

Abstract
Epigenetic modifications such as cytosine methylation and histone modification are linked to the pathology of ischemic brain injury. Recent research has implicated 5-hydroxymethylcytosine (5hmC), a DNA base derived from 5-methylcytosine (5mC) via oxidation by ten-eleven translocation (Tet) enzymes, in DNA methylation-related plasticity. Here we show that 5hmC abundance was increased after ischemic injury, and Tet2 was responsible for this increase; furthermore, inhibiting Tet2 expression abolished the increase of 5hmC caused by ischemic injury. The decrease in 5hmC modifications from inhibiting Tet2 activity was accompanied by increased infarct volume after ischemic injury. Genome-wide profiling of 5hmC revealed differentially hydroxymethylated regions (DhMRs) associated with ischemic injury, and DhMRs were enriched among the genes involved in cell junction, neuronal morphogenesis and neurodevelopment. In particular, we found that 5hmC modifications at the promoter region of brain-derived neurotrophic factor (BDNF) increased, which was accompanied by increased BDNF mRNA, whereas the inhibition of Tet2 reduced BDNF mRNA and protein expression. Finally, we show that the abundance of 5hmC in blood samples from patients with acute ischemic stroke was also significantly increased. Together, these data suggest that 5hmC modification could serve as both a potential biomarker and a therapeutic target for the treatment of ischemic stroke.
Introduction
DNA methylation in mammals occurs at cytosine in the CpG dinucleotide sequence and is considered one of the major forms of epigenetic regulation (1). Methylation of CpG islands in gene promoter regions alters chromatin structure and gene transcription, resulting in the transcriptional repression of associated genes (2). Previous studies have shown that DNA methylation plays a very important role in cerebral ischemic injury (3–5). DNA methyltransferases (DNMTs) are responsible for the conversion of unmodified cytosine to 5-methylcytosine (5mC), levels of which are dramatically increased after ischemic insults, contributing to ischemic brain injury (3), and the pharmacological inhibition of DNMTs or DNMT1 knockout causes the brain to resist ischemic insults and promotes brain functional recovery (3,4). Therefore, DNA methylation is an important factor that can affect the outcome after brain injury.
Recently, 5-hydroxymethylcytosine (5hmC) has emerged as a new epigenetic modification; it results from the oxidation of 5mC by ten-eleven translocation (Tet) enzymes (6,7). Unlike 5mC, 5hmC is relatively enriched in the gene bodies of actively transcribed genes, especially at the 3′ end, and is nearly exclusively in CpGs with low-to-medium GC content (8). As a mechanism of demethylation, 5hmC located in gene bodies is associated with higher levels of gene transcription (8–11) . Because 5hmC is highly abundant in neurons in the central nervous system and changes with age (12), 5hmC is believed to play an important role in neurodevelopment and aging (10,11). Furthermore, mounting evidence demonstrates that 5hmC is involved in the pathology of various neurological diseases, among them Alzheimer's disease, fragile X-associated tremor/ataxia syndrome and Huntington's disease (13–15). A recent study also revealed alterations of 5hmC after kidney ischemia (16); however, it remained unclear whether targeting 5hmC would serve as a good therapeutic strategy for the treatment of ischemic brain injury.
In this study, we characterized the alterations of 5hmC after ischemic brain injury and found that 5hmC abundance could be increased by ischemic injury, which is mediated mainly by Tet2 protein. Genome-wide profiling of 5hmC identified differentially hydroxymethylated regions (DhMRs) associated with ischemic injury, which included genes involved in cell junction, neuronal morphogenesis and neurodevelopment. Interestingly, we also observed a significant increase in 5hmC in blood samples from patients with acute ischemic stroke. Our work suggests that 5hmC modification could serve as a potential therapeutic target to treat ischemic stroke.
Results
Ischemic injury increases 5hmC in mouse brain
Previous studies have shown that DNA methylation contributes to neuronal cell death and brain damage after ischemic brain injury (3–5); however, the role of 5hmC modification, a novel epigenetic marker, as a mechanism of demethylation in ischemic brain injury is still unknown. To explore this, we first examined 5hmC levels in ischemic tissues in a middle cerebral artery occlusion (MCAO) mouse model by dot blotting. Proximal occlusion of the middle cerebral artery (MCA) via the intraluminal suture technique (the so-called filament or suture model) is the model most frequently used in experimental stroke research, offering the advantage of inducing reproducible transient or permanent ischemia of the MCA territory. In this MCAO mouse model, at different time points (24, 36 and 48 h and 7 days) after ischemia–reperfusion (I/R) injury, 5hmC levels were increased at 24 h and reached their peak at 48 h (Fig. 1A), but 5mC levels were increased at 24 h and reached peaks at 36 h after I/R injury (Fig. 1B). This was confirmed by quantitative analyses (P < 0.05). At the same time, fluorescent staining demonstrated an increase in 5hmC and a decrease in 5mC at 48 h after I/R injury in ischemic brain areas compared with sham brain areas (Fig. 1C and D). These results suggest that ischemic injury alters the dynamics of cytosine methylation.
Figure 1.

Dynamics of 5hmC and 5mC induced by cerebral I/R injury. The top panel shows the representative dot blot staining of 5hmC (A) and 5mC (B) for the DNA extracted from sham and IR brains, respectively. Methylene blue staining to validate an equal loading amount of DNA. The bottom panel shows statistical analysis of the relative intensity of 5hmC (A) and 5mC (B) in dot blot analysis. The changes of anti-5hmC (C) and anti-5mC (D) from sham (control, Con) and I/R brains are shown. *P < 0.05 and **P < 0.01.
Tet2 expression is upregulated in I/R-insulted mouse brain
Because 5hmC is produced via oxidation of 5mC by Tet proteins, including Tet1, Tet2 and Tet3, we further examined the mRNA levels of Tet1, Tet2 and Tet3 by quantitative polymerase chain reaction (qPCR) at 24 h after I/R injury and determined which Tet gene is involved in the increase of 5hmC modification induced by cerebral I/R injury. We found that Tet2 mRNA levels were significantly increased after I/R injury, but Tet1 mRNA levels were not changed, and Tet3 mRNA levels decreased (Fig. 2A). Western blot further confirmed the upregulation of Tet2 expression (Fig. 2B). Therefore, increased expression of Tet2 correlates with the increase in 5hmC after ischemic injury.
Figure 2.

Expression of Tet2 specifically upregulated after cerebral I/R injury. (A) qPCR analysis of the mRNA levels of Tet1/2/3 in sham and I/R mouse brains at 24 h after I/R injury. GAPDH was used as a control. (B) Anti-Tet2 western blot for the proteins extracted from sham and I/R brains, respectively. Versus sham group, *P < 0.05.
Inhibition of Tet2 activity enhances infarct volume after ischemic injury
To reveal whether the alteration of 5hmC contributes to brain injury after cerebral I/R injury, we used a previously reported Tet2 inhibitor, SC1 (Pluripotin), and administered it intracerebroventricularly in mice (16). Consistent with an earlier report, SC1 reduced both Tet2 mRNA levels and 5hmC abundance in ischemic brain tissues (Fig. 3A and Supplementary Material, Fig. S1) (16). The administration of SC1 worsened brain injury and increased infarct volume, as shown in Figure 3B (P < 0.05). To further examine the role of Tet2 in ischemic brain injury, we looked at the infarct volume of Tet2 knockout mice. Similar to our SC1 results, Tet2 knockout mice also displayed an enlarged infarct volume after I/R injury (Fig. 3C). These findings indicated that Tet2-mediated 5hmC modification plays an important role in cerebral ischemic injury.
Figure 3.

Tet2 inhibition reduces 5hmC levels and increased infarct volume after MCAO. (A) The mice were administered (i.c.v.) with the Tet2 inhibitor SC1 30 min before MCAO. Dot blot analysis was performed with anti-5hmC in ischemic tissues. (B) SC1 pre-treatment increased infarct volume after MCAO. (C) Tet2 KO increased infarct volume after MCAO. Representative TTC-stained coronal sections are shown. *P < 0.05.
I/R injury causes genome-wide 5hmC alteration
To determine the genome-wide 5hmC distribution in both I/R and sham groups, we employed a previously established chemical labeling and affinity purification method (14), coupled with high-throughput sequencing. We used ischemic tissues or matched control tissues from three pairs of I/R and sham mice for the analyses. Genome-wide patterns of 5hmC levels were evaluated by counting 5hmC-mapped reads in each 10 kb bin from the sham and I/R samples and then normalized to the total sequencing coverage. Consistent with our dot blot data, most of the I/R bins contained more 5hmC reads than the sham samples (Fig. 4A). Figure 4B shows the overlapping features of normalized densities of 5hmC reads in sham and I/R groups with known genomic features on annotated gene bodies and defined CGI (CpG islands) obtained from the UCSC Tables for NCBI37v1/mm9. Irrespective of treatment conditions, 5hmC-mapped reads were enriched mostly on intragenic regions. Interestingly, the enrichment of 5hmC at intragenic CGI was significantly less after I/R injury (stroke), whereas there were more 5hmC reads mapped to the exons after I/R injury.
Figure 4.
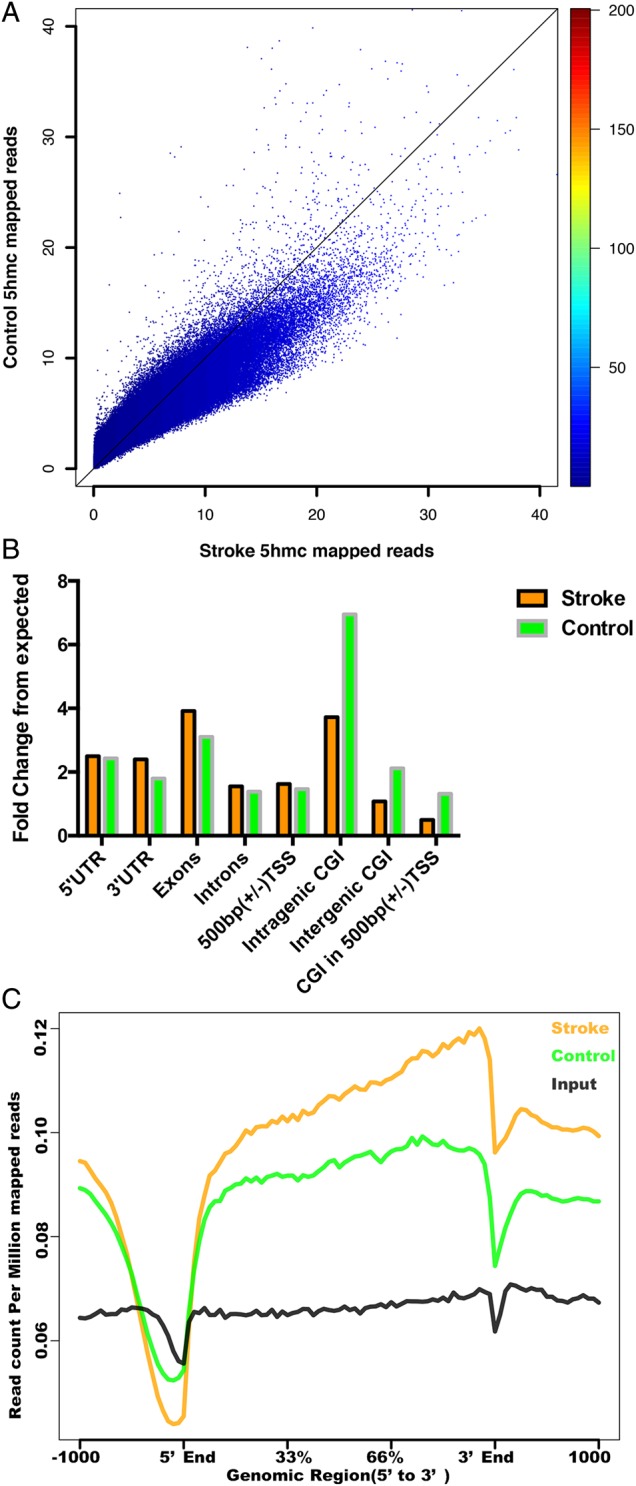
Dynamic changes of 5hmC induced by stroke. (A) Genome-wide 5hmC read density is increased in the stroke mouse model. Genome-wide 5hmC reads were counted within each 10 kb bin in control and stroke mouse genome. 5hmC levels were higher in more stroke bins than control. (B) Normalized 5hmC densities overlapping with various genomic features. (C) Normalized 5hmC read densities across TSS, TES and RefSeq gene bodies. Gene bodies were normalized to 0–100% as relative positions.
As the overall 5hmC level in I/R injury was increased significantly, we sought to visualize the distribution of 5hmC on the defined genomic features. Figure 4C shows the 5hmC reads from sham and I/R mice and non-enriched DNA input mapped to 5 kb up- and downstream of gene bodies, transcription start sites (TSSs) and transcription end sites (TESs) by ngsplot, respectively. We obtained the mean and standard errors of 5hmC reads in each bin for group t-test analysis, and the average P-value was 0.0001, indicating that the intragenic 5hmC is significantly different in sham and I/R samples. The 5hmC levels in both sham and I/R mice were largely depleted on TSSs, consistent with previous results (9,10). The distributions of 5hmC on TES showed the same depletion pattern as on TSS, albeit to a lesser extent. The intragenic 5hmC levels in I/R mice were significantly higher than those in sham mice, in contrast to the mild differences on TSS or TES. We found this particularly interesting as our previous data suggested that the intragenic 5hmC peaks are correlated with brain-specific gene expression (10).
Identification and characterization of DhMRs after I/R injury
We next proceeded to identify and characterize I/R (stroke)- and sham (control)-specific DhMRs across the genome, aiming to pinpoint specific loci that displayed altered 5hmC profiles as a result of I/R injury. We found a total of 2658 sham-specific DhMRs and 4311 stroke-specific DhMRs, which were distributed across the genome (Fig. 5A). To investigate the defined genomic features associated with the identified DhMRs, we annotated both sham- and stroke-specific DhMRs of the mouse genome using HOMER (Hypergeometric Optimization of Motif EnRichment) software. HOMER annotations revealed that more stroke-specific DhMRs were located on promoters, 5′ UTR and 3′ UTR (Fig. 5B–D). To further explore the biological significance of these particular DhMRs, we used GREAT (Genomic Regions Enrichment of Annotations Tool) to perform gene ontology (GO) analyses for the sham- and stroke-specific DhMRs. Several GO biological processes associated with cell junction and neuronal development were found to be highly enriched among stroke-specific DhMRs, whereas other pathways were associated with sham-specific DhMRs, albeit with less significant P-values (Fig. 5E). We then searched for the motifs among both sham- and stroke-specific DhMRs. Among stroke-specific DhMRs, we saw a significant enrichment of Iroquois homeobox protein 4 (IRX4)-binding motifs (Fig. 5E). Intriguingly, a single nucleotide polymorphism (SNP) associated with IRX4 was linked to ischemic stroke in a previous genome-wide association study (17). These data together indicate that specific pathways could be modulated by 5hmC in response to I/R injury, which may contribute to ischemic injury.
Figure 5.

Identification and characterization of DhMRs in stroke. (A) Chromosome circular map shows a genome-wide dynamic change of normalized 5hmC read count ratio between control and stroke in genome-wide DhMRs. The orange bar represents the normalized 5hmC read count ratio in control-specific DhMRs. The green bar represents the normalized 5hmC read count ratio in stroke-specific DhMRs. (B) Control- and stroke-specific DhMRs overlapping with various genomic features. (C and D) DhMR-associated genes showed differential 5hmC signals. (E) Gene oncology analyses for stroke-specific DhMRs. GO analysis was performed using GREAT with stroke-specific DhMRs. Most significant GO biological processes are indicated in the bar graph. Enriched IRX4-binding motif among stroke-specific DhMRs is shown.
SC1 pre-treatment decreases the expression of BDNF after I/R injury
Although unchanged or decreased 5hmC modification in genes was not consistent with changes in their mRNA levels in earlier literature, our previous data revealed that the increased 5hmC modification in genes was accompanied by an increase in the mRNA levels of genes (18). To explore this further, we examined the relationship between the 5hmC modification of brain-derived neurotrophic factor (BDNF), a critical neuroprotective neurotrophic factor after cerebral ischemia, and BDNF expression. We found that the 5hmC modification increased in BDNF promoter and transcriptional end regions after cerebral I/R injury (Fig. 6A); at the same time, the mRNA level of BDNF was significantly increased (Fig. 6B). On the contrary, SC1 treatment reduced BDNF mRNA (Fig. 6C) and protein levels (Fig. 6D).
Figure 6.

Ischemic injury increased both 5hmC level at the BDNF promoter region and BDNF expression. (A) Differential 5hmC distributions on the BDNF gene were shown in the IGV browser. (B) BDNF mRNA increased after cerebral I/R injury. (C) When the mice were treated with the Tet2 inhibitor SC1, SC1 decreased BDNF mRNA after cerebral I/R injury. GAPDH was used as a loading control. (D) Representative graph of BDNF proteins is shown. SC1 treatment reduced BDNF expression after cerebral I/R injury. Beta-tubulin was used as a loading control. Versus sham group, *P < 0.05 and versus I/R group, ##P < 0.05.
To further determine the 5hmc alteration of genes between I/R and sham mice, a total of 33 protein-coding genes already linked to stroke were analyzed; the correlation rate was 75.8%. Sixteen stroke-related genes encoding long-non-coding RNAs (lncRNAs) were also analyzed, and the correlation rate was 43.8% [Supplementary Material, Tables S1 (19–32) and S2 (33) and Fig. S2].
5hmC level is higher in blood samples from patients with acute ischemic stroke
To examine whether 5hmC modification is a marker of ischemic stroke, we checked 5hmC levels in blood samples from patients with acute ischemic stroke and age-matched controls by dot blotting. Compared with healthy controls, 5hmC levels were significantly increased in blood samples from patients with ischemic stroke (Fig. 7A and B).
Figure 7.

5hmC level is increased in blood cell DNA from patients with acute ischemic stroke. (A) Dot blotting analysis with anti-5hmC in blood samples from controls and patients with acute ischemic stroke. (B) Statistical analysis of the relative intensity of 5hmC in controls and patients with acute ischemic stroke. N = 12. Versus control group, *P < 0.05.
Discussion
Epigenetic modification is emerging as a critical mechanism in the molecular pathogenesis of various human diseases. DNA methylation has been studied widely in ischemic brain injury in past years, but the role played by 5hmC, a new epigenetic modification, after cerebral ischemia has been unclear. In this study, we showed that overall 5hmC abundance was significantly increased in ischemic brain tissues; the enzyme Tet2 was responsible for this increase. The reduction of Tet2 by either a chemical (SC1) or knockout worsened ischemic brain injury in an MCAO mouse model. Genome-wide 5hmC profiling led to the identification of DhMRs (both gain and loss) that are enriched on genes involved in cell junction and neuronal development after stroke. Finally, we showed that alterations of 5hmC on the BDNF gene influenced the mRNA and protein levels of BDNF. These findings indicate that 5hmC modifications could potentially serve as a new therapeutic target in the treatment of stroke.
5hmC abundance increases after ischemic brain injury in mice
Until recently, 5hmC was known to be abundant in certain cell types, such as Purkinje neurons and embryonic stem (ES) cells (6,12). As the brain has high levels of 5hmC, it is believed to play a significant role in neural differentiation and development (11,12,34–36). Mounting evidence demonstrates that modification of 5hmC can contribute to many neurological disorders, among them Huntington's disease, Alzheimer's disease and fragile X-associated tremor/ataxia syndrome (13–15). Despite the recent growing interest in 5hmC modifications, whether 5hmC is involved in cerebral ischemia had gone unexplored. Our results here indicate that overall 5hmC abundance is significantly increased in ischemic brain regions, and reducing 5hmC increases the infarct volume after MCAO, suggesting a regulatory role for 5hmC modification on stroke-related genes. We thus speculated that 5hmC may play an important role in regulating gene expression after ischemic brain injury. In contrast to ischemic brain injury, overall 5hmC abundance was reduced in the mouse kidney after I/R (16). This discrepancy may be due to tissue differences when exposed to ischemic stimuli. More importantly, in this study, the increase of 5hmC modifications in blood cells was also found in patients with acute ischemic stroke, consistent with results in mouse brains after ischemic injury. This suggests that the alteration of 5hmC could be a response of the whole body after ischemic brain injury.
Tet2 is responsible for the increase of 5hmC after cerebral ischemic stimuli
5hmC is generated by Tet proteins, which transfer molecular oxygen to a hydroxyl group to 5mC (6). So far, there are three known Tet protein family members: Tet1, Tet2 and Tet3 (37). Tet1–3 proteins have specific distributions in different tissues and are most abundant in zygotes, ES cells and the brain. Tet2 proteins are highly expressed in ES cells, especially hematopoietic cells, and are believed to play key roles in hematopoiesis because Tet2 knockout mice have severe hematopoietic defects (38–40). In this study, we showed that Tet2, but not Tet1 or Tet3, was responsible for the increase in 5hmC modifications and played an important role in ischemic brain injury. Because Tet2 knockout increased the infarct volume after MCAO (Fig. 3), this points to a specific role of Tet2 for ischemic insults. Our results also demonstrated that the inhibition of Tet2 expression by SC1 decreased both 5hmC (Fig. 3) and BDNF expression (Fig. 6) in mouse brains upon I/R injury. Because previous studies revealed that loss of 5mC causes the brain to resist ischemic insults and promotes brain functional recovery (3,4), the increase in Tet2 after I/R injury in this study (Fig. 2) may lead to a reduction of 5mC and an increase in gene demethylation, which could be an important mechanism for protecting the brain from ischemic brain injury. Therefore, increasing Tet2 expression could be a therapeutic strategy for the treatment of ischemic brain injury.
Dynamics in response to ischemic injury
Previous research has shown that 5hmC can be very dynamic during neurodevelopment, as well as in response to disease states and environmental stimuli. The data presented here are consistent with those findings. Our genome-wide 5hmC profiling studies suggest that most 5hmC changes occurred around the 5′-UTR, 3′-UTR and TSSs. The identification of wild-type and stroke-specific DhMRs allowed us to focus on unique genomic loci with altered 5hmC levels. GO analyses of these loci suggest that DhMRs are highly enriched among genes that are involved in cell junction organization, neuronal morphogenesis and neurodevelopment. The discovery of this enrichment among genes involved in cell junction organization is intriguing. Indeed, synaptic plasticity is well known to require synchronization and coordination of neurons and glial cells via various mechanisms of intercellular communication. Genes involved in cell junction play important roles in that process. Moreover, a growing body of evidence has revealed that intercellular communication could be critical in the spread of protective and/or deleterious signals during stroke. Our findings suggest that the epigenetic modulation of these cell junction organization genes may play significant roles in stroke.
Further sequence analyses of the DhMRs associated with stroke also revealed an enrichment of the IRX4-binding motif. In an earlier study, the SNP rs4975709 in IRX4 (odds ratio = 5.06; 95% confidence interval 2.66–9.62; P = 7.7 × 10−7) appeared to be associated with ischemic stroke (17). IRX4 is among the mammalian orthologs of the Iroquois complex in Drosophila melanogaster, which includes the highly homologous homeobox genes caupolican, araucan and mirror that act as pre-pattern molecules in neurogenesis. The IRX4 gene appears to be an important mediator of ventricular differentiation during cardiac development. IRX4 might play a causal role in the development of congenital heart disease, particularly ventricular septal defect (41). It would be interesting to explore how changes in 5hmC could alter IRX4 binding activity in the context of ischemic stroke.
Modification and BDNF expression
Earlier work uncovered no clear correlation between 5hmC modification and gene expression, at least in ES cells. BDNF, a member of the neurotrophin family, is known to play a key role in the development and survival of neurons in the central nervous system (42). A previous study indicated that the neuronal activity-dependent activation of the BDNF gene is mediated by decreased CpG methylation of the BDNF promoter IV and the release of a chromatin repressor complex containing MeCP2 methyl-binding protein (43). Therefore, we examined the demethylation role of 5hmC on the BDNF promoter. We found a change of 5hmC modification in the BDNF promoter region after MCAO by high-throughput sequencing (Fig. 6A). At the same time, the increased expression of BDNF was confirmed by western blot and reverse transcription PCR (RT-PCR). When mice were treated with SC1, the expression of BDNF decreased significantly, suggesting that the expression of BDNF is regulated by 5hmC modification at BDNF promoters. Thus it is possible that 5hmC modification could regulate the expression of other stroke-related genes. This is why we chose to examine 49 stroke-related genes, including 16 genes encoding lncRNA. By comparing 5hmC alterations with gene expression changes reported in earlier literature, we found that, among 49 stroke-related genes, 75.8% of the protein-coding genes and 43.8% of the genes encoding stroke-related lncRNA displayed 5hmC alterations that were consistent with their expression changes. This suggests that 5hmC modification may play a more important role in regulating the expression of the protein-coding genes.
In summary, we show that ischemic injury increases the abundance of 5hmC, which is mediated by Tet2 enzyme, and our manipulation of Tet2 activity could modulate the severity of ischemic injury. Genome-wide analyses suggest an enrichment of 5hmC alterations among the genes involved in cell junction and neurodevelopment, highlighting that the key role cell junction organization could play in response to ischemic injury. Our data suggest that 5hmC may serve as a biomarker or therapeutic target for the treatment of ischemic stroke.
Materials and Methods
Animals
Male ICR mice weighing ∼23–25 g were purchased from the SLAC Company (China). Tet2 KO mice were reported previously and obtained from the Jackson Laboratory (38). All animal procedures were approved by the University Committee on Animal Care of Soochow University and conducted in accordance with the guidelines of the Animal Use and Care of the National Institutes of Health (NIH) and the ARRIVE (Animal Research: Reporting In Vivo Experiments).
MCAO model
The MCAO method was described previously (44). Mice were anesthetized by 4% chloral hydrate, and then the right common carotid artery (CCA), the external carotid artery (ECA) and the internal carotid artery were isolated through a ventral midline neck incision. The ECA was ligated and cut at 2–3 mm distal from the ECA–CCA branch. At about 1.5 mm distal from the ECA–CCA branch, a small incision was made on the ECA. A 6-0 nylon filament (Ethicon Inc., USA) with silicone resin-coated tip (0.23 mm in diameter and 4 mm in length) was inserted through the incision into the internal carotid artery about 9–11 mm. The reperfusion of the ischemic area was carried out by removing the filament after 45 or 75 min of occlusion of the artery. Sham mice underwent the same experimental procedures, but the nylon filament was only advanced about 5 mm. Body temperature was kept at 36.5–37.5°C from the start of the surgery until the animals recovered from anesthesia. Cerebral blood flow was monitored as described previously (45).
Intracerebroventricular administration of SC1
The Tet2 inhibitor SC1 (Pluripotin, Cayman Chemical, USA) was administered into the right lateral ventricle about 2.5 mm deep by a 28-G needle through a small hole on the right parietal region (0.5 mm posterior and 1.0 mm lateral to the Bregma). Two microliters of SC1 (10 mm) or saline was injected.
2,3,5-Triphenyltetrazolium chloride staining
After the mice were deeply anesthetized at 24 h after reperfusion, brains were removed quickly from the skull and then cut into 1 mm slices. Slices were incubated with 0.2% 2,3,5-triphenyltetrazolium chloride (TTC, Sigma-Aldrich Company, USA) at 37°C for 30 min, then fixed in 4% paraformaldehyde and photographed at 24 h after fixation. The stained slices were photographed by a camera. The infarct area in each brain slice was analyzed by AlphaEase Image Analysis Software V3.1.2 (Alpha Innotech Corp., USA). The percentage of hemispheric infarction volume was calculated as described in our previous study (46).
Dot blot analysis
After 45 min of ischemia followed by 24, 36 and 48 h and 7 days of reperfusion, the right hemisphere tissue was collected and grounded in liquid nitrogen and stored at −80°C. The genomic DNA was extracted for dot blot analysis by a DNA extraction kit (Version 4.0) from Takara Company (China). The dot blot assay was performed as described previously (47). Briefly, DNA samples were pre-treated with 2 N NaOH/10 mm Tris–HCl (pH 8.5) and spotted on a nitrocellulose membrane at room temperature for 15 min. After incubation at 80°C in an oven for 30 min, the membrane was blocked with 5% milk in phosphate-buffered saline with 0.1% Tween 20 (PBST) for 1 h. After washing, the membrane was incubated with the primary antibody (anti-5mC mouse antibody, Millipore, USA or anti-5hmC rabbit antibody, Active Motif, USA) at 4°C overnight. On the second day, the membrane was incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, USA) for 1 h at room temperature. The membrane was washed and developed with the ECL chemiluminescence system (Thermo Company, USA). The immunoreactive dots were captured on autoradiographic films. The densitometry of the dots was analyzed with Alpha Ease Image Analysis Software.
Methylene blue staining
The dot blot membrane was hybridized with 0.02% methylene blue (Sigma-Aldrich Company) in 0.3 m sodium acetate (pH 5.2) to stain DNA for 10 min (48). After washing and photography, the densitometry of methylene blue staining was analyzed with Alpha Ease Image Analysis Software, and we used a loading control to evaluate the densitometries of 5mC and 5hmC.
Immunohistochemistry analysis
At 24 or 48 h after cerebral reperfusion, mice were deeply anesthetized and brains were perfused through the heart with 4% paraformaldehyde. Brains were post-fixed for 24 h and then were dehydrated by 15% and 30% sucrose. After being embedded with tissue freezing medium (O.C.T., SAKURA Tissue-Tek, USA), brain tissues were cut into 15 μm thickness frozen sections. After sections were incubated with blocking buffer for 1 h at room temperature, sections were incubated with primary antibodies (anti-5mC or anti-5hmC) overnight at 4°C. Immunofluorescence secondary antibodies (Jackson ImmunoResearch Laboratories) were then applied and incubated at 37°C for 1 h. Microphotographs were taken under a fluorescent microscope.
Western blot
After 45 min of MCAO and 48 h of reperfusion, the right hemisphere tissue was collected and grounded in liquid nitrogen and stored at −80°C. Brain tissues were solubilized in lysis buffer by sonication on ice. The tissue samples were then centrifuged and the supernatants collected for protein concentration determination. The same amount of total proteins (about 30–50 μg) was separated by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes. After blocking with 5% dry milk in PBST for 2 h, the blots were incubated with primary antibody in PBST overnight at 4°C and then washed and incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. The blots were developed with the ECL chemiluminescence system, and the densitometry of immunoreactive bands captured on autoradiographic films was analyzed with Alpha Ease Image Analysis Software. Anti-BDNF rabbit antibody (Epitomics, USA), anti-Tet2 rabbit antibody (Abcam, USA) and anti-β-tubulin mouse antibody (Sigma-Aldrich Company) were used.
Quantitative RT-PCR
Total RNA was extracted from tissues. Reverse transcription of cDNA was performed using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Germany), and RT-PCR was conducted with SYBR Green (Roche, Germany) on the 7500 Real-Time PCR system (Applied Biosystems, USA). GAPDH was used as an endogenous control. Primer sequences are listed in Supplementary Material, Table S3.
Genomic DNA isolation for 5hmC profiling
Ischemic brain area samples and sham samples were collected at 48 h after I/R and homogenized on ice by tissue grinder in 600 μl lysis buffer [100 mm Tris–HCl (pH 8.5), 5 mm EDTA, 0.2% SDS, 200 mm NaCl]. The samples were incubated with 5 μl Proteinase K (20 mg/ml) at 55°C overnight. After a quick spin, 2 μl RNase A (10 mg/ml) was added to each sample and kept at room temperature for 30 min. An equal volume of phenol:chloroform:isoamylalcohol (25:24:1) was mixed with each sample and vortexed for 20 s. After centrifuging at 12 000 rpm for 10 min, the upper phase of the samples was transferred to new tubes. The same volume of isopropanol was mixed with the samples to precipitate DNA. DNA pellets were washed with 70% ethanol and dissolved in sterile water. DNA samples were used for 5hmC profiling analysis.
5hmC-specific. chemical labeling, affinity purification and sequencing
5hmC enrichment was performed using a previously described procedure with an improved selective chemical labeling method (10). DNA libraries were generated following the Illumina protocol for ‘Preparing Samples for ChIP Sequencing of DNA’ (Part no. 111257047 Rev. A) using 25–50 ng of input genomic DNA or 5hmC-captured DNA to initiate the protocol.
Bioinformatic data analyses
Processing of sequencing data was performed as described previously (10). Briefly, FASTQ sequence files from biological replicates were concatenated and aligned to the Mus musculus reference genome (NCBI37v1/mm9) using Bowtie 0.12.6, keeping only unique non-duplicate genomic matches with no more than two mismatches within the first 25 bp. Unique, non-duplicate reads from non-enriched input genomic DNA of ischemic brain regions and each 5hmC-enriched sequence set were counted in 100, 1000 and 10 000 bp bins genome-wide and subsequently normalized to the total number of non-duplicate reads in millions. Input-normalized values were then subtracted from 5hmC-enriched values per bin to generate normalized 5hmC signals. To determine genomic regions with altered 5hmC profiles, we identified true 5hmC-enriched regions or ‘peaks’ using the Model-based Analysis of ChIP-Seq (MACS) algorithm.
Correlation analyses between 5hmC abundance and gene expression after stroke
To demonstrate the relationship between 5hmC abundance and gene expression after stroke, we searched the literature and examined 49 stroke-related genes, including 16 genes encoding lncRNA that were reported in other studies. All of these gene views of 5hmC relative enrichment intensity were generated using the software Integrative Genomics Viewer (IGV 2.0.10). We further analyzed the changes of the areas of DhMRs in these stroke-related genes and then compared the changes in 5hmC abundance with the changes in gene expression from earlier reports.
Patients with acute ischemic stroke and controls
This study was approved by the Ethics Committee of Soochow University. Twelve patients with first-ever acute ischemic stroke (age 61.8 ± 3.10) and 12 age-matched control subjects (age 60.8 ± 3.45) were recruited for this study and provided informed consent to participate. These selected patients were admitted within 3 days after onset of stroke symptoms and were confirmed with diffusion-weighed image sequence of magnetic resonance imaging. Control subjects were collected from outpatients without cerebral infarction during the same period. Patients and controls with tumors, leukemia and neurodegenerative diseases, including Alzheimer's disease and Parkinson's disease, were excluded.
Statistical analysis
All data are expressed as means ± SEM. GraphPad Prism 4 (GraphPad Software Inc., USA) was used for statistical analysis. Student's t-test was used to determine the differences between two groups for infarct volume; one-way analysis of variance followed by Tukey's multiple-comparison test was used to compare the differences among groups if there were significant differences. P < 0.05 was considered statistically significant.
Accession number
Sequencing data have been deposited to GEO with accession number GSE67188.
Supplementary Material
Funding
This study was supported by grants from the National Natural Science Foundation of China (81071095, 81120108011 and 81361128010 to X.X.), Ministry of Science and Technology of China (2013CB945400 to M.S.), National Institutes of Health (NS079625 to P.J.), Canadian Institutes of Health Research (CIHR, CCI-132567 to J.K.) and the Priority Academic Program Development of Jiangsu Higher Education Institutions of China.
Supplementary Material
Acknowledgement
The authors would like to thank C. Strauss for critical reading of the manuscript.
Conflict of Interest statement. None declared.
References
- 1.Jones P.A., Takai D. (2001) The role of DNA methylation in mammalian epigenetics. Science, 293, 1068–1070. [DOI] [PubMed] [Google Scholar]
- 2.Jaenisch R., Bird A. (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet., 33, 245–254. [DOI] [PubMed] [Google Scholar]
- 3.Endres M., Meisel A., Biniszkiewicz D., Namura S., Prass K., Ruscher K., Lipski A., Jaenisch R., Moskowitz M.A., Dirnagl U. (2000) DNA methyltransferase contributes to delayed ischemic brain injury. J. Neurosci., 20, 3175–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qureshi I.A., Mehler M.F. (2010) Emerging role of epigenetics in stroke: part 1: DNA methylation and chromatin modifications. Arch. Neurol., 67, 1316–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westberry J.M., Prewitt A.K., Wilson M.E. (2008) Epigenetic regulation of the estrogen receptor alpha promoter in the cerebral cortex following ischemia in male and female rats. Neuroscience, 152, 982–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tahiliani M., Koh K.P., Shen Y.H., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L. et al. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science, 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito S., D'Alessio A.C., Taranova O.V., Hong K., Sowers L.C., Zhang Y. (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature, 466, 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu M., Hon G.C., Szulwach K.E., Song C.X., Zhang L., Kim A., Li X., Dai Q., Shen Y., Park B. et al. (2012) Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell, 149, 1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song C.X., Szulwach K.E., Fu Y., Dai Q., Yi C., Li X., Li Y., Chen C.H., Zhang W., Jian X. et al. (2011) Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol., 29, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szulwach K.E., Li X., Li Y., Song C.X., Wu H., Dai Q., Irier H., Upadhyay A.K., Gearing M., Levey A.I. et al. (2011) 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci., 14, 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang T., Pan Q., Lin L., Szulwach K.E., Song C.X., He C., Wu H., Warren S.T., Jin P., Duan R. et al. (2012) Genome-wide DNA hydroxymethylation changes are associated with neurodevelopmental genes in the developing human cerebellum. Hum. Mol. Genet., 21, 5500–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kriaucionis S., Heintz N. (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science, 324, 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chouliaras L., Mastroeni D., Delvaux E., Grover A., Kenis G., Hof P.R., Steinbusch H.W., Coleman P.D., Rutten B.P., van den Hove D.L. (2013) Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients. Neurobiol. Aging, 34, 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao B., Lin L., Street R.C., Zalewski Z.A., Galloway J.N., Wu H., Nelson D.L., Jin P. (2014) Genome-wide alteration of 5-hydroxymethylcytosine in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet., 23, 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang F.L., Yang Y.R., Lin X.W., Wang J.Q., Wu Y.S., Xie W.J., Wang D.D., Zhu S., Liao Y.Q., Sun Q.M. et al. (2013) Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum. Mol. Genet., 22, 3641–3653. [DOI] [PubMed] [Google Scholar]
- 16.Huang N., Tan L., Xue Z.G., Cang J., Wang H. (2012) Reduction of DNA hydroxymethylation in the mouse kidney insulted by ischemia reperfusion. Biochem. Biophys. Res. Commun., 422, 697–702. [DOI] [PubMed] [Google Scholar]
- 17.Schurks M., Buring J.E., Ridker P.M., Chasman D.I., Kurth T. (2011) Genetic determinants of cardiovascular events among women with migraine: a genome-wide association study. PloS ONE, 6, e22106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao B., Jin P. (2014) Unlocking epigenetic codes in neurogenesis. Genes Dev., 28, 1253–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X., Luo Y., Sun H., Feng J., Ma S., Liu J., Huang B. (2015) Dynamic expression changes of Bcl-2, caspase-3 and Hsp70 in middle cerebral artery occlusion rats. Brain Inj., 29, 93–97. [DOI] [PubMed] [Google Scholar]
- 20.Raghavendra Rao V.L., Bowen K.K., Dhodda V.K., Song G., Franklin J.L., Gavva N.R., Dempsey R.J. (2002) Gene expression analysis of spontaneously hypertensive rat cerebral cortex following transient focal cerebral ischemia. J. Neurochem., 83, 1072–1086. [DOI] [PubMed] [Google Scholar]
- 21.Li J., Zhao Y.D., Zeng J.W., Chen X.Y., Wang R.D., Cheng S.Y. (2014) Serum brain-derived neurotrophic factor levels in post-stroke depression. J. Affect. Disord., 168, 373–379. [DOI] [PubMed] [Google Scholar]
- 22.Miyazaki H., Nagashima K., Okuma Y., Nomura Y. (2001) Expression of glial cell line-derived neurotrophic factor induced by transient forebrain ischemia in rats. Brain Res., 922, 165–172. [DOI] [PubMed] [Google Scholar]
- 23.Talwar T., Srivastava M.V. (2014) Role of vascular endothelial growth factor and other growth factors in post-stroke recovery. Ann. Ind. Acad. Neurol., 17, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin T.N., Wang P.Y., Chi S.I., Kuo J.S. (1998) Differential regulation of ciliary neurotrophic factor (CNTF) and CNTF receptor alpha (CNTFR alpha) expression following focal cerebral ischemia. Brain Res., 55, 71–80. [DOI] [PubMed] [Google Scholar]
- 25.Slevin M., Krupinski J., Mitsios N., Perikleous C., Cuadrado E., Montaner J., Sanfeliu C., Luque A., Kumar S., Kumar P. et al. (2008) Leukaemia inhibitory factor is over-expressed by ischaemic brain tissue concomitant with reduced plasma expression following acute stroke. Eur. J. Neurol., 15, 25–33. [DOI] [PubMed] [Google Scholar]
- 26.Wacker B.K., Perfater J.L., Gidday J.M. (2012) Hypoxic preconditioning induces stroke tolerance in mice via a cascading HIF, sphingosine kinase, and CCL2 signaling pathway. J. Neurochem., 123, 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei D., Xiong X., Zhao H. (2015) Tim-3 cell signaling and iNOS are involved in the protective effects of ischemic postconditioning against focal ischemia in rats. Metab. Brain Dis., 30, 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenglet S., Montecucco F., Mach F., Schaller K., Gasche Y., Copin J.C. (2014) Analysis of the expression of nine secreted matrix metalloproteinases and their endogenous inhibitors in the brain of mice subjected to ischaemic stroke. Thromb. Haemost., 112, 363–378. [DOI] [PubMed] [Google Scholar]
- 29.Bergeron M., Yu A.Y., Solway K.E., Semenza G.L., Sharp F.R. (1999) Induction of hypoxia-inducible factor-1 (HIF-1) and its target genes following focal ischaemia in rat brain. Eur. J. Neurosci., 11, 4159–4170. [DOI] [PubMed] [Google Scholar]
- 30.Boscia F., Gala R., Pignataro G., de Bartolomeis A., Cicale M., Ambesi-Impiombato A., Di Renzo G., Annunziato L. (2006) Permanent focal brain ischemia induces isoform-dependent changes in the pattern of Na+/Ca2+ exchanger gene expression in the ischemic core, periinfarct area, and intact brain regions. J. Cereb. Blood Flow Metab., 26, 502–517. [DOI] [PubMed] [Google Scholar]
- 31.Pal G., Lovas G., Dobolyi A. (2014) Induction of transforming growth factor beta receptors following focal ischemia in the rat brain. Plos ONE, 9, e106544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clancy P., Lincz L.F., Maguire J., McEvoy M., Koblar S.A., Golledge J. (2014) Tenascin-C is increased in atherothrombotic stroke patients and has an anti-inflammatory effect in the human carotid artery. Biofactors, 40, 448–457. [DOI] [PubMed] [Google Scholar]
- 33.Dharap A., Nakka V.P., Vemuganti R. (2012) Effect of focal ischemia on long noncoding RNAs. Stroke, 43, 2800–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ficz G., Branco M.R., Seisenberger S., Santos F., Krueger F., Hore T.A., Marques C.J., Andrews S., Reik W. (2011) Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature, 473, 398–402. [DOI] [PubMed] [Google Scholar]
- 35.Pastor W.A., Pape U.J., Huang Y., Henderson H.R., Lister R., Ko M., McLoughlin E.M., Brudno Y., Mahapatra S., Kapranov P. et al. (2011) Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature, 473, 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruzov A., Tsenkina Y., Serio A., Dudnakova T., Fletcher J., Bai Y., Chebotareva T., Pells S., Hannoun Z., Sullivan G. et al. (2011) Lineage-specific distribution of high levels of genomic 5-hydroxymethylcytosine in mammalian development. Cell Res., 21, 1332–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iyer L.M., Tahiliani M., Rao A., Aravind L. (2009) Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle, 8, 1698–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ko M., Bandukwala H.S., An J., Lamperti E.D., Thompson E.C., Hastie R., Tsangaratou A., Rajewsky K., Koralov S.B., Rao A. (2011) Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl Acad. Sci. USA, 108, 14566–14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Z., Cai X., Cai C.L., Wang J., Zhang W., Petersen B.E., Yang F.C., Xu M. (2011) Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood, 118, 4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quivoron C., Couronne L., Della Valle V., Lopez C.K., Plo I., Wagner-Ballon O., Do Cruzeiro M., Delhommeau F., Arnulf B., Stern M.H. et al. (2011) TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell, 20, 25–38. [DOI] [PubMed] [Google Scholar]
- 41.Cheng Z., Wang J., Su D., Pan H., Huang G., Li X., Li Z., Shen A., Xie X., Wang B. et al. (2011) Two novel mutations of the IRX4 gene in patients with congenital heart disease. Hum. Genet., 130, 657–662. [DOI] [PubMed] [Google Scholar]
- 42.Bibel M., Barde Y.A. (2000) Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev., 14, 2919–2937. [DOI] [PubMed] [Google Scholar]
- 43.Zhou Z.L., Hong E.J., Cohen S., Zhao W.N., Ho H.Y.H., Schmidt L., Chen W.G., Lin Y.X., Savner E., Griffith E.C. et al. (2006) Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron, 52, 255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu X., Chua C.C., Gao J., Hamdy R.C., Chua B.H. (2006) Humanin is a novel neuroprotective agent against stroke. Stroke, 37, 2613–2619. [DOI] [PubMed] [Google Scholar]
- 45.Wang C., Pei A.J., Chen J., Yu H.L., Sun M.L., Liu C.F., Xu X.S. (2012) A natural coumarin derivative esculetin offers neuroprotection on cerebral ischemia/reperfusion injury in mice. J. Neurochem., 121, 1007–1013. [DOI] [PubMed] [Google Scholar]
- 46.Xu X.S., Chua C.C., Gao F.P., Chua K.W., Wang H., Hamdy R.C., Chua B.H.L. (2008) Neuroprotective effect of humanin on cerebral ischemia/reperfusion injury is mediated by a PI3 K/Akt pathway. Brain Res., 1227, 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S.H., Ito S., Yang C., Wang P., Xiao M.T. et al. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell, 19, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Minor E.A., Court B.L., Young J.I., Wang G.F. (2013) Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem., 288, 13669–13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.