

Abstract
Many genes involved in brain development have been associated with human neurodevelopmental disorders, but underlying pathophysiological mechanisms remain undefined. Human genetic and mouse behavioral analyses suggest that ENGRAILED-2 (EN2) contributes to neurodevelopmental disorders, especially autism spectrum disorder. In mouse, En2 exhibits dynamic spatiotemporal expression in embryonic mid-hindbrain regions where monoamine neurons emerge. Considering their importance in neuropsychiatric disorders, we characterized monoamine systems in relation to forebrain neurogenesis in En2-knockout (En2-KO) mice. Transmitter levels of serotonin, dopamine and norepinephrine (NE) were dysregulated from Postnatal day 7 (P7) to P21 in En2-KO, though NE exhibited the greatest abnormalities. While NE levels were reduced ∼35% in forebrain, they were increased 40–75% in hindbrain and cerebellum, and these patterns paralleled changes in locus coeruleus (LC) fiber innervation, respectively. Although En2 promoter was active in Embryonic day 14.5–15.5 LC neurons, expression diminished thereafter and gene deletion did not alter brainstem NE neuron numbers. Significantly, in parallel with reduced NE levels, En2-KO forebrain regions exhibited reduced growth, particularly hippocampus, where P21 dentate gyrus granule neurons were decreased 16%, suggesting abnormal neurogenesis. Indeed, hippocampal neurogenic regions showed increased cell death (+77%) and unexpectedly, increased proliferation. Excess proliferation was restricted to early Sox2/Tbr2 progenitors whereas increased apoptosis occurred in differentiating (Dcx) neuroblasts, accompanied by reduced newborn neuron survival. Abnormal neurogenesis may reflect NE deficits because intra-hippocampal injections of β-adrenergic agonists reversed cell death. These studies suggest that disruption of hindbrain patterning genes can alter monoamine system development and thereby produce forebrain defects that are relevant to human neurodevelopmental disorders.
Introduction
Regulation of brain development requires finely tuned interactions among hundreds of genes, while its dysregulation can lead to pervasive neurodevelopmental disorders (1,2). Numerous genetic risk factors have now been identified that contribute to vulnerability to multiple disorders including autism spectrum disorder (ASD) and schizophrenia (3), but the underlying pathophysiology is still poorly understood. Many of these disease-associated genes are expressed during brain development and contribute to a variety of cellular functions, from brain region patterning, to cell proliferation and migration, to synapse formation and efficacy (4). Studying these genes in preclinical models provides the opportunity to identify affected neurobiological pathways that may be relevant to human neurodevelopmental disorders.
Genetic variants of ENGRAILED-2 (EN2) were found to be genetically associated with autistic disorder and ASD in 167 nuclear families, and this association was independently confirmed in 222 families as well as another 129 families collected by different investigators (5,6). This association was also replicated by six other groups in different ethnicities (7–10), whereas association with schizophrenia remains uncertain (11). The disease-associated alleles for ASD introduce new transcription factor binding sites that elicit increased gene expression levels (12,13). EN2 mRNA levels are increased in the cerebellum of postmortem autism brain samples, evidence reported by us (12,13) and others (14,15). With regard to clinical symptoms of various neurodevelopmental disorders, En2-KO mice exhibit behavioral abnormalities in social interaction, depression-related tasks, contextual-fear conditioning, spatial memory, seizure threshold and sensory-motor gating (16–18), suggesting that mediating neurobiological mechanisms may be defined using this model. Perhaps surprising is the fact that major En2 gene expression is in the hindbrain (19), whereas many of these behaviors depend heavily on forebrain circuits. How then might this hindbrain gene impact forebrain functions?
Both human and mouse have two ENGRAILED homologs, EN1/En1 and EN2/En2 (20,21). In E8.5–E12.5 mouse embryos, En2 is expressed broadly in the midbrain–hindbrain junction region of the brainstem, where it serves as a patterning gene to determine antero-posterior cell fate identity (22). Significantly, this early En2 expression domain (19,23) includes the precursors for neurons whose axons project to the forebrain to release monoamine neurotransmitters, including dopamine (DA), serotonin (5HT) and norepinephrine (NE) (24). These long projecting pathways have well established roles in the development of target forebrain populations, regulating cell proliferation, survival, differentiation and neural circuits (25–31). While both En1 and En2 have been shown to be expressed by DA and 5-HT neurons during development, to our knowledge, only En1 has been shown in NE neurons of the locus coeruleus (LC) (32,33), with En2 expression remaining undefined. Genetic studies indicate that both Engrailed genes are required for normal development of raphe neurons (32,34), and they both contribute to mature DA neuron survival in normal (35,36) and Parkinson's disease mouse models (37). While En1 has major effects on monoamine neuron development, En2 effects were detected only in the En1/En2 double KO (23,33,38). With development, En2 expression localizes dorsally to the colliculi and cerebellum where it regulates ingrowing afferents, cell proliferation and foliation (39–44). Recently, in adults, very low levels of En2 mRNA have been detected in specific forebrain regions including the hippocampus and somato-sensory cortex (17,18), though specific cellular localization seems uncertain (45).
Interestingly, the behaviors altered in En2-KO mice (16,18), particularly depression-related tasks and memory deficits, are typical of those affected by disrupting monoamine neurotransmitter systems through pharmacological or genetic approaches (29,46,47). Our previous studies found that these adult behavioral deficits could be reversed by treatment with NE re-uptake inhibitor, desipramine (48), which raises the question of whether monoamine systems are abnormal in this mutant. Monoamine systems are of major importance in neuropsychiatric disorders since they are targeted by the principle families of drugs used to treat these patients (49). In animal models, treatment with monoamine-related drugs, such as those that increase NE and 5HT signaling, can rescue abnormal behaviors as well as stimulate adult hippocampal neurogenesis (50,51), though causal relationships between neurogenesis and behavior remain controversial (51–54). However, from the perspective of development, the perinatal and juvenile periods require special attention, since during the first three postnatal weeks, axonal fibers from hindbrain monoamine neurons grow into the forebrain, establishing target innervation and adult neurotransmitter levels (55,56). On the other hand, interfering with neurotransmitter system development can cause deficits similar to neurodevelopmental disorders (57,58). Furthermore, monoamine abnormalities have been found in neurodevelopmental disorders: for example, in autism, blood 5-HT levels are increased (59,60), 5-HT synthesis is increased in cerebellar dentate nucleus, (61) and 5-HT system development is dysregulated (62,63), whereas NE levels exhibit exaggerated responses (59,64). Monoamines are also dysregulated in patients with schizophrenia, where there is evidence of decreased DA innervation of the neocortex, alterations in DA metabolism and changes in D1 receptor distribution (65–67).
If monoamine systems are affected in ASD and other neurodevelopmental disorders, it remains unclear which neurobiological pathways underlie these deficits. Brain growth and neurogenesis are two biological pathways that change in parallel with monoamine system activation and, interestingly, are dysregulated in autism and other neurodevelopmental disorders (26,30,63,68–72). Brain imaging studies in autism, schizophrenia and depression have revealed changes in brain growth and volume (63,73,74), some of which correlate with presumptive alterations in monoamine system functions (75,76), but underlying mechanisms remain undefined.
Given EN2's genetic association with neurodevelopmental disorders and the behavioral phenotypes in the En2-KO, it is therefore logical to consider monoamine neurons as one potential system targeted by gene dysfunction to contribute to disease phenotypes. To elucidate the function of En2 in monoamine system development, we explored in En2-KO mice the possibility of changes in development of the forebrain, a major site to which brainstem monoamine neurons project during the latter half of gestation and early postnatal life (77). We now report that En2 deletion reduces monoamine innervation of target forebrain structures, affecting forebrain structural growth, postnatal neurogenesis and related behavior. Conversely, increasing NE signaling by direct agonist injection into the hippocampus partially reverses the abnormal neurogenesis phenotype. These results provide insight into how changes in a hindbrain patterning gene can have far ranging phenotypic effects through monoamine systems and may be relevant to neuropsychiatric disorders.
Results
Neurotransmitter levels, especially NE, are reduced in the forebrain but elevated in the hindbrain in Postnatal day 21 En2-KO mice
En2 could contribute to monoamine system development since its embryonic expression domain includes monoamine neuronal progenitors (23,32,33). To determine whether En2 deletion has effects on forebrain monoamines, we performed initial studies at Postnatal day 21 (P21, adolescence), an age when major developmental events are nearing completion and specific regions can be accurately isolated to obtain abundant material. To investigate monoamine neurotransmitter systems, we quantified the absolute levels of NE, 5-HT and DA and metabolites of 5-HT (5-hydroxyindole acetic acid, 5-HIAA) and DA (3,4-Dihydroxyphenylacetic acid, DOPAC) in homogenates of frontal cortex, striatum, amygdala, hippocampus, midbrain, pons/medulla and cerebellum by using high-performance liquid chromatography (HPLC). At P21, the most prominent change was in NE, which was increased by ∼40% in the En2-KO midbrain and pons/medulla and by 75% in cerebellum (Fig. 1A and B). In contrast, in the forebrain, NE was reduced by ∼35% in hippocampus and frontal cortex and by 13% in amygdala, with no change in striatum compared with wild-type (WT) mice (Fig. 1A and B). Smaller changes in other monoamines were also detected (see below). These data in the juvenile En2-KO reveal pronounced phenotypes of reduced monoamines. These deficits were more prominent than those found in previous studies of the adult En2-KO which reported changes only in cerebellum, specifically 5-HT (16). Thus we considered the possibility that alterations were developmental stage specific.
Figure 1.

Levels of monoamine neurotransmitters are altered in multiple brain regions during development of En2-KO mice. (A) Illustration of percent change in NE levels of P21 En2-KO mice compared with WT. Red represents 100% elevation and blue represents 100% reduction with yellow indicating no change. (B) Quantification of absolute NE levels in dissected brain regions at P21, as determined by HPLC and expressed as µg/g tissue. (C) NE levels in En2-KO hippocampus are reduced at multiple ages, from P7 to P60. (D) NE levels in En2-KO cerebellum are increased at P7, P14 and P21. (E) Absolute levels of 5-HT (lower) and 5-HIAA (upper) are increased in En2-KO cerebellum at multiple ages before P60. Quantification of levels of (F) GABA and (G) glutamate reveals no differences at P21 in multiple brain regions, as determined by HPLC and expressed as µmol/l. Amyg: amygdala, Cereb: cerebellum, Hippo: hippocampus, Midbr: midbrain, Pons/Med: pons and medulla. (10–15 animals/genotype, from 2 to 5 litters). Student's t-test: *P < 0.05, **P < 0.01, ***P < 0.001.
Changes in NE occur primarily in early development and partially recover with time
To define the time course of altered transmitter development, we examined monoamine levels from birth (P0) to adulthood (P60) (Table 1). Already at birth, when consistent regional dissections could not be performed, NE was reduced by 46% in whole forebrain of En2-KO (N = 10/genotype, 2–3 litters; P = 0.002), whereas the transmitter was increased by 18% in the entire hindbrain (P = 0.05). From P7 to P60, when region-specific dissections were possible, we focused on the hippocampus and the cerebellum, the two regions that showed the largest changes at P21 (Fig. 1A and B). In hippocampus, the reduction in NE was already observed at P7, and the deficits were even greater at P14 (41%, P < 0.001) and P21 (33%, P < 0.001) (Fig. 1C). However, by P60, the deficit in NE level had partially recovered (23%, P = 0.047). In contrast, cerebellar NE levels were increased from P7 through P21, but no difference was detected in the P60 adult (Fig. 1D). Thus both hippocampus and cerebellum displayed abnormal and opposing patterns of monoamine levels that partially normalized with developmental age.
Table 1.
Postnatal developmental changes of absolute levels of NE in brain regions of En2-KO and WT mice
| µg/g tissue |
Pons/Med | Cereb | Mid Br | Amyg | Hippo | Striatum | Fr. Cortex | |
|---|---|---|---|---|---|---|---|---|
| P14 | WT | 1.03 ± 0.05 | 0.42 ± 0.03 | 0.69 ± 0.03 | 0.41 ± 0.02 | 0.51 ± 0.02 | 0.26 ± 0.02 | 0.27 ± 0.03 |
| KO | 1.29 ± 0.04 | 0.56 ± 0.02 | 0.86 ± 0.04 | 0.33 ± 0.02 | 0.30 ± 0.02 | 0.20 ± 0.02 | 0.18 ± 0.02 | |
| Percent change | 25% | 34% | 24% | −19% | −41% | −25% | −33% | |
| P-value | 0.0003 | 0.0004 | 0.002 | 0.004 | 2.80E−07 | 0.050 | 0.011 | |
| P21 | WT | 1.20 ± 0.03 | 0.48 ± 0.03 | 0.86 ± 0.02 | 0.57 ± 0.01 | 0.62 ± 0.02 | 0.22 ± 0.03 | 0.36 ± 0.02 |
| KO | 1.68 ± 0.04 | 0.84 ± 0.05 | 1.23 ± 0.05 | 0.49 ± 0.02 | 0.42 ± 0.02 | 0.18 ± 0.05 | 0.23 ± 0.03 | |
| Percent change | 40% | 75% | 43% | −13% | −33% | −35% | ||
| P-value | 4.90E−10 | 3.95E−07 | 1.70E−08 | 0.008 | 1.27E−07 | 0.451 | 0.002 | |
| P60 | WT | 1.45 ± 0.09 | 0.80 ± 0.05 | 1.35 ± 0.10 | 0.85 ± 0.03 | 1.04 ± 0.08 | 0.70 ± 0.05 | |
| KO | 1.69 ± 0.12 | 0.85 ± 0.08 | 1.38 ± 0.08 | 0.71 ± 0.02 | 0.80 ± 0.08 | 0.60 ± 0.07 | ||
| Percent change | −16% | −23% | ||||||
| P-value | 0.122 | 0.528 | 0.787 | 0.005 | 0.047 | 0.235 | ||
NE levels, expressed as µg/g tissue, were assessed using HPLC on brain regions at P14, 21 and 60. Amyg, amygdala; Cereb, cerebellum; Fr. Cortex, frontal cortex; Hippo, hippocampus; Midbr, midbrain; Pons/Med, pons and medulla. (10–15 animals/genotype/age, from 2 to 5 litters). Student's t-test significant changes are presented in bold.
Interestingly, developmental patterns that were observed in the hippocampus and cerebellum were also detected in neighboring structures. Thus deficits in forebrain NE were also observed at select ages in amygdala, striatum and frontal cortex, whereas NE increases in hindbrain were also detected in midbrain and pons/medulla (Table 1). However, the changes in NE levels in the En2-KO mainly recovered by adulthood (P60), leaving only reductions of 16% in the amygdala and 23% in the hippocampus. Overall, En2 deletion caused major changes during the first 3 weeks of postnatal development, increasing NE levels in mid/hindbrain regions and reducing levels in the forebrain, with residual differences remaining in the amygdala and hippocampus of the adult.
Changes in 5-HT and DA levels are similar to NE but more limited
In the En2-KO, changes in the levels of 5-HT (Table 2) and DA (Table 3) in the hindbrain and forebrain were in the same direction as NE, though they were regionally more limited and of smaller magnitudes. Over the course of postnatal development, only the cerebellum exhibited persistent differences, with increases in 5-HT as well as its metabolite, 5-HIAA (Fig. 1E). For example, in P21 cerebellum, 5-HT was increased by ∼50% and 5-HIAA by 42%. However, differences were no longer detected at P60. Significant changes were also observed in forebrain regions. At birth (P0), 5-HT levels were increased by ∼13% in both forebrain and mid/hindbrain regions (N = 10/genotype, 2–3 litters; P < 0.01). At P21, 5-HT was reduced by ∼20% in the striatum and frontal cortex (Table 2). These data suggest that there was early dysregulation of 5-HT transmitter system development.
Table 2.
Absolute amount of 5-HT during postnatal development in brain regions of En2-KO and WT mice
| µg/g tissue |
Pons/Med | Cereb | Mid Br | Amyg | Hippo | Striatum | Fr. Cortex | |
|---|---|---|---|---|---|---|---|---|
| P14 | WT | 0.85 ± 0.05 | 0.05 ± 0.01 | 1.01 ± 0.08 | 0.62 ± 0.04 | 0.38 ± 0.03 | 0.21 ± 0.02 | 0.17 ± 0.02 |
| KO | 0.87 ± 0.07 | 0.07 ± 0.01 | 1.04 ± 0.09 | 0.66 ± 0.02 | 0.37 ± 0.01 | 0.21 ± 0.01 | 0.14 ± 0.01 | |
| Percent change | 42% | |||||||
| P-value | 0.880 | 0.030 | 0.784 | 0.422 | 0.661 | 0.769 | 0.177 | |
| P21 | WT | 1.04 ± 0.04 | 0.11 ± 0.01 | 1.34 ± 0.06 | 0.90 ± 0.02 | 0.52 ± 0.02 | 0.45 ± 0.01 | 0.32 ± 0.02 |
| KO | 1.07 ± 0.06 | 0.16 ± 0.02 | 1.43 ± 0.11 | 0.96 ± 0.04 | 0.51 ± 0.01 | 0.37 ± 0.02 | 0.25 ± 0.03 | |
| Percent change | 51% | −18% | −21% | |||||
| P-value | 0.653 | 0.035 | 0.444 | 0.234 | 0.740 | 0.006 | 0.043 | |
| P60 | WT | 1.26 ± 0.04 | 0.20 ± 0.02 | 0.20 ± 0.02 | 1.63 ± 0.06 | 1.33 ± 0.05 | 1.00 ± 0.07 | |
| KO | 1.17 ± 0.04 | 0.18 ± 0.02 | 0.18 ± 0.02 | 1.55 ± 0.07 | 1.23 ± 0.06 | 0.98 ± 0.05 | ||
| Percent change | ||||||||
| P-value | 0.150 | 0.632 | 0.829 | 0.384 | 0.229 | 0.883 | ||
5-HT levels, expressed as µg/g tissue, were assessed using HPLC on brain regions at P14, 21 and 60. Amyg, amygdala; Cereb, cerebellum; Fr. Cortex, frontal cortex; Hippo, hippocampus; Mid Br, midbrain; Pons/Med, pons and medulla. (10–15 animals/genotype/age, from 2 to 5 litters) Student's t-test significant changes are presented in bold.
Table 3.
Limited changes of DA absolute levels were observed in the midbrain and amygdala of En2-KO compared with WT mice
| µg/g tissue |
Mid Br | Amyg | Striatum | Fr. Cortex | |
|---|---|---|---|---|---|
| P14 | WT | 0.20 ± 0.01 | 0.16 ± 0.01 | 3.46 ± 0.43 | |
| KO | 0.26 ± 0.01 | 0.15 ± 0.01 | 3.57 ± 0.57 | ||
| Percent change | 30% | ||||
| P-value | 1.5E−05 | 0.583 | 0.878 | ||
| P21 | WT | 0.21 ± 0.01 | 0.32 ± 0.03 | 6.03 ± 0.11 | 0.12 ± 0.04 |
| KO | 0.27 ± 0.01 | 0.4 ± 0.08 | 6.33 ± 0.16 | 0.14 ± 0.04 | |
| Percent change | 29% | ||||
| P-value | 1.8E−06 | 0.354 | 0.140 | 0.762 | |
| P60 | WT | 0.25 ± 0.03 | 0.83 ± 0.16 | 0.33 ± 0.09 | |
| KO | 0.28 ± 0.03 | 0.44 ± 0.03 | 0.45 ± 0.13 | ||
| Percent change | −47% | ||||
| P-value | 0.489 | 0.050 | 0.422 | ||
DA levels, expressed as µg/g tissue, were assessed using HPLC on brain regions at P14, 21 and 60. Amyg, amygdala; Fr. Cortex, frontal cortex; Mid Br, midbrain; Pons/Med, pons and medulla. (10–15 animals/genotype/age, from 2 to 5 litters) Student's t-test significant changes are presented in bold.
More restricted changes were observed in DA levels (Table 3), which were unaccompanied by changes in metabolite, DOPAC. DA levels were only increased in the early postnatal midbrain, by 30% at P14 and by 29% at P21 (N = 10–15/genotype, 2–5 litters; P < 0.001). In contrast, a deficit in DA was observed only in the P60 adult, in which the transmitter was decreased only on the amygdala by 48% (N = 10/genotype, 2–3 litters; P < 0.05). DA was easily detected in striatum and frontal cortex but exhibited no genotype differences, whereas the transmitter was undetectable in hippocampus.
Locally synthesized neurotransmitters GABA and glutamate are not affected
While the above neurotransmitter abnormalities may reflect changes in hindbrain monoamine neurons and their projecting axons, it is also possible they simply reflect broad developmental changes occurring within each region. To examine this issue, we measured levels of gamma-aminobutyric acid (GABA), a transmitter produced by local inhibitory neurons, and glutamate, reflecting both local neurons and glia. Using the same regional homogenates that were assessed for monoamines, we detected no differences in levels of either GABA or glutamate at P21 (Fig. 1F and G), consistent with the possibility that abnormalities reflect changes in hindbrain monoamine projecting neurons contained within the En2 expression domain.
The adult male En2-KO exhibits sex-dependent deficits in the forced swim task
The above data indicate that En2-KO mice exhibit developmental deficits in forebrain monoamine systems, especially NE. Abnormalities in NE have been associated with depression-related phenotypes (78). Significantly, we previously found that adult En2-KO mice exhibited depression-related behaviors (18) that were reversed by chronic treatment with NE reuptake inhibitor desipramine (48), suggesting NE deficits may contribute to these abnormalities. Since our past studies were performed by our collaborators at national institute of mental health (18,48), it was important to show that mice bred in our laboratory recapitulated this phenotype that was reversed by modulating NE. Replicating the same behavioral abnormality on a depression-related task at two sites would contribute to the strength of the noradrenergic hypothesis.
We performed behavioral studies as before, on adult (P60) animals, when most developmental changes in monoamines had resolved (Tables 1–3), though minor reductions remained for NE in hippocampus and amygdala. Interestingly, since monoamine systems exhibit sex-dependent development (79,80) and neuropsychiatric diseases exhibit sex-dependent patterns, we reanalyzed the HPLC data according to sex. NE, 5-HT and 5-HIAA were all reduced by 17–27% in the frontal cortex of the adult male En2-KO mice, whereas no significant differences were observed in females (Table 4). (No sex-dependent differences in monoamines were detected in any brain region at P21; data not shown). We conducted two standardized preclinical behavioral assays used to evaluate depression-related phenotypes, the Porsolt forced swim and tail suspension tests. To control for possible developmental variability, all testing was performed on adult littermates derived from En2 heterozygote (Het) crosses (Het × Het). Two separate cohorts of mice were tested. En2-KO males exhibited 2-fold higher immobility in the forced swim test compared with WT males (t(1,20) = 2.86 P = 0.009), a behavioral change that coincided with the sex-dependent deficits in forebrain monoamines (Table 4). In contrast, En2-KO females were not significantly different from WT females (t(1,23) = 0.02 P = 0.98). On the other hand, there were no genotype differences on the tail suspension test (t(1,35) = 1.15 P = 0.26). A second cohort of mice yielded similar results with significantly higher immobility times in males (t(1,34) = 2.44 P = 0.02). Since behavioral studies required dedicated cohorts, a separate group of WT and En2-KO were bred through Het matings to repeat assessment of neurotransmitter levels. Neurochemical analyses by HPLC demonstrated replication of the neurotransmitters changes reported in Figure 1 and Tables 1 and 2. Thus the abnormalities in En2-KO monoamine systems observed during development are associated with strong behavioral phenotypes in the forced swim test.
Table 4.
Changes in monoamine neurotransmitters in frontal cortex of P60 male and female En2-KO and WT mice
| Male |
Female |
|||||
|---|---|---|---|---|---|---|
| NE | 5-HT | 5-HIAA | NE | 5-HT | 5-HIAA | |
| Percent change | −26.5 | −17.4 | −19.8 | −9.5 | 18.7 | −10.0 |
| P-value | 0.03 | 0.03 | 0.04 | 0.28 | 0.13 | 0.18 |
(Four to five animals/sex/genotype, from 2 to 3 litters). Student's t-test significant changes are presented in bold.
Markers of NE fiber innervation in the En2-KO are diminished in hippocampus and increased in cerebellum
Given the developmental changes in tissue neurotransmitters, we wondered whether alterations in monoamine fiber innervation may be involved in the En2-KO phenotype. While En2 deletion has no effects on the numbers of NE and 5-HT neurons at birth (32,33,35), there is little information on possible effects in forebrain target fields. The major source of NE in the brain derives from fibers originating in the LC located in the dorsal pons (81,82). The rate-limiting enzyme for transmitter biosynthesis is tyrosine hydroxylase (TH), which acts locally in terminals to generate NE. One possible mechanism for reduced forebrain NE would be a deficit in TH, which we assessed by measuring total protein levels using western blotting at P21. TH levels were decreased by 50% in the hippocampus whereas the enzyme was increased by 40% in cerebellum (Fig. 2A and B). These region-specific changes in TH parallel those for NE levels (Fig. 1B) and suggest that alterations in neurotransmitter biosynthesis contribute to the observed abnormalities. Since TH protein levels often correlate with tissue innervation, we characterized TH immunoreactive fibers. In WT hippocampus, numerous branching fibers bearing varicosities were observed in the dentate gyrus molecular layer at three different ages, P7, P21 and adult P60. Fiber density increased from P7 to P14 and then remained stable into adulthood (Fig. 2C and E). In contrast, the En2-KO exhibited a fiber deficit at all ages. While there was a 43% reduction at P7, as WT fiber innervation increased with age, the KO deficit grew to 73% at P21. (Fig. 2C and E). This deficit in the KO persisted but partially recovered by adulthood, as the P60 hippocampus exhibited a 60% reduction in fibers (Fig. 2E; P60 was significantly different from P21, P = 0.006). On the other hand, in the cerebellum, TH immunoreactive fibers were increased by 97% in the P21 En2-KO (P = 0.002), an increase that parallels total protein levels (Fig. 2A) and the changes in NE (Fig. 1A and D). These parallel changes in biochemical and morphological markers of NE innervation support a model of genotype specific abnormalities in establishing the transmitter system during development, that partially recover over time.
Figure 2.

Markers of NE innervation in the En2-KO are altered in parallel with monoamine neurotransmitters. (A) Representative immunoblots of protein levels of TH, 5-HTT and TrOH in hippocampus and cerebellum of En2-KO and WT at P21. (B) Relative density of TH protein levels in hippocampus and cerebellum of WT and En2-KO mice, normalized to actin and expressed as percent WT signal (N = 4–9/genotype, from 2 to 3 litters). (C) Immunofluorescent labeling of TH (left) and NE transporter (NET, right) proteins reveals axonal fibers in P21 WT and En2-KO hippocampal dentate gyrus molecular layer. Images were obtained from molecular layer (inset; red box) above dorsal blade and below ventral blade of granule cell layer. Branching TH fibers bearing varicosities that were observed in the WT were diminished in En2-KO. Similar deficits were observed for NET fibers in the En2-KO. Bar = 50 µm (D) Immunofluorescent labeling of galanin (Gal) reveals axonal fibers in P21 WT and En2-KO hippocampal dentate gyrus molecular layer (Left). Bar = 50 µm. At low magnification (×20; Right), total galanin immunofluorescent signal was localized above the granule cell layer somas (DAPI). OML, outer molecular layer; IML, inner molecular layer; GCL, granule cell layer. Bar = 50 µm. (E) Quantification of nerve fibers expressing TH, NET and galanin immunofluorescence in dentate gyrus molecular layer at P7, P21 and P60. Number of fibers crossing a pre-set grid matrix was assessed on two images/section, four sections per hippocampus, three animals/genotype. Cereb, cerebellum; Hippo, hippocampus. Student's t-test: *P < 0.05, **P < 0.01, ***P < 0.001.
To further explore the range of NE system alterations specifically in forebrain regions beyond En2's expression domain, we examined another marker of LC fibers, the norepinephrine transporter (NET) which takes NE back into axon terminals following release (83). At P21, NET-positive fibers were reduced by 65% in the molecular layer of the En2-KO dentate gyrus (Fig. 2C and E), a deficit similar to that of TH. Furthermore, ∼70–80% of LC neurons co-express the 29 amino acid neuropeptide, galanin, with NE and/or TH (84). Unlike NE, which is synthesized locally in axon synaptic terminals by TH, galanin is produced in the neuronal soma and transported axonally to target regions. Galanin innervation was also reduced in the En2-KO dentate gyrus (Fig. 2D and E), when quantified as immunoreactive fibers at high magnification (reduced by 41%; Fig. 2E; P = 0.0003) or as total molecular layer fluorescence at lower magnification (Fig. 2D; reduced by 25%; WT = 2031 ± 36, KO = 1527 ± 166; N = 3/genotype; P = 0.007). The parallel changes in these three markers of LC neurons suggest that axonal fibers are reduced in the dentate gyrus of En2-KO mice at P21.
Since consistent though smaller changes were also observed for 5-HT in the En2-KO, we also examined levels of its rate-limiting biosynthetic enzyme, tryptophan hydroxylase (TrOH), using western blotting. However, we observed no changes in TrOH protein levels at P21 in either the hippocampus or cerebellum (Fig. 2A), when significant changes in 5-HT transmitter levels were observed (Table 2). Similarly, we observed no changes in the 5-HT re-uptake transporter (5-HTT) protein (Fig. 2A), suggesting that En2 deletion may alter 5-HT transmitter levels and metabolism independently of serotonergic innervation.
En2 expression in the LC during embryonic development
Since we detected changes in NE transmitter levels and fiber innervation in the En2-KO forebrain, we wondered whether En2 was expressed in the LC during development because localization has not been specifically reported (32). Due to the absence of specific antibodies to detect En2 protein, we took advantage of the En2-tauLacZ reporter gene transgenic mouse created by Joyner and colleagues, which replaces the En2 coding sequence with that of the LacZ enzyme, allowing easy detection of En2 promoter activity (85). Levels of LacZ protein are thought to reflect En2 gene expression. To define En2 expression in the LC, we performed double immunostaining for LacZ and TH. In the E14.5 embryo (Fig. 3A), LacZ immunoreactivity was detected in a pattern similar to that previously described for En2 detected by in situ hybridization (19,23): in the mid-hindbrain isthmus, extending as a gradient both anteriorly and posteriorly. TH immunostaining of the LC was localized to the dorsal pons, just lateral to the mesencephalic fifth cranial nucelus (Me5; Fig. 3A). LacZ signal was more broadly expressed in the dorsal pons, and appeared to colocalize with TH in the majority of cells, as shown at higher magnification (Fig. 3B). TH signal was also visible in the fourth ventricle choroid plexus, which is known to be highly innervated by fibers of peripheral sympathetic neurons (86). One day later at E15.5, double-labeled LacZ+-LC neurons were still detected. However, by age E17.5, LacZ expression (Fig. 3C and D) was no longer detectable in the region of the LC. These observations suggest that En2 is at least expressed by LC neurons at E14.5 and E15.5, with expression ceasing by E17.5. This pattern of expression is similar to that described for a neighboring monoamine nucleus, the dorsal raphe (32,34). The expression of En2 in the embryonic LC is consistent with it playing roles in establishing cell identity as well as regulating early fiber outgrowth and targeting.
Figure 3.
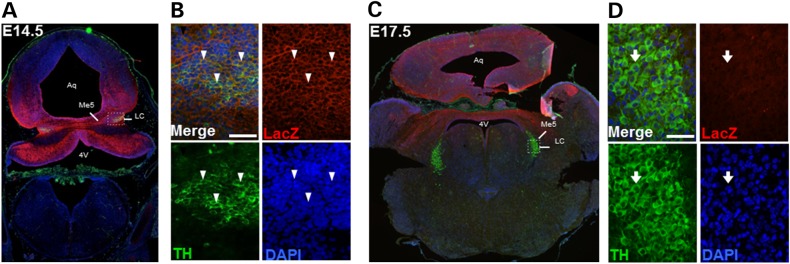
Expression of the En2-tauLacZ reporter gene in LC progenitors during embryogenesis. Images show double immunolabeling for LacZ (red) and TH (green) with DAPI counterstaining (blue) at E14.5 (A) and E17.5 (C). (A) En2 expression appears in the dorsal pons and cerebellum, including in TH+ neurons in the LC at E14.5. (B) High magnification of LC neurons shows colocalization of TH and LacZ signals (arrowheads). Bar = 50 µm. (C) At E17.5, LacZ staining is visible in cerebellum and midbrain, but does not colocalize with the LC, (D) High magnification shows LC TH+ neurons that are LacZ− (arrow) at E17.5. Bar = 50 µm. Analyses were performed on three embryos at each age.
NE neuron numbers in the brainstem are unaffected in the En2-KO
One possible explanation for reduced forebrain monoamine fibers may be a deficiency in NE-containing neurons in the hindbrain LC or subcoeruleus nuclei, the two major sources of forebrain innervation (87). Significantly, however, we detected no differences in the numbers of LC (Fig. 4A) or subcoeruleus neurons (Fig. 4B) at P21, using stereological assessment of TH-positive cells in the pons of WT and En2-KO mice (Fig. 4C), consistent with previous studies in the embryo and at birth (32). There were also no differences in the number of TH+ neurons in the medullary nuclei, A1/C1 and A2/C2, in the adult animal (A1/C1: WT = 2641 ± 351, KO = 2842 ± 337 cells; N = 3/genotype; P = 0.70) and (A2/C2: WT = 3559 ± 345, KO = 3250 ± 251 cells; N = 3/genotype; P = 0.51). In sum, the changes in NE fiber innervation do not seem to depend on alterations in brainstem NE neuron populations.
Figure 4.

Numbers and size of NE producing neurons. Representative images of TH immunostaining in the WT and En2-KO for stereology of (A) LC and (B) SubCoeruleus (SubC) neurons. Bar = 200 µm. (C) Stereological counting of LC and SubC shows no differences in En2-KO compared with WT mice at P21. Three animals/genotype. Bar = 200 µm. (D) Photomicrograph of TH staining in WT and KO neurons of the LC. (E) Quantification of the average size of LC cell bodies was performed using two methods including the Contour measurement of cell circumference (left) as well as the Nucleator Program of the MBF Stereology platform (right). Measurements were performed only on cells exhibiting cytoplasmic TH signal surrounding DAPI positive nuclei. Every fifth section from the entire rostrocaudal extent of the LC was assessed for each animal, assessing all cells present in the sections. Five animals/genotype. Student's t-test: **P < 0.01, ***P < 0.001.
En2 deletion alters the size of LC neurons
Even though the numbers of LC neurons was not affected by En2 deletion, in a model of another neurodevelopmental disorder, Rett syndrome, the size and function of these neurons is affected (88,89). Thus we estimated the average cell body volume in the P60 LC by measuring the area of cells presenting a nucleus within a TH+ cell body on all tissue sections containing the LC (Fig. 4D). En2-KO LC neurons had a reduced cell body area (Fig. 4E; P = 0.008). We also used the Nucleator Program of the StereoInvestigator platform (MBF Bioscience) on the same sections, that calculates volumes based on the intersection of six lines, centered in the nucleus, that extend to the plasma membrane. With this approach, we detected a 22% reduction in cell body size in the En2-KO LC (Fig. 4E; P = 5 × 10−5). Thus En2 expression is necessary for normal development of LC neurons, a finding similar to that of MeCP2 KO (88) where reductions in LC neuron size and volume correlate with changes in neurophysiological properties, forebrain TH levels and social interactions.
En2 deletion leads to structural changes in multiple brain regions
Monoamine neurotransmitters are known to influence multiple aspects of neural development, including precursor proliferation, cell survival, axonal growth and synapse formation (63,70,71). To examine whether altered monoamines in the En2-KO are associated with changes in growth, we analyzed the weights of the different brain regions at P21 (Fig. 5A). Growth reductions in brainstem and cerebellum, where En2 is highly expressed, are well-documented consequences of mutating this regulatory gene (32,33), and our analysis confirmed such effects. However, there were also changes in the weights of regions outside En2's expression domain, including reductions of 11.6% of the hippocampus, 12.7% of striatum and 3.9% of total forebrain (Fig. 5A), whereas total body weights were unchanged (WT = 10.9 ± 0.2, KO = 10.4 ± 0.9; mean g ± SEM; P = 0.61, N = 6/genotype). Given parallel reductions in both weight and monoamine systems in P21 hippocampus, we also quantified DNA content, which serves as a proxy for total cell number. DNA content was reduced by 15.6% in En2-KO hippocampus compared with WT control (Fig. 5B, P = 0.024), suggesting that there were fewer total cells. To further define this issue, we employed stereological methods. The total volume of the P21 hippocampus was not altered (WT = 7.2 ± 1.2, KO = 8.07 ± 1.4; mean volume (mm3) ± SEM; P > 0.05; N = 5/genotype). In addition, the absolute number of pyramidal neurons in Ammon's horn (CA 1–3), a population generated in the embryo prior to major monoamine innervation (77), was also unchanged. In contrast, the dentate gyrus granule cell layer, whose neurons are generated following monoamine innervation, exhibited a 16.2% decrease in absolute neuron number (Fig. 5C). Thus in the absence of En2, developmental deficits in forebrain growth occur that correlate with reductions in NE levels and innervation.
Figure 5.

Structural changes occur in multiple brain regions in the En2-KO. (A) Weights (mg) of brain regions are reduced in the En2-KO compared with WT mice at P21. Six animals/genotype from 3 litters for forebrain, mid-hindbrain and cerebellum and 19 animals from 6 litters for hippocampus and striatum. (B) Measurement of DNA content of the hippocampus shows a reduction in the En2-KO. Six animals/genotype. (C) Stereological assessment of the hippocampus indicates a 16% reduction in the number of dentate gyrus granule layer neurons (DG) in the En2-KO but no change in CA1-3 pyramidal neurons. Five animals/genotype. CA1-3, cornu ammonis 1–3; Cereb, cerebellum; DG, dentate gyrus; FrB, forebrain; Hippo, hippocampus; Mid/hindB, midbrain and hindbrain. Student's t-test: *P < 0.05, **P < 0.01, ***P < 0.001.
Forebrain neurogenesis is dysregulated in the En2-KO
The reduced growth of multiple brain regions in the En2-KO raises the possibility that developmental neurogenesis is dysregulated. Changes in monoamines may contribute to reduced brain growth in the KO, since they are known stimulators of neurogenesis (30) a mechanism that may underlie their therapeutic benefits in adults (78). However, their roles and functions during development are less well defined (25,90). Given the reductions in forebrain growth as well as total cells and granule neurons in the hippocampus, we examined cell death and proliferation, expecting they would be increased and reduced, respectively.
To define cell apoptosis, we assessed in P21 dentate gyrus two markers of cell death, including cells immunolabeled for cleaved caspase-3 or exhibiting pyknotic nuclei. Cleaved caspase-3-labeled cells were increased by 77% in En2-KO dentate gyrus (Fig. 6A and D; P = 0.004) and pyknotic cells were increased 61% (Fig. 6E; P = 0.002), indicating that cell death is enhanced in the absence of En2. In contrast, in the CA1-3 region, where pyramidal neuron numbers were not affected, cleaved caspase-3-labeled cells did not differ between genotypes (WT = 30.0 ± 5.7, KO = 30.0 ± 5.5; P = 1.000; N = 3/genotype). Thus excessive cell death in the En2-KO dentate gyrus may be one mechanism that accounts for the reduced numbers of granule neurons detected by stereology.
Figure 6.

The neurogenic regions of the En2-KO dentate gyrus and SVZ exhibit changes in markers of both proliferation and cell death at P21. BrdU immunostaining in WT and En2-KO mice was performed 2 h post BrdU injection. (A) There were more cleaved caspase-3 (cCasp3) labeled cells in the En2-KO dentate gyrus (arrows) than in WT. The neural precursor markers BrdU (B) and PCNA (C) labeled more cells in the En2-KO than WT mice in the dentate gyrus (arrows). Quantification shows an increase in cell death markers cCaspase3 (D) and Pyknotic nuclei (E) as well as the markers of cell proliferation BrdU (F) and PCNA (G). Quantifications are expressed as cells/section. In the SVZ, (H) BrdU immunolabeling and (I) quantification show increased cells in the En2-KO. (J) Cleaved caspase-3 immunolabeling and (K) quantification reveal increased cells in the SVZ of the En2-KO. Cells were quantified on three sections/animal and 4–5 animals/genotype. cCasp3, cleaved caspase3; Hippo, hippocampus; SVZ, subventricular zone. Student's t-test: *P < 0.05, **P < 0.01.
Alternatively, reduced granule neurons in the En2-KO may reflect a deficit in precursor cell proliferation. To assess proliferation, we quantified the number of cells labeled with thymidine analog, BrdU, an S phase marker, 2 h after peripheral injection, and PCNA, a marker of cells engaged in the cell cycle (91). Contrary to expectations, there was a >2-fold increase in BrdU-labeled cells in the En2-KO dentate gyrus compared with WT (Fig. 6B and F; P = 0.009), suggesting that precursor proliferation may be increased. In addition, there was an 87% increase in the number PCNA-labeled cells in the dentate gyrus hilus and subgranular zone (SGZ) (Fig. 6C and G; P = 0.011), suggesting that the total pool of precursors was also increased in the absence of En2.
In addition to hippocampus, growth deficits were also observed in the striatum and the overall forebrain. To determine whether dysregulation of neurogenesis at P21 was a phenotype exclusive to the hippocampus, we examined the neighboring subventricular zone (SVZ) of the lateral ventricles. We found that BrdU labeling was increased by 65.6% (Fig. 6H and I; P = 0.027) and cleaved caspase-3-labeling was increased by 46.2% (Fig. 6J and K; P = 0.046), suggesting that the absence of En2 produced more generalized effects on brain growth. However, on the other hand, there were no differences in BrdU labeling in the amygdala (WT = 35.3 ± 4, KO = 36.9 ± 1; P = 0.73), a region reported to exhibit postnatal neurogenesis under certain conditions (92). Thus at a time when NE levels and innervation are perturbed in the hippocampus and forebrain, there are abnormalities in both cell death and precursor proliferation that may contribute to the observed reductions in brain growth.
Hippocampal stem cell pool size and proliferation are increased in early and intermediate neural progenitors of the En2-KO
The foregoing data indicate that in the P21 En2-KO, granule neuron numbers are reduced, an outcome associated with increases in both cell proliferation and cell death. In turn, we speculated that proliferation may be increased as a compensatory mechanism to overcome enhanced cell death. To begin addressing this issue, we first characterized the size of precursor cell pools and their entry into mitotic S phase. As described above, it should be noted that hippocampal volume was not different between genotypes, though granule neurons were selectively reduced in the En2-KO (Fig. 5). In the dentate gyrus, proliferating stem cells progress through a sequence of stages that can be identified by immunolabeling with Sox2 for early precursors, Tbr2 for intermediate precursors and Dcx for more restricted neuroblasts (93). In the absence of En2, the P21 dentate gyrus exhibited 17% more Sox2+ cells in the hilus and SGZ (Fig. 7A, and B; P = 0.040). Furthermore, there was a 2-fold increase in Tbr2+ intermediate precursors (P = 0.007), the most affected cell population, whereas there was no significant change in Dcx+ cells (Fig. 7A and B). These data suggest that En2 deletion causes an expansion of the early hippocampal precursor pools but not later neuroblasts. This population enlargement may account for the increase in the numbers of cells exhibiting proliferation marker PCNA (Fig. 6). However, in addition to stem cell pool size, is there also a change in S phase entry that contributes to excess proliferation? To examine this question, stem cells in each stage were double labeled with BrdU, to define the percent of cells engaged in S phase, termed the BrdU-labeling index. Indeed, the proportion of Sox2+ cells engaged in S phase (Sox2 + BrdU+/total Sox2) was more than doubled, increasing from 1.5% in the WT to 3.2% in the En2-KO (Fig. 7C and D; P = 0.006), suggesting more cells were proliferating. On the other hand, while the total population of intermediate Tbr2+ cells had doubled, there was no change in their rate of proliferation (Fig. 7D). However, due to the combined changes in stem cell pool size and S phase entry, there was an overall increase in proliferative cells in the early stem cell pools in the absence of En2 (Fig. 7E; Sox2: P = 0.002; Tbr2: P = 0.039). These observations indicate that excess proliferation in the En2-KO hippocampus reflects increases in early stem cells and intermediate precursors but not neuroblast progenitors.
Figure 7.

En2-KO animals exhibit increased numbers and proliferation of hippocampal neural stem cells expressing Sox2 and Tbr2 at P21. (A) Representative images of immunostaining of neural progenitor cells for Sox2, Tbr2 and Dcx. Arrowheads identify multi-labeled cells. (B) Quantification of Sox2 and Tbr2 expressing cells in the dorsal hippocampal dentate gyrus shows an increase while later Dcx progenitors are unchanged in the En2-KO. (C) Double immunostaining for BrdU and progenitor markers Sox2, Tbr2 or Dcx in WT and En2-KO hippocampus. (D) Proliferative activity is increased in the Sox2+ cell population of the En2-KO hippocampus, assessed as BrdU-labeling index. There was no change in the Tbr2 or Dcx populations of the En2-KO. (E) The increase in the absolute number of proliferative cells (BrdU+) in the En2-KO hippocampus consists of neural stem cells from the Sox2 (Sox2+/BrdU+) and Tbr2+ (Tbr2+/BrdU+) populations. Data were obtained from three hippocampal sections/animal, and four animals/genotype. Student's t-test: *P < 0.05, **P < 0.01.
Neuroblast progenitor cells undergo increased cell death in the En2-KO hippocampal dentate gyrus
Since excess proliferation in the KO hippocampus was accompanied by increased cell death, we defined the affected stem cell pools using double immunolabeling with cleaved caspase-3. Neither the early Sox2+ stem cells nor Trb2+ intermediate precursors exhibited a genotype specific difference in the apoptosis marker (Fig. 8). On the other hand, Dcx+ neuroblasts exhibited a 2-fold increase in cleaved caspase-3 double-immunolabeling, suggesting this later stage progenitor was most vulnerable to enhanced cell death in the absence of En2. Interestingly, there was no change in apoptosis of the post-mitotic neurons expressing NeuN+ (Fig. 8). These data suggest that apoptosis is specifically dysregulated in En2-KO hippocampal neuroblast progenitor cells but not in early progenitors or later post-mitotic neurons.
Figure 8.

Cell death is enhanced in Dcx+ neuroblast cells of the En2-KO hippocampus at P21. (A) The arrowheads point to double-immunostaining of cleaved caspace-3 with markers of neural progenitors, Sox2, Tbr2 and Dcx and post-mitotic neuron marker, NeuN. No cells exhibited double labeling for Tbr2 and cleaved caspase-3. (B) The number of colabeled cells for activated caspase-3 and Dcx (Dcx+/cCasp3+) is increased in the En2-KO but there is no change in Sox2 or NeuN-labeled cells. Data were obtained from three hippocampal sections/animal, and five animals/genotype. cCasp3, cleaved caspase-3. Student's t-test: *P < 0.05.
Despite excess proliferation, enhanced cell death impairs survival of newly born neurons in the En2-KO hippocampus
If excessive cell death at P21 actually prevents population expansion of neuroblast cells and mature neurons, one might expect that newly born cells would display a reduced rate of survival. To examine this question, P21 WT and En2-KO mice were injected with BrdU to label cells in S phase, and half of the animals were killed at 2 h while the balance were maintained for another 3 weeks, until P42, to compare the numbers of BrdU+ cells that survived (Fig. 9A and B). While there were twice as many BrdU-labeled cells in the En2-KO dentate gyrus compared with the WT on P21, a much larger fraction was lost by 3 weeks in the En2-KO. Indeed, compared with the WT, the En2-KO exhibited a 4-fold greater cell loss (Fig. 9C). Consequently, although more stem cells were proliferating in the En2-KO, specifically early Sox2 and Tbr2 populations (Fig. 7), the final outcome was that the remaining cell numbers were similar between genotypes. Furthermore, there was no apparent change in the differentiation potential of the newly born cells in the En2-KO, as similar proportions of BrdU+ cells exhibited the neuronal marker NeuN (Fig. 9D). These findings are consistent with an increase of apoptosis affecting neuroblast progenitor cells, a mechanism that likely contributes to the reduction in the number of dentate gyrus granule neurons in the En2-KO mice (Fig. 5).
Figure 9.

Reduced survival of new born neurons in the En2-KO mice. (A) BrdU-labeled cells in the dentate gyrus of WT and En2-KO mice, 2 h or 3 weeks after BrdU injection at P21. (B) Quantification of the number of BrdU-labeled cells 2 h and 3 weeks after BrdU injection. While there are twice as many labeled cells in the En2-KO at 2 h than the WT, there are no genotype differences in surviving cells at 3 weeks. (C) Four-fold more BrdU-labeled cells are lost over 3 weeks in the KO compared with the WT animal. (D) There is no genotype difference in the percentage of BrdU cells that are double labeled with NeuN 3 weeks after BrdU injection at P21. (A and B) data were obtained from the entire dentate gyrus, every tenth section, N = 5 animals/genotype Student's t-test: **P < 0.01, Two-way ANOVA ***P< 0.001. (D) 60–100 cells were counted over three sections/animal, five animals/genotype, from 2 to 3 litters.
Intra-hippocampal injection of β-adrenergic receptor agonists restores normal cell death in the En2-KO
The correlation of reduced NE levels in the postnatal hippocampus to diminished brain growth and dysregulated neurogenesis at P21 raises the possibility that increasing NE signaling may reverse developmental abnormalities in cell death and proliferation, both of which are increased (Fig. 6). In previous studies of the adult En2-KO, we found that a chronic (3 week) increase in NE signaling produced by supplying NE reuptake inhibitor desipramine in the water was able to reverse depression-related behaviors and social interaction deficits (48). However, in the current studies, to define a role for deficient NE signaling in increased apoptosis and proliferation in the P21 En2-KO, we chose acute intra-hippocampal injections of β-adrenergic receptor agonists for two reasons. First, we chose to employ direct adrenergic agonists instead of re-uptake inhibitors, because NE transporter expression is incomplete at early postnatal ages (55). Second, we wanted to assess relatively direct action of the drugs on the population of interest, the neurogenic stem cells of the dentate gyrus, hopefully avoiding both complex mechanisms and interpretations. We chose the general β-adrenergic agonist isoproterenol (IsoP) as well as the selective and molecularly distinct β3 subtype specific agonist BRL37344, as both have been found to affect adult hippocampal neurogenesis (30), though effects in the immature brain are undefined (55). To diminish possible confounding effects of brain injections, we compared effects of injected agonist on one side to injection of saline vehicle on the other, in a separate experiment for each drug. We stereotaxically injected IsoP and saline or BRL37344 and saline into WT and En2-KO mouse hippocampus at P21 (Fig. 10A). Twenty-two hours later we injected intraperitoneally thymidine analog Edu (to assess S phase entry) and perfused and fixed mouse brains at 24 h to assess cleaved capsase-3 immunoreactivity and Edu labeling of cells in S phase. Significantly, injection of IsoP into the En2-KO hippocampus reduced apoptosis 24 h later compared with the contralateral saline injected side (Fig. 10B, P = 0.023), suggesting that deficits in NE signaling may contribute to enhanced cell death. Moreover, injection of BRL37344 also returned apoptosis to levels similar to that of the WT animal (Fig. 10C, P = 0.041). Indeed, the levels of apoptosis in the En2-KO mice following both agonists did not differ from those of WT animals injected with saline vehicle.
Figure 10.

Pharmacological activation of β-adrenergic receptors reverses excessive cell death in the En2-KO hippocampus at P21. (A) Representative sites of bilateral intra-hippocampal injections into the dentate gyrus are shown on a DAPI counterstained section. Either IsoP or BRL37344 was injected alternatively in the left or right hemisphere, while saline was injected into the contralateral hemisphere. (B) Quantification of the number of cleaved caspase-3+ cells. IsoP significantly reduces the number of apoptotic cells in the En2-KO hippocampus, whereas it has no effect in the WT hippocampus. (C) IsoP has no effect on Edu labeling 24 h after injection in either genotype. (D) BRL37344 injection significantly reduces the number of apoptotic cells in the En2-KO but has no effect in the WT hippocampus. (E) BRL37344 has no effect on Edu labeling 24 h after injection in either genotype. 6–7 animals/condition, Student's t-test: *P < 0.05.
In contrast to effects on apoptosis, neither drug affected Edu labeling in the hippocampal dentate gyrus at 24 h in the WT or KO mice (Fig. 10C and E). If increased proliferation in the En2-KO reflects a compensatory response to enhanced cell death, the 24 h period may not be sufficient time for a responsive change in stem cell proliferation. Alternatively, enhanced proliferation may reflect potential cell autonomous effects of En2 in the forebrain unrelated to NE signaling, a mechanism that warrants further study.
Discussion
The deletion mutant of En2 exhibits a variety of behavioral deficits relevant to neurodevelopmental disorders such as ASD, schizophrenia, depression, epilepsy and learning disabilities (16–18). Considering the roles of monoamine systems and the value of related drugs in these disorders, we characterized monamine system development and found abnormalities especially during the first three postnatal weeks, with NE being the most adversely affected. Monoamine abnormalities of a lesser degree persisted into adulthood in the male En2-KO, which exhibited depression-related behavior. During development, while En2 was expressed in the early embryonic LC, its deletion did not alter NE neuron numbers. Rather, fiber innervation patterns of LC neurons, the major source of brain NE, showed changes that paralleled abnormalities in NE levels, suggesting a new mechanism responsible for the neurotransmitter deficits. Consistent with known monoamine neurodevelopmental activities (26,30,63,68–72), we found that reduced forebrain NE levels correlated with decreased forebrain weight and reduced hippocampal cell and neuron numbers. Furthermore, in the hippocampal neurogenic zone, sequential increases in cell proliferation and cell death resulted in reduced survival of newly born but not post-mitotic neurons. These abnormalities in hippocampal neurogenesis may reflect the deficits in NE resulting from reduced LC innervation, because intra-hippocampal injection of β-adrenergic receptor agonists reversed the elevated cell death in the En2-KO dentate gyrus. These studies indicate that hindbrain genes and disorders may secondarily impact forebrain development and function by altering the efficacy of long distance neural connections.
Importance of monoamine functions during perinatal development
The expression of monoamine neurotransmitters and respective receptors and transporters progressively increases during the early postnatal period to attain adult levels (55,56). Importantly, interfering with their development causes long-term structural and functional abnormalities that are similar to human neurodevelopmental disorders (57,58). Furthermore, experimental alterations of monoamines during the perinatal period can elicit stage-specific or even opposite outcomes from those produced in the adult (56), including effects on neurogenesis (30,90,94,95). For all these reasons, we performed our analyses during development of the En2-KO, and the results stand in marked contrast to previous adult studies that show only modest changes of 5-HT in cerebellum (16). In the current study, we measured the three monoamines both within and outside the classical hindbrain expression domain of En2, and found unexpected neurotransmitter changes in the forebrain. NE levels were most affected in the cerebellum and forebrain regions including hippocampus, amygdala and cortex, all regions implicated in the pathology of ASD and a variety of other disorders (1,74,96–98). Significantly, proper monoamine signaling is necessary for the normal function of these brain regions (27).
En2 is expressed in the LC and is necessary for normal innervation of the brain
Using the En2-tau-LacZ transgenic mouse, we detected abundant gene expression in the hindbrain and particularly the LC at E14.5 and 15.5, but expression diminished thereafter. This early expression of En2 in the LC suggests that it plays significant roles in cellular development and early fiber outgrowth. Consistent with this proposal is our finding that in the En2 mutant, LC neuron size was reduced by 15–22%, a change that likely alters electrophysiological properties of the neurons, as reported in the Rett KO model (88). Furthermore, fiber projections to the forebrain were markedly reduced whereas those to the cerebellum were increased. Changes in LC fibers were detected using three distinct markers of noradrenergic neurons, specifically TH, NET and galanin, and changes were found as early as P7 and some persisted into adulthood (P60). Abnormalities in prenatal axonal targeting would indeed serve as one explanation for the changes in monoamine neurotransmitters levels, which we detected already in the perinatal period. Similar changes in LC neuron size, function and forebrain projections have been described in a preclinical model of Rett syndrome, a neurodevelopmental disorder associated with ASD and intellectual disability (88). Are alterations in NE levels and fibers due to changes in the numbers of neurons in the LC or other noradrenergic nuclei in the brainstem? Using stereology, we did not detect differences in neuron numbers in the P21 LC, consistent with previous studies in embryonic and newborn mice (32) and the limited human neuropathology in ASD and schizophrenia (99,100). Moreover, we did not identify any changes in NE neuron numbers in the subcoeruleus and A5 located in the pons, nor A1/C1 and A2/C2 in the medulla. Since NE neuron numbers were unchanged, these data suggest that either the LC fibers were misrouted and/or LC neuron anterior–posterior identity was shifted. As a transcription factor, En2 is known to regulate nuclear expression of a number of axon growth and pathfinding genes (101,102) which may contribute to changes in fiber growth. However, recent studies also indicate that outside the nucleus, indeed outside the cell, extracellular En2 protein can regulate neuronal outgrowth (41,103). In culture, En2 can regulate axonal growth cone turning, whereas in vivo, extracellular protein can determine the spatial distribution of axon terminals, specifically guiding where retinal axons synapse in their colliculus target tissue, another possible mechanism for the dysregulation we observe. Changes in monoamine neurotransmitters in the En2-KO are likely due to a developmental disturbance of monoamine axon growth and pathfinding, leading to altered target innervation.
Relationship of monoamine levels to forebrain growth
Outside the classical hindbrain domain of En2 expression, reductions in monoamines in the En2-KO paralleled regional deficits in brain growth. Growth deficiencies included the total forebrain, the striatum and especially the hippocampus, which exhibited reductions in brain region weight, DNA content and granule neurons specifically. With respect to development, roles for monoamines in regulation of developmental processes have been proposed for both 5-HT and NE, with increases correlated with increased brain growth (26,30,63,68–72). Conversely, in the En2-KO, reductions in forebrain monoamines correlated with reduced brain region size, further suggesting that 5HT and NE regulate brain growth. One locus of monoamine regulation of brain growth would be developmental neurogenesis.
In the absence of En2, cell production was dysregulated in both major regions of postnatal neurogenesis, the dentate gyrus and SVZ. At first glance, the increases in hippocampal stem cell proliferation may seem surprising, since both NE and 5-HT are well-known to stimulate hippocampal neurogenesis in adult animals (30,94). However, recent developmental studies suggest monoamines in fact inhibit perinatal neurogenesis (90). In turn, when monoamines are reduced, neurogenesis would be expected to increase, as we observed in the En2-KO. In addition, a number of studies suggest that monoamines promote the survival of newly born neurons in the hippocampus (104–107) and may act via β-adrenergic receptors (28,30,108). For example, in the vesicular monoamine transporter deletion mutant, in which all monoamines are reduced, there is also enhanced neuronal cell death (25). Thus we speculate that reductions in monoamine signaling lead to disinhibition of precursor proliferation as well as enhanced precursor cell death.
To test the role of NE signaling in dysregulated neurogenesis, we performed intra-hippocampal injections of two distinct β-adrenergic agonists and found that both were able to reverse the enhanced cell death at 24 h, suggesting that reductions of NE contributed to the apoptosis. However, the agonists had no effect on increased proliferation. We speculate that increased proliferation of Sox2/Tbr2 precursors was a compensatory response of the stem cell pool to the enhanced apoptosis of the later Dcx neuroblasts. If this is the case, 24 h may be too short time interval for the stem cells to register changes in the Dcx cell pool. Overall, these observations support a role for LC–NE signaling in developmental neurogenesis and suggest that diminished innervation and NE lead to enhanced cell death to contribute to growth reductions in the En2-KO forebrain.
We may also consider an alternative mechanism contributing to enhanced proliferation and cell death. NE could affect neurogenesis indirectly through α1a adrenergic receptors (109) on interneurons. GABA signaling can both inhibit proliferation of neural stems cells as well as prevent apoptosis of differentiating hippocampal neurons (110). Thus, in the En2-KO, decreased NE signaling could reduce interneuron inhibition of proliferation and apoptosis, resulting in increases of cell proliferation and cell death.
In contrast to the forebrain, brain growth deficits in the hindbrain did not correlate with monoamine levels. We suggest that growth reductions in the En2-KO hindbrain and cerebellum reflect the absence of the transcription factor in these cells themselves, a cell autonomous function, as supported by many previous studies (39–44). Further, we recently reported that En2 functions cell autonomously in cerebellar granule neuron precursors of the external granule layer to promote cell cycle exit and neuronal differentiation (44).
In the current studies, we propose that the absence of En2 in the hindbrain LC neurons leads to their decreased size, reduced innervation of forebrain structures and diminished NE regulation of hippocampal neurogenesis, a plausible example of abnormal long distance neural connectivity in neurodevelopmental disorders (111–114). A cell autonomous role for En2 regulation of forebrain-projecting monoamine neurons has been proposed for the serotonin–raphe system (34). However, several other models warrant consideration. As En2 is also expressed widely in the brainstem, developmental abnormalities of LC neurons may reflect changes in the local environment in which neurons mature and extend axonal fibers. Alternatively, we and others have reported very low (1000-fold lower) levels of En2 mRNA in the forebrain including hippocampus, thalamus, striatum and hypothalamus (17,18), suggesting that cell autonomous or local actions of En2 may possibly contribute to the increased proliferation we observed. However, cellular localization of En2 expression remains elusive. In a recent study using an antibody that recognizes both En1 and En2 proteins, immunoreactive signal appears to be in multiple cell types in the cortex and hippocampus, and the signal sometimes co-localizes with GABA interneuron markers (45), though the nature of the other cells remains undefined. Variable levels of signal are also present in the hippocampal dentate gyrus, including many pyramidal neurons, though there is no clear signal in the SGZ where neural stem cells reside. Unfortunately antibody immunostaining in the En2-KO animal was not reported. In contrast, in our studies using the sensitive En2-tauLacZ reporter mouse, we do not detect any signal in the hippocampus, though robust LacZ immunostaining and reaction product are detected in ventral structures such as thalamus and striatum. This lack of hippocampal signal is consistent with in situ hybridizations by The Allen Brain Atlas. Thus En2 does not seem to be expressed in hippocampal cells affected in the En2-KO. Thus we favor a non-cell autonomous model to explain the defect of hippocampal neurogenesis and its reversal by β-adrenergic receptors agonists.
Relationships of deficits in hippocampal neurogenesis and forebrain monoamines to hippocampal-dependent behaviors
Abnormalities in monoamines and neurogenesis are correlated with several behavioral disorders in mice and humans. While changes in NE systems are associated with human depression, a common comorbidity with neuropsychiatric disorders (115,116), pharmacotherapies for depression are known to increase monoamine signaling and enhance hippocampal neurogenesis (53). Our behavioral assays found that adult male mice alone exhibited monoamine deficits and increased immobility times in the forced swim test, similar to previous reports (18) while females were not affected and had normal transmitter levels. These observations suggested that the depression-related behavior was due to reduced monoamine levels and that increasing NE might reverse behavioral deficits. Indeed, our recent studies using NE reuptake inhibitor, desipramine, (48) support this contention. Our replication of a depression-related behavior in an independent cohort of the En2-KO mouse indicates the robustness of this phenotype and adds additional support to the noradrenergic hypothesis.
The reductions we detect in NE levels, dentate gyrus cell number and neurogenesis are likely to impair the En2-KO performance on a variety of hippocampal-dependent behaviors (18), such as depression like tasks, Morris water maze performance, paired pulse inhibition, contextual-fear conditioning and social interactions (117–120). More broadly, abnormal hippocampal neurogenesis is a common feature of many models of neuropsychiatric disorders including addiction, depression, schizophrenia and epilepsy (96), and potentially autism (121). On the other hand, many mouse mutants with behavioral abnormalities relevant to ASD or schizophrenia also exhibit alterations in hippocampal neurogenesis (122–124). Thus, with regard to developing new therapies, one target for effective treatments may be the normalization of hippocampal neurogenesis, which may be a pathogenetic pathway and/or a biomarker for positive pharmacological response.
Relevance of abnormal monoamine systems to human neurodevelopmental disorders and ASD
The developmental changes we observe in NE and its regulators, TH and NET, in the En2-KO may represent a model by which genes regulating monoamines, including their synthesis, storage and catabolism, contribute to genetic risk of neurodevelopmental disorders including ASD (125), schizophrenia (126,127), depression (128,129) and bipolar disorder (130). Developmental disruption of LC neuron innervation and forebrain monoamine levels may represent a common disease pathway because similar changes are reported in other disease models including Rett syndrome, schizophrenia and fragile × mental retardation (88–90,131–133). Significantly, in individuals with autism who have gene alleles expected to increase levels of 5-HT (5-HTT transporter) or all three monoamines (MAO-A catabolic enzyme), brain imaging has demonstrated macrocephaly (75,76) a relationship that is also found in the related animal models (134). The direct correlation of forebrain monoamine levels and forebrain growth may suggest regulatory relationships. Furthermore, from a slightly different perspective, our studies indicate that En2 function is required for normal connectivity between the hindbrain and the forebrain (135). Abnormal long distance connections between specific brain regions, including those running from the hindbrain to the forebrain, have long been described as fundamental to many ASD phenotypes (135,136). Indeed, the LC stands as one of the first long distance projecting transmitter systems to be defined, whose function was required for normal brain development and adult cognitive performance (81,137). In addition to developmental effects, monoamine abnormalities likely contribute to many aspects of the mature phenotype, including changes in attention, mood, aggression, anxiety, sleep and repetitive and obsessive behaviors (138).
Although neurobiological abnormalities were prominent during early development, some improvements in neurotransmitter levels and target innervation occurred with age. While adult hippocampal innervation improved only modestly with age, NE levels showed greater recovery, suggesting the action of other compensatory processes, such as increased TH expression per neuron, or changes in release/re-uptake mechanisms or catabolic enzymes. Interestingly, there was greater improvement in the En2-KO female cerebral cortex than in the male. Developmental patterns of sex differences are also well described in ASD (2,139) that exhibits a 4:1 ratio of male:female prevalence (1,2) as well as schizophrenia with a 1.4:1 ratio (140). Given the pronounced sexual dimorphism of the NE and 5-HT systems (79,80), it may not be surprising that there are distinct sex-dependent recoveries of neurotransmitter levels and behaviors. Our findings may recommend the value of exploring differences in monoamine systems as factors in sex-dependent disease prevalence.
Our studies may also shed light on mechanisms by which altered monoamines contribute to ASD. Abnormal activation of the β2-adrenergic receptor, through the use of terbutaline during the third trimester of pregnancy to suppress preterm delivery, is associated with a 4-fold increased risk of ASD (141,142). Therapeutic levels of NE agonists during gestation may dysregulate brain neurogenesis producing macrocephaly, a well-described phenotype (74). Moreover, monoamines are dysregulated in children with ASD, who exhibit abnormal 5-HT systems, including disrupted temporal development, altered patterns of 5-HT synthesis between cerebellum and frontal cortex and left and right hemispheres, and dysmorphic axonal fibers on forebrain neuropathology (61–63). The increases in 5-HT and 5-HIAA in En2-KO cerebellum may be relevant to the increased 5-HT synthesis detected in the cerebellar dentate nucleus on PET scans in autism (61). Our animal studies suggest that defining NE metabolism in individuals with ASD might be informative, and the recent development of PET ligands that bind NET to define LC neurons may be a fruitful approach for future studies (143).
In sum, our study of the En2-KO mouse identifies unexpected roles for En2 in forebrain development. This model provides critical insights into how a hindbrain regulatory gene affects monoamine neurotransmitter system development, forebrain growth and neurogenesis. In turn, these observations lay a foundation to understand the cellular and molecular defects that underlie altered behaviors of the En2-KO, which may be relevant to human neurodevelopmental pathology. As the autism associated EN2 allele is a common variant in the human population (5,6), these insights suggest that changes in monoamines lay a platform on which other genetic and environmental factors interact to produce complex structural and behavioral phenotypes.
Materials and Methods
Animals
Engrailed-2 Het KO mice (39) on a C57BL6 J/129S2SV PAS background were obtained from The Jackson Laboratory (#: 002657; Bar Harbor, ME) as well as the En2tau-lacZ (En2tm5.1Alj/J). Since homozygous En2-KO are viable and fertile, we created separate WT and En2-KO lines that were used for most biochemical and morphological experiments. There is minimal genetic difference between these lines given their similar genetic heterogeneity as previously reported (16). All behavioral studies, however, were performed on WT and En2-KO littermates that were obtained from heterozygous crosses Het × Het using our WT and En2-KO lines. Siblings of animals tested behaviorally were also assessed biochemically by HPLC for neurotransmitter levels (see below) producing results similar to those performed on the separate WT and En2-KO lines. All mice were maintained in our colony on a 12:12 light:dark cycle. All animal procedures were assessed and approved by the Robert Wood Johnson Medical School Institutional Animal Care and Use Committee. Animals were managed by Robert Wood Johnson Animal Facility, and maintenance, husbandry, transportation, housing and use were in compliance with the Laboratory Animal Welfare Act (PL 89-544; PL-91-579) and National Institutes of Health guidelines (Manual Chapter 4206). The first day post coitum was determined by the presence of a vaginal plug and recorded at E0.5. The day of birth was reported as postnatal day 0 (P0). For select experiments, BrdU (Sigma, Saint Louis, MO) in sterile 0.9% NaCl, 7 mm NaOH solution was injected intraperitoneally at 100 mg/kg of body weight.
Drug treatment
Following isoflurane anesthetization, the surgery for the intra-hippocampal injection of IsoP (0.5 μl at 20 μm, Sigma) or BRL37344 (0.5 μl at 10 μm, Sigma) was performed at the stereotaxic coordinates (AP −1.3, Latreal +/1.7, DV 1–.8) as previously described (144,145). Drug injections were performed at P21 alternatively in either the left or right hemisphere with the contralateral hemisphere injected with saline. Then 22 h after hippocampal injection, mice received a dose of peripheral injection of EdU and brain tissue was fixed 2 h later. EdU staining was performed as recommended by the manufacturer (Life Technologies). Any section containing a dentate gyrus injured by the injection was discarded from the analysis. The three closest sections to the site of injection were analyzed for each hemisphere. All counting was performed blind of conditions.
HPLC
Brain regions were rapidly weighed and then frozen immediately after dissection and stored at −80°C until use. For each genotype, groups of 10–15 mice were obtained from 2 to 3 litters for neurotransmitter studies. Tissue was sonicated in phosphate-buffered saline (PBS), pH 7.4 at a wt/vol of 50 mg/ml and used for western blot. Aliquots for HPLC were diluted immediately to 10 mg/ml to a final concentration of 0.2 N perchloric acid. Homogenates were centrifuged and the supernatants were collected. The levels of NE, DA, 3,4-dihydroxyphenylacetic acid (DOPAC), 5-HIAA and 5-HT were measured by HPLC with electrochemical detection (Decade II, Antec Leyden, the Netherlands). The levels of GABA and glutamate were measured by HPLC with electrochemical detection (ALEXYS, Antec Leyden, the Netherlands) of the o-phthalaldehyde adduct. Concentrations were determined by comparison to peak heights of known standards run in parallel (146).
Western blot
Brain regions were frozen immediately after dissection and stored at −80°C until use. Protein extracts were obtained by homogenizing tissues in lysis buffer containing 50 mm Tris–HCl, pH 7.5, 150 mmm NaCl, 10 mm ethylenediaminetetraacetic acid, 2 mm ethylene glycol tetraacetic acid, 1% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate), 0.5% NP-40, 1% Triton X-100, and a mix of protease and phosphatase inhibitors consisting of 10 g/ml leupeptin, 10 g/ml aprotinin, 20 g/ml trypsin inhibitor, 50 mm NaF, 1 mm PMSF, 0.5 M microcystin, and 1 mm o-vanadate. Total protein was measured using Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL). Protein samples (50 µg/lane) were electrophoresed on 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis, transferred to a PVDF membrane, incubated 1 h in blocking solution [5% non-fat dry milk in tris buffer saline, 0.05% tween-20 (TBST) and immunoblotted using TH (Chemicon—Millipore, Billerica, MA), TrOH (1:1000, EMD Millipore, Billerica, MA) and 5-HTT (1:2000, Calbiochem–Merck, Darmstadt, Germany) antibodies in blocking solution. After 1 h incubation with horseradish peroxidase-conjugated anti-mouse or rabbit antibody (GE Healthcare, Pittsburgh, PA) in 1% milk/TBST, binding was revealed using chemiluminescence as we described previously (147).
Tissue fixation, histology and immunohistochemistry
Postnatal animals were anesthetized by IP injection of xylazine (1 mg/kg) and ketamine (10 mg/kg). Fixation of the brain was performed by transcardial perfusion with phosphate buffer (PB) and then 4% paraformaldehyde in PBS (4% PFA). Brains were dissected and further fixed in 4% PFA overnight at 4°C. For pregnant mice, following maternal CO2 exposure, embryos were removed and fixed by overnight immersion in 4%PFA at 4°C. After fixation, all samples were washed with PBS (three times for 10 min) and dehydrated/cryoprotected in 30% sucrose in PBS until they sank at 4°C. Then tissue was embedded in OCT and stored at −80°. Frozen sectioning was performed on a cryostat (Leica, Nussloch, Germany) to collect 12 µm sections for double immunostaining or 50 µm free floating sections for single immunostaining. We used 20 µm sections for BrdU immunostaining in a 1:5 series. LC and SubC stereology was performed on 50 µm sections in a 1:3 series. After three washes, sections were blocked in 5% serum, 0.5% triton in PBS then stained with primary antibodies to TH, NET, TrOH (1:500, Chemicon–Millipore, Billerica, MA), Galanin (1:4000, Peninsula Lab. San Carlos, CA), LacZ (1:200, Z3781, Promega, Madison, WI), cleaved-caspase3 (1:2000, Cell Signaling, Boston, MA), BrdU (mouse 1:100, BD Bioscience, San Jose, CA; rat 1:200, AbD Serotec, Kidlington, UK), Sox2 (1:200, Sigma), Tbr2 (1:200, AbCam, Cambridge, MA), Dcx (1:200, Santa Cruz, Dallas, TX), PCNA (1:1000, Santa Cruz) or NeuN (1:500, Chemicon) overnight at 4°C under gentle agitation in antibody solution (5% goat or donkey serum, 0.3% Triton-X in PBS) using previous methods (148). For BrdU and cleaved-caspase3 single immunostaining, a biotinylated secondary antibody anti-mouse or rabbit IgG (1:200, Vector, Burlingame, CA) was incubated for 1 h and then with ABC kit as recommended by manufacturer (Vector labs, Burlingame, CA) and revealed with 3,3′-diaminobenzidine (Sigma). Sections were counterstained with toluidine blue, dehydrated in ethanol, then in histoclear (National Diagnostics, Atlanta, GA) and mounted with Cytoseal (Thermo Scientific, Rockford, IL). For hippocampal neurogenesis markers (Sox2, Tbr2, Dcx, NeuN and PCNA) and double immunostaining, after incubation with the primary antibody, sections were washed then incubated 1 h with secondary Alexa 488- or 594-conjugated goat or donkey anti-rabbit, anti-mouse or anti-goat IgG (1:200 in PBS, Invitrogen, Carlsbad, CA) antibody at room temperature in 0.3% triton/PBS. Then the second immunostaining was performed. After washes, sections were then counterstained with propidium iodide (1:100 in 0.3% Triton-X/PBS) or DAPI (1:1000 in PBS) and washed again. Floating sections were mounted on slides and dried before coverslipping.
Image analysis and fiber and cell counting
The quantification of fiber densities within defined brain regions was performed blind on a Zeiss fluorescent microscope Axiovert 200 M (Zeiss, Oberkochen, Germany) with an Apotome module to obtain Z-stack images throughout the section thickness. Four pre-set vertical grid lines evenly distributed were superimposed onto the fluorescent, condensed Z-stack images and the numbers of immunofluorescent fibers crossing the grid lines were counted. Grid matrix crossings were assessed on images from the dentate gyrus molecular layer, one above and below the dorsal and ventral blades, respectively, from four 50 µm sections of the central region of the hippocampus in a 1:5 series from each animal, for each genotype. Fibers were analyzed in P21 immature sexually males and females or at P60 only adult male were study.
The SGZ of the dentate gyrus was defined as the region spanning from two cells bodies inside the granular cell layer (GCL) to two cell bodies inside the hilus. However, at P21 the entire hilus region was assessed because at this age, the SGZ has not yet reached maturity and new born neurons emerge from the whole region (149,150). For markers of proliferation and cell death, all labeled cells on 1:10 series section through the entire dentate gyrus (7–8 sections/animal) were quantified directly under the Aristoplan microscope (Leica, Wetzlar, Germany). For analysis of stem cells markers (Sox2, Tbr2 and Dcx) and double-labeling experiments, quantification was performed on 3–4 consecutives 1:10 series sections in the dorsal dentate gyrus, which region exhibits robust and highly reproducible neurogenesis on coronal sections (151). For PCNA, Sox2, Dcx and NeuN quantification, images of optical sections were acquired with a Zeiss Axiovert fluorescent microscope and its apotome module. Every image was acquired with identical exposure time and without post-acquisition exposure or background correction. The photomerge function of Photoshop (Adobe) was used after multiple fluorochrome images were acquired for each section. Assessments were performed blind to genotype under the microscope, and when applying Axiovision (Zeiss) and ImagePro Plus (MediaCybernetics, Rockville, MD) software. The BrdU-labeling index (LI) in neural progenitor populations was calculated for each neural progenitor marker from two immunostaining one to measure N, the total number of neural progenitors cells (Sox2, Tbr2 or Dcx), a second to measure n, the number neurals progenitors marker that are BrdU+. BrdU LI was calculated as LI = n/N.
Unbiased stereology for total cell analysis and soma volume
Cell numbers of the P21 dentate gyrus and CA1-3 regions were estimated by using the optical fractionator function of the MBF StereoInvestigator (MicroBrightField, Williston, VT). Color video images were obtained with a Zeiss Axio-imager M2 microscope (Zeiss). Outlines of the brain regions were drawn at low magnification (×20 objective) according to the Mouse Brain Atlas (152), and nuclei were counted under ×63 objective in oil. Cells counted in the first cell layer of the SGZ were included in the ‘GCL’. At each counting frame section thickness was measured to correct the estimation of cell population by the number weighted section thickness with the StereoInvestigator software (MicroBrightField). The counting parameters were: counting frame area 100 µm2, depth 10 µm2 and sampling grid area 10 000 µm2 (148,151). Cell counting was performed on every 10th section through the entire extent of the hippocampus, and data were obtained from 7 to 8 sections per animal. Sections were 50 µm each and stained with toluidine blue, facilitating the visibility of individual cells. Quantification of cells observed on bilateral hippocampi was performed for five animals/group. LC and subcoeruleus neurons were estimated on TH stained 50 µm hindbrain sections in a 1:3 series. For the LC stereology, the parameters were: counting frame area 2500 µm2, depth 10 µm2 and sampling grid area 10 000 µm2. Subcoeruleus neurons were estimated by counting all TH neurons in the proper atlas location ventral to the LC on each slide in the 1:3 series (range 4–6 sections/animal) and then multiplying by 3 to obtain total number.
Estimation of LC soma volume was performed on images of TH immunostained serial sections (every 3rd) of 40 µm thickness over the entire LC, corresponding typically to three or four sections. Images of the bilateral LC were taken as a single optical section acquired with a Zeiss Axiovert 200 M microscope using an Apotome module using ×40 objective with fixed exposure and post-acquisition parameters. On each image containing the LC, the surface of the TH immunoreactive cell soma containing a nucleus was estimated by drawing around the cell contour using Axiovision (Zeiss). The same sections were also analyzed using the Nucleator Probe (MicroBrightField) where the origin of the nucleator was placed at the center of the nucleus and the software systematically applies six radial lines. Then the observer clicks at the intersection of these lines with the cell membrane.
DNA content assay
Tissue was homogenized in ice cold distilled water. DNA was precipitated with 10% trichloroacetic acid (TCA). After centrifugation, the pellet was hydrolyzed with 1 N KOH and neutralized, DNA was resedimented with 5% TCA and denatured at 90°C. Samples were quantified with diphenylamine reagent that reacts proportionally with DNA to generate a colored reaction product that is detected by spectrophotometry. Sample values were converted to total DNA based on standard curves run in parallel for each assay, as previously described (151).
Tail suspension test
The tail suspension test was conducted as previously described (18). Mice were securely fastened by the distal end of the tail to a flat metallic surface and suspended in a visually isolated area (40′ 40′ 40 cm white Plexiglas box). The presence or absence of immobility, defined as the absence of limb movement, was sampled every 5 s over a 6 min test session by a highly trained observer who remained blind to genotype. Data collected were normally distributed and homogenous for variance. Genotype differences between adult En2-KO and WT littermates were analyzed by a two-tailed Student's unpaired t-test, using GraphPad Prism 4.0 statistical software (GraphPad Software, La Jolla, CA).
Forced swim test
The Porsolt forced swim test was conducted as previously described (REF). Mice were gently placed in a transparent Plexiglas cylinder (20 cm in diameter) filled with water (25 ± 2°C). Filling the cylinder to a depth of 12 cm prevented mice from using their tails to support themselves in the water. Immobility was defined as the cessation of limb movements except minor movement necessary to keep the mouse afloat. Immobility was sampled every 5 s during the last 4 min of a 6 min test session by a highly experienced observer who remained blind to genotype. Data collected were normally distributed and homogenous for variance. Sex and genotype differences in immobility scores for adult En2-KO and WT littermates were analyzed by a two-tailed Student's unpaired t-test, using GraphPad Prism 4.0 statistical software (GraphPad, La Jolla, CA).
Statistical analysis
Data are expressed as means or percentages ± SEM. Statistical analyses were performed using Student's t-test for two-group comparison and ANOVA followed by Dunnett's post hoc test for multiple group comparison, using GraphPad Instat 3.05 software (GraphPad Software).
Funding
This work was supported by the NJ Governor's Council for Medical Research and Treatment of Autism (E.D.-B.); the National Institutes of Health (MH076624 to J.H.M., NS052733 to P.K.S.); and the Mindworks Charitable Lead Trust.
Acknowledgements
We thank Janet Alder and David P. Crockett for their support for the intra-hippocampal injection procedure.
Conflict of Interest statement. None declared.
References
- 1.DiCicco-Bloom E., Lord C., Zwaigenbaum L., Courchesne E., Dager S.R., Schmitz C., Schultz R.T., Crawley J., Young L.J. (2006) The developmental neurobiology of autism spectrum disorder. J. Neurosci., 26, 6897–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lord C.J., Spence S.J. (2006) In Moldin S.O., Rubenstein J.L.R. (eds), Understanding Autism: From Basic Neuroscience to Treatment. CRC Press, Boca Raton, FL, US, pp. 1–23. [Google Scholar]
- 3.Sullivan P.F., Daly M.J., O'Donovan M. (2012) Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat. Rev. Genet., 13, 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purcell S.M., Moran J.L., Fromer M., Ruderfer D., Solovieff N., Roussos P., O'Dushlaine C., Chambert K., Bergen S.E., Kahler A. et al. (2014) A polygenic burden of rare disruptive mutations in schizophrenia. Nature, 506, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gharani N., Benayed R., Mancuso V., Brzustowicz L.M., Millonig J.H. (2004) Association of the homeobox transcription factor, ENGRAILED 2, with autism spectrum disorder. Mol. Psychiatry, 9, 474–484. [DOI] [PubMed] [Google Scholar]
- 6.Benayed R., Gharani N., Rossman I., Mancuso V., Lazar G., Kamdar S., Bruse S.E., Tischfield S., Smith B.J., Zimmerman R.A. et al. (2005) Support for the homeobox transcription factor gene ENGRAILED 2 as an autism spectrum disorder susceptibility locus. Am. J. Hum. Genet., 77, 851–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petit E., Herault J., Martineau J., Perrot A., Barthelemy C., Hameury L., Sauvage D., Lelord G., Muh J.P. (1995) Association study with two markers of a human homeogene in infantile autism. J. Med. Genet., 32, 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brune C.W., Korvatska E., Allen-Brady K., Cook E.H. Jr, Dawson G., Devlin B., Estes A., Hennelly M., Hyman S.L., McMahon W.M. et al. (2008) Heterogeneous association between engrailed-2 and autism in the CPEA network. Am. J. Med. Genet. B Neuropsychiatr. Genet., 147B, 187–193. [DOI] [PubMed] [Google Scholar]
- 9.Wang L., Jia M., Yue W., Tang F., Qu M., Ruan Y., Lu T., Zhang H., Yan H., Liu J. et al. (2008) Association of the ENGRAILED 2 (EN2) gene with autism in Chinese Han population. Am. J. Med. Genet. B Neuropsychiatr. Genet., 147B, 434–438. [DOI] [PubMed] [Google Scholar]
- 10.Yang P., Lung F.W., Jong Y.J., Hsieh H.Y., Liang C.L., Juo S.H. (2008) Association of the homeobox transcription factor gene ENGRAILED 2 with autistic disorder in Chinese children. Neuropsychobiology, 57, 3–8. [DOI] [PubMed] [Google Scholar]
- 11.Malhotra D., McCarthy S., Michaelson J.J., Vacic V., Burdick K.E., Yoon S., Cichon S., Corvin A., Gary S., Gershon E.S. et al. (2011) High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron, 72, 951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi J., Ababon M.R., Soliman M., Lin Y., Brzustowicz L.M., Matteson P.G., Millonig J.H. (2014) Autism associated gene, engrailed2, and flanking gene levels are altered in post-mortem cerebellum. PLoS One, 9, e87208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi J., Ababon M.R., Matteson P.G., Millonig J.H. (2012) Cut-like homeobox 1 and nuclear factor I/B mediate ENGRAILED2 autism spectrum disorder-associated haplotype function. Hum. Mol. Genet., 21, 1566–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.James S.J., Shpyleva S., Melnyk S., Pavliv O., Pogribny I.P. (2013) Complex epigenetic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl. Psychiatry, 3, e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James S.J., Shpyleva S., Melnyk S., Pavliv O., Pogribny I.P. (2014) Elevated 5-hydroxymethylcytosine in the Engrailed-2 (EN-2) promoter is associated with increased gene expression and decreased MeCP2 binding in autism cerebellum. Transl. Psychiatry, 4, e460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheh M.A., Millonig J.H., Roselli L.M., Ming X., Jacobsen E., Kamdar S., Wagner G.C. (2006) En2 knockout mice display neurobehavioral and neurochemical alterations relevant to autism spectrum disorder. Brain Res., 1116, 166–176. [DOI] [PubMed] [Google Scholar]
- 17.Tripathi P.P., Sgado P., Scali M., Viaggi C., Casarosa S., Simon H.H., Vaglini F., Corsini G.U., Bozzi Y. (2009) Increased susceptibility to kainic acid-induced seizures in Engrailed-2 knockout mice. Neuroscience, 159, 842–849. [DOI] [PubMed] [Google Scholar]
- 18.Brielmaier J., Matteson P.G., Silverman J.L., Senerth J.M., Kelly S., Genestine M., Millonig J.H., DiCicco-Bloom E., Crawley J.N. (2012) Autism-relevant social abnormalities and cognitive deficits in engrailed-2 knockout mice. PLoS One, 7, e40914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis C.A., Noble-Topham S.E., Rossant J., Joyner A.L. (1988) Expression of the homeo box-containing gene En-2 delineates a specific region of the developing mouse brain. Genes Dev., 2, 361–371. [DOI] [PubMed] [Google Scholar]
- 20.Zec N., Rowitch D.H., Bitgood M.J., Kinney H.C. (1997) Expression of the homeobox-containing genes EN1 and EN2 in human fetal midgestational medulla and cerebellum. J. Neuropathol. Exp. Neurol., 56, 236–242. [DOI] [PubMed] [Google Scholar]
- 21.Hanks M.C., Loomis C.A., Harris E., Tong C.X., Anson-Cartwright L., Auerbach A., Joyner A. (1998) Drosophila engrailed can substitute for mouse Engrailed1 function in mid-hindbrain, but not limb development. Development, 125, 4521–4530. [DOI] [PubMed] [Google Scholar]
- 22.Joyner A.L. (1996) Engrailed, Wnt and Pax genes regulate midbrain–hindbrain development. Trends Genet., 12, 15–20. [DOI] [PubMed] [Google Scholar]
- 23.Davis C.A., Joyner A.L. (1988) Expression patterns of the homeo box-containing genes En-1 and En-2 and the proto-oncogene int-1 diverge during mouse development. Genes Dev., 2, 1736–1744. [DOI] [PubMed] [Google Scholar]
- 24.Goridis C., Rohrer H. (2002) Specification of catecholaminergic and serotonergic neurons. Nat. Rev. Neurosci., 3, 531–541. [DOI] [PubMed] [Google Scholar]
- 25.Stankovski L., Alvarez C., Ouimet T., Vitalis T., El-Hachimi K.H., Price D., Deneris E., Gaspar P., Cases O. (2007) Developmental cell death is enhanced in the cerebral cortex of mice lacking the brain vesicular monoamine transporter. J. Neurosci., 27, 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narboux-Neme N., Angenard G., Mosienko V., Klempin F., Pitychoutis P.M., Deneris E., Bader M., Giros B., Alenina N., Gaspar P. (2013) Postnatal growth defects in mice with constitutive depletion of central serotonin. ACS Chem. Neurosci., 4, 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh C., Bortolato M., Bali N., Godar S.C., Scott A.L., Chen K., Thompson R.F., Shih J.C. (2013) Cognitive abnormalities and hippocampal alterations in monoamine oxidase A and B knockout mice. Proc. Natl. Acad. Sci. USA, 110, 12816–12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masuda T., Nakagawa S., Boku S., Nishikawa H., Takamura N., Kato A., Inoue T., Koyama T. (2012) Noradrenaline increases neural precursor cells derived from adult rat dentate gyrus through beta2 receptor. Prog. Neuropsychopharmacol Biol. Psychiatry, 36, 44–51. [DOI] [PubMed] [Google Scholar]
- 29.Lesch K.P., Waider J. (2012) Serotonin in the modulation of neural plasticity and networks: implications for neurodevelopmental disorders. Neuron, 76, 175–191. [DOI] [PubMed] [Google Scholar]
- 30.Jhaveri D.J., Mackay E.W., Hamlin A.S., Marathe S.V., Nandam L.S., Vaidya V.A., Bartlett P.F. (2010) Norepinephrine directly activates adult hippocampal precursors via beta3-adrenergic receptors. J. Neurosci., 30, 2795–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frederick A.L., Stanwood G.D. (2009) Drugs, biogenic amine targets and the developing brain. Dev. Neurosci., 31, 7–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simon H.H., Scholz C., O'Leary D.D. (2005) Engrailed genes control developmental fate of serotonergic and noradrenergic neurons in mid- and hindbrain in a gene dose-dependent manner. Mol. Cell Neurosci., 28, 96–105. [DOI] [PubMed] [Google Scholar]
- 33.Simon H.H., Saueressig H., Wurst W., Goulding M.D., O'Leary D.D. (2001) Fate of midbrain dopaminergic neurons controlled by the engrailed genes. J. Neurosci., 21, 3126–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fox S.R., Deneris E.S. (2012) Engrailed is required in maturing serotonin neurons to regulate the cytoarchitecture and survival of the dorsal raphe nucleus. J. Neurosci., 32, 7832–7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sgado P., Alberi L., Gherbassi D., Galasso S.L., Ramakers G.M., Alavian K.N., Smidt M.P., Dyck R.H., Simon H.H. (2006) Slow progressive degeneration of nigral dopaminergic neurons in postnatal Engrailed mutant mice. Proc. Natl. Acad. Sci. USA, 103, 15242–15247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alavian K.N., Sgado P., Alberi L., Subramaniam S., Simon H.H. (2009) Elevated P75NTR expression causes death of engrailed-deficient midbrain dopaminergic neurons by Erk1/2 suppression. Neural. Dev., 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alvarez-Fischer D., Fuchs J., Castagner F., Stettler O., Massiani-Beaudoin O., Moya K.L., Bouillot C., Oertel W.H., Lombes A., Faigle W. et al. (2011) Engrailed protects mouse midbrain dopaminergic neurons against mitochondrial complex I insults. Nat. Neurosci., 14, 1260–1266. [DOI] [PubMed] [Google Scholar]
- 38.Wurst W., Auerbach A.B., Joyner A.L. (1994) Multiple developmental defects in Engrailed-1 mutant mice: an early mid-hindbrain deletion and patterning defects in forelimbs and sternum. Development, 120, 2065–2075. [DOI] [PubMed] [Google Scholar]
- 39.Joyner A.L., Herrup K., Auerbach B.A., Davis C.A., Rossant J. (1991) Subtle cerebellar phenotype in mice homozygous for a targeted deletion of the En-2 homeobox. Science, 251, 1239–1243. [DOI] [PubMed] [Google Scholar]
- 40.Wilson S.L., Kalinovsky A., Orvis G.D., Joyner A.L. (2011) Spatially restricted and developmentally dynamic expression of engrailed genes in multiple cerebellar cell types. Cerebellum, 10, 356–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wizenmann A., Brunet I., Lam J.S., Sonnier L., Beurdeley M., Zarbalis K., Weisenhorn-Vogt D., Weinl C., Dwivedy A., Joliot A. et al. (2009) Extracellular Engrailed participates in the topographic guidance of retinal axons in vivo. Neuron, 64, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng Y., Sudarov A., Szulc K.U., Sgaier S.K., Stephen D., Turnbull D.H., Joyner A.L. (2010) The Engrailed homeobox genes determine the different foliation patterns in the vermis and hemispheres of the mammalian cerebellum. Development, 137, 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sillitoe R.V., Vogel M.W., Joyner A.L. (2010) Engrailed homeobox genes regulate establishment of the cerebellar afferent circuit map. J. Neurosci., 30, 10015–10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossman I.T., Lin L., Morgan K.M., Digiovine M., Van Buskirk E.K., Kamdar S., Millonig J.H., Dicicco-Bloom E. (2014) Engrailed2 modulates cerebellar granule neuron precursor proliferation, differentiation and insulin-like growth factor 1 signaling during postnatal development. Mol. Autism., 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sgado P., Genovesi S., Kalinovsky A., Zunino G., Macchi F., Allegra M., Murenu E., Provenzano G., Tripathi P.P., Casarosa S. et al. (2013) Loss of GABAergic neurons in the hippocampus and cerebral cortex of Engrailed-2 null mutant mice: implications for autism spectrum disorders. Exp. Neurol., 247, 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wise R.A. (2004) Dopamine, learning and motivation. Nat. Rev. Neurosci., 5, 483–494. [DOI] [PubMed] [Google Scholar]
- 47.Sara S.J. (2009) The locus coeruleus and noradrenergic modulation of cognition. Nat. Rev. Neurosci., 10, 211–223. [DOI] [PubMed] [Google Scholar]
- 48.Brielmaier J., Senerth J.M., Silverman J.L., Matteson P.G., Millonig J.H., DiCicco-Bloom E., Crawley J.N. (2014) Chronic desipramine treatment rescues depression-related, social and cognitive deficits in Engrailed-2 knockout mice. Genes Brain Behav., 13, 286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castren E., Elgersma Y., Maffei L., Hagerman R. (2012) Treatment of neurodevelopmental disorders in adulthood. J. Neurosci., 32, 14074–14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Samuels B.A., Mendez-David I., Faye C., David S.A., Pierz K.A., Gardier A.M., Hen R., David D.J. (2014) Serotonin 1A and serotonin 4 receptors: essential mediators of the neurogenic and behavioral actions of antidepressants. Neuroscientist, pii: 1073858414561303. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sahay A., Hen R. (2007) Adult hippocampal neurogenesis in depression. Nat. Neurosci., 10, 1110–1115. [DOI] [PubMed] [Google Scholar]
- 52.Pascual-Brazo J., Baekelandt V., Encinas J.M. (2014) Neurogenesis as a new target for the development of antidepressant drugs. Curr. Pharm. Des., 20, 3763–3775. [DOI] [PubMed] [Google Scholar]
- 53.Eisch A.J., Petrik D. (2012) Depression and hippocampal neurogenesis: a road to remission? Science, 338, 72–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.David D.J., Samuels B.A., Rainer Q., Wang J.W., Marsteller D., Mendez I., Drew M., Craig D.A., Guiard B.P., Guilloux J.P. et al. (2009) Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron, 62, 479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bylund D.B., Reed A.L. (2007) Childhood and adolescent depression: why do children and adults respond differently to antidepressant drugs? Neurochem. Int., 51, 246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olivier J.D., Blom T., Arentsen T., Homberg J.R. (2011) The age-dependent effects of selective serotonin reuptake inhibitors in humans and rodents: a review. Prog. Neuropsychopharmacol. Biol. Psychiatry, 35, 1400–1408. [DOI] [PubMed] [Google Scholar]
- 57.Park M.J., Guest C.B., Barnes M.B., Martin J., Ahmad U., York J.M., Freund G.G. (2008) Blocking of beta-2 adrenergic receptors hastens recovery from hypoglycemia-associated social withdrawal. Psychoneuroendocrinology, 33, 1411–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bortolato M., Chen K., Godar S.C., Chen G., Wu W., Rebrin I., Farrell M.R., Scott A.L., Wellman C.L., Shih J.C. (2011) Social deficits and perseverative behaviors, but not overt aggression, in MAO-A hypomorphic mice. Neuropsychopharmacology, 36, 2674–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson G.M., Freedman D.X., Cohen D.J., Volkmar F.R., Hoder E.L., McPhedran P., Minderaa R.B., Hansen C.R., Young J.G. (1987) Whole blood serotonin in autistic and normal subjects. J. Child Psychol. Psychiatry, 28, 885–900. [DOI] [PubMed] [Google Scholar]
- 60.Cook E.H. Jr, Leventhal B.L., Freedman D.X. (1988) Free serotonin in plasma: autistic children and their first-degree relatives. Biol. Psychiatry, 24, 488–491. [DOI] [PubMed] [Google Scholar]
- 61.Chugani D.C., Muzik O., Rothermel R., Behen M., Chakraborty P., Mangner T., da Silva E.A., Chugani H.T. (1997) Altered serotonin synthesis in the dentatothalamocortical pathway in autistic boys. Ann. Neurol., 42, 666–669. [DOI] [PubMed] [Google Scholar]
- 62.Chugani D.C., Muzik O., Behen M., Rothermel R., Janisse J.J., Lee J., Chugani H.T. (1999) Developmental changes in brain serotonin synthesis capacity in autistic and nonautistic children. Ann. Neurol., 45, 287–295. [DOI] [PubMed] [Google Scholar]
- 63.Whitaker-Azmitia P.M. (2001) Serotonin and brain development: role in human developmental diseases. Brain Res. Bull., 56, 479–485. [DOI] [PubMed] [Google Scholar]
- 64.Lam K.S., Aman M.G., Arnold L.E. (2006) Neurochemical correlates of autistic disorder: a review of the literature. Res. Dev. Disabil., 27, 254–289. [DOI] [PubMed] [Google Scholar]
- 65.Akil M., Pierri J.N., Whitehead R.E., Edgar C.L., Mohila C., Sampson A.R., Lewis D.A. (1999) Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am. J. Psychiatry, 156, 1580–1589. [DOI] [PubMed] [Google Scholar]
- 66.Okubo Y., Suhara T., Suzuki K., Kobayashi K., Inoue O., Terasaki O., Someya Y., Sassa T., Sudo Y., Matsushima E. et al. (1997) Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature, 385, 634–636. [DOI] [PubMed] [Google Scholar]
- 67.Bruder G.E., Keilp J.G., Xu H., Shikhman M., Schori E., Gorman J.M., Gilliam T.C. (2005) Catechol-O-methyltransferase (COMT) genotypes and working memory: associations with differing cognitive operations. Biol. Psychiatry, 58, 901–907. [DOI] [PubMed] [Google Scholar]
- 68.Berger-Sweeney J., Hohmann C.F. (1997) Behavioral consequences of abnormal cortical development: insights into developmental disabilities. Behav. Brain Res., 86, 121–142. [DOI] [PubMed] [Google Scholar]
- 69.Boylan C.B., Blue M.E., Hohmann C.F. (2007) Modeling early cortical serotonergic deficits in autism. Behav. Brain Res., 176, 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gaspar P., Cases O., Maroteaux L. (2003) The developmental role of serotonin: news from mouse molecular genetics. Nat. Rev. Neurosci., 4, 1002–1012. [DOI] [PubMed] [Google Scholar]
- 71.Herlenius E., Lagercrantz H. (2004) Development of neurotransmitter systems during critical periods. Exp. Neurol., 190 Suppl 1, S8–S21. [DOI] [PubMed] [Google Scholar]
- 72.Hoglinger G.U., Rizk P., Muriel M.P., Duyckaerts C., Oertel W.H., Caille I., Hirsch E.C. (2004) Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat. Neurosci., 7, 726–735. [DOI] [PubMed] [Google Scholar]
- 73.Andreasen N.C. (2010) The lifetime trajectory of schizophrenia and the concept of neurodevelopment. Dialogues Clin. Neurosci., 12, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Amaral D.G., Schumann C.M., Nordahl C.W. (2008) Neuroanatomy of autism. Trends Neurosci., 31, 137–145. [DOI] [PubMed] [Google Scholar]
- 75.Davis L.K., Hazlett H.C., Librant A.L., Nopoulos P., Sheffield V.C., Piven J., Wassink T.H. (2008) Cortical enlargement in autism is associated with a functional VNTR in the monoamine oxidase A gene. Am. J. Med. Genet. B Neuropsychiatr. Genet., 147B, 1145–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wassink T.H., Hazlett H.C., Epping E.A., Arndt S., Dager S.R., Schellenberg G.D., Dawson G., Piven J. (2007) Cerebral cortical gray matter overgrowth and functional variation of the serotonin transporter gene in autism. Arch. Gen. Psychiatry, 64, 709–717. [DOI] [PubMed] [Google Scholar]
- 77.Moudy A.M., Kunkel D.D., Schwartzkroin P.A. (1993) Development of dopamine-beta-hydroxylase-positive fiber innervation of the rat hippocampus. Synapse, 15, 307–318. [DOI] [PubMed] [Google Scholar]
- 78.Santarelli L., Saxe M., Gross C., Surget A., Battaglia F., Dulawa S., Weisstaub N., Lee J., Duman R., Arancio O. et al. (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science, 301, 805–809. [DOI] [PubMed] [Google Scholar]
- 79.Pinos H., Collado P., Rodriguez-Zafra M., Rodriguez C., Segovia S., Guillamon A. (2001) The development of sex differences in the locus coeruleus of the rat. Brain Res. Bull., 56, 73–78. [DOI] [PubMed] [Google Scholar]
- 80.Dominguez R., Cruz-Morales S.E., Carvalho M.C., Xavier M., Brandao M.L. (2003) Sex differences in serotonergic activity in dorsal and median raphe nucleus. Physiol. Behav., 80, 203–210. [DOI] [PubMed] [Google Scholar]
- 81.Foote S.L., Bloom F.E., Aston-Jones G. (1983) Nucleus locus ceruleus: new evidence of anatomical and physiological specificity. Physiol. Rev., 63, 844–914. [DOI] [PubMed] [Google Scholar]
- 82.Oleskevich S., Descarries L., Lacaille J.C. (1989) Quantified distribution of the noradrenaline innervation in the hippocampus of adult rat. J. Neurosci., 9, 3803–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blakely R.D., Bauman A.L. (2000) Biogenic amine transporters: regulation in flux. Curr. Opin. Neurobiol., 10, 328–336. [DOI] [PubMed] [Google Scholar]
- 84.Melander T., Hokfelt T., Rokaeus A., Cuello A.C., Oertel W.H., Verhofstad A., Goldstein M. (1986) Coexistence of galanin-like immunoreactivity with catecholamines, 5-hydroxytryptamine, GABA and neuropeptides in the rat CNS. J. Neurosci., 6, 3640–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sgaier S.K., Millet S., Villanueva M.P., Berenshteyn F., Song C., Joyner A.L. (2005) Morphogenetic and cellular movements that shape the mouse cerebellum; insights from genetic fate mapping. Neuron, 45, 27–40. [DOI] [PubMed] [Google Scholar]
- 86.Lindvall M., Owman C. (1981) Autonomic nerves in the mammalian choroid plexus and their influence on the formation of cerebrospinal fluid. J. Cereb. Blood Flow Metab., 1, 245–266. [DOI] [PubMed] [Google Scholar]
- 87.Szabadi E. (2013) Functional neuroanatomy of the central noradrenergic system. J. Psychopharmacol., 27, 659–693. [DOI] [PubMed] [Google Scholar]
- 88.Taneja P., Ogier M., Brooks-Harris G., Schmid D.A., Katz D.M., Nelson S.B. (2009) Pathophysiology of locus ceruleus neurons in a mouse model of Rett syndrome. J. Neurosci., 29, 12187–12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Roux J.C., Panayotis N., Dura E., Villard L. (2010) Progressive noradrenergic deficits in the locus coeruleus of Mecp2 deficient mice. J. Neurosci. Res., 88, 1500–1509. [DOI] [PubMed] [Google Scholar]
- 90.Cheng A., Scott A.L., Ladenheim B., Chen K., Ouyang X., Lathia J.D., Mughal M., Cadet J.L., Mattson M.P., Shih J.C. (2010) Monoamine oxidases regulate telencephalic neural progenitors in late embryonic and early postnatal development. J. Neurosci., 30, 10752–10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Taupin P. (2007) BrdU immunohistochemistry for studying adult neurogenesis: paradigms, pitfalls, limitations, and validation. Brain Res. Rev., 53, 198–214. [DOI] [PubMed] [Google Scholar]
- 92.Bernier P.J., Bedard A., Vinet J., Levesque M., Parent A. (2002) Newly generated neurons in the amygdala and adjoining cortex of adult primates. Proc. Natl. Acad. Sci. USA, 99, 11464–11469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.von Bohlen und Halbach O. (2011) Immunohistological markers for proliferative events, gliogenesis, and neurogenesis within the adult hippocampus. Cell Tissue Res., 345, 1–19. [DOI] [PubMed] [Google Scholar]
- 94.Lledo P.M., Alonso M., Grubb M.S. (2006) Adult neurogenesis and functional plasticity in neuronal circuits. Nat. Rev. Neurosci., 7, 179–193. [DOI] [PubMed] [Google Scholar]
- 95.Kirshenbaum G.S., Lieberman S.R., Briner T.J., Leonardo E.D., Dranovsky A. (2014) Adolescent but not adult-born neurons are critical for susceptibility to chronic social defeat. Front Behav. Neurosci., 8, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eisch A.J., Cameron H.A., Encinas J.M., Meltzer L.A., Ming G.L., Overstreet-Wadiche L.S. (2008) Adult neurogenesis, mental health, and mental illness: hope or hype? J. Neurosci., 28, 11785–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Penzes P., Cahill M.E., Jones K.A., VanLeeuwen J.E., Woolfrey K.M. (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci., 14, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wegiel J., Flory M., Kuchna I., Nowicki K., Ma S.Y., Imaki H., Wegiel J., Cohen I.L., London E., Brown W.T. et al. (2014) Brain-region-specific alterations of the trajectories of neuronal volume growth throughout the lifespan in autism. Acta. Neuropathol. Commun., 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marner L., Soborg C., Pakkenberg B. (2005) Increased volume of the pigmented neurons in the locus coeruleus of schizophrenic subjects: a stereological study. J. .Psychiatr Res., 39, 337–345. [DOI] [PubMed] [Google Scholar]
- 100.Martchek M., Thevarkunnel S., Bauman M., Blatt G., Kemper T. (2006) Lack of evidence of neuropathology in the locus coeruleus in autism. Acta Neuropathol., 111, 497–499. [DOI] [PubMed] [Google Scholar]
- 101.Logan C., Wizenmann A., Drescher U., Monschau B., Bonhoeffer F., Lumsden A. (1996) Rostral optic tectum acquires caudal characteristics following ectopic engrailed expression. Curr. Biol., 6, 1006–1014. [DOI] [PubMed] [Google Scholar]
- 102.Shigetani Y., Funahashi J.I., Nakamura H. (1997) En-2 regulates the expression of the ligands for Eph type tyrosine kinases in chick embryonic tectum. Neurosci. Res., 27, 211–217. [DOI] [PubMed] [Google Scholar]
- 103.Brunet I., Weinl C., Piper M., Trembleau A., Volovitch M., Harris W., Prochiantz A., Holt C. (2005) The transcription factor Engrailed-2 guides retinal axons. Nature, 438, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Madrigal J.L., Feinstein D.L., Dello Russo C. (2005) Norepinephrine protects cortical neurons against microglial-induced cell death. J. Neurosci. Res., 81, 390–396. [DOI] [PubMed] [Google Scholar]
- 105.Patel N.J., Chen M.J., Russo-Neustadt A.A. (2010) Norepinephrine and nitric oxide promote cell survival signaling in hippocampal neurons. Eur. J. Pharmacol., 633, 1–9. [DOI] [PubMed] [Google Scholar]
- 106.Klempin F., Babu H., De Pietri Tonelli D., Alarcon E., Fabel K., Kempermann G. (2010) Oppositional effects of serotonin receptors 5-HT1a, 2, and 2c in the regulation of adult hippocampal neurogenesis. Front Mol. Neurosci., 3, doi:10.3389/fnmol.2010.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Takamura N., Nakagawa S., Masuda T., Boku S., Kato A., Song N., An Y., Kitaichi Y., Inoue T., Koyama T. et al. (2014) The effect of dopamine on adult hippocampal neurogenesis. Prog. Neuropsychopharmacol. Biol. Psychiatry, 50, 116–124. [DOI] [PubMed] [Google Scholar]
- 108.Jhaveri D.J., Nanavaty I., Prosper B.W., Marathe S., Husain B.F., Kernie S.G., Bartlett P.F., Vaidya V.A. (2014) Opposing effects of alpha2- and beta-adrenergic receptor stimulation on quiescent neural precursor cell activity and adult hippocampal neurogenesis. PLoS One, 9, e98736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hillman K.L., Lei S., Doze V.A., Porter J.E. (2009) Alpha-1A adrenergic receptor activation increases inhibitory tone in CA1 hippocampus. Epilepsy Res., 84, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Song J., Zhong C., Bonaguidi M.A., Sun G.J., Hsu D., Gu Y., Meletis K., Huang Z.J., Ge S., Enikolopov G. et al. (2012) Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature, 489, 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kana R.K., Libero L.E., Moore M.S. (2011) Disrupted cortical connectivity theory as an explanatory model for autism spectrum disorders. Phys. Life Rev., 8, 410–437. [DOI] [PubMed] [Google Scholar]
- 112.Geschwind D.H., Levitt P. (2007) Autism spectrum disorders: developmental disconnection syndromes. Curr. Opin. Neurobiol., 17, 103–111. [DOI] [PubMed] [Google Scholar]
- 113.Wolff J.J., Gu H., Gerig G., Elison J.T., Styner M., Gouttard S., Botteron K.N., Dager S.R., Dawson G., Estes A.M. et al. (2012) Differences in white matter fiber tract development present from 6 to 24 months in infants with autism. Am. J. Psychiatry, 169, 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McPartland J.C., Jeste S.S. (2015) Connectivity in context: emphasizing neurodevelopment in autism spectrum disorder. Biol. Psychiatry, 77, 772–774. [DOI] [PubMed] [Google Scholar]
- 115.Matson J.L., Nebel-Schwalm M.S. (2007) Comorbid psychopathology with autism spectrum disorder in children: an overview. Res. Dev. Disabil., 28, 341–352. [DOI] [PubMed] [Google Scholar]
- 116.Siris S.G., Addington D., Azorin J.M., Falloon I.R., Gerlach J., Hirsch S.R. (2001) Depression in schizophrenia: recognition and management in the USA. Schizophr. Res., 47, 185–197. [DOI] [PubMed] [Google Scholar]
- 117.Guo N., Yoshizaki K., Kimura R., Suto F., Yanagawa Y., Osumi N. (2013) A sensitive period for GABAergic interneurons in the dentate gyrus in modulating sensorimotor gating. J. Neurosci., 33, 6691–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Samuels B.A., Hen R. (2011) Neurogenesis and affective disorders. Eur. J. Neurosci., 33, 1152–1159. [DOI] [PubMed] [Google Scholar]
- 119.Kempermann G., Krebs J., Fabel K. (2008) The contribution of failing adult hippocampal neurogenesis to psychiatric disorders. Curr. Opin. Psychiatry, 21, 290–295. [DOI] [PubMed] [Google Scholar]
- 120.Wei L., Meaney M.J., Duman R.S., Kaffman A. (2011) Affiliative behavior requires juvenile, but not adult neurogenesis. J Neurosci, 31, 14335–14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wegiel J., Kuchna I., Nowicki K., Imaki H., Wegiel J., Marchi E., Ma S.Y., Chauhan A., Chauhan V., Bobrowicz T.W. et al. (2010) The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol., 119, 755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Guo W., Allan A.M., Zong R., Zhang L., Johnson E.B., Schaller E.G., Murthy A.C., Goggin S.L., Eisch A.J., Oostra B.A. et al. (2011) Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat. Med., 17, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Amiri A., Cho W., Zhou J., Birnbaum S.G., Sinton C.M., McKay R.M., Parada L.F. (2012) Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis. J. Neurosci., 32, 5880–5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hiramoto T., Kang G., Suzuki G., Satoh Y., Kucherlapati R., Watanabe Y., Hiroi N. (2011) Tbx1: identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum. Mol. Genet., 20, 4775–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tassone F., Qi L., Zhang W., Hansen R.L., Pessah I.N., Hertz-Picciotto I. (2011) MAOA, DBH, and SLC6A4 variants in CHARGE: a case-control study of autism spectrum disorders. Autism Res., 4, 250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wei Y.L., Li C.X., Li S.B., Liu Y., Hu L. (2011) Association study of monoamine oxidase A/B genes and schizophrenia in Han Chinese. Behav. Brain Funct., 7, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Simons C.J., van Winkel R. and Group. (2013) Intermediate phenotype analysis of patients, unaffected siblings, and healthy controls identifies VMAT2 as a candidate gene for psychotic disorder and neurocognition. Schizophr. Bull., 39, 848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang S.Y., Lin M.T., Lin W.W., Huang C.C., Shy M.J., Lu R.B. (2009) Association of monoamine oxidase A (MAOA) polymorphisms and clinical subgroups of major depressive disorders in the Han Chinese population. World J. Biol. Psychiatry, 10, 544–551. [DOI] [PubMed] [Google Scholar]
- 129.Yu Y.W., Tsai S.J., Hong C.J., Chen T.J., Chen M.C., Yang C.W. (2005) Association study of a monoamine oxidase a gene promoter polymorphism with major depressive disorder and antidepressant response. Neuropsychopharmacology, 30, 1719–1723. [DOI] [PubMed] [Google Scholar]
- 130.Kunugi H., Ishida S., Kato T., Tatsumi M., Sakai T., Hattori M., Hirose T., Nanko S. (1999) A functional polymorphism in the promoter region of monoamine oxidase-A gene and mood disorders. Mol. Psychiatry, 4, 393–395. [DOI] [PubMed] [Google Scholar]
- 131.Bayles R., Baker E., Eikelis N., El-Osta A., Lambert G. (2010) Histone modifications regulate the norepinephrine transporter gene. Cell Cycle, 9, 4600–4601. [DOI] [PubMed] [Google Scholar]
- 132.Gruss M., Braun K. (2001) Alterations of amino acids and monoamine metabolism in male Fmr1 knockout mice: a putative animal model of the human fragile X mental retardation syndrome. Neural Plast., 8, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sekiguchi H., Iritani S., Habuchi C., Torii Y., Kuroda K., Kaibuchi K., Ozaki N. (2011) Impairment of the tyrosine hydroxylase neuronal network in the orbitofrontal cortex of a genetically modified mouse model of schizophrenia. Brain Res., 1392, 47–53. [DOI] [PubMed] [Google Scholar]
- 134.Altamura C., Dell'Acqua M.L., Moessner R., Murphy D.L., Lesch K.P., Persico A.M. (2007) Altered neocortical cell density and layer thickness in serotonin transporter knockout mice: a quantitation study. Cereb. Cortex, 17, 1394–1401. [DOI] [PubMed] [Google Scholar]
- 135.Murcia C.L., Gulden F., Herrup K. (2005) A question of balance: a proposal for new mouse models of autism. Int. J. Dev. Neurosci., 23, 265–275. [DOI] [PubMed] [Google Scholar]
- 136.Schipul S.E., Keller T.A., Just M.A. (2011) Inter-regional brain communication and its disturbance in autism. Front Syst. Neurosci., 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Usher M., Cohen J.D., Servan-Schreiber D., Rajkowski J., Aston-Jones G. (1999) The role of locus coeruleus in the regulation of cognitive performance. Science, 283, 549–554. [DOI] [PubMed] [Google Scholar]
- 138.Berridge C.W., Waterhouse B.D. (2003) The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res. Brain Res. Rev., 42, 33–84. [DOI] [PubMed] [Google Scholar]
- 139.Werling D.M., Geschwind D.H. (2013) Sex differences in autism spectrum disorders. Curr. Opin. Neurol., 26, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Abel K.M., Drake R., Goldstein J.M. (2010) Sex differences in schizophrenia. Int. Rev. Psychiatry, 22, 417–428. [DOI] [PubMed] [Google Scholar]
- 141.Croen L.A., Connors S.L., Matevia M., Qian Y., Newschaffer C., Zimmerman A.W. (2011) Prenatal exposure to beta2-adrenergic receptor agonists and risk of autism spectrum disorders. J. Neurodev. Disord., 3, 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Connors S.L., Crowell D.E., Eberhart C.G., Copeland J., Newschaffer C.J., Spence S.J., Zimmerman A.W. (2005) beta2-adrenergic receptor activation and genetic polymorphisms in autism: data from dizygotic twins. J. Child Neurol., 20, 876–884. [DOI] [PubMed] [Google Scholar]
- 143.Pietrzak R.H., Gallezot J.D., Ding Y.S., Henry S., Potenza M.N., Southwick S.M., Krystal J.H., Carson R.E., Neumeister A. (2013) Association of posttraumatic stress disorder with reduced in vivo norepinephrine transporter availability in the locus coeruleus. JAMA Psychiatry, 70, 1199–1205. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 144.Alder J., Fujioka W., Lifshitz J., Crockett D.P., Thakker-Varia S. (2011) Lateral fluid percussion: model of traumatic brain injury in mice. J. Vis. Exp., doi:10.3791/3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rocha M.A., Crockett D.P., Wong L.Y., Richardson J.R., Sonsalla P.K. (2008) Na(+)/H(+) exchanger inhibition modifies dopamine neurotransmission during normal and metabolic stress conditions. J. Neurochem., 106, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zeevalk G.D., Manzino L., Hoppe J., Sonsalla P. (1997) In vivo vulnerability of dopamine neurons to inhibition of energy metabolism. Eur. J. Pharmacol., 320, 111–119. [DOI] [PubMed] [Google Scholar]
- 147.Mairet-Coello G., Tury A., DiCicco-Bloom E. (2009) Insulin-like growth factor-1 promotes G(1)/S cell cycle progression through bidirectional regulation of cyclins and cyclin-dependent kinase inhibitors via the phosphatidylinositol 3-kinase/Akt pathway in developing rat cerebral cortex. J. Neurosci., 29, 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Falluel-Morel A., Sokolowski K., Sisti H.M., Zhou X., Shors T.J., Dicicco-Bloom E. (2007) Developmental mercury exposure elicits acute hippocampal cell death, reductions in neurogenesis, and severe learning deficits during puberty. J. Neurochem., 103, 1968–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wagner J.P., Black I.B., DiCicco-Bloom E. (1999) Stimulation of neonatal and adult brain neurogenesis by subcutaneous injection of basic fibroblast growth factor. J. Neurosci., 19, 6006–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sokolowski K., Obiorah M., Robinson K., McCandlish E., Buckley B., DiCicco-Bloom E. (2013) Neural stem cell apoptosis after low-methylmercury exposures in postnatal hippocampus produce persistent cell loss and adolescent memory deficits. Dev. Neurobiol., 73, 936–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cheng Y., Black I.B., DiCicco-Bloom E. (2002) Hippocampal granule neuron production and population size are regulated by levels of bFGF. Eur. J. Neurosci., 15, 3–12. [DOI] [PubMed] [Google Scholar]
- 152.Paxinos G., Franklin K.B.J., Franklin K.B.J. (2001) The Mouse Brain in Stereotaxic Coordinates. Academic Press, San Diego. [Google Scholar]