

Abstract
Myostatin is a secreted signaling molecule that normally acts to limit muscle growth. As a result, there is extensive effort directed at developing drugs capable of targeting myostatin to treat patients with muscle loss. One potential concern with this therapeutic approach in patients with muscle degenerative diseases like muscular dystrophy is that inducing hypertrophy may increase stress on dystrophic fibers, thereby accelerating disease progression. To investigate this possibility, we examined the effect of blocking the myostatin pathway in dysferlin-deficient (Dysf−/−) mice, in which membrane repair is compromised, either by transgenic expression of follistatin in skeletal muscle or by systemic administration of the soluble form of the activin type IIB receptor (ACVR2B/Fc). Here, we show that myostatin inhibition by follistatin transgene expression in Dysf−/− mice results in early improvement in histopathology but ultimately exacerbates muscle degeneration; this effect was not observed in dystrophin-deficient (mdx) mice, suggesting that accelerated degeneration induced by follistatin transgene expression is specific to mice lacking dysferlin. Dysf−/− mice injected with ACVR2B/Fc showed significant increases in muscle mass and amelioration of fibrotic changes normally seen in 8-month-old Dysf−/− mice. Despite these potentially beneficial effects, ACVR2B/Fc treatment caused increases in serum CK levels in some Dysf−/− mice, indicating possible muscle damage induced by hypertrophy. These findings suggest that depending on the disease context, inducing muscle hypertrophy by myostatin blockade may have detrimental effects, which need to be weighed against the potential gains in muscle growth and decreased fibrosis.
Introduction
Myostatin is a transforming growth factor-β (TGF-β) family member that acts as a negative regulator of skeletal muscle mass (1). Genetic studies in numerous species have shown that loss of myostatin results in dramatic increases in muscle mass (2–7), and pharmacological agents capable of blocking myostatin signaling have been shown to cause significant muscle hypertrophy when given systemically to mice (8). As a result, there has been considerable effort directed at developing myostatin inhibitors as potential therapeutic agents to increase muscle growth, and there are currently multiple such agents being tested in clinical trials in a wide range of indications characterized by muscle loss.
One indication for which myostatin inhibition is being tested as a potential therapy is muscular dystrophy. Muscular dystrophy encompasses a group of disorders characterized by progressive muscle degeneration and weakness (9). Many forms of muscular dystrophy result from inherited mutations in genes encoding proteins involved in maintaining the structural integrity of muscle fibers. The most common form affects dystrophin, which is a vital component of a protein complex that connects the cytoskeleton of a muscle fiber to the surrounding extracellular matrix; disruption of this link renders the muscle fibers susceptible to contraction-induced injury (10). In patients with severe forms of muscular dystrophy, the regenerative capacity of the muscle to respond to injury is unable to compensate for the degenerative process, leading to progressive loss of muscle function.
The identification of myostatin has raised the possibility that blocking this signaling pathway may be a therapeutic strategy to prevent or reverse the loss of muscle mass and strength in patients with muscular dystrophy, and numerous preclinical studies in mdx mice, which carry mutations in the dystrophin gene, have demonstrated beneficial effects of myostatin inhibition on muscle mass and function (11–18). One theoretical concern with this therapeutic approach is the possibility that inducing muscle hypertrophy may cause additional membrane stress to already fragile muscle fibers, thereby further inducing damage. If so, myostatin inhibition in patients with muscular dystrophy may provide short-term benefits in terms of increasing muscle growth but may have the long-term effect of accelerating disease progression. To investigate this issue, we examined the effect of blocking myostatin signaling in dysferlin-deficient (Dysf−/−) mice, which is a model for limb-girdle type 2B and Miyoshi muscular dystrophies. Our rationale for these studies was that, if myostatin inhibition results in increased membrane damage, these effects would be revealed specifically in Dysf−/− mice, in which membrane repair is compromised (19). Here, we show that inhibition of myostatin signaling indeed can result in an exacerbation of muscle degeneration in the setting of dysferlinopathy.
Results
Follistatin overexpression accelerates muscle loss in Dysf−/− mice
To investigate the effect of blocking myostatin signaling in the setting of dysferlinopathy, we used both genetic and pharmacological approaches in mice. For the genetic approach, we overexpressed follistatin (Fst), which is a secreted protein capable of binding and inhibiting various members of the TGF-β superfamily, including myostatin (20). For these studies, we utilized a transgenic mouse line (F66) in which we had expressed Fst under the control of a skeletal muscle-specific myosin light chain promoter/enhancer (20). As shown in Figure 1A–D and Supplementary Material, Table S1, and as we reported previously, F66 transgenic mice exhibit dramatic increases in skeletal muscle mass throughout the body. We examined the effect of crossing the F66 transgene into Dysf−/− mice. At early ages, F66 transgene expression in Dysf−/− mice (i.e. F66;Dysf−/− mice) caused increased muscle mass to a similar extent as F66 did in a wild-type (wt) background (Fig. 1A–D; Supplementary Material, Table S1). Hence, blockade of the myostatin signaling pathway by Fst is capable of inducing muscle hypertrophy in the absence of functional dysferlin. These early gains in muscle weights, however, were not maintained as the mice aged, with muscle masses in F66;Dysf−/− mice decreasing sharply beginning at ∼4 months of age; in fact, by 8 months of age, F66;Dysf−/− mice showed even lower muscle weights than Dysf−/− mice.
Figure 1.

Hypertrophy induced by Fst transgene expression accelerates muscular degeneration in Dysf−/− mice. (A–D) Effect of F66 on weights of pectoralis, triceps, quadriceps and gastrocnemius muscles in Dysf−/− mice as a function of age. Graphs were made from the data shown in Supplementary Material, Table S1. Note the dramatic loss of muscle mass induced by F66 in Dysf−/− mice beginning at 4 months of age. *P < 0.05 versus wt; **P < 0.01 versus wt; ***P < 0.001 versus wt; #P < 0.05 versus Dysf−/−; ##P < 0.01 versus Dysf−/−; ###P < 0.001 versus Dysf−/−; †P < 0.05 versus F66; ††P < 0.01 versus F66; †††P < 0.001 versus F66. (E–H) Effect of F66 on weights of pectoralis, triceps, quadriceps and gastrocnemius muscles in mdx mice as a function of age. Note that F66 did not induce muscle loss in mdx mice. Graphs were made from the data shown in Supplementary Material, Table S2. **P < 0.01 versus wt; ***P < 0.001 versus wt; ##P < 0.01 versus mdx; ###P < 0.001 versus mdx; †P < 0.05 versus F66; ††P < 0.01 versus F66; †††P < 0.001 versus F66. (I) Sections were stained with H&E, von Kossa (to highlight calcified fibers) and Masson's trichrome (to highlight areas of fibrosis). The F66;Dysf−/− mice showed the pathologic changes including fibrosis and centrally nucleated muscle fibers, even at 10 weeks of age, when Dysf−/− mice showed no distinct muscular degeneration. The F66;mdx mice showed the pathologic changes similar to mdx mice. Scale bar represents 50 µm.
A previous study using mdx mice had reported that gains in muscle mass due to loss of myostatin activity were maintained as the mice aged (21). Because that study had utilized the Mstn loss-of-function mutation to eliminate myostatin function, we sought to determine whether the effects that we had observed in Dysf−/− mice were somehow specific to the F66 transgene by crossing the F66 transgene onto an mdx background. In contrast to what we observed in F66;Dysf−/− mice, F66;mdx mice exhibited significantly increased muscle weights compared with mdx mice, which were maintained at all ages that we examined (Fig. 1E–H; Supplementary Material, Table S2). Hence, accelerated muscle loss induced by Fst transgene expression was specific to mice lacking dysferlin.
Effect of Fst overexpression on muscle histopathology and function in Dysf−/− mice
The detrimental effect of F66 in the absence of dysferlin was clearly seen upon histological examination of the muscles. As previously reported, histological analysis of muscles from Dysf−/− mice showed signs of a slowly progressive muscular degeneration (19); by 4 months of age, a few necrotic fibers and centrally located nuclei were detected, and by 8 months of age, most of pathological characteristics of muscular dystrophy were observed (Fig. 1I; Supplementary Material, Fig. S1). F66;Dysf−/− mice, however, showed more severe muscular degeneration than age-matched Dysf−/− mice. As early as 10 weeks of age, when increases in muscle weights due to the F66 transgene were comparable in wt and Dysf−/− mice and when Dysf−/− muscles showed no distinct histological signs of degeneration, F66;Dysf−/− muscles showed dystrophic features, including central nucleation, fibrosis and calcified fibers (Fig. 1I). In contrast, although dystrophic changes were evident in muscles of mdx mice, the presence of the F66 transgene did not exacerbate degeneration in the mdx background and even appeared to improve the histopathology as a function of age (Fig. 1I; Supplementary Material, Fig. S1); this improvement in histopathology resulting from Fst expression in mdx mice was also consistent with what had been reported in mdx mice lacking myostatin (11–15,17).
We also examined the muscles in F66;Dysf−/− mice by electron microscopy (EM). As shown in Figure 2A, EM analysis of quadriceps muscles of Dysf−/− mice at 10 weeks of age generally showed regular cross-striations with well-arranged myofibrils, although some fibers in Dysf−/− mice showed membrane-bound spaces surrounded by swollen mitochondria, and inflammatory cell infiltration between fibers was also seen. In F66;Dysf−/− mice, however, in addition to the inflammatory cell infiltration, mitochondrial degeneration was more prominent and severe, and necrotic fibers surrounded by fibroblasts were more frequently observed. These changes were not observed in F66 muscles, which showed regular cross-striations with little evidence of muscle fiber degeneration, although we did observe occasional fibers in F66 mice with randomly fragmented myofibrils.
Figure 2.

Effect of Fst transgene expression on muscle fiber integrity and function in Dysf−/− mice. (A) Electron microscope images of quadriceps muscles at 10 weeks of age. While wt mice showed the regular sarcomeric pattern with no degenerative change in mitochondrial morphology, Dysf−/− mice showed membrane-bound spaces surrounded by swollen mitochondria (arrows in Dysf−/− image). F66 muscles showed randomly fragmented myofibrils in regular sarcomeric pattern, representing Type IIb fast-twitch fibers. F66;Dysf−/− muscles showed accumulation of vacuoles and mitochondria aggregation in subsarcolemmal area, necrotic fiber with complete loss of myofibrils (arrow in F66;Dysf−/− image) and fibroblast next to necrotic fiber (arrowhead). Scale bar represents 2 µm. (B) Serum CK (U/L) levels as a function of age. Note the sharp increase in CK levels in F66;Dysf−/− mice at 10 weeks of age. Graphs were made from the data shown in Supplementary Material, Table S3. *P < 0.05 versus wt; **P < 0.01 versus wt; ***P < 0.001 versus wt; #P < 0.01 versus Dysf−/−; ##P < 0.001 versus Dysf−/−. (C) EBD uptake by abdominal, quadriceps, gastrocnemius/soleus, gluteus/hamstring and triceps muscles. Note the increased EBD uptake in F66;Dysf−/− mice compared with the other groups. Graphs were made from the data shown in Supplementary Material, Table S5. *P < 0.05 versus wt; **P < 0.01 versus wt; ***P < 0.001 versus wt; ##P < 0.01 versus Dysf−/−; ###P < 0.001 versus Dysf−/−. (D) Specific force of EDL muscles from wt, Dysf−/−, F66 and F66;Dysf−/− mice at 4 months of age. F66;Dysf−/− mice showed a significant decrease in specific force compared with others. *P < 0.05 versus wt; **P < 0.01 versus wt; #P < 0.05 versus Dysf−/−; †P < 0.05 versus F66.
The harmful effect of the F66 transgene in Dysf−/− mice was also evident when we measured serum creatine kinase (CK, U/L) levels, which are routinely used as an indicator of muscle damage. As described previously (22), CK levels of Dysf−/− mice were elevated by 6 months of age, reflecting the ongoing muscle degeneration resulting from dysferlin deficiency (Fig. 2B; Supplementary Material, Table S3). F66;Dysf−/− mice, however, showed a sharp increase in CK levels at 10 weeks of age, which was observed neither in F66 mice nor in Dysf−/− mice (P < 0.001 versus Dysf−/−). As mice aged, CK levels gradually declined in F66;Dysf−/− mice, presumably reflecting the loss of functional muscle as a result of the enhanced muscle degeneration. As reported previously (23), mdx mice had greatly elevated CK levels with high variability, and these elevations did not appear to be worsened by the presence of the F66 transgene (Supplementary Material, Table S4).
We also evaluated the integrity of the plasma membrane by membrane-impermeable Evans blue dye (EBD) uptake. At 10 weeks of age, significant elevations in EBD uptake were observed in Dysf−/− mice compared with wt and F66 mice in all muscle groups that were analyzed (Fig. 2C; Supplementary Material, Table S5), presumably resulting from the membrane repair defect in the absence of dysferlin. These elevations in EBD uptake, however, were exacerbated in F66;Dysf−/− mice, consistent with the elevated serum CK levels in these mice. Finally, we examined the effect of the F66 transgene on muscle contractile function. Although no significant differences were seen at 10 weeks of age, F66;Dysf−/− mice showed a significant decrease in specific force of the EDL muscle (P < 0.01) at 4 months of age (Fig. 2D) compared with wt, F66 and Dysf−/− mice. Hence, overexpression of Fst in Dysf−/− mice had a clear detrimental effect not only on muscle mass, histopathology and membrane integrity, but also on muscle contractile function.
Effect of pharmacological blockade of myostatin signaling in Dysf−/− mice
As a complementary approach to these genetic studies, we also examined the effect of blocking myostatin signaling pharmacologically by direct administration of a myostatin inhibitor to Dysf−/− mice. We showed previously that myostatin is capable of binding the two activin type II receptors, ACVR2 and ACVR2B (20), and that a soluble form of ACVR2B in which the extracellular domain of the receptor is fused to an Fc domain can cause significant muscle hypertrophy when given systemically to wt mice (24). We examined the effect of administering this soluble receptor (ACVR2B/Fc) to Dysf−/− mice. As shown in Figure 3A and Supplementary Material, Table S6, Dysf−/− mice given four weekly intraperitoneal (i.p.) injections of ACVR2B/Fc beginning at either 6 weeks or 7 months of age showed significant increases in muscle mass comparable to wt mice injected with ACVR2B/Fc. Moreover, fibrotic changes seen in 8-month-old Dysf−/− mice appeared to be significantly ameliorated by ACVR2B/Fc treatment (Fig. 3B); these histological improvements were consistent with what had been reported for ACVR2B/Fc treatment of mdx mice (18). EM analysis also showed that membrane-bound spaces surrounded by swollen mitochondria normally seen in Dysf−/− myofibers were less prominent in muscles of ACVR2B/Fc-treated Dysf−/− mice (Fig. 3C). Despite these potentially beneficial effects, ACVR2B/Fc treatment caused increases in serum CK levels in some Dysf−/− mice at both 10 weeks and 8 months of age (Fig. 3D; Supplementary Material, Table S7), suggesting that hypertrophy induced by ACVR2B/Fc administration to Dysf−/− mice may have some adverse effects as well, though not nearly to the degree seen with Fst overexpression.
Figure 3.

Effect of ACVR2B/Fc administration on muscle weights, muscle histopathology and serum CK levels in Dysf−/− mice. (A) Effect of ACVR2B/Fc on muscle weights. Bars indicate percent increase in muscle weights in ACVR2B/Fc-injected wt and Dysf−/− mice compared with PBS-injected wt and Dysf−/− mice, respectively. ACVR2B/Fc and PBS were administered by weekly i.p. injections over a span of 4 weeks at 10 mg kg−1 starting at age 6 weeks and 7 months. All calculations were made from the data shown in Supplementary Material, Table S6. (B) Sections of the quadriceps muscles stained with H&E and Masson's trichrome. Note that ACVR2B/Fc-injected Dysf−/− mice showed significant increases in muscle fiber diameters comparable to ACVR2B/Fc-injected wt mice and decreases in the extent of fibrosis compared with the PBS-injected Dysf−/− mice even at age 8 months. Scale bar represents 50 µm. (C) Electron microscope images. Note the membrane-bound spaces surrounded by swollen mitochondria in Dysf−/− myofiber (arrows). No specific changes were found in ACVR2B/Fc-injected wt muscles. However, ACVR2B/Fc-injected Dysf−/− myofiber showed reduction of membrane-bound spaces and swollen mitochondria compared with Dysf−/− myofiber. Scale bar represents 1 µm. (D) Effect of ACVR2B/Fc on serum CK levels. Note the increases in serum CK levels in ACVR2B/Fc-injected Dysf−/− mice compared with PBS-injected Dysf−/− mice at both 10 weeks and 8 months of age (P < 0.01). The graph was made from the data shown in Supplementary Material, Table S7.
Effect of myostatin blockade on global gene expression patterns in wt and Dysf−/− mice
Finally, we used microarray analysis to examine global transcriptional changes in the quadriceps muscles from wt, Dysf−/−, F66, F66;Dysf−/− mice and ACVR2B/Fc-injected wt and Dysf−/− mice at 10 weeks of age. By principal components analysis (PCA) mapping, the F66;Dysf−/− and F66 groups were well segregated from each other as well as the other groups (Fig. 4A). The differences of global gene expression patterns among the six groups were also seen in their heat maps (Fig. 4B). As expected, two classes of genes that showed clear differences among the various groups were genes related to dysferlin function and genes encoding components of the myostatin/TGF-β signaling pathway (Fig. 4C and D). Seven genes related to dysferlin function, Dysf, Myof, Anxa1, Anxa2, S100a4, S100a10 and Capn3, were visualized in Volcano plots in five different comparisons (Supplementary Material, Fig. S2); Myof, Anxa1, Anxa2, S100a4 and S100a10 were significantly upregulated by Fst overexpression while Capn3 was significantly downregulated by Fst overexpression. Mstn, Gdf11, Inha and Inhba genes encode proteins that have been shown to be bound and inhibited by Fst and ACVR2B/Fc. While there was no significant change in Dysf−/− compared with wt, Fst overexpression and ACVR2B/Fc administration induced the dramatic changes of expression levels of these genes in both Dysf−/− and wt (Supplementary Material, Fig. S3); Gdf11 was upregulated slightly in ACVR2B/Fc-injected wt and ACVR2B/Fc-injected Dysf−/− and significantly in F66 and F66;Dysf−/−; Mstn was downregulated slightly in ACVR2B/Fc-injected Dysf−/− compared with Dysf−/− and significantly in F66;Dysf−/− compared with Dysf−/−. The full list of genes with fold changes is given in Supplementary Material, Table S8.
Figure 4.
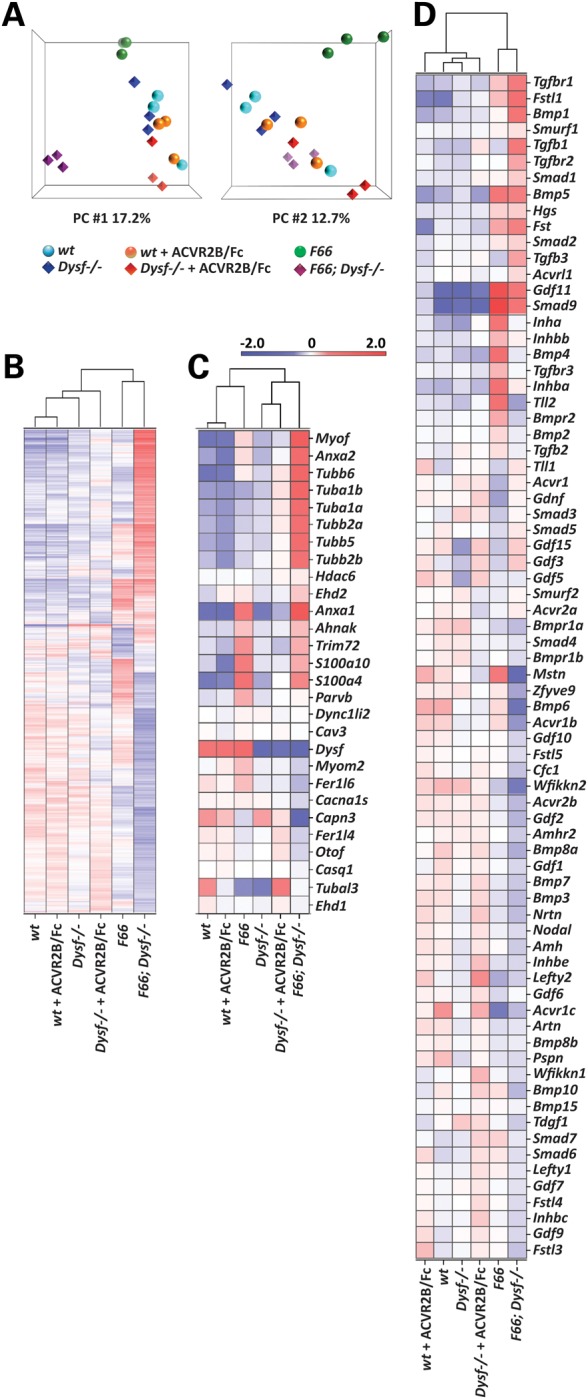
Analysis of global gene expression patterns in wt, Dysf−/−, F66, F66; Dysf−/− and ACVR2B/Fc-injected wt and Dysf−/− mice. (A) PCA mapping analysis. Affymetrix Mouse Exon 1.0 ST arrays were hybridized in three biologically independent experiments with RNA from quadriceps muscles of wt (blue circle), Dysf−/− (blue diamond), F66 (green circle), F66;Dysf−/− (purple diamond) mice and ACVR2B/Fc-injected wt (orange circle) and Dysf−/− (red diamond) mice (three replicates in six different groups: 18 samples). Note that F66;Dysf−/− and F66 groups are well segregated from each other and from the other remaining groups in PCA mappings. First component (PC # 1) and second component (PC # 2) data are shown. (B) Heat map corresponding to differentially expressed genes (P < 0.01). Data represent averages of three independent experiments in red (enriched) or blue (depleted). (C) Heat map corresponding to genes related to dysferlin function. Relative expression levels (fold change compared to wt) are shown in Supplementary Material, Table S8. (D) Heat map corresponding to genes encoding components of the myostatin/TGF-β signaling pathway. Relative expression levels (fold change compared to wt) are shown in Supplementary Material, Table S8.
Biological processes altered by either Fst overexpression or ACVR2B/Fc treatment were detected by Gene ontology (GO) analysis (Supplementary Material, Table S9). The differential expression patterns of genes involved in processes related to response to stress were observed and shown in their heat maps (Fig. 5; for additional details, see Supplementary Material, Dataset). Fst overexpression in both wt and Dysf−/− background dramatically changed the expression pattern of genes related to response to stress (Fig. 5A). Hypertrophy induced by either Fst overexpression or ACVR2B/Fc treatment in Dysf−/− background resulted in similar expression patterns of genes related to inflammatory responses (Fig. 5B) and response to wounding (Fig. 5C), consistent with the important membrane repair function of dysferlin. Hence, the differences that we observed on muscle mass and histopathology among the various experimental groups were clearly reflected in the differential expression patterns of genes related to altered biological processes.
Figure 5.

Heat maps of enriched top biological processes from GO analysis. (A) Heat map corresponding to response to stress (205 genes). Note that F66 and F66;Dysf−/− are grouped together. (B) Heat map corresponding to inflammatory response (73 genes). Note that ACVR2B/Fc-injected Dysf−/− and F66;Dysf−/− are grouped together. (C) Heat map corresponding to response to wounding (73 genes). Note that Dysf−/−, ACVR2B/Fc-injected Dysf−/− and F66;Dysf−/− are grouped together. Data represent averages of three independent experiments in red (enriched) or blue (depleted). GO analysis was conducted for significantly differentially expressed genes (fold change >2.0 and P < 0.01), and enriched top biological processes are shown in Supplementary Material, Table S9. List of genes in each biological process with relative expression levels (fold change compared with wt) is shown in Supplementary Material, Dataset.
Discussion
The identification of myostatin as a negative regulator of skeletal muscle mass has generated considerable interest in the possibility that targeting the myostatin pathway may be an effective strategy to enhance muscle growth and function in patients with muscle loss. Indeed, the potential benefits of myostatin inhibition have been documented in numerous models of muscle wasting and degeneration in mice (for review, see ref. 8), and there are now several studies that have reported that myostatin inhibition can increase lean body mass and improve muscle function in humans as well (25–27). In this respect, a number of myostatin inhibitors are currently being tested in Phase II/III clinical trials for a variety of indications characterized by muscle loss, including cancer, hip replacement surgery, age-related sarcopenia, end-stage renal disease, chronic obstructive pulmonary disease and inclusion body myositis (see clinicaltrials.gov).
Although increasing muscle growth in these disease settings may provide short-term benefit, there are potential long-term concerns about the use of this therapeutic strategy. One theoretical concern is that inducing muscle hypertrophy may promote muscle damage by increasing stress on myofibers. Although this effect may be inconsequential under normal conditions, the increased damage to fibers might accelerate disease progression when muscle fibers are already fragile, for example, as a result of genetic loss of key structural components in the case of muscular dystrophy. Here, we have presented results of studies in which we have attempted to address this possibility by analyzing the effect of blocking myostatin signaling in mice lacking dysferlin. Because studies have shown that dysferlin is likely involved in Ca2+-dependent membrane repair of damaged skeletal muscle fibers (19,28,29), we reasoned that potential detrimental effects of myostatin inhibition in terms of increasing muscle fiber stress and damage might be more pronounced in Dysf−/− mice. Our results show that myostatin inhibition does, in fact, accelerate muscle degeneration in Dysf−/− mice and suggest that the anabolic effects of myostatin inhibition may come at the expense of additional damage to muscle fibers.
It will be important to weigh these potential adverse effects against the beneficial effects seen with myostatin inhibition not only on muscle growth but also on histopathology, such as decreased fibrosis. In mice in which myostatin signaling was blocked by Fst transgene expression, the massive initial muscle hypertrophy was clearly outweighed by the subsequent detrimental effects leading to accelerated muscle degeneration. In mice treated with ACVR2B/Fc, however, the beneficial effects on muscle mass and histopathology were significant, with the only evidence of adverse effects being the elevated CK levels. In this regard, the beneficial effects of ACVR2B/Fc administration to Dysf−/− mice were observed even in mice at 14 months of age (Supplementary Material, Table S6). Further studies using a variety of different myostatin inhibitors, treatment timing/durations, and doses will be essential in determining whether the beneficial effects of myostatin inhibition can be achieved while minimizing the potential detrimental effects. It will also be critical to conduct these types of studies not only in the setting of chronic muscle degenerative diseases but also in clinical settings characterized by more acute muscle loss, in which the fragility and structural integrity of the muscle fibers may not be as important an issue.
Materials and Methods
Mice
F66 transgenic mice that express Fst under the myosin light chain promoter have been described previously (20). Because the F66 transgene is located on the Y chromosome, we focused all of our analysis on male mice. To analyze the effect of F66 in Dysf−/− mice, F66 transgenic mice were mated with Dysf−/− mice. F66;Dysf+/− males from this cross were mated to Dysf+/− females obtain F66;Dysf−/− and F66;Dysf+/+ males. As the mdx mutation in mdx mouse is X chromosome-linked, F66;mdx male mice were generated from matings between F66 males and mdx females. All mice were maintained on a C57BL/6 background.
ACVR2B/Fc purification and injection
ACVR2B/Fc is a soluble form of the activin type IIB receptor (ACVR2B) in which the extracellular ligand binding domain was fused to the mouse IgG2a Fc domain (ACVR2B/Fc) to enhance stability in vivo (24). ACVR2B/Fc protein expression from the stable Chinese hamster ovary (CHO) cells was induced with 100 nm Cadmium in serum-free EX-CELL™ 325 PF CHO media (Sigma, St Louis, MO, USA), and ACVR2B/Fc protein was purified from the conditioned medium using protein A Sepharose. Male C57BL/6 (wt) and Dysf−/− mice beginning at either 6 weeks or 7 months of age were given four weekly intraperitoneal (i.p.) injections of either ACVR2B/Fc (10 mg kg−1, i.e. 200 μg per injection for 6-week-old mice and 250 μg per injection for 7-month-old mice) or PBS.
Muscle weight and histopathologic analysis
For measurement of muscle weights, pectoralis, triceps, quadriceps and gastrocnemius muscles from both sides of mice aged from 4 weeks to 8 months were dissected, and the average weight was used for each muscle. For histopathologic analysis, the harvested quadriceps muscles were snap frozen in isopentene cooled in liquid nitrogen, and 10 µm frozen cross-sections were taken from the right quadriceps and subjected to hematoxylin/eosin (H&E), von Kossa (to highlight calcified fibers) and Masson's trichrome staining (to highlight areas of fibrosis). Images were acquired with a ×20 objective using a Zeiss AxioCam MRc5 microscope in combination with AxioVision 4.8 software.
Evans blue dye uptake
To detect plasma membrane disruption, 10 µl per 1 g of body weight of sterile EBD (5 mg/ml in PBS) was injected intraperitoneally into 10-week-old mice 2 days before harvesting. Abdominal, quadriceps, gastrocnemius/soleus, gluteus/hamstring and triceps muscles were dissected, minced and incubated in formamide at 55°C for 2 h, and the supernatant absorbances were measured at 620 nm in a spectrophotometer.
Isolated muscle mechanics
Mice were anesthetized with ketamine/xylazine, and muscles were removed and prepared as previously described (30). Isolated muscle mechanics, including isometric tetanic force as well as susceptibility to eccentric contractile injury, were performed on the extensor digitorum longus (EDL). Specific force was measured as force per cross-section area (n = 3 per age and genotype).
Transmission electron microscopy
Quadriceps muscles of 10-week-old mice were fixed with 2% glutaraldehyde and 2% formaldehyde in 0.1 m cacodylate buffer (pH 7.4), and post-fixed with 1% osmium tetroxide in 0.1 m cacodylate buffer. Fixed samples were en bloc stained with 2% uranyl acetate, dehydrated through series of ethanol and propylene oxide, and embedded in EMbed-812 resin. Ultrathin sections were cut and stained with uranyl acetate and lead citrate. Photographs were taken through the Philips BioTwin CM120 electron microscope.
Microarray analysis
Total RNA was extracted from quadriceps muscles of 10-week-old wt, Dysf−/−, F66, F66; Dysf−/−, and ACVR2B/Fc-injected wt and Dysf−/− mice (six different groups), and three biological replicates were set up for each group. Affymetrix Mouse Exon 1.0 ST arrays were hybridized with extracted RNA (three replicates in six different groups, 18 samples) at the Johns Hopkins Microarray Core Facility. Microarray data were visualized by PCA mapping, volcano plots and heat maps. GO (www.geneontology.org) analysis was conducted for significantly differentially expressed genes (fold change >2.0 and P < 0.01) using the Spotfire analytic platform (31). Raw microarray data have been deposited in the Gene Expression Omnibus database (GSE62945).
Statistical analysis
All values are presented as mean ± SEM from at least three independent experiments unless otherwise stated. An unpaired two-tailed Student's t-test and one-way ANOVA were performed for statistical analyses with two groups and more than two groups, respectively. P < 0.05 was considered significant.
Study approval
Mice were handled and housed according to the approved Institutional Animal Care and Use Committee (IACUC) protocols MO12M07 and MO13M283 of the Johns Hopkins Medical Institutions. All animal studies were approved by the IACUC of the Johns Hopkins Medical Institutions.
Supplementary Material
Funding
This work was supported by the National Institutes of Health (R01AR059685 to S.-J.L., R01AR060636 to S.-J.L., P01NS0720027 to E.M.M.) and the Jain Foundation (to S.-J.L.). S.-J.L. was supported by Lawrence Ellison Foundation Senior Scholar in Aging Award AG-SS-2678-11 and by generous gifts from Michael and Ann Hankin, Partners of Brown Advisory, and James and Julieta Higgins. Funding to pay the Open Access publication charges for this article was provided by Lawrence Ellison Foundation Senior Scholar in Aging Award AG-SS-2678-11.
Supplementary Material
Acknowledgements
We thank Mohamed Lehar, Michael Delannoy and Barbara Smith for assistance with electron microscopy, Nadine Forbes-McBean for serum CK measurement, and Douglas E. Albrecht at the Jain Foundation for helpful advice.
Conflict of Interest statement. None declared.
References
- 1.McPherron A.C., Lawler A.M., Lee S.J. (1997) Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature, 387, 83–90. [DOI] [PubMed] [Google Scholar]
- 2.McPherron A.C., Lee S.J. (1997) Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA, 94, 12457–12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kambadur R., Sharma M., Smith T.P., Bass J.J. (1997) Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res., 7, 910–916. [DOI] [PubMed] [Google Scholar]
- 4.Schuelke M., Wagner K.R., Stolz L.E., Hubner C., Riebel T., Komen W., Braun T., Tobin J.F., Lee S.J. (2004) Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med., 350, 2682–2688. [DOI] [PubMed] [Google Scholar]
- 5.Clop A., Marcq F., Takeda H., Pirottin D., Tordoir X., Bibe B., Bouix J., Caiment F., Elsen J.M., Eychenne F. et al. (2006) A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet., 38, 813–818. [DOI] [PubMed] [Google Scholar]
- 6.Mosher D.S., Quignon P., Bustamante C.D., Sutter N.B., Mellersh C.S., Parker H.G., Ostrander E.A. (2007) A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet., 3, e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuentes E.N., Pino K., Navarro C., Delgado I., Valdes J.A., Molina A. (2013) Transient inactivation of myostatin induces muscle hypertrophy and overcompensatory growth in zebrafish via inactivation of the SMAD signaling pathway. J. Biotechnol., 168, 295–302. [DOI] [PubMed] [Google Scholar]
- 8.Lee S.J., Hill J.A., Olson E.N. (eds). (2012) MUSCLE Fundamental Biology and Mechanisms of Disease. Elsevier, London, UK, Vol. 2, pp. 1077–1084. [Google Scholar]
- 9.Rahimov F., Kunkel L.M. (2013) The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J. Cell Biol., 201, 499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohn R.D., Campbell K.P. (2000) Molecular basis of muscular dystrophies. Muscle Nerve, 23, 1456–1471. [DOI] [PubMed] [Google Scholar]
- 11.Wagner K.R., McPherron A.C., Winik N., Lee S.J. (2002) Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol., 52, 832–836. [DOI] [PubMed] [Google Scholar]
- 12.Bogdanovich S., Krag T.O., Barton E.R., Morris L.D., Whittemore L.A., Ahima R.S., Khurana T.S. (2002) Functional improvement of dystrophic muscle by myostatin blockade. Nature, 420, 418–421. [DOI] [PubMed] [Google Scholar]
- 13.Bogdanovich S., Perkins K.J., Krag T.O., Whittemore L.A., Khurana T.S. (2005) Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J., 19, 543–549. [DOI] [PubMed] [Google Scholar]
- 14.Nakatani M., Takehara Y., Sugino H., Matsumoto M., Hashimoto O., Hasegawa Y., Murakami T., Uezumi A., Takeda S., Noji S. et al. (2008) Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J., 22, 477–487. [DOI] [PubMed] [Google Scholar]
- 15.Qiao C., Li J., Jiang J., Zhu X., Wang B., Xiao X. (2008) Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum. Gene. Ther., 19, 241–254. [DOI] [PubMed] [Google Scholar]
- 16.Murphy K.T., Ryall J.G., Snell S.M., Nair L., Koopman R., Krasney P.A., Ibebunjo C., Holden K.S., Loria P.M., Salatto C.T. et al. (2010) Antibody-directed myostatin inhibition improves diaphragm pathology in young but not adult dystrophic mdx mice. Am. J. Pathol., 176, 2425–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morine K.J., Bish L.T., Selsby J.T., Gazzara J.A., Pendrak K., Sleeper M.M., Barton E.R., Lee S.J., Sweeney H.L. (2010) Activin IIB receptor blockade attenuates dystrophic pathology in a mouse model of Duchenne muscular dystrophy. Muscle Nerve, 42, 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.George Carlson C., Bruemmer K., Sesti J., Stefanski C., Curtis H., Ucran J., Lachey J., Seehra J.S. (2011) Soluble activin receptor type IIB increases forward pulling tension in the mdx mouse. Muscle Nerve, 43, 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bansal D., Miyake K., Vogel S.S., Groh S., Chen C.C., Williamson R., McNeil P.L., Campbell K.P. (2003) Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature, 423, 168–172. [DOI] [PubMed] [Google Scholar]
- 20.Lee S.J., McPherron A.C. (2001) Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA, 98, 9306–9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner K.R., Liu X., Chang X., Allen R.E. (2005) Muscle regeneration in the prolonged absence of myostatin. Proc. Natl. Acad. Sci. USA, 102, 2519–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Millay D.P., Maillet M., Roche J.A., Sargent M.A., McNally E.M., Bloch R.J., Molkentin J.D. (2009) Genetic manipulation of dysferlin expression in skeletal muscle: novel insights into muscular dystrophy. Am. J. Pathol., 175, 1817–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McArdle A., Edwards R.H., Jackson M.J. (1994) Time course of changes in plasma membrane permeability in the dystrophin-deficient mdx mouse. Muscle Nerve, 17, 1378–1384. [DOI] [PubMed] [Google Scholar]
- 24.Lee S.J., Reed L.A., Davies M.V., Girgenrath S., Goad M.E., Tomkinson K.N., Wright J.F., Barker C., Ehrmantraut G., Holmstrom J. et al. (2005) Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc. Natl. Acad. Sci. USA, 102, 18117–18122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krivickas L.S., Walsh R., Amato A.A. (2009) Single muscle fiber contractile properties in adults with muscular dystrophy treated with MYO-029. Muscle Nerve, 39, 3–9. [DOI] [PubMed] [Google Scholar]
- 26.Attie K.M., Borgstein N.G., Yang Y., Condon C.H., Wilson D.M., Pearsall A.E., Kumar R., Willins D.A., Seehra J.S., Sherman M.L. (2013) A single ascending-dose study of muscle regulator ACE-031 in healthy volunteers. Muscle Nerve, 47, 416–423. [DOI] [PubMed] [Google Scholar]
- 27.Amato A.A., Sivakumar K., Goyal N., David W.S., Salajegheh M., Praestgaard J., Lach-Trifilieff E., Trendelenburg A.U., Laurent D., Glass D.J. et al. (2014) Treatment of sporadic inclusion body myositis with bimagrumab. Neurology, 83, 2239–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bansal D., Campbell K.P. (2004) Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol., 14, 206–213. [DOI] [PubMed] [Google Scholar]
- 29.Han R., Campbell K.P. (2007) Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol., 19, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barton E.R., Wang B.J., Brisson B.K., Sweeney H.L. (2010) Diaphragm displays early and progressive functional deficits in dysferlin-deficient mice. Muscle Nerve, 42, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao J., Jones-Brando L., Talbot C.C. Jr., Yolken R.H. (2011) Differential effects of three canonical toxoplasma strains on gene expression in human neuroepithelial cells. Infect. Immun., 79, 1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.