of individuals with null alleles for each locus are indicated.
a TDMS are from (Anderson et al. 2002), TDAK (http:/www.ncbi.nlm.nih.gov/nuccore/ACCESSIONNUMBER).
Fragment Analysis
The fsa data files were viewed using GeneMapper version 3.7 (Applied Biosystems, Foster City, CA) to identify and classify the alleles present per locus and per insect.
Statistical Methods
Summary Statistics for Each Locus and Village
For each locus, the allele size range, average number of alleles per village, and percent of individuals with missing data were summarized. For each village, genetic diversity was estimated using Nei’s unbiased estimator of HS (Nei and Chesser 1983) and unbiased allelic richness (El Mousadik and Petit 1996) using FSTAT (Goudet 1995, 2002).
Estimating Population Genetic Structure
Two approaches were used to estimate the genetic structure. First because the villages and houses represent discrete physical units, multilocus versions of Wright’s F statistics (Wright 1978) were used in a hierarchical AMOVA (Arlequin ver. 3.51, Excoffier and Lischer 2010) to test for significant population structure among villages (FRT) and among houses within villages (FSR). FIS values are not presented because they cannot be accurately estimated given the small number of individuals per house; however, relatedness (Lynch and Ritland 1999) of insects within houses and within villages was calculated using GenAlEx version 6.3 (Peakall and Smouse 2006). To facilitate interpretation of F statistics based on microsatellite loci, we calculated the maximum possible differentiation among villages FRT(max) (Hedrick 2005). This allows standardization of F values because for loci with more than two alleles, the magnitude is dependent on the amount of genetic variation (FRT < 1 − HR, where 1 − HR is the average within-subpopulation homozygosity [Hedrick 2005]); the maximum value may be much lower than 1 for highly variable loci. Letting pij be the frequency of allele i in subpopulation j, the maximum F value is:
= FRT/FRT(max). In addition, isolation-by-distance was tested (GENEPOP ver. 4.1.4, Watts et al. 2007, Rousset 2008). The statistic ê was used to estimate the neighborhood size (Watts et al. 2007) as the slope of the regression of ê on ln(distance).
The second approach to assessing genetic population structure used Bayesian Markov chain Monte Carlo (MCMC) simulation (Pritchard et al. 2000) to estimate the number of genetic clusters and then determined the geographic distribution of the genetic clusters among villages and among houses within a village. The number of genetic clusters among the sample of all 178 insects was determined with the software STRUCTURE version 2.3.2 (Pritchard et al. 2000) using the recommended conservative settings: correlated allele frequencies (migration or shared ancestry may lead to similar frequencies in different populations) and admixture (allows that some insects might be progeny of migrants). A single run of the algorithm starts with an initial random association of alleles in K clusters and iteratively rearranges the alleles so that variation within clusters is minimized and variation among clusters is maximized. Each replicate compares the simulated clusters to a random clustering and estimates the likelihood of the data Ln(K) for the specified K (number of clusters). We simulated 10 independent replicates for each value of K from 2 to 6. Each replicate included a burn-in period to ensure the results did not vary depending on the starting configuration. Previous studies (Evanno et al. 2005) recommend a burn in of at least 20,000 iterations followed by 20,000 iterations. We used a burn in of 100,000 iterations followed by 100,000 iterations, although preliminary analyses showed values >20,000 did not significantly change the results. The appropriate number of clusters was determined with the statistic ΔK (Evanno et al. 2005); however, previous work has shown that with a hierarchical island population structure (i.e., houses within villages), STRUCTURE will detect the uppermost level of structure, in this case villages (Evanno et al. 2005). The distribution of genetic clusters within and among villages was visualized using CLUMPP ver. 1.1.2 and DISTRUCT ver. 1.1 (Rosenberg 2004, Jakobsson and Rosenberg 2007) to clarify relationships.
Test for Biased Dispersal and Variation in Infection Prevalence
We tested for biased dispersal between males and females and between Tr. cruzi-infected and uninfected insects. Two parameters for biased dispersal were analyzed with FSTAT Version 2.9.3.2 (Goudet et al. 2002) using 10,000 randomizations. First, FST, which should be lower in the group with greater dispersal, was compared between the pairs described above. The second test compares the variance of assignment index, corrected for among-population variation in gene diversity (vAIc). Within a village, the dispersing group will include both residents (with common genotypes) and immigrants (with rare genotypes), so that vAIc will be larger for the dispersing group (Goudet et al. 2002, Prugnolle and de Meeus 2002). In addition to testing for biased dispersal, we used a nested ANOVA to test for a difference in the prevalence of insect infection with Tr. cruzi among houses within villages and among villages.
Results
Summary Statistics for Each Locus and Village
As expected for microsatellite loci, all seven loci are highly polymorphic (6–14 alleles per locus, per village, Table 2). The genetic variability of these loci is quite similar among the six villages; allelic richness varies from 9–11 and Nei’s HS ranges from 0.74 to 0.85 (Fig. 2).
Fig. 2.

Similarity of the genetic variability of seven microsatellite loci in T. dimidiata from six villages in Jutiapa, Guatemala, based on allelic richness and Nei’s HS.
Estimating Population Genetic Structure
Hierarchical AMOVA (Table 3 ) indicates small, but statistically significant, differences among villages (FRT = 0.049) and among houses within villages (FSR = 0.110). Standardization of FRT by FRT(max) = 0.217 gives a value of = FRT/ FRT(max) = 0.226. Average relatedness among insects was at the level of second cousins (r = 0.031) within four villages and higher in two villages (close to the level of first cousins, r = 0.125; Fig. 3). Some migration among houses within villages is apparent because relatedness was significantly greater than zero in 60% of the houses with three or more insects (12/20). In eight of these houses, the relatedness was at the level of half-sibs (r = 0.25) or higher. In only one of the 20 houses is there evidence that all the insects could be from the same parents (i.e., standard error of the mean includes r = 0.5, the level of full sibs). The isolation-by-distance analysis (Fig. 4) indicates that related individuals are spatially near each other; however, the correlation is not very strong: ê = 0.0719 + 0.0165 × ln(distance), 95% confidence interval for slope [0.0061–0.0292]. The estimate of the neighborhood size is Nb = 60.
Table 3.
Hierarchical AMOVA shows significant population structure among villages (FRT) and among houses within villages (FSR)
| Source of variation | df | Sum of squares | Variance components | Fixation Index | P |
|---|---|---|---|---|---|
| Among villages (R) | 5 | 53.16 | 0.13 | FRT = 0.049 | < 0.001 |
| Among houses within villages (S) | 24 | 127.30 | 0.29 | FSR = 0.110 | < 0.001 |
| Total | 213 | 582.98 | 2.77 |
Fig. 3.
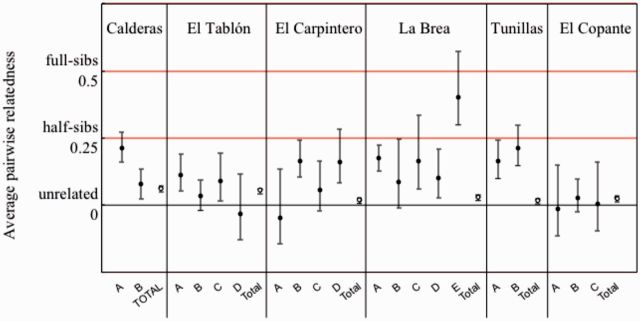
Variability in average pairwise relatedness among houses with >2 insects (A–E,•) and for the 28–30 insects from each village (Village, ○). Values are ± SEM. Note values can be negative due to sampling error.
Fig. 4.

Linear regression (…….) showing that genetic differentiation between insects increases with geographical distance. Data are pairwise genetic distances among six villages (28–30 insects per village. Linear regression: ê = 0.0719 + 0.0165 * ln(distance); 95% confidence interval for slope [0.0061, 0.292]).
With hierarchical island structure, such as houses within villages, we expect the Bayesian MCMC simulations from STRUCTURE to detect the highest level of structure i.e., villages (Evanno et al. 2005). The STRUCTURE results (Fig. 5) found five genetic clusters, one less than the number of villages (Supporting Information Supp Fig. 1 [online only]). Although a single cluster dominates many villages, migration of T. dimidiata is inferred because all of the clusters are found in multiple villages and all villages have more than one cluster. The replicate STRUCTURE runs nearly always grouped the adults into the same clusters (correlation among the 10 structure runs = 98.9% for K = 5 [i.e., five clusters]). Many individual insects are of one cluster (i.e., more than half of most of the vertical lines are one color; Fig. 5), especially in Calderas, yet many insects from Tunillas and El Copante are genetic admixtures. Although each cluster is present in all six villages, within a village often one cluster (Calderas: green, El Tablón: purple, and Tunillas: orange) or sometimes two or more clusters (El Carpintero: red and purple, La Brea: blue and orange, and El Copante: red blue and purple) tend to predominate. Most houses had insects from more than one cluster (e.g., Calderas) while in two houses all the insects were from the same genetic cluster (La Brea orange and El Copante red). We take this evidence of multiple clusters per house and multiple houses per cluster to indicate migration and gene flow among houses and among villages.
Fig. 5.
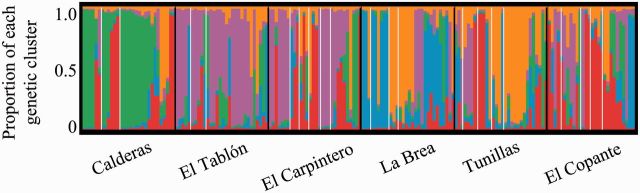
Distribution of five genetic clusters (indicated by colors) among villages, based on Bayesian MCMC simulation. Vertical black lines separate the six villages, indicated below the figure. White lines separate the houses. Each insect is represented by a thin vertical line partitioned into colored segments indicating one of K = 5 genetic clusters. For example, the first house in Calderas contained four insects from the green cluster, an orange–green–blue admixture, and an orange–purple admixture. Only houses with more than two insects are delineated; the remaining insects are pooled in the rightmost partition for each village.
Testing Biased Dispersal and Variation in Infection Prevalence
There was no evidence of biased dispersal based on Tr. cruzi infection status (uninfected vs infected: FST 0.08 vs 0.07, P > 0.05; vAIC 15.38 vs 14.85, P > 0.05) or sex (female vs male: FST 0.07 vs 0.05, P > 0.05; vAIC 16.52 vs 19.30, P > 0.05). Further, the overall prevalence of Tr. cruzi infection was 34%; however, neither villages nor houses within villages differ in infection prevalence (likelihood ratio chi-square: villages = 8.88, df = 4, P > 0.05; houses within villages = 24.38; df = 19, P > 0.05).
Discussion
In this study, we show that in southern Guatemala, populations of the major Chagas disease vector, T. dimidiata, contain distinct genetic clusters that are found in multiple houses within the same village, suggesting migration and gene flow among houses. This conclusion is based on both multilocus versions of Wright's F statistics (among houses within villages: FSR = 0.110, P < 0.001, Table 3) and Bayesian maximum likelihood clustering (Fig. 5). Gene flow is also supported because average relatedness among insects was on the order of full or half-sibs in over half the houses with more than three insects (Fig. 3). In addition, the lack of aggregation of infected insects within households or villages supports the conclusion that insects are migrating, but tests for biased dispersal indicated that migration is not influenced by Tr. cruzi infection and is similar among males and females.
Although cryptic species are one possible explanation for the observed genetic differentiation (i.e., five clusters), there is no evidence of cryptic species in this hyperendemic area of southern Guatemala (Panzera et al. 2006, Dorn et al. 2007, Bargues et al. 2008, Dorn et al. 2009). The single lineage of T. dimidiata in this region is found exclusively in domestic and peri-domestic habitats, often with high house infestation (Monroy et al. 2003b, Bargues et al. 2008). A more likely explanation for the observed genetic structure is migration among houses, which leads to several clusters within a house. This explanation would include cases with a mated female moving to a house and producing several offspring.
Three lines of evidence support population structure among villages. First F statistics calculated from AMOVA show statistically significant genetic differentiation among villages (FRT = 0.05, standardized = 0.23, P < 0.001, Table 3). Second we detected weak, but statistically significant, isolation by distance (Fig. 4). Third Bayesian MCMC simulation detected five genetic clusters and although all six villages contained all five genetic clusters, one or two clusters predominate in most villages (Fig. 5).
All six villages had similar estimates of allelic diversity (Fig. 2), showing the villages do not differ dramatically in factors that affect allele frequencies and distribution, including T. dimidiata population size and gene flow, or that differences in these factors cancel each other out. For most loci, there were few if any samples with null alleles (i.e., no amplification, Table 2); five of the seven loci had null allele frequencies ≤2.2%. Although null alleles can cause individuals to be incorrectly assigned in Bayesian clustering methods, simulations have shown the bias to be small, lowering the proportion of incorrectly assigned individuals to only about one percent (Carlsson 2008). Null alleles can also inflate estimates of genetic differentiation by detecting excess homozygotes (Carlsson 2008); however, analysis of our data both with and without the locus with the highest frequency of null alleles produced very similar estimates of FRT (0.049 and 0.051).
Although our results cannot be directly compared with previous studies for which standardized F statistics are not provided, these results are similar to previous studies in southern Guatemala that show weak, but statistically significant, population structure (Dorn et al. 2003). Microsatellite markers have revealed less structure (maximum pairwise FST < 0.05, range 0.01–0.05) among 14 villages up to 250 km apart in Yucatán, Mexico (Dumonteil et al. 2007). The degree of genetic differentiation among villages detected in this study (FRT = 0.05, standardized = 0.23; Table 3) is higher. This higher among-village differentiation in southern Guatemala compared with Yucatán may be due to the insects occupying sylvatic as well as domestic and peri-domestic ecotopes in Yucatán, but only domestic and peri-domestic ecotopes in southern Guatemala. Higher genetic differentiation in southern Guatemala was also suggested in a study characterizing microsatellite diversity in T. dimidiata (Anderson et al. 2002). In Jutiapa, 7 of 30 genotypes were heterozygotes; in contrast, 18 of 32 genotypes from Yucatán were heterozygotes, supporting the idea of more population structure in southern Guatemala.
The insects in Yucatán and Jutiapa are very different, and some in Yucatán are thought to be members of a cryptic species (Dorn et al. 2007, Bargues et al. 2008). Yucatán populations are sylvan, with seasonal entry into houses and high migration. Individuals from sylvatic populations in northern Guatemala are similar to those in Yucatán; they rarely enter houses, show seasonal migration, and disperse as both nymphs and adults, as evidenced by infestation of experimental chicken coops (Monroy et al. 2003c). Our results show that the basically domestic population in southern Guatemala also migrates among houses and villages but to a lesser extent. The utility of microsatellite-based population genetic studies is revealed by the ability to detect lower gene flow among houses and villages in a largely domestic population.
So although it is domesticated, our results show that T. dimidiata is mobile in southern Guatemala. As a domestic vector in this region, rather than moving between sylvan and domestic ecotopes, it moves among houses. Even though migration between houses may incur a fitness cost, we can think of two possible advantages to migration. First, if individuals remained in the same house, inbreeding would ensue; migration effectively avoids endogamy. Evidence in support of this comes from a nearby southern Guatemalan village where a single house had 41 T. dimidiata families. There were few individuals per family (average family size of 2.17 individuals), and 46.3% of the families were represented by just one individual (Melgar et al. 2007). Second, flight is associated with poor nutritional status (Lehane et al. 1992). If higher bug densities within a house result in lower nutritional status, increased migration can prevent the number of bugs in a house from becoming too high.
Population genetics theory evaluates drift, migration, mutation, natural selection, and nonrandom mating, as possible explanations for population structure. With finite population size, drift should lead to fixation of different alleles in different subpopulations (houses or villages). The presence of the same genetic cluster in different houses and different villages indicates migration among houses within villages and among villages. The amount of genetic structure within villages (FSR = 0.110) is significant with respect to population genetics and can lead to population differentiation (Wright 1978); however, there is still sufficient gene flow and migration to explain the observed house recolonization (Tabaru et al. 1999).
Source-sink dynamics may help understand the pattern of variation. At the time the insects were collected, Tunillas and El Carpintero had high numbers of houses and high infestation indices (203 houses, 48% infestation and 147 houses, 37% infestation, respectively), and thus may be a source of insects. El Tablón, with 58 houses and 25% infestation, may show low variation because of genetic drift from the small size of the insect population; however, Copante with 38% of 34 houses infested did not show similar low genetic variation and may be a sink. Calderas, with 43% of 104 houses infested, may show low genetic variation because it is more isolated, many of the families are related to law enforcement and anecdotally, it appears the general population interacts with them less. La Brea with 124 houses and 33% infestation supports the conclusion that the insects travel long distances between La Brea and/or Tunillas and Carpintero. However, these possibilities suggested by the data require further study.
Control Implications
The pattern of genetic variability detected in this study suggests that use of insecticides alone is not a sustainable control method for T. dimidiata for this region because reinfestation is probable. A previous study in this region reported reinfestation rates of 50% only 20–45 mo after insecticide spraying and that fumigation increased migration (Hashimoto et al. 2006). Because our study indicates gene flow and migration among houses and villages occurs, if fumigation increases migration further, it might increase the number of houses infested.
In Jutiapa and southern Guatemala, house improvements combined with insecticide spraying are quite effective in controlling T. dimidiata in spite of the more limited dispersal capabilities of these domesticated populations (Monroy et al. 2009). In contrast, in Yucatán and northern Guatemala, where house improvements are not effective, alternative strategies, such as creating a more effective barrier between the inside and outside environments using screens and mosquito nets and reducing the amount of clutter and bug refugia, are necessary to reduce infestation rates (Dumonteil et al. 2004, Zeledon and Rojas 2006, Bustamante et al. 2009).
Our results on the population genetic structure of T. dimidiata help to illuminate the role of migration and gene flow in Chagas disease transmission and provide important information for the design of effective control strategies in southern Guatemala.
Supplementary Data
Supplementary data are available at Journal of Medical Entomology online.
Acknowledgments
We thank the residents of the villages and the personnel of the Ministry of Health of Guatemala for help collecting the insects. This work was made possible by support from grant R15 A1079672-01A1 from the National Institute of Allergy and Infectious Diseases (NIAID) at the National Institutes of Health (NIH) to P.D., grant TDR –WHO ID# A10249 from the World Health Organization (Tropical Disease Research-World Health Organization) to M.C.M., and National Science Foundation (NSF) grant BCS-1216193 as part of the joint NSF-NIH-USDA (United States Department of Agriculture) Ecology and Evolution of Infectious Diseases program.
References Cited
- Anderson J. M., Lai J. E., Dotson E. M., Cordon-Rosales C., Ponce C., Norris D. E., Beard C. B. 2002. Identification and characterization of microsatellite markers in the Chagas disease vector Triatoma dimidiata (Hemiptera: Reduviidae). Infect. Genet. Evol. 1: 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargues M. D., Klisiowicz D. R., Gonzalez-Candelas F., Ramsey J. M., Monroy C., Ponce C., Salazar-Schettino P. M., Panzera F., Abad-Franch F., Sousa O. E., et al. 2008. Phylogeography and genetic variation of Triatoma dimidiata, the main Chagas disease vector in Central America, and its position within the genus Triatoma. PLoS Negl. Trop. Dis. 2: e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandon-Naranjo M., Zuriaga M. A., Azofeifa G., Zeledon R., Bargues M. D. 2010. Molecular evidence of intraspecific variability in different habitat-related populations of Triatoma dimidiata (Hemiptera: Reduviidae) from Costa Rica. Parasitol. Res. 106: 895–905. [DOI] [PubMed] [Google Scholar]
- Brinkmann B., Klintschar M., Neuhuber F., Huhne J., Rolf B. 1998. Mutation rate in human microsatellites: influence of the structure and length of the tandem repeat. Am. J. Human Genet. 62: 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante D. M., Monroy C., Pineda S., Rodas A., Castro X., Ayala V., Quinones J., Moguel B., Trampe R. 2009. Risk factors for intradomiciliary infestation by the Chagas disease vector Triatoma dimidiata in Jutiapa, Guatemala. Cad. Saúde Pública 25: S83–S92. [DOI] [PubMed] [Google Scholar]
- Calderon C. I., Dorn P. L., Melgar S., Chavez J. J., Rodas A., Rosales R., Monroy C. M. 2004. A preliminary assessment of genetic differentiation of Triatoma dimidiata (Hemiptera: Reduviidae) in Guatemala by random amplification of polymorphic DNA-polymerase chain reaction. J. Med. Entomol. 41: 882–887. [DOI] [PubMed] [Google Scholar]
- Carlsson J. 2008. Effects of microsatellite null alleles on assignment testing. J. Hered. 99: 616–623. [DOI] [PubMed] [Google Scholar]
- Dorn P. L., Monroy C., Curtis A. 2007. Triatoma dimidiata (Latreille, 1811): a review of its diversity across its geographic range and the relationship among populations. Infect. Genet. Evol. 7: 343–352. [DOI] [PubMed] [Google Scholar]
- Dorn P. L., Melgar S., Rouzier V., Gutierrez A., Combe C., Rosales R., Rodas A., Kott S., Salvia D., Monroy C. M. 2003. The Chagas vector, Triatoma dimidiata (Hemiptera: Reduviidae), is panmictic within and among adjacent villages in Guatemala. J. Med. Entomol. 40: 436–440. [DOI] [PubMed] [Google Scholar]
- Dorn P. L., Calderon C., Melgar S., Moguel B., Solorzano E., Dumonteil E., Rodas A., Rua N. de la, Garnica R., Monroy C. 2009. Two distinct Triatoma dimidiata (Latreille, 1811) taxa are found in sympatry in Guatemala and Mexico. PLoS Negl. Trop. Dis. 3: e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumonteil E., Gourbiere S., Barrera-Perez M., Rodriguez-Felix E., Ruiz-Pina H., Banos-Lopez O., Ramirez-Sierra M. J., Menu F., Rabinovich J. E. 2002. Geographic distribution of Triatoma dimidiata and transmission dynamics of Trypanosoma cruzi in the Yucatán peninsula of Mexico. Am. J. Human Genet. 67: 176–183. [DOI] [PubMed] [Google Scholar]
- Dumonteil E., Ruiz-Pina H., Rodriguez-Felix E., Barrera-Perez M., Ramirez-Sierra M. J., Rabinovich J. E., Menu F. 2004. Re-infestation of houses by Triatoma dimidiata after intra-domicile insecticide application in the Yucatán peninsula, Mexico. Mem. Inst. Oswaldo Cruz 99: 253–256. [DOI] [PubMed] [Google Scholar]
- Dumonteil E., Tripet F., Ramirez-Sierra M. J., Payet V., Lanzaro G., Menu F. 2007. Assessment of Triatoma dimidiata dispersal in the Yucatán Peninsula of Mexico by morphometry and microsatellite markers. Am. J. Human Genet. 76: 930–937. [PubMed] [Google Scholar]
- El Mousadik A., Petit R. J. 1996. High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor. Appl. Genet. 92: 832–839. [DOI] [PubMed] [Google Scholar]
- Evanno G., Regnaut S., Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- Excoffier L., Lischer H. E. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10: 564–567. [DOI] [PubMed] [Google Scholar]
- Goudet1995. FSTAT (Version 1.2): A Computer Program to Calculate F-Statistics. J Hered 86: 485–486. [Google Scholar]
- Goudet J., Perrin N., Waser P. 2002. Tests for sex-biased dispersal using biparentally inherited genetic markers. Mol. Ecol. 11: 1103–1114. [DOI] [PubMed] [Google Scholar]
- Hardy O. J., Charbonnel N., Freville H., Heuertz M. 2003. Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163: 1467–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K., Cordon-Rosales C., Trampe R., Kawabata M. 2006. Impact of single and multiple residual sprayings of pyrethroid insecticides against Triatoma dimidiata (Reduviiade; Triatominae), the principal vector of Chagas disease in Jutiapa, Guatemala. Am. J. Trop. Med. Hyg. 75: 226–230. [PubMed] [Google Scholar]
- Hedrick P. W. 2005. A standardized genetic differentiation measure. Evolution 59: 1633–1638. [PubMed] [Google Scholar]
- Hotez P. J., Bottazzi M. E., Franco-Paredes C., Ault S. K., Periago M. R. 2008. The neglected tropical diseases of Latin America and the Caribbean: a review of disease burden and distribution and a roadmap for control and elimination. PLoS Negl. Trop. Dis. 2: e300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson M., Rosenberg N. A. 2007. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806. [DOI] [PubMed] [Google Scholar]
- Lehane M. J., McEwen P. K., Whitaker C. J., Schofield C. J. 1992. The role of temperature and nutritional status in flight initiation by Triatoma infestans. Acta Trop. 52: 27–38. [DOI] [PubMed] [Google Scholar]
- Lent H., Wygodzinsky P. W. 1979. Revision of the Triatominae (Hemiptera, Reduviidae), and their significance as vectors of Chagas' disease. Bull. Am. Mus. Nat. Hist. 163: 127–520. [Google Scholar]
- Lynch M., Ritland K. 1999. Estimation of pairwise relatedness with molecular markers. Genetics 152: 1753–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melgar S., Chavez J. J., Landaverde P., Herrera F., Rodas A., Enriquez E., Dorn P., Monroy C. 2007. The number of families of Triatoma dimidiata in a Guatemalan house. Mem. Inst. Oswaldo Cruz 102: 221–223. [DOI] [PubMed] [Google Scholar]
- Monroy C., Rodas A., Mejia M., Rosales R., Tabaru Y. 2003a. Epidemiology of Chagas disease in Guatemala: infection rate of Triatoma dimidiata, Triatoma nitida and Rhodnius prolixus (Hemiptera, Reduviidae) with Trypanosoma cruzi and Trypanosoma rangeli (Kinetoplastida, Trypanosomatidae). Mem. Inst. Oswaldo Cruz 98: 305–310. [DOI] [PubMed] [Google Scholar]
- Monroy C., Bustamante D. M., Rodas A. G., Enriquez M. E., Rosales R. G. 2003b. Habitats, dispersion and invasion of sylvatic Triatoma dimidiata (Hemiptera: Reduviidae: Triatominae) in Petén, Guatemala. J. Med. Entomol. 40: 800–806. [DOI] [PubMed] [Google Scholar]
- Monroy C., Bustamante D. M., Rodas A., Rosales R., Mejia M., Tabaru Y. 2003c. Geographic distribution and morphometric differentiation of Triatoma nitida Usinger 1939 (Hemiptera: Reduviidae: Triatominae) in Guatemala. Mem. Inst. Oswaldo Cruz 98: 37–43. [DOI] [PubMed] [Google Scholar]
- Monroy C., Bustamante D. M., Pineda S., Rodas A., Castro X., Ayala V., Quinones J., Moguel B. 2009. House improvements and community participation in the control of Triatoma dimidiata re-infestation in Jutiapa, Guatemala. Cad. Saúde Pública 25: S168–S178. [DOI] [PubMed] [Google Scholar]
- Nakagawa J., Cordón-Rosales C., Juárez J., Itzep C., Nonami T. 2003. Impact of residual spraying on Rhodnius prolixus and Triatoma dimidiata in the department of Zacapa in Guatemala. Mem. Inst. Oswaldo Cruz 98: 277–282. [DOI] [PubMed] [Google Scholar]
- Nei M., Chesser R. K. 1983. Estimation of fixation indices and gene diversities. Ann. Human Genet. 47: 253–259. [DOI] [PubMed] [Google Scholar]
- Panzera F., Ferrandis I., Ramsey J., Ordonez R., Salazar-Schettino P. M., Cabrera M., Monroy C., Bargues M. D., Mas-Coma S., O'Connor J. E., et al. 2006. Chromosomal variation and genome size support existence of cryptic species of Triatoma dimidiata with different epidemiological importance as Chagas disease vectors. Trop. Med. Int. Health 11: 1092–1103. [DOI] [PubMed] [Google Scholar]
- Peakall R., Smouse P. E. 2006. GENALEX 6: Genetic Analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro J. C., Lucero D. E., Stevens L. 2007. PCR reveals significantly higher rates of Trypanosoma cruzi infection than microscopy in the Chagas vector, Triatoma infestans: high rates found in Chuquisaca, Bolivia. BMC Infect. Dis. 7: 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro J. C., Gilligan L. M., Stevens L. 2008. Microsatellites reveal a high population structure in Triatoma infestans from Chuquisaca, Bolivia. PLoS Negl. Trop. Dis. 2: e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard J. K., Stephens M., Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prugnolle F., Meeus T. de. 2002. Inferring sex-biased dispersal from population genetic tools: a review. Heredity 88: 161–165. [DOI] [PubMed] [Google Scholar]
- Ramirez C. J., Jaramillo C. A., del Pilar Delgado M., Pinto N. A., Aguilera G., Guhl F. 2005. Genetic structure of sylvatic, peridomestic and domestic populations of Triatoma dimidiata (Hemiptera: Reduviidae) from an endemic zone of Boyaca, Colombia. Acta Trop. 93: 23–29. [DOI] [PubMed] [Google Scholar]
- Rosenberg N. A. 2004. DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes 4: 137–138. [Google Scholar]
- Rousset F. 2008. GENEPOP '007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Res. 8: 103–106. [DOI] [PubMed] [Google Scholar]
- Schweigmann N., Vallve S., Muscio O., Ghillini M., Alberti A., Wisnivesky-Colli C. 1988. Dispersal flight by Triatoma infestans in an arid area of Argentina. Med. Vet. Entomol. 2: 401–404. [DOI] [PubMed] [Google Scholar]
- Stevens L., Dorn P. L., Schmidt J. O., Klotz J. H., Lucero D., Klotz S. A. 2011. Kissing Bugs: The Vectors of Chagas. Adv. Parasitol. 75: 165–188. [DOI] [PubMed] [Google Scholar]
- Tabaru Y., Monroy M. C., Rodas A., Mejia M., Rosales R. 1999. The geographical distribution of vectors of Chagas' disease and populations at risk of infection in Guatemala Med. Entomol. Zool. 50: 9–17. [Google Scholar]
- Watts P. C., Rousset F., Saccheri I. J., Leblois R., Kemp S. J., Thompson D. J. 2007. Compatible genetic and ecological estimates of dispersal rates in insect (Coenagrion mercuriale: Odonata: Zygoptera) populations: analysis of ‘neighbourhood size' using a more precise estimator. Mol. Ecol. 16: 737–751. [DOI] [PubMed] [Google Scholar]
- Wright S. 1978. Evolution and the Genetics of Populations, Volume 4, Variability within and among populations, The University of Chicago Press, Chicago. [Google Scholar]
- Zeledon R., Rojas J. C. 2006. Environmental management for the control of Triatoma dimidiata (Latreille, 1811), (Hemiptera: Reduviidae) in Costa Rica: a pilot project. Mem. Inst. Oswaldo Cruz 101: 379–386. [DOI] [PubMed] [Google Scholar]
- Zeledon R., Solano G., Zuniga A., Swartzwelder J. C. 1973. Biology and ethology of Triatoma dimidiata (Latreille, 1811). 3. Habitat and blood sources. J. Med. Entomol. 10: 363–370. [DOI] [PubMed] [Google Scholar]
- Zeledon R., Calvo N., Montenegro V. M., Lorosa E. S., Arevalo C. 2005. A survey on Triatoma dimidiata in an urban area of the province of Heredia, Costa Rica. Mem. Inst. Oswaldo Cruz 100: 507–512. [DOI] [PubMed] [Google Scholar]