

Abstract
Histamine (HA) acts as a neurotransmitter in the brain, which participates in the regulation of many biological processes including inflammation, gastric acid secretion and neuromodulation. The enzyme histamine N-methyltransferase (HNMT) inactivates HA by transferring a methyl group from S-adenosyl-l-methionine to HA, and is the only well-known pathway for termination of neurotransmission actions of HA in mammalian central nervous system. We performed autozygosity mapping followed by targeted exome sequencing and identified two homozygous HNMT alterations, p.Gly60Asp and p.Leu208Pro, in patients affected with nonsyndromic autosomal recessive intellectual disability from two unrelated consanguineous families of Turkish and Kurdish ancestry, respectively. We verified the complete absence of a functional HNMT in patients using in vitro toxicology assay. Using mutant and wild-type DNA constructs as well as in silico protein modeling, we confirmed that p.Gly60Asp disrupts the enzymatic activity of the protein, and that p.Leu208Pro results in reduced protein stability, resulting in decreased HA inactivation. Our results highlight the importance of inclusion of HNMT for genetic testing of individuals presenting with intellectual disability.
Introduction
Intellectual disability (ID) is a neurodevelopmental disorder, characterized by considerable limitation of intellectual functioning, adaptive behavior, or daily living skills, and with an onset before 18 years of age. It is one of the most important challenges in healthcare, with significant life-long socio-economic burden. ID is genetically heterogeneous and may result from chromosomal aberrations, or from either autosomal recessive (AR), autosomal dominant, X-linked or mitochondrial mutations. With the prevalence of ∼1% of children worldwide (1), ID can be divided into two main groups: nonsyndromic (NS) ID, where it might present as the sole clinical feature, whereas in syndromic ID additional clinical or dysmorphological features may also be present. Over the past few years, next-generation sequencing technologies have led to the identification of a number of ID-associated genes, emphasizing the considerable genetic heterogeneity of ID (2). Studies into the molecular basis of autosomal recessive forms of ID (ARID) are lagging some way behind studies of X-linked ID, in part because the larger families needed for gene mapping are rare in North American and European populations. However, a recent review suggests that ARID is not rare, and in outbred populations as many as 13–24% of ID may be due to AR genes (2).
Histamine (HA), a biogenic amine, plays a key role in the regulation of gastric acid secretion (3), and is a neurotransmitter in the central nervous system (CNS) (4). HA is produced and stored in airway mast cells, basophils and in the synaptic vesicles of HAergic neurons. In response to immune allergens, HA releases from storage granules and rapidly diffuses into surrounding tissues. Released HA is rapidly inactivated and disappears from the bloodstream within minutes [reviewed in Schwartz et al. (4)].
Histamine N-methyltransferase (HNMT; MIM 605238) is a cytoplasmic protein that belongs to the methyltransferase superfamily and is one of two enzymes involved in the metabolism of HA. N-methylation catalyzed by HNMT and oxidative deamination catalyzed by diamine oxidase (DAO; encoded by amino oxidase, copper containing 1 (AOC1)) are the two major pathways for HA biotransformation in mammals (5,6). HNMT catalyzes the methylation of HA in the presence of S-adenosylmethionine (SAM), forming N-tele-methylhistamine (7,8). HNMT is widely expressed in human tissues; with the greatest expression in kidney and liver, followed by spleen, colon, prostate, ovary, spinal cord cells, bronchi and trachea. HNMT is the key enzyme for HA degradation in the bronchial epithelium (9). Since DAO is not expressed in the CNS, N-methylation through HNMT is the main pathway responsible for termination of the neurotransmitter actions of HA in the brain (4).
Here, we report the identification of HNMT as a novel gene responsible for ID and discuss the consequences of the identified missense mutations on the protein function.
Results
Family A (Iranian family)
Ascertainment and clinical evaluation
We ascertained a consanguineous family with Turkish background from the Avaj area within Qazvin province in Iran, in which the first-cousin parents had nine children, four of them were affected with NS ID; two males and two females (Fig. 1). The study was approved by the Research and Ethics Board of Qazvin Medical University and appropriate written informed consent was obtained from the parents. The affected family members were assessed by an experienced neurologist and standard clinical assessment forms were used to document the findings. The clinical descriptions of the patients are summarized in Table 1. The affected females showed profound to severe ID and their speech was limited to just a few words, whereas in affected males the condition was milder. A mild degree of regression after about 5 years of age was reported for affected members. The patients did not have any neurological problems, autistic features, congenital malformations or facial dysmorphisms. Body height, weight and head circumference were normal in all patients. Wechsler Intelligence Scales for Children (WISC) were used to assess the IQ in patients. For patient IV:I, we performed a magnetic resonance imaging scan which revealed no morphological brain abnormalities.
Figure 1.

Analysis of Family A (Iranian family): (A) Pedigree. Black-shaded symbols indicate affected individuals (IV:1, IV:4, IV:6 and IV:9). (B) Photos of affected individuals: from left to right: IV:1, IV:4, IV:6 and IV:9. (C) Homozygosity mapping data analysis indicates peaks (LOD = 3.0) on chromosomes 2 and 13. (D) Electropherograms from Sanger confirmation in family members showing NM_006895.2 (HNMT): c.179G>A; p.Gly60Asp WT, heterozygous and homozygous sequence. (E) In silico modeling of p.Leu208Pro within HNMT predicted protein structure for p.Gly60Asp WT and mutant using PDB file 2AOT and Pymol software. The red arrow indicates the location of residue 60 within the protein.
Table 1.
Clinical and biometric features for the Iranian family (Family A) and the Kurdish family (Family B)
| Family A: IV:1 | Family A: IV:4 | Family A: IV:6 | Family A: IV:8 | Family B: III:1 | Family B: III:3 | Family B: III:4 | |
|---|---|---|---|---|---|---|---|
| HNMT mutation (NM_006895.2) | c.179G>A (p.Gly60Asp) | c.179G>A (p.Gly60Asp) | c.179G>A (p.Gly60Asp) | c.179G>A (p.Gly60Asp) | c.632T>C (p.Leu208Pro) | c.632T>C (p.Leu208Pro) | c.632T>C (p.Leu208Pro) |
| Gender | F | M | F | M | M | F | M |
| Parental consanguinity | First cousins | First cousins | First cousins | First cousins | First cousins | First cousins | First cousins |
| Ethnic origin | Turkish | Turkish | Turkish | Turkish | Kurdish | Kurdish | Kurdish |
| Age at examination (years) | 35 | 33 | 31 | 27 | 18 | 15 | 13 |
| Height (cm) (SD) | 151 (−2.5 SD) | 172 (−0.8 SD) | 153 (−2.2 SD) | 170 (−1.2 SD) | 165 (−1.8 SD) | 151 (−2 SD) | 138 (−2.1 SD) |
| Weight (kg) (SD) | 65 (+1.5 SD) | 71 (+0.7 SD) | 63 (+1.2 SD) | 69 (+0.6 SD) | 56 (+0.2 SD) | 44 (+0.2 SD) | 40 (+0.8 SD) |
| Head circumference (cm) (SD) | 55 (Mean) | 54.5 (−1.46 SD) | 55 (Mean) | 54 (−1.46 SD) | 54 (−1.5 SD) | 52 (−1.7 SD) | 52 (−1.7 SD) |
| ID | IQ: 28 (severe) | IQ: 49 (moderate) | IQ: 25 (severe) | IQ: 54 (mild) | IQ: 20–34 (severe) | IQ: 20–34 (severe) | IQ: 20-34 (severe) |
Height and head circumference are given in cm; weight in kg. Standard deviation (SD) from mean values is given in parentheses.
Homozygosity-by-descent (HBD) mapping and mutation identification
HBD mapping led to the identification of a 14-Mb autozygous locus on 2q21.3 (single-nucleotide polymorphisms, SNPs: rs1869829–rs7573156), and a 3-Mb autozygous within region 13q33.1 (SNPs: rs1336666–rs1475276) with a significant LOD (logarithm (base 10) of odds) score of 3.13 (Fig. 1C). Additionally, existence of copy number variations (CNVs) exclusive to the affected individuals was also ruled out. Exome target enrichment was performed within the linkage intervals by using a custom Agilent SureSelect array, followed by sequencing 8.3 Gb of 101 bp paired-end reads using the Illumina Genome Analyzer II platform with 100% coverage and average depth of 202 reads. After filtering the variants with dbSNP130, and 1000 Genome data, we annotated the remaining mutations with the RefSeq gene model. Analysis of prospective changes from the critical regions indicated DNA variants in three genes: METTL21C [chr13:103346806C>G, NM_001010977.1:c.43G>C; p.Gly60Arg], ZRANB3 [chr2:136107663T>C, NM_032143.2:c.482A>G; p.Tyr161Cys] and HNMT [chr2:138727776G>A; NM_006895.2:c.179G>A; p.Gly60Asp] (coordinates used in hg19). The evolutionary conservation of the relevant nucleotides, as defined by the PhyloP44 score and the pathogenicity of these variants, as predicted by PolyPhen2 and SIFT were calculated (Table 2). Analysis with Condel—an integrated analysis that uses prediction using five different algorithms, including PolyPhen2, SIFT and Mutation Assessor (10), was also performed (Table 2). The co-segregation pattern of the three variants was checked in the family, and only the HNMT variant segregated correctly. The parents of the patients were both heterozygous for the HNMT missense variant, which was not found in either in a homozygous or heterozygous form among 100 unrelated healthy Iranian and 200 Pakistani individuals.
Table 2.
Analysis of variants identified by targeted exome sequencing in the Iranian and Kurdish families (A and B, respectively)
| Family | Gene (accession #) | Variant | SNP? | Polyphen2 | SIFT | PROVEAN | PhyloP44 (Mean) | Condel | Segregates with phenotype, Y/N |
|---|---|---|---|---|---|---|---|---|---|
| Family A (Iranian) | HNMT (NM_006895.2) | Gly60Asp | 1 (probably damaging) | 0.003 (damaging) | −6.303 (damaging) | 3.092 | 1 (deleterious) | Y | |
| METTL21C (NM_001010977.1) | Gly15Arg | rs2390760 | 0.002 (benign) | 0.429 (tolerated) | 0.523 (neutral) | −0.444 | 0 (neutral) | N | |
| ZRANB3 (NM_032143.2) | Tyr161Cys | rs181335970 | 1 (probably damaging | 0.001 (damaging) | −6.847 (damaging) | 2.268 | 0.990 (deleterious) | N | |
| Family B (Kurdish) | HNMT (NM_006895.2) | Leu208Pro | 1 (probably damaging) | 0.001 (damaging) | −6.534 (damaging) | 2.475 | 1 (deleterious) | Y |
Scores and predicted effect are indicated from Polyphen2, SIFT, PROVEAN, PhyloP44 and Condel (which is itself an integration of five separate prediction algorithms).
We identified a potentially pathogenic missense mutation (HNMT c.179G>A [p.Gly60Asp]) in HNMT (RefSeqNC_000002.11), which encodes a two-domain protein; MTase, which is the larger domain, is composed of a seven-stranded β-sheet and is mainly responsible for interaction with cofactor (SAM) and substrate (HA), and the S domain of HNMT, which may interact with other proteins for its in vivo function. The homozygous c.179G>A (p.Gly60Asp) variant occurs in the conserved MTase region I (Ile56-Gly64), which is part of the SAM-binding pocket (Fig. 3), highlighting the role of Gly60 in the interaction of HNMT with its cofactor. The predicted 3D structure of HNMT appears to be altered by the substitution (Fig. 1E). This variant was not present in either dbSNP138, nor the Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA, USA (http://evs.gs.washington.edu/EVS/; accessed January 2014), nor in 1000 Genomes (http://browser.1000genomes.org/index.html), but was present in 2 of 125 604 alleles in the Exome Aggregation Consortium database (ExAC; Cambridge, MA, USA; http://exac.broadinstitute.org; October 2014). Importantly, it was shown to be absent from a panel of 200 ethnically matched control chromosomes, as well as an in-house database of 521 exomes/genomes from unrelated individuals of Middle Eastern origin. The c.179G>A (p.Gly60Asp) mutation showed complete segregation with the disease in all affected family members; parents were heterozygous carriers and unaffected children available for genetic screening were either carriers or did not harbor the mutation.
Figure 3.

Three-dimensional Protein structure and ClustalW2 analysis: structures of HNMT (pdb 1jqd) at the catalytic domain for (A) Gly60, (B) Asp60, with HA in yellow and S-adenosyl homocysteine (SAH) in pink, and Gly60 and Asp60 in light pink, and (C) ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2) alignment/comparison of HNMT across vertebrate species showing conservation at Gly60Asp (highlighted in pink). Structures at the hydrophobic pocket around residue 208 (labeled pink) for (D) Leu208 and (E) Pro208. Leu155, Leu204, Leu211 and Leu213 were labeled in gray. Ile288 and Ile290 were labeled in yellow. Tyr215 and Gly212 were labeled in blue. (F) ClustalW2 alignment of HNMT for the hydrophobic pocket surrounding Leu208, with Leu208 highlighted in pink, Leu155, Leu204, Leu211 and Leu213 labeled in gray, Ile288 and Ile290 labeled in yellow. Tyr215 and Gly212 labeled in blue. Sequences used for the HNMT alignment included human (NP_008826.1), mouse (NP_536710.1), opossum (from N-SCAN and Genscan gene predictions, UCSC browser), chicken (NP_001264802.1), Xenopus laevis (NP_001080614.1), zebrafish (NP_001003636.1), Tetraodon nigroviridis (Q4SBY6.1), lancelet (Branchiostoma floridae: predicted from mRNA XM_002613293.1) and sea urchin (Strongylocentrotus purpuratus: from mRNAs CX698504, CD312314 and CX689147).
A known C-to-T SNP (rs11558538) in HNMT changes the amino acid at position 105 from threonine to isoleucine, with the frequencies of the Thr105 and Ile105 alleles being ∼90 and 10%, respectively. The Ile105 allele is correlated with diminished HNMT enzymatic activity, which could result in reduced HA inactivation and increased sensitivity to this amine (11,12). We checked this polymorphism in Family A and verified that Ile105 allele is not present in the patients.
Family B (Kurdish family)
Ascertainment and clinical evaluation
We ascertained a three-generation consanguineous Kurdish family (G016), originally from Iraq, recruited in Germany. The first-cousin parents had seven children, three of them presented with NS ID; the pedigree is shown in Figure 2. The probands were examined by experienced clinical geneticists who assessed their physical and mental status. The study was approved by the local institutional ethics committee, and appropriate informed consent was obtained from the parents. To exclude chromosomal abnormalities, karyotype analysis by G-banding was performed in all affected individuals; karyotypes were found to be normal. Clinical descriptions of the patients are summarized in Table 1. On assessment, the 18-year-old boy (III:1) showed severe ID. He started to walk at 1 year of age. His speech was severely delayed and he attended special school for intellectually disabled children. His sister, III:3, a 15-year-old, was also severely intellectually disabled. She had a normal motor development, but active speech started at the age of about 2 years and she attended, as with the older brother, a special school for intellectually disabled children. The younger affected male (III:4), age 13 years, was more delayed than his older siblings. The mother reported that at birth the child was thin and hypotonic. He began walking at 2½ years. He started to speak at 4–5 years of age. He presented with hyperactive behavior. At the age of 12 years, he was diagnosed with myelodysplastic syndrome. The patients did not present with congenital malformations or facial dysmorphisms. Physical measurements are also reported in Table 1.
Figure 2.

Analysis of Family B (Kurdish family): (A) Pedigree. Black-shaded symbols indicate affected individuals (III:1, III:3 and III:4). (B) Photos of affected individuals: from left to right: III:1, III:3 and III:4. (C) Homozygosity mapping data analysis indicates peaks (LOD = 2.4) including a large interval (30 Mb) on chromosome 2. (D) Electropherograms from Sanger confirmation in family members showing NM_006895.2 (HNMT):c.632T>C; p.Leu208Pro WT, heterozygous and homozygous sequence. (E) In silico modeling of p.Leu208Pro within HNMT. Modeling was performed with PDB file 2AOT and PyMol software. The red arrow indicates the location of residue 208 within the protein.
HBD mapping and mutation identification
Analysis of genotypes for the available individuals (III:1, III:2, III:3 and II:1) of family G016 revealed a large interval of autozygosity on 2q21.2–q24.3. This 30 Mb interval was flanked by the heterozygous SNPs, rs10928469 and rs6705268. Four additional autozygous loci were identified: a 3.4-Mb interval on 10q26 (rs11244548–rs4751029), a 1.7-Mb HBD on 11p12–p13 (rs672597–rs10836780), a 12.6-Mb region on 11q22.3–q23.3 (rs10895742–rs521609), and finally an 8.5-Mb locus in 18q12.1–q12.3 (rs1021598–rs12971263; Fig. 2C).
We performed targeted next-generation sequencing by enrichment of the exonic regions within the linkage intervals. After filtering the variants, a single homozygous variant in HNMT [chr2: 138771444T>C; NM_006895.2:c.623T>C; p.Leu208Pro] (coordinates used in hg19) was ranked as potentially pathogenic. The mutation has not been reported in dbSNP 138, 1000 Genomes or the NHLBI Exome Variant Server, and was absent in our in-house databases of 521 exomes/genomes from unrelated individuals of Middle Eastern origin, but was present in 1 of 126 358 alleles in the ExAC database. Direct Sanger sequencing analysis was performed for all available family members, and demonstrated co-segregation with the disease in the family according to a recessive mode of inheritance. The predicted 3D structure of HNMT does not appear to be significantly altered by the substitution (Fig. 2E). The mutation affects a highly conserved amino acid (Fig. 3) and, in line with this, the PhyloP score (13) was found to be 4425, Grantham score 98 (14). In silico analyses with SIFT, PolyPhen-2, ConDel and others all predicted the amino acid substitution to be damaging (Table 2).
ID and ASD cohorts from outbred population
Cohorts of N = 991 ID and N = 1000 autism spectrum disorder (ASD) subjects were screened using a pooled targeted sequencing approach. No potentially damaging rare homozygous or compound heterozygous variants were identified. A rare heterozygous variant, NM_006895.2:c.430-1G>A; Chr2:138762701G>A, which would potentially alter splicing of exon 5, was identified in one ASD and one ID individual. This variant was not present in the ExAC database (∼65 000 exomes), nor in a database of 521 Middle Eastern exomes. Although potentially damaging heterozygous single-nucleotide variants (SNVs) and CNVs are reported elsewhere for schizophrenia, bipolar disorder, ID and autism spectrum disorder (Supplementary Material, Fig. S1), loss of function (LoF) variants are reported for HNMT in more than 40 individuals in the ExAC control data, and thus, it seems unlikely that heterozygous LoF variants are associated with these disorders.
In silico modeling of HNMT Leu208Pro predicts functional impairment
To gain insights into the structural consequence of the p.Leu208Pro mutation in HMNT, we performed structure-based in silico three-dimensional modeling of the mutant protein (Fig. 2E). The non-conservative leucine to proline change at position 208 occurs in one of the six regions with an alpha helix conformation (α-E) which flanks the seven-stranded β-sheet within the MTase fold (15). The analysis indicated that although the mutation is not proximal to the catalytic site, the rigidity introduced by the proline residue would likely alter the helical conformation, destabilizing the protein and therefore affecting the enzymatic function. Proline is well known as an alpha helix breaker due to its side chain and steric constraints. In fact, the hydrogen bonding network and conformation of the helix would be disrupted by the side chain forced into the space occupied by the helix backbone, and by the methyl group at the position normally occupied by an amide proton (16). We have quantified this effect by performing in silico energy calculations with PoPMuSiC 2.1 and IMutant 2.0 (17), which calculate stability changes upon mutation. In both cases, the simulation revealed a decrease in stability, 3.73 and 2.09 kcal/mol, respectively. An additional analysis using FoldX (18) revealed a more stark decrease in stability of 8.98 kcal/mol. A leucine to proline substitution is causing the most destabilizing effect when all permutations are considered. Thus, we hypothesize that the p.Leu208Pro mutant protein is either unstable or the function is severely impaired.
Gly60Asp and Leu208Pro alterations do not affect HNMT protein localization
To check if Gly60Asp and Leu208Pro changes disturb HNMT protein localization, COS-7 cells were transfected with mutant and wild-type (WT) HNMT constructs with GFP Tag. Visualization of the fusion protein was performed using a confocal laser scanning system (data not shown). HNMT-Gly60Asp HNMT-GFP, like WT, is localized to the cytoplasm of the cells, suggesting that there is no difference in cellular localization between mutant and WT form of the HNMT protein. For Leu208Pro, the transformed cells appear to show a punctuate distribution of HNMT-GFP, possibly indicative of the formation of protein aggregates; however, consistent results were not achieved (data not shown). We conclude that Gly60Asp does not impair the proper cellular localization of HNMT.
Patients' lymphoblasts are considerably more vulnerable to HA than the controls
We examined the vulnerability of patients' lymphoblasts (available for Family A, but not Family B) to HA when compared with lymphoblasts from unrelated healthy controls. Cells were first treated with different concentrations of HA and then using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay, the viability of cells was determined. Cells in both groups were found to rapidly undergo cell death upon culture in vitro after treating with high concentrations of HA (at 500 µM), which is in line with findings suggesting that HA induces neutrophil apoptosis at the sites of allergic inflammation (19). While control cells were able to survive at lower concentration of HA (125 µM), patients' lymphoblast cells were still not tolerant to the cytotoxic effects of HA, supporting our hypothesis that HNMT, as the defense mechanism against HA, is defective in the patients (P-value, 1 × 10−5) (Fig. 4).
Figure 4.

Cell viability using the MTT assay. Lymphoblast cells from 4 patients from Family A and 12 healthy unrelated controls were treated with 125 µM concentration of HA for 2 h. Reconstituted MTT in an amount equal to 10% of the culture medium volume was added to the cells. The plate was read at 570 nm. Values are mean ± SD for three independent experiments, and averaged across all the individuals per group.
No significant difference in HA and N-tele-methylhistamine levels between patients carrying Gly60Asp variant and healthy unrelated controls
The primary goal of HA inactivation is its conversion to metabolites that will not activate HA receptors, and this is achieved either by methylation or by oxidation. HNMT catalyzes the transfer of a methyl group from SAM to the secondary amino group of the imidazole ring of HA, forming N-tele-methylhistamine. Therefore, if HNMT is defective, we would expect to detect high levels of HA and low levels of methylhistamine. We measured the HA and methylhistamine levels in patients' and controls' lymphoblast cell lines using an ELISA assay. However, we could not detect any significant difference between them (data not shown).
Gly60Asp alteration does not affect HNMT expression at the RNA and protein level
To examine whether the Gly60Asp alteration disrupts the expression of HNMT, quantitative RT-PCR and western blotting analysis were performed using patients' and controls' lymphoblast cell lines. We did not detect any significant difference in HNMT expression between patients and controls at both the RNA and protein level (data not shown).
Gly60Asp variant affects thermal stability of HNMT protein
Melting temperatures are the best descriptor of thermal stability. A difference in the melting temperature corresponds to a difference in energy between the pair of proteins. As indicated in Figure 5, glycine to aspartic acid replacement decreases the melting temperature of the HNMT protein from 62.3 ± 1.7 to 58.3 ± 1.1°C for WT and the Gly60Asp variant, respectively, suggesting that amino acid change at position 60 will lead to reduced thermal stability of HNMT.
Figure 5.
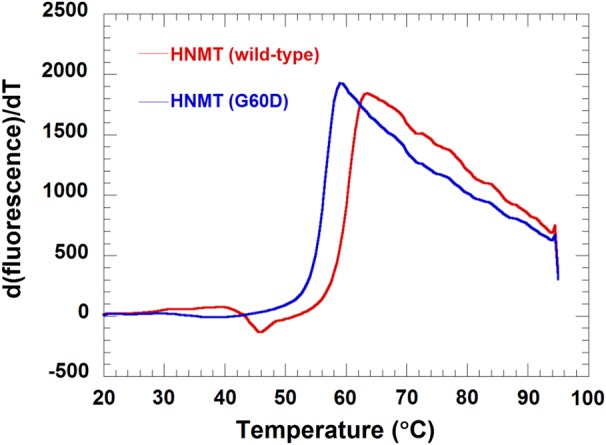
Thermal stability of HNMT (WT) and HNMT (Gly60Asp) using Thermofluor®. The experiment was performed by mixing 5 μl of 1 mg/mL of HNMT (WT) or HNMT (Gly60Asp) with 5 μl 200-fold diluted SYPRO Orange and a buffer containing 50 mm Tris–HCl and 100 mm NaCl, pH 8.0, in a 98-well RT-PCR plate. Reaction mixtures were heated at 0.5°C/min from 20 to 95°C. A plot between d(fluorescence)/dT versus temperature of HNMT (WT) (red line) and HNMT (Gly60Asp) (blue line) is shown. A melting temperature of 62.3 ± 1.7 and 58.3 ± 1.1°C for WT and the Gly60Asp variant, respectively, was obtained.
Gly60Asp variant disturbs the affinity of HNMT protein for binding to SAM
The dissociation constant (Kd) is commonly used to describe the affinity and binding property between a ligand and a protein, i.e. how tightly a ligand binds to a particular protein. Figure 6 presents the calorimetric titration of HNMT (WT) and the Gly60Asp variant with SAM by isothermal titration calorimetry. Figure 6A indicates the isothermal binding curve for the titration of 30 μm HNMT (WT) with 0.42 mm SAM. The isothermal binding curve (Fig. 6B) yields a Kd of 49.2 ± 2.0 μm. Figure 6C demonstrates a significant decrease in affinity of the Gly60Asp variant for SAM, providing another line of evidence for the effects of the amino acid substitution at Gly60.
Figure 6.

Isothermal calorimetric titration of HNMT (WT) and Gly60Asp variant with SAM and HA. The experiments for HNMT (WT) consisted of 20 consecutive injections of 15 μl of 0.42 mm SAM into 30 μm HMNT (WT) at 25°C. (A) The released heats of injection after baseline correction by subtracting the heat of the reference measurement. (B) Integrated data and data analysis using non-linear least square fitting in Origin 7. The dissociation constant of HNMT (WT) for SAM was 49.2 ± 2.0 μm. (C) Released heats of 20 consecutive injections of 15 μl of 2 mm SAM into 30 μm HNMT (Gly60Asp) at 25°C.
Gly60Asp variant significantly disrupts catalytic activity of HNMT protein
The isothermal titration calorimetric (ITC) method was used to assess the interaction between WT and Gly60Asp variant HNMT with SAM. As indicated in Table 3, the Km (substrate concentration at which the reaction rate is half its maximum value) of WT HNMT lies in the normal range of most enzymes. However, this value was not measurable for Gly60Asp variant (Fig. 7). This can be explained by the failure of mutant HNMT to bind with SAM due to the dramatic change of its substrate-binding pocket.
Table 3.
Apparent Km for HA from HNMT reaction
| Enzyme | Km (μm) | Vmax (μm s−1) | kcat (s−1) |
|---|---|---|---|
| HNMT (WT) | 5.47 ± 1.41 | 8.21 ± 0.16 × 10−3 | 8.21 ± 0.16 × 10−3 |
WT and Gly60Asp HNMT constructs were used; however, only results for WT are given, as the catalytic activity of HNMT-Gly60Asp was too low to record using the VP-ITC system (MicroCal, GE Healthcare).
Figure 7.

Catalytic activity of HNMT (WT) and HNMT (Gly60Asp) as determined by isothermal titration calorimetry. (A) Raw calorimetric data for the methylation of HA catalyzed by HNMT (WT) using SAM as a methyl group donor in 50 mm Tris–HCl buffer, pH 8.0, containing 100 mm NaCl at 25°C. The experiment was performed by injection of 2 mm HA (2 μl/injection; 20 injections). The cell contained 1 μm HNMT (WT) and 100 μm SAM. A Michaelis–Menten plot for methylation by HNMT (WT) is shown in the inset of A. (B) Raw calorimetric data for the reaction with the Gly60Asp variant (same conditions as for WT).
Leu208Pro destabilizes HNMT
Attempts were made to generate the Pro208 variant of HNMT; however, soluble protein was not detectable from the constructs, suggesting that misfolding occurs, leading to rapid degradation of the protein. Addition of N-terminal GST tag, or expression in the presence of chaperones, was not successful, and the variant protein was only detectable in the inclusion body fraction (data not shown). Interestingly, constructs for additional variants at Leu208 (Leu208Val, Leu208Phe, Leu208Arg, Leu208Thr, Leu208Asp and Leu208Asn) have also been generated in Escherichia coli, and only substitutions with the first two (apolar) amino acids, valine and phenylalanine, and to a much lower extent the polar, positively charged arginine (i.e. 5 mg/20 g cells instead of 20 mg/20 g cells), lead to soluble and active HNMT protein (P. Macheroux and C. Tongsook, personal communication). For this reason, we were unable to perform parallel experimentation to the Gly60Asp mutation for thermal stability, SAM-binding affinity and ITC measurements, and would also be a plausible explanation for the punctate cellular distribution of Leu208Pro-HNMT-GFP.
Discussion
HA in adult brain acts as a neurotransmitter in several biological processes. Moreover, it has been reported that, during the development of the rat brain in the fetus, HA concentration reaches its maximum level throughout the period where neuronal differentiation takes place in several brain regions, suggesting that HA, acting as a neurogenic factor, is an important modulator in the developing brain (20,21). Even at very low concentrations, HA has strong pharmacological activity (22). HA-induced apoptosis is mediated by caspase activation and PKC-γ signaling (23). Therefore, synthesis, release and degradation of HA have to be carefully regulated to avoid undesirable reactions. HAergic neurons synthesize substantial quantities of HA and store it in special storage granula inside the cell (24). Basal plasma HA concentrations of 0.3–1.0 ng/ml are considered normal and exceeding these HA levels give rise to concentration-dependent HA-mediated symptoms (25). The level of HA in brain is slightly lower than that of other biogenic amines; however, its turnover is considerably faster and once released, it must be inactivated within a few minutes (19). HNMT is the sole enzyme in the CNS, inactivating the neurotransmitter actions of HA (26).
We identified two homozygous missense mutations in the HNMT gene segregating with NS ID in an Iranian family of Turkish origin (Family A) and in a Kurdish family (Family B). For Family A, the condition had a relatively similar pattern during the childhood of the patients, starting at around 5 years of age and gradually worsening. Males were relatively mildly affected, but the condition in females was profound. This could be at least in part due to gender differences in neurotransmitter activity of HA. It has been reported that in certain areas of the brain, neurotransmitter synthesis, content and metabolism are sexually differentiated and under the influence of sex steroids in development and adulthood (27,28). As mentioned above, once HA is released in the brain, it would typically be cleared from the intracellular space within a few minutes by HNMT. Therefore, malfunctioning HNMT would be expected to result in a high residual level of HA in the region. Using ELISA, we tested to see if there is any difference in the level of HA between patients' and controls' lymphoblast cell lines. However, no significant difference was detected. Since the alternative pathway of HA inactivation, i.e. DAO is active in the lymphoid lineage (but not in the CNS), therefore we measured HA level after blocking the DAO pathway and again no difference was detected (data not shown). As discussed above, during rat embryonic development, HA concentration reaches its maximum level at embryo days 14–16 when neuronal differentiation takes place and then steadily decreases until birth, yielding the concentration present in the adult organism. This may explain why differences in the level of HA among the patients' and controls' lymphoblast cell lines were not detected.
HNMT has a two-domain structure—the MTase domain and the S domain. The MTase domain primarily carries out cofactor binding and probably catalysis, while both domains contribute to HA binding (11,26). The Gly60Asp variant occurs within the conserved MTase region I (Ile56-Gly64), which is part of the SAM-binding pocket, highlighting the role of Gly60 in the interaction of HNMT with its cofactor. From our protein modeling analysis, we predict that the Gly60Asp variant does not bind SAM. Inspection of the active site also shows that the aspartate side chain would interfere with the ribose ring of SAM, rationalizing the lack of binding. Therefore, Gly60Asp does not affect overall protein stability, but compromises SAM in the active site of the enzyme. This interpretation is fully supported by the results obtained with the recombinant HNMT-Gly60Asp variant.
Leu208, as part of a very compact hydrophobic pocket, has several contacts with the residues in the same α-helix, the adjacent α-helix and the two β-strands. As a result, the leucine-to-proline substitution has a disruptive effect on the hydrophobic pocket, and it is likely that the substitution breaks the α-helix, and this in turn disrupts the hydrophobic packing in this area leading to exposure of hydrophobic residues, and thus favoring misfolding and aggregation of the protein.
Expression studies of the Gly60Asp variant revealed that the mutation does not disrupt the expression of HNMT at either the mRNA or protein level, suggesting that HNMT malfunction is not a result of reduced enzyme concentration. Moreover, observations from protein localization studies of HNMT proved that Gly60Asp and Leu208Pro variants do not disrupt the normal distribution of protein throughout the cytoplasm of cell. Results from thermal stability, binding affinity and catalytic activity investigations of HNMT WT and the Gly60Asp variant, and failed attempts to stabilize and solubilize Leu208Pro, support the deleterious effects of these substitutions on HNMT function in these two ID families.
A short isoform of HNMT has also been reported that includes the N-terminal 126 amino acids (Genbank ID: NM_001024075.1; NP_001019246.1 (Q8IU56)). The function of this isoform is unknown. The mutation identified in Family A (Gly60Asp) would potentially affect this as well as the long isoform, whereas the mutation identified in Family B (Leu208Pro) would only impact the longer isoform. Also, it is predicted that the Leu208Pro substitution is extremely destabilizing for the protein. The effects of different mutations on protein stability and function, as well as on different isoforms, could account for different clinical aspects and severities for the two families.
Decreased HA levels have been found in the brain of patients with Alzheimer disease (AD) (29), including reduction in the neuronal pool of hippocampus, hypothalamus and temporal cortex of AD patients (30), but, in contradicting reports, increased HA levels in cerebrospinal fluid and brain tissue were demonstrated (31,32). HA measurements per se may be, however, unreliable due to many confounding factors as, e.g. HA levels increase with postmortem interval (30), but may be valuable when accompanied by determination of activity or levels of HA-related enzymes.
While we can postulate about the effect of these mutations on levels of HA in the CNS, and possible detrimental effects on neurodevelopment, it cannot be ignored that a decrease in levels of the catabolite and product of HNMT activity, N-tele-methylhistamine may also play an important role in the disease etiology in these families. It has been suggested that there are HA/glutamate functional interactions in the brain (33), and it has already been shown that HA can potentiate NR2B-type N-methyl-d-aspartate (NMDA) receptors in hippocampal neurons, and that N-tele-methylhistamine also produces an equipotent enhancement of NMDA currents (34). In addition, N-tele-methylhistamine has been reported to be an agonist for β3 γ-butyric acid (GABA) receptors (35). HA has recently been reported to increase neural differentiation to FOXP2 neurons in cultured cells (36). Mutation of the FOXP2 gene has been reported as the cause of a rare speech/language disorder (MIM 602081) (37). Given the potential for N-tele-methylhistamine to act as an analog of HA, it would be interesting to see whether it would have a similar effect, in which case one could postulate that some of the deficits identified in the patients reported here could be the result of such a mechanism.
Conclusion
In the current study, we have shown that mutations resulting in HNMT LoF are associated with a NS form of ARID. We also confirmed that the p.Gly60Asp substitution results in complete loss of HNMT enzymatic activity, resulting in reduced HA inactivation and increased sensitivity to this amine. For p.Leu208Pro, although the mutation does not lie in the catalytic region of the protein, the rigidity introduced by the proline residue would most likely alter the helix conformation, destabilizing the protein and therefore affecting protein stability and the substrate-binding site.
Collectively, these findings indicate that HNMT plays an important role in human neurodevelopment. Our results indicate that HNMT should be included in genetic testing of individuals presenting with ID in consanguineous populations and, given estimates of a role for AR genes in 13–24% of ID in non-consanguineous populations (2), in outbred populations.
Materials and Methods
This study was approved by the Research Ethics Committees of the Qazvin University of Medical Sciences, Qazvin, Iran, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran, Max Planck Institute of Molecular Genetics, Berlin, Germany, the Centre for Addiction and Mental Health in Toronto, Canada, and Queen's University, Kingston, Canada. Informed written consent was obtained for all participating subjects.
Gene mapping
Family A (Iranian family)
Genomic DNA was extracted from peripheral blood leukocytes by standard methods. We used the Affymetrix GeneChip Mapping SNP 6.0 array (950K SNPs and 950 K CNV markers) to analyze DNA samples of all affected individuals, parents and one healthy sibling. Approximately 200 000 markers with good quality genotypes (based on their array hybridization confidence score) were selected for linkage analysis. Appropriate input files for the linkage analysis programs Merlin (38) and Allegro (39) were generated by ALOHOMORA software (40) with subsets of 300–500 markers in a sliding window mode based on mapping information from DeCode and Caucasian allele frequencies. Quality control checks such as gender check and verifying the relationships between individuals within the family were performed (38). Mendelian inconsistencies and unlikely genotypes were detected by the PedCheck (41) and Merlin (38) programs, respectively, and they were excluded from genotyping data prior to linkage analysis. Parametric linkage analysis was carried out based on an AR mode of inheritance, and assuming complete penetrance.
Family B (Kurdish family)
Genomic DNA was extracted from blood samples using standard protocols. Genotyping (SNP analysis) was performed using the Affymetrix 250k Genome-Wide Human SNP Array (Affymetrix, Santa Clara, CA, USA) for available individuals. We used ALOHOMORA software (40) for SNP array quality controls, as described previously (42). The program Merlin was applied for parametric multipoint linkage analysis, consistent with an AR mode of inheritance, a disease allele frequency of 10−3 and complete penetrance.
Exon enrichment and high-throughput sequencing
Custom-made Agilent SureSelect DNA Capture Arrays (Agilent Technologies, Inc., Santa Clara, CA, USA) were used for the enrichment of exons from homozygous intervals, including, on average, 60 bp of flanking sequences on either side of the exon. Enriched exons were sequenced on an Illumina Genome Analyzer II, generating 76-bp single reads with 98.9% coverage.
Sequence alignment, variant calling, annotation and verification
Raw sequence reads were prescreened to remove low-quality reads, and then aligned to the human reference genome (hg19, GRCh37) with SOAP (version 2.20). Aligned and unaligned reads were used to call the SNVs and Indels, respectively. Variant lists were filtered against dbSNP137, whole genomes from 185 healthy individuals (1000 Genomes Project), and 200 exomes from Danish individuals, and 6500 exomes present in the Exome Variant Server (NHLBI GO Exome Sequencing Project, Seattle, WA, USA; http://evs.gs.washington.edu/EVS/; 30 August 2013; v.0.0.21). In addition, variants were compared with an in-house database containing more than 521 exomes from individuals of Middle Eastern origin. Variants were ranked as potential candidates as previously described (43), using an improved version of Medical Re-sequencing Analysis Pipeline (MERAP) (44). The OMIM catalog (http://www.ncbi.nlm.nih.gov/omim) and the Human Gene Mutation Database (HGMD, http://www.hgmd.org/) were used as a filter to identify all previously described pathogenic changes. Sanger sequencing was used to confirm the co-segregation of the final candidate variants in the family.
Targeted exome sequencing for HNMT
Ninety-nine genes were included in the custom design: 64 for known or suspected NS ARID genes, including HNMT; 4 for known or suspected NS AR autism genes; 7 for known or suspected NS X-linked ID or ASD genes; 7 genes for known syndromic ARID, also reported in ASD cohorts and 17 known or suspected NS autosomal dominant ID or ASD genes. N = 1000 ASD and N = 991 ID-unrelated individuals from an outbred population (Canada) were included. Samples were measured using the RNaseP assay by quantitative PCR, using ViiA™7 Real-Time PCR (Life Technologies, Carlsbad, CA, USA), and pooled at equimolar concentrations in pools of 20. Seven pools were barcoded and run using the custom Ampliseq (Life Technologies) primer pools on a single Proton P1v2 chip. Samples were analyzed using the Ion Torrent software (Life Technologies), and Bam files generated and runs visualized using the Integrated Genome Viewer (Broad Institute: http://www.broadinstitute.org/igv/), and for the purpose of this study, focusing just on HNMT coding regions.
Expression studies of HNMT
Quantitative reverse transcriptase-PCR
Primers were designed to amplify the coding sequence of the HNMT gene. Coding sequence primers for β-Actin (ACTB) and HPRT were used as an internal reference for all the runs. Reverse transcriptase (RT)-PCR was performed in quadruplicates using a 384-well optical plate, with a final reaction volume of 16 µl. Universal SYBR Green PCR conditions were used, consisting of 95°C for 2 min and 30 s and 40 cycles at 95°C for 4 s and 60°C for 20 s. Each reaction contained 2 µl of cDNA in a 16-µl volume run in 384-well optical plates on a ViiA™7 (Life Technologies). For each gene analyzed, all samples were run on one plate, to avoid interplate variability, and in quadruplicate. Furthermore, each plate contained H2O, RT-minus and RNA-minus as negative controls. The Ct for all reactions was calculated automatically by the ViiA™7 (Life Technologies) software. Gene expression analysis was calculated using the comparative Ct method.
Western blot analysis
Protein samples from patients and controls were separated by SDS page (4–20% Mini-PROTEAN TGX polyacrylamide gel, Bio-Rad Laboratories, Hercules, CA, USA) and electrically transferred to a nitrocellulose membrane (BioTrace NT nitrocellulose membrane, PALL Life Sciences, Ann Arbor, MI, USA). The membrane was blocked for 1 h using 5% skimmed milk in TBS-Tween, incubated overnight with the primary rabbit anti-HNMT (1 : 1000, Bethyl Laboratories, Inc., Montgomery, TX, USA) and then incubated with the secondary antibody horseradish peroxidase-conjugated donkey anti-rabbit IgG (1 : 1000, GE Healthcare UK Limited, Little Chalfont, UK). The immunoblots were developed by an enhanced chemiluminescence western blot detection system (GE Healthcare).
Heterologous expression of WT HNMT, p.Gly60Asp and p.Leu208Pro variants in E. coli
The open reading frames encoding the target proteins (WT HNMT; p.Gly60Asp and p.Leu208Pro variants) were amplified by PCR from the pcDNATM3.3-TOPO TA vector using primers 5′-TCTCATATGATGGCATCTTCCATGAGGAGC-3′ (sense strand) and 5′-TCAGCGGCCGCTTAATGATGATGATGATGATGTGCCTCAATCACTATGAAACTCAGA G-3′ (anti-sense strand) using Phusion High-Fidelity DNA polymerase (Thermo Scientific, Waltham, MA, USA). The amplified genes were then cloned into the pET-21a vector using the NdeI and NotI restriction sites for expression as C-terminally hexa-histidine tagged proteins. Similarly, glutathione S-transferase (GST) fusions were generated by cloning the open reading frames into pGEX-6p2 using the BamHI and NotI restriction sites. Induction of expression of HNMT WT, and Gly60Asp, and Leu208Pro variants with C-terminal His6 or N-terminal GST tag was carried out at an OD600 ∼1, by addition of 0.1 mm IPTG (final concentration). The culture was maintained at 18°C for 14 h prior to harvest, resulting in a yield of 5 g of cell paste per liter of cell culture.
Purification of HNMT WT and the Gly60Asp variant
Frozen cell paste (∼20 g) was thawed and resuspended in lysis buffer (50 mm Tris–HCl, pH 8.0 containing 100 mm NaCl, 10 mm imidazole, 1 mm DTT and 100 μm PMSF). Cells were disrupted by ultrasonication with 50% amplitude for 15 min. After ultrasonication, the suspension was centrifuged at 18 000 rpm at 4°C for 1 h and the pellet was discarded. The clear crude extract was filtered (0.22 µm) prior to loading onto a 5-ml Ni-Sepharose™ High-Performance HisTrap™ HP column equilibrated with lysis buffer. The column was washed with 100 ml of 50 mm Tris–HCl buffer, pH 8.0, containing 100 mm NaCl and 20 mm imidazole. Elution of HNMT was initiated using 50 mm Tris–HCl buffer, pH 8.0, containing 100 mm NaCl and 300 mm imidazole. Fractions containing HNMT were pooled, concentrated using a Centricon YM10 and loaded onto a Superdex 200 prep grade column previously equilibrated with 50 mm Tris–HCl buffer, pH 8.0, containing 100 mm NaCl. Fractions containing target protein were pooled and concentrated as before. The purified protein was stored at −80°C.
Determination of melting temperature of HNMT (WT) and the Gly60Asp variant
To investigate the thermal stability of HNMT (WT) and the Gly60Asp variant, the melting temperature was determined using a fluorescence-based thermal shift assay. Experiments were performed in a real-time PCR detection system (Bio-Rad) with a 96-well plate in a FRET scan mode. In the experiments, 5 µl of 1 mg/ml HNMT (WT) or the Gly60Asp variant was mixed with 5 µl of 200-fold diluted SYPRO Orange solution and 15 µl of a buffer containing 50 mm Tris–HCl, pH 8.0 and 100 mm NaCl to a final volume of 25 µl in a 96-well plate. Thermal unfolding of the protein was monitored using a temperature gradient from 20 to 95°C, measuring fluorescence emission at 0.5°C increments with a 60-s hold for signal stabilization. The melting temperature of the protein was derived from the peak of the derivatives of the experimental data.
Determination of dissociation constant (Kd) of HNMT (WT) and Gly60Asp variant
Dissociation constants for binding of SAM and HA to HNMT (WT) and Gly60Asp variant were determined using the VP-ITC system (MicroCal, GE Healthcare). Experiments were performed at 25°C in 50 mm Tris–HCl buffer, pH 8.0, containing 100 mm NaCl. All solutions were degassed before measurements. Titration experiments for SAM and HA were performed by using 20 injections of 15 µl of 0.42 mm SAM or 4 mm HA (duration time 29.9 s and spacing time 250 s) into the cell containing 30 µM HNMT (WT). In the case of the Gly60Asp variant, 30 µM was titrated with SAM (2 mm, 20 injection, 15 µl/injection, duration time 29.9 s and spacing time 250 s) or HA (200 µM, 20 injection, 15 μl/injection, duration time 29.9 s and spacing time 250 s). To determine Kd values of HNMT (WT) and Gly60Asp variant, one set of sites fitting with Origin version 7.0 (MicroCal) for ITC data analysis was used.
Determination of catalytic activity of HNMT WT and Gly60Asp variant
Kinetic parameters for HNMT (WT) and the Gly60Asp variant were determined using the VP-ITC system (MicroCal) at 25°C in 50 mm Tris–HCl buffer, pH 8.0, containing 100 mm NaCl in multi-injection mode. All solutions were degassed before the measurements. The HNMT analysis consisted of 20 injections of 2 µl (duration time 4 s and spacing time 250 s) of 2 mm HA into the cell containing 1 µM of HNMT (WT) or Gly60Asp variant and 100 µM SAM. The experimental data were fitted with Origin version 7.0 (MicroCal) for ITC data analysis to obtain kinetic parameters.
In vitro toxicology assay
The cytotoxic effects of HA on patients' lymphoblast cells carrying the p.Gly60Asp variation and lymphoblasts from unrelated healthy controls were determined using the MTT assay. This method measures the metabolic reduction of MTT to a colored water-insoluble formazan salt by mitochondrial dehydrogenases. Lymphoblast cells were available for all four affected individuals from Family A. For comparison, lymphoblast cell lines from 12 unrelated unaffected individuals were selected, and matched for age, gender and number of passages for the cells. Cells for each line were seeded at 10 000 cells per well in 96-well plates (in triplicates for each line) and cultured in serum-free DMEM for 2 h. Cells were subsequently incubated for 2 h with 125 µM HA after which reconstituted MTT in an amount equal to 10% of the culture medium volume was added. After incubation of the plates at 37°C for 1 h, the cells were then washed with phosphate-buffered saline and the formazan salts dissolved in 200 ml of dimethyl sulfoxide with gentle shaking for 10 min at room temperature. The plates were read at 570 nm using a Tecan Spectra Fluor plate reader, and data were averaged across replicates and across both the affected and unaffected group.
HNMT protein localization
We generated two different sets of genetic constructs for HNMT protein localization. RNA was extracted from one of the patients' (Family A) fibroblast cells, and then HNMT full-length cDNA (CCDS2181.1) was PCR amplified and cloned into the pcDNA3.1/CT-GFP-TOPO expression vector (Invitrogen, Carlsbad, CA, USA). Using the Q5® Site-Directed Mutagenesis Kit (New England BioLabs, Ipswich, MA, USA), an A>G change was introduced to the HNMT mutant plasmid (c.179G>A) in order to generate the HNMT WT construct. Subsequently, a T>C change was introduced to the WT construct to generate a mutant HNMT construct for the c.623T>C, (p.Leu208Pro) substitution. Thus, we generated three constructs that are identical except for the mutation changes, in order that we would be able to exclude any potential effects of intervening SNPs, as it has been well documented that genetic variation among individuals can result in as much as 5-fold differences in HNMT activity (20). Orientation of the inserts and correct sequences were finally confirmed by Sanger sequencing. To examine the consequences of mutations on cellular localization of the HNMT protein, we transiently transfected COS-7 cells with 2 µg of purified constructs (p.Gly60Asp, p.Leu208Pro and WT) with Polyfect (Qiagen, Germantown, MD, USA). Twenty-four h after transfection, we visualized the HNMT-GFP fusion protein in transfected cells using a Zeiss Axioplan 2 imaging microscope (Carl Zeiss AG, Oberkochen, Germany), equipped with the LSM510 array confocal laser scanning system, and the Zeiss LSM510 version 3.2 SP2 software package.
Measurement of HA/methylhistamine levels in patients’ and controls’ lymphoblast cells’ by ELISA
Cell-free supernatants were used to measure the HA and N-tele-methylhistamine levels in patients' lymphoblast cell lines and in a group of unrelated healthy controls using an ELISA assay, performed according to the manufacturer's instructions (GenWay Biotech, Inc., San Diego, CA, USA) through a Fluoroskan Ascent microplate reader (Thermo Scientific) at 450 nm. Standard reagents were employed to draw a calibration curve. The samples OD values and the calibration curve were used to calculate the HA and N-tele-methylhistamine concentrations in cases and controls.
Bioinformatic analysis of the HNMT protein
Protein structure modeling was conducted with the Phyre2 server with the 3D structure image realized by JMOL. In addition, mutant protein structures were modeled with the FoldX package Version 3.0 Beta3 (18). Models were built based on the WT crystal structure [PDB 2AOT (26); available in the Protein Data Bank PDB] as the template using the BuildModel function after energy minimization of the WT structure using the RepairPDB function. Figures were generated with PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.1, Schrodinger, LLC).
Supplementary Material
Funding
This research was supported by a grant from the Canadian Institutes of Health Research (#MOP-102758) and Max Planck Society and EU FP 7 project GENCODYS, grant # 241995.
Supplementary Material
Acknowledgments
We thank all family members and affected individuals for participating in this study. We also thank the Qazvin University of Medical Sciences and Qazvin Rehabilitation Organization for their support in this study. We also thank the Austrian Academic Exchange Service (ÖAD) for a fellowship to C.T. and the Austrian Science Fund (FWF) for the support through the doctoral school “Molecular Enzymology” (W901).
Conflict of Interest statement. None declared.
References
- 1.Maulik P.K., Mascarenhas M.N., Mathers C.D., Dua T., Saxena S. (2011) Prevalence of intellectual disability: a meta-analysis of population based studies. Res. Dev. Disabil., 32, 419–436. [DOI] [PubMed] [Google Scholar]
- 2.Musante L., Ropers H.H. (2014) Genetics of recessive cognitive disorders. Trends Genet., 30, 32–39. [DOI] [PubMed] [Google Scholar]
- 3.Loiselle J., Wollin W. (1993) Mucosal histamine elimination and its effect on acid secretion in rabbit gastric mucosa. Gastroenterology, 104, 1013–1020. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz J.C., Arrang M.M., Garbarg M., Pollaed H., Ruat M. (1991) Histaminergic transmission in the mammalian brain. Physiol. Rev., 71, 1–51. [DOI] [PubMed] [Google Scholar]
- 5.Maslinski C. (1975) Histamine and its metabolism in mammals. Part II. Catabolism of histamine and histamine liberation. Agents Actions, 5, 183–225. [DOI] [PubMed] [Google Scholar]
- 6.Hough L.B., Green J.P. (1984) Histamine and its receptors in the nervous system. In Lajtha A. (ed.), Handbook of Neurochemistry, Vol. 6: Receptors in the Nervous System, Springer, USA, pp. 145–211. [Google Scholar]
- 7.Lindahl K.M. (1960) The histamine methylating enzyme system in liver. Acta Physiol. Scand., 49, 114–138. [DOI] [PubMed] [Google Scholar]
- 8.Brown D.D., Tomchick R., Axelrod J. (1959) The distribution and properties of a histamine-methylating enzyme. J. Biol. Chem., 234, 2948–2950. [PubMed] [Google Scholar]
- 9.Galinsky R.E., Bernstein J.A., Liggett S.B., Weinshilboum R.M. (2000) Histamine N-methyl transferase pharmacogenetics: association of a common functional polymorphism with asthma. Pharmacogenetics, 10, 261–266. [DOI] [PubMed] [Google Scholar]
- 10.González-Pérez A., López-Bigas N. (2011) Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet., 88, 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pang Y.P., Zheng X.E., Weinshilboum R.M. (2001) Theoretical 3D model of histamine N-methyltransferase: insights into the effects of a genetic polymorphism on enzymatic activity and thermal stability. Biochem. Biophys. Res. Commun., 287, 204–208. [DOI] [PubMed] [Google Scholar]
- 12.Preuss C.V., Wood T.C., Szumlanski C.L., Raftogianis R.B., Otterness D.M., Girard B., Scott M.C., Weinshilboum R.M. (1998) Human histamine N-methyltransferase pharmacogenetics: common genetic polymorphisms that alter activity. Mol. Pharmacol., 53, 708–717. [DOI] [PubMed] [Google Scholar]
- 13.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res., 20, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grantham R. (1974) Amino acid difference formula to help explain protein evolution. Science, 185, 862–864. [DOI] [PubMed] [Google Scholar]
- 15.Horton J.R., Sawada K., Nishibori M., Cheng X. (2005) Structural basis for inhibition of histamine N-methyltransferase by diverse drugs. J. Mol. Biol., 353, 334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piela L., Nemethy G., Scheraga H.A. (1987) Proline-induced constraints in alpha-helices. Biopolymers, 26, 1587–1600. [DOI] [PubMed] [Google Scholar]
- 17.Dehouck Y., Grosfils A., Folch B., Gilis D., Bogaerts P., Rooman M. (2009) Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC. Bioinformatics, 25, 2537–2543. [DOI] [PubMed] [Google Scholar]
- 18.Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L. (2005) The FoldX web server: an online force field. Nucl. Acids Res., 33, W382–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin C.Y., Kalimo H., Panula P. (2002) The histaminergic system in human thalamus: correlation of innervation to receptor expression. Eur. J. Neurosci., 15, 1125–1138. [DOI] [PubMed] [Google Scholar]
- 20.Panula P., Lintunen M., Karlstedt K. (2000) Histamine in brain development and tumors. Semin. Cancer Biol., 10, 11–14. [DOI] [PubMed] [Google Scholar]
- 21.Molina-Hernández A., Díaz N.F., Arias-Montaño J.A. (2012) Histamine in brain development. J. Neurochem., 122, 872–882. [DOI] [PubMed] [Google Scholar]
- 22.Maslinski C., Fogel W.A. (1991) Catabolism of histamine. In Uvnäs B. (ed.), Handbook of Experimental Pharmacology. Histamine and Histamine Antagonists. Springer, Berlin-Heidelberg, Vol. 97. pp. 165–189. [Google Scholar]
- 23.Hur J., Kang M.K., Park J.Y., Lee S.Y., Bae Y.S., Lee S.H., Park Y.M., Kwak J.Y. (2003) Pro apoptotic effect of high concentrations of histamine on human neutrophils. Int. Immunopharmacol., 3, 1491–1502. [DOI] [PubMed] [Google Scholar]
- 24.Darvas Z., Falus A. (2004) Histidine decarboxylase (HDC) enzyme and gene. In Falus A. (ed.), Histamine: Biology and Medical Aspects. SpringMed Publishing, Budapest, pp. 31–42. [Google Scholar]
- 25.Maintz L., Novak N. (2007) Histamine and histamine intolerance. Am. J. Clin. Nutr., 85, 1185–1196. [DOI] [PubMed] [Google Scholar]
- 26.Horton J.R., Sawada K., Nishibori M., Zhang X., Cheng X. (2001) Two polymorphic forms of human histamine methyltransferase: structural, thermal and kinetic comparisons. Structure, 9, 837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaccari A. (1980) Sexual differentiation of monoamine neurotransmitters. In Parvez H. (ed.), Biogenic Amines in Development. Elsevier, North-Holland, Amsterdam, pp. 327–352. [Google Scholar]
- 28.De Vries G.J., Buijs R.M., Van Leeuwen F.W. (1984) Sex differences in vasopressin and other neurotransmitter systems in the brain. Prog. Brain Res., 61, 185–203. [DOI] [PubMed] [Google Scholar]
- 29.Mazurkiewicz-Kwilecki I.M., Nsonwah S. (1989) Changes in the regional brain histamine and histidine levels in postmortem brains of Alzheimer patients. Can. J. Physiol. Pharmacol., 67, 75–78. [DOI] [PubMed] [Google Scholar]
- 30.Panula P., Rinne J., Kuokkanen K., Eriksson K.S., Sallmen T., Kalimo H., Relja M. (1998) Neuronal histamine deficit in Alzheimer's disease. Neuroscience, 82, 993–997. [DOI] [PubMed] [Google Scholar]
- 31.Cacabelos R., Yamatodani A., Niigawa H., Hariguchi S., Tada K., Nishimura T. (1989) Brain histamine in Alzheimer's disease. Methods Find. Exp. Clin. Pharmacol., 11, 353–360. [PubMed] [Google Scholar]
- 32.Pérez A., Albarran M.A., Cacabelos R. (1988) Biochemical studies of body fluids in senile dementia of the Alzheimer type (SDAT) and multiinfarct dementia (MID). Correlation analysis between histamine and vasopressin in CSF and plasma. Acta Neurol. Scand., 77, 129. [Google Scholar]
- 33.Faucard R., Armand V., Héron A., Cochois V., Schwartz J.C., Arrang J.M. (2006) N-methyl-d-aspartate receptor antagonists enhance histamine neuron activity in rodent brain. J. Neurochem., 98, 1487–1496. [DOI] [PubMed] [Google Scholar]
- 34.Burban A., Faucard R., Armand V., Bayard C., Vorobjev V., Arrang J.M. (2010) Histamine potentiates N-methyl-d-aspartate receptors by interacting with an allosteric site distinct from the polyamine binding site. J. Pharmacol. Exp. Ther., 332, 912–921. [DOI] [PubMed] [Google Scholar]
- 35.Saras A., Gisselmann G., Vogt-Eisele A.K., Erlkamp A.S., Kletke O., Pusch H., Hatt H. (2008) Histamine action on vertebrate GABAA receptors: direct channel gating and potentiation of GABA responses. J. Biol. Chem., 283, 10470–10475. [DOI] [PubMed] [Google Scholar]
- 36.Molina-Hernández A., Rodríguez-Martínez G., Escobedo-Ávila I., Velasco I. (2013) Histamine up-regulates fibroblast growth factor receptor 1 and increases FOXP2 neurons in cultured neural precursors by histamine type 1 receptor activation: conceivable role of histamine in neurogenesis during cortical development in vivo. Neural Dev., 8, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lai C.S., Fisher S.E., Hurst J.A., Vargha-Khadem F., Monaco A.P. (2001) A forkhead-domain gene is mutated in a severe speech and language disorder. Nature, 413, 519–523. [DOI] [PubMed] [Google Scholar]
- 38.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet., 30, 97–101. [DOI] [PubMed] [Google Scholar]
- 39.Gudbjartsson D.F., Jonasson K., Frigge K.L., Kong A. (2000) Allegro, a new computer program for multipoint linkage analysis. Nat. Genet., 25, 12–13. [DOI] [PubMed] [Google Scholar]
- 40.Rüschendorf F., Nürnberg P. (2005) ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics, 9, 2123–2125. [DOI] [PubMed] [Google Scholar]
- 41.O'Connell J.R., Weeks D.E. (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet., 63, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garshasbi M., Motazacker M.M., Kahrizi K., Behjati F., Abedini S.S., Nieh S.E., Firouzabadi S.G., Becker C., Rüschendorf F., Nürnberg P. et al. (2006) SNP array-based homozygosity mapping reveals MCPH1 deletion in family with autosomal recessive mental retardation and mild microcephaly. Hum. Genet., 118, 708–715. [DOI] [PubMed] [Google Scholar]
- 43.Najmabadi H., Hu H., Garshasbi M., Zemojtel T., Abedini S.S., Chen W., Hosseini M., Behjati F., Haas S., Jamali P. et al. (2011) Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature, 478, 57–63. [DOI] [PubMed] [Google Scholar]
- 44.Hu H., Wienker T.F., Musante L., Kalscheuer V.M., Kahrizi K., Najmabadi H., Ropers H.H. (2014) Integrated sequence analysis pipeline provides one-stop solution for identifying disease-causing mutations. Hum. Mutat., 35, 1427–1435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.