

Abstract
Patient: Female, 21
Final Diagnosis: Unresectable liver adenomatosis associated with congenital absence of portal vein
Symptoms: —
Medication: —
Clinical Procedure: Living donor liver transplantation
Specialty: Transplantology
Objective:
Rare disease
Background:
Abernethy malformation (AM), or congenital absence of portal vein (CAPV), is a very rare disease which tends to be associated with the development of benign or malignant tumors, usually in children or young adults.
Case Report:
We report the case of a 21-year-old woman diagnosed with type Ib AM (portal vein draining directly into the inferior vena cava) and unresectable liver adenomatosis. The patient presented mild liver dysfunction and was largely asymptomatic. Living donor liver transplantation was performed using a left hemiliver graft from her mother. Postoperatively, the patient attained optimal liver function and at 9-month follow-up has returned to normal life.
Conclusions:
We consider that living donor liver transplantation is the best therapeutic solution for AM associated with unresectable liver adenomatosis, especially because compared to receiving a whole liver graft, the waiting time on the liver transplantation list is much shorter.
MeSH Keywords: Adenoma, Liver Cell; Congenital Abnormalities; Liver Transplantation
Bakground
Congenital absence of the portal vein (CAPV), which presently is synonymous with the term Abernethy malformation, is a very rare anomaly of the abdominal splanchnic venous system and was first described in 1793 by John Abernethy in a 10-month-old girl during an autopsy [1]. Howard and Davenport [2] coined the term ‘Abernethy malformation’, thus describing any congenital extrahepatic portosystemic shunt, diverting portal blood from the liver. CAPV is usually (80%) diagnosed in children [3]; age at diagnosis ranges from the prenatal period [4] to a 64-year-old patient [5]. Upon presentation, there is a wide range of clinical signs and symptoms: various degrees of encephalopathy due to hyperammonemia, jaundice, fatigue, right upper-quadrant pain, and platypnea provoked by orthodeoxia associated with hepatopulmonary syndrome (HPS) [6]. Cyanosis as the only complaint, also associated with HPS, was reported by Alvarez et al. in a 5-year-old boy with CAPV [7]. Early detection of CAPV, which has been increasingly accomplished in recent years due to the improved imaging techniques, is of paramount importance in treating these patients, especially because a significant subset of patients with CAPV develop benign or malignant liver tumors.
There is currently no standardized approach for the management of CAPV; however, all patients in whom encephalopathy and poor liver function are not controlled, as well as those who develop liver tumors, should be referred either for deceased donor liver transplantation (DDLT) or living donor liver transplantation (LDLT).
Here, we report a case of CAPV (Abernethy malformation type Ib) associated with unresectable hepatocellular adenoma in a young adult, successfully managed by LDLT with a left hemiliver graft. To the best of our knowledge, this is the 14th reported case of living donor liver transplantation for CAPV (Table 1), and the first reported case in which the indication for LDLT in the setting of CAPV was unresectable hepatocellular adenomatosis.
Table 1.
Reported cases of LTx for Abernethy malformation.
| Year | Reporting Authors | Age | Gender | Type of shunt | Indication for Ltx | Type of liver graft | Outcome |
|---|---|---|---|---|---|---|---|
| 1989 | Barton JW, Keller MS | 11 | F | Ia | Hepatoblastoma | OLT | Follow-op 18 Mo doing well |
| 1990 | Woodle ES et al. | 10 | F | Ia | BA | OLT | NA |
| 1994 | Morgan G, Superina R | 9w | F | Ib | BA | Split-Reduced Size | Unsuccessful due to bowel necrosis secondary to intestinal edema |
| 1997 | Howard ER et al. | 9 | F | Ib | BA | NA | NA |
| 1999 | Taoube KA et al. | 13 mo | M | NA | BA | OLT | NA |
| 2000 | Andreani et al. | 1.8 | M | NA | BA | OLT reduced-size | 20-month follow-up, good graft function |
| 2001 | Shinkai M et al. | 13 mo | F | I | PSE | OLT | NA |
| 2004 | Charre L et al. | 21 | M | I – portal vein collector draining in the internal iliac vein | Hematochezia | NA | NA |
| 2004 | Wojcicki M et al. | 45 | M | Ia | PSE | OLT | Follow-up 2.5 years, doing well |
| 2005 | Hibi M et al. | 3 | M | NA | FNH | NA | NA |
| 2005 | Ohnishi Y et al. | 27d | M | SMV draining in azygos vein; absent IVC | Intrapulmonary shunt and multiple brain abscess | LDLT-REDUCED LLS | Follow-up 5 months, doing well |
| 2005 | Takeichi T et al. | 35 | F | Ib | PSE | DOMINO | 10-month po, doing well |
| 2005 | Ogita K et al. | 2 | M | NA | PSE | LDLT | NA |
| 2005 | Suda et al. | 35 | F | NA | PSE | LDLT | NA |
| 2006 | Soejima Y et al. | 2 | M | Ib | Persistent hyperammonemia, hypergalactosemia | APOLT LDLT-LLS | Alive |
| 2006 | Sumida W et al. | 19mo | F | Ia | PSE | LDLT-LLS | Alive 19 Mo |
| 2007 | Witters et al. | 42 | F | II | HCC | OLT | NA |
| 2007 | Emre S et al. | 9 | F | Ib | HPS | APOLT- LLS from deceased donor | Alive 15 Mo |
| 2009 | Singhal A et al. | 4 | M | Ib - 4 mm portal vein entering liver | BA + HPS + hepatic nodules | LDLT-LHL | Alive 18 Mo |
| 2009 | Taku I et al. | 7 | M | II | Pulmonary hypertension partially controlled by shunt ligation | LDLT-LHL | Alive |
| 2009 | Kasahara M et al. | 16 | F | PSS between SMV+SV and right renal vein | Recurrent hyperammonemia | LDLT-LLS | Alive |
| 2010 | Matsuura T et al. | 18 | F | PSS between SMV-RIV via IMV | Mild encephalopathy and general fatigue due to persistent hyperammonemia | APOLT LDLT-EXTENDED LHL | Alive 3 Mo |
| 2010 | Hori T et al. | 4.9 | M | Ib | Pulmonary hypertension | LDLT- EXTENDED LLS | Alive |
| 2011 | Osorio MJ et al. | 7 | M | Ib | HPS, unresectable FNH | OLT | Alive 6 Mo |
| 2011 | Law YM et al. | 5 | F | PSS between convergence of SMV and sv and azygos vein | Severe pulmonary hypertension due to the shunt | SPLIT-LLS | Intrahepatic biliary strictures- >retransplantation->chronic graft rejection-> death after 2 years |
| 2012 | Uchida et al. | 14 | M | Ia | HPS | LDLT-LHL | 3-year follow-up, doing well |
| 2014 | Gordon-Burroughs et al. | 61 | F | Ib | Recurrent HCC post left hemihepatectomy | OLT | 3-year follow-up, free from HCC |
| 2015 | Present case | 21 | F | Ib | Unresectable hepatocellular adenoma | LDLT-LHL | Alive at 9 Mo |
APOLT – auxiliary partial orthotopic liver transplantation; BA – biliary atresia; FNH – focal nodular hyperplasia; HPS – hepatopulmonary syndrome; IMV – inferior mesenteric vein; IVC – inferior vena cava, HCC – hepatocellular carcinoma, LDLT – living donor liver transplantation; LHL – left hemiliver; mo, months; LLS – left lateral section; OLT – orthotopic liver transplantation; PSE – portosystemic encephalopathy; PSS – portosystemic shunt; RIV – right internal iliac vein; SMV – superior mesenteric vein; SV – splenic vein.
Case Report
The 21-year-old patient was diagnosed with a large unresectable tumor in the right hemiliver (110/100 mm, S5–8) and a type Ib Abernethy malformation during a bioptic laparotomy in 2011. Immunohistochemical study showed liver adenoma. The patient had no signs of portal hypertension and throughout the pretransplant period showed no encephalopathy or hyperammonemia. No hepatopulmonary syndrome or congenital cardiac disease was diagnosed in the patient; echocardiogram performed with agitated saline as contrast confirmed intra-pulmonary shunting. Moreover, pretransplant CT showed no additional abdominal congenital anomalies. She was placed on the DDLT waiting list, maintaining satisfactory liver function throughout the waiting period, while the right hemiliver mass remained stable in dimensions. A second large liver mass (68/66 mm, S2–3) was diagnosed by computed tomography in the left hemiliver, as well as 9 smaller ones in both hemilivers (S4: 20/21 mm; S5–S6: 32/20 mm, 14/14 mm, 16/6 mm, 7/6 mm, 9/8 mm, 8/8 mm, 7/7 mm, 19/16 mm) (Figures 1–4). Therefore, a decision was made to transplant the patient with a graft from a living related donor.
Figure 1.

(A) CT arterial phase. (B) Portal phase. Large hypervascular mass in the right hemiliver (arrowheads), with a central hypoattenuating scar (arrow), and little washout in portal phase. Another isoattenuating mass is seen in the left hemiliver (star), with washout in portal phase (B). CT protocol: Unenhanced CT and early phase helical. CT was performed with the administration of an IV bolus injection of 1.5 mL/kg of nonionic contrast material (Omnipaque [iohexol], Daiichi Pharmaceutical) of 350 mg I/mL), and then late phase helical CT was performed 60 s after the start of the injection. The scanning delay for the early phase scan was approximately 35 s.
Figure 2.
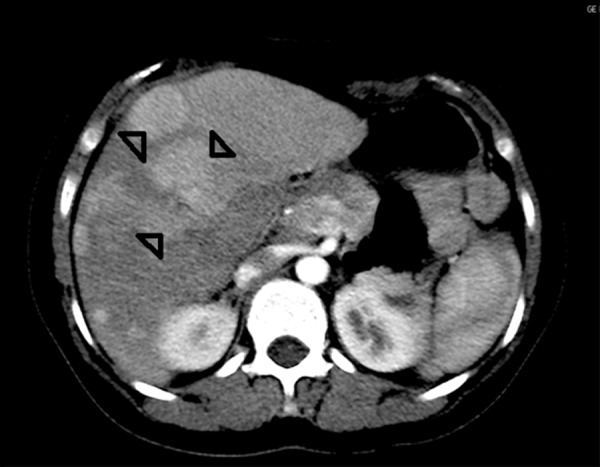
CT arterial phase. Several other smaller hypervascular masses are seen in the right hemiliver (arrowheads).
Figure 3.
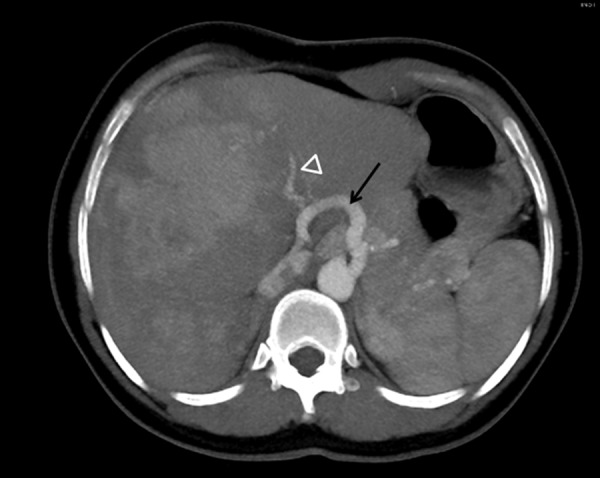
CT arterial phase (axial MIP). Important hypertrophy of the common hepatic artery (arrow) and intrahepatic branches (white arrowhead).
Figure 4.

CT portal phase. (A, B). MPR oblique reconstruction. A venous branch formed after the confluence of the SMV (arrow) and splenic vein (star) is seen draining in the IVC (arrowhead). (C) Portal phase. No intrahepatic portal branches are seen.
Preoperative laboratory findings were: total bilirubin, 0.5 mg/dL (0.3–1.2); aspartate aminotransferase, 112 IU/L (0–35); alanine aminotransferase, 32 IU/L (0–45); alkaline phosphatase 316 IU/L (42–128), total serum proteins 7 g/dL (6.4–8.3), serum albumin 3.8 g/dL (3.5–5.2); hemoglobin 11.6 g/dL (11.5–17); and prothrombin time (international normalized ratio), 0.97 (0.9–1.27). Testing for hepatitis B or C viral infection was negative.
A left hemiliver graft was transplanted from her mother. Both the donor and the recipient are ABO group and Rh-compatible (blood type A+). Transection of the donor liver parenchyma was performed on the right side of the middle hepatic vein with Sonopet (Stryker, Kalamazoo, Michigan) and electrocautery; no vascular inflow interruption was caused on either side of the plane of transection. Procedure duration was 445 min; intraoperative bleeding was approximately 500 mL; the graft weighed 550 g; recipient body weight was 57 kg; the graft weight on recipient weight ratio (GRWR) was 1.1%; the liver was perfused with 4000 mL of HTK solution on the back-table; warm ischemia time was 109 min.
Intraoperative examination confirmed the absence of a portal vein in the hepatoduodenal ligament, as well as the presence of a type Ib portosystemic shunt (Figure 5). There were pre-operative concerns that the portal conduit would be too short and that, in turn, would necessitate interposition of a vascular graft or a synthetic one. However, careful dissection of the portal conduit and its detachment from the IVC with a small (2 mm) circular cuff allowed the surgical team to perform the anastomosis between the splenomesenteric trunk and the donor left portal vein in an end-to-end manner.
Figure 5.
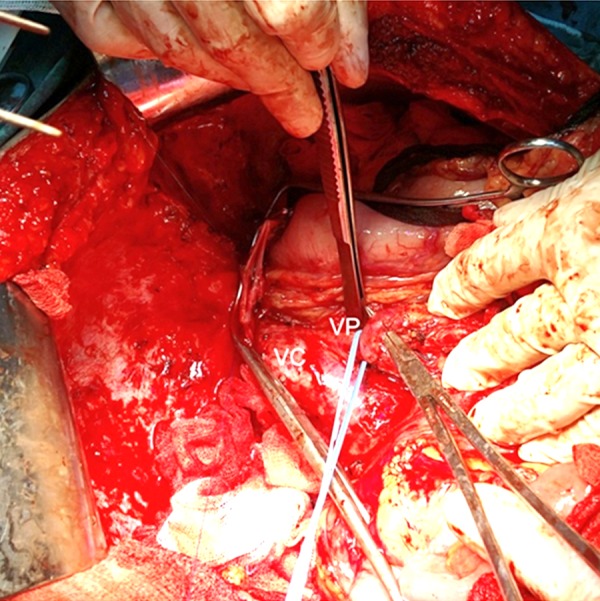
Intraoperative aspect: VP – portosystemic shunt (common trunk of SMV and splenic vein); VC – inferior vena cava.
The total time during which the portal vein was clamped was only 15 min, with no intestinal edema. The other anastomoses were as follows: common trunk of donor middle hepatic vein and left hepatic vein to the recipient common trunk of the middle and left hepatic veins; donor left hepatic artery (replaced from the left gastric artery) to recipient hepatic artery proper; donor left biliary duct to recipient main biliary duct. Even though the preoperative CT scan showed hypertrophy of the recipient hepatic artery, there was no mismatch between the anastomotic partners. The histopathological examination of the explanted liver showed an aspect of liver adenomatosis (Figure 6).
Figure 6.
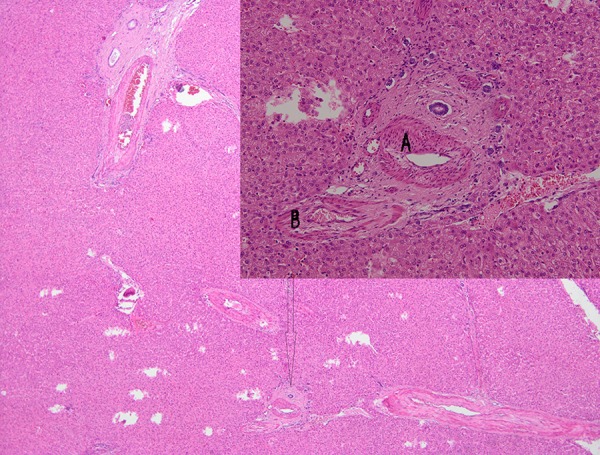
Hematoxylin-eosin stain ×100 – portal tract of the explanted liver (arrow). The portal tract lacks portal venules; arteries present a hypertrophic wall (A); biliary ducts have normal aspect (B).
The immunosuppressive regimen consisted in 2 doses of basiliximab as induction, while maintenance was achieved with tacrolimus and mycophenolate. The evolution of the graft was uneventful throughout the postoperative course; there was no early allograft dysfunction and peak ALT and AST were 266 IU/L and 167 IU/L, respectively. The postoperative course was marked by myoclonic convulsions due to multiple small ischemic strokes in the parietooccipital region, occurring on POD 3, successfully treated with anticonvulsants. No other complications appeared. The patient was discharged after 72 days and has currently returned to her normal life, with normal liver function.
Discussion
In the 4-week embryo, the venous system consists of 3 pairs of veins with distinct ontogenetic origin: cardinal veins of embryonic origin, umbilical veins from the chorion, and vitelline veins from the yolk sac [8]. The peri-intestinal vitelline venous loop evolves into the portal vein during weeks 4 to 10 antepartum, with some atresic and some progressive venous connections. It has been proposed that type I extrahepatic portosystemic shunts are a result of exaggerated involution of the paraduodenal vitelline veins during the weeks 4–8 of gestation [9].
Morgan and Superina classified extrahepatic shunts into 2 types: type I – liver not perfused with portal blood because of complete shunt (so-called CAPV), and type II – liver perfused with portal blood with the presence of a partial shunt (IIa – congenital shunt and IIb – acquired shunt) [10]. CAPV can be further subclassified into type Ia (SMV and splenic vein do not join to form confluence) and type Ib (SMV and splenic vein join to form confluence). If the portal vein presents a lack of complete development, the portocaval shunt is either the result of the persistence of the right vitelline vein (in which case the shunt connects to the retrohepatic IVC), or of the persistence of the left vitelline vein (the shunt is connected to the IVC or the right atrium above the confluence of the hepatic veins) [3].
The portosystemic shunt can communicate with the IVC at any level or even the left renal vein, the azygos vein, left hepatic vein, right internal iliac vein, the right atrium, and coronary sinus [11]. Type I Abernethy malformation seems to occur more frequently in females, while type II tends to appear more often in males [12].
In terms of terminology, type Ia Abernethy malformation is the only proper congenital absence of the portal vein, since the portosystemic shunt in the type Ib malformation carries portal blood; therefore, it can be called a portal vein. Other authors [8] consider that the correct definition of CAPV is the complete absence of intrahepatic portal venules histopathologically confirmed in the explanted liver. However, various studies used different terminology, many of them equating the Abernethy malformation of any type with CAPV [13].
Extrahepatic portosystemic shunts are extremely rare, presenting the highest incidence in Japan; this might be caused by the mass screening of newborns for hypergalactosemia, thus identifying cases of Abernethy malformation very early [12]. Most patients with CAPV are female (c. 65%) and are diagnosed during childhood (approximately 30% of them by the age of 5 years) [3].
A minority of the CAPV patients, due to the absence of portal hypertension and with a relatively high tolerance level of the central nervous system (CNS) to hyperammonemia, are usually only diagnosed as adults.
In 1990, Woodle et al. reported the first pediatric case of CAPV successfully transplanted [14], while the first DDLT for CAPV in an adult was reported in 2004 by Wojcicki et al. [15]. It is not the CAPV in itself that represents an indication for liver transplantation, but its serious, very often life-threatening associated conditions.
AM can be associated with hepatopulmonary syndrome, which is the clinical manifestation of intrapulmonary arterio-venous shunts. A possible explanation for this phenomenon is that it constitutes the effect of an upregulation in the circulating levels of nitric oxide, which by-passes the liver metabolism through the shunt. This determines vasodilatation of the pulmonary bed with an alteration of the ventilation/perfusion ratio, with the consequent platypnea caused by orthodeoxia.
Abernethy malformation type II can be safely approached by ligating the extrahepatic porto-caval shunts or by interventional radiology methods: coil embolization or catheter-device closure, and subsequently monitoring the portal pressure, as well as monitoring for pulmonary hypertension, which can be caused by intrahepatic portosystemic shunts that can spontaneously develop after the procedure [16].
However, most patients with CAPV type I should be considered for liver transplantation (LT) if they either develop uncontrollable hyperammonemia or in the presence of HPS or liver masses [17]. Moreover, given the high risk of developing severe HPS and pulmonary hypertension, which may preclude LT, Soejima et al. reported that prophylactic liver transplantation should be considered in children diagnosed with CAPV [18].
A complete resolution of HPS was noted after auxiliary partial orthotopic liver transplantation (APOLT) with a left lateral segment graft, indicating that HPS is provoked by inability of the liver to perform the clearance of vasoactive substances [19]. Hypothetically, there is a risk of tumor development in the remnant native liver in case of APOLT. Therefore, the choice between the types of liver grafts should be weighed carefully, with some teams clearly favoring LDLT with complete removal of the native liver [20].
Resection of the tumor may be considered if we expect tumor-free margins of the specimen, as well as normal synthetic capability and a satisfactory size of the remnant liver, as reported by Witjes et al. [21]. Major hepatectomies (right hemihepatectomy extended to segment 4) [22] and right hemihepatectomy [23] have been performed successfully; however, we consider that potential tumors could arise in the remaining liver parenchyma.
An important preoperative finding in imaging studies is a dilated hepatic artery, which compensates the decreased inflow provoked by CAPV [24]. Hypertrophy of the recipient hepatic artery could be a potential pitfall during the performing of the anastomosis due to the mismatch of the anastomotic partners; this could be overcome considering 2 important aspects: an experienced microsurgeon should perform the anastomosis; and oblique section of the donor anastomotic partner, carefully tailored so that it matches the recipient one. Since CAPV is not associated with portal hypertension, there is no collateral circulation, and this may pose an intraoperative risk of important intestinal congestion during the portal anastomosis. In the present case, given the short period of shunt clamping (15 min), there was no portal hypertension and, consequently, no intestinal edema. Sumida et al. devised an anastomotic technique using an ovarian vein from the living donor, which avoids reconstructing the portal vein with total clamp, thus completely excluding the risk of intestinal congestion [25]. A short portal conduit could render the performance of the portal anastomosis extremely difficult. There are multiple ways of approaching this situation: interposing a venous graft (harvested from the donor’s saphena magna or from ABO-compatible deceased donors) or a synthetic one; and preserving a small cuff on the portal conduit when detaching it from the IVC.
In a review of 136 cases of congenital extrahepatic portosystemic shunts, Kobayashi et al. [26] showed that 55 of them (43%) presented with tumors, most of them (83.6%) benign.
The absence of portal hepatotrophic factors (such as insulin and glucagon [27,28] and epidermal growth factor [29]) due to CAPV is probably a risk factor for the development of nodular liver lesions: benign tumors such as hepatocellular adenomas, focal nodular hyperplasia (FNH) [30], and nodular regenerative hyperplasia (NRH) [31], as well as malignant ones, such as hepatoblastoma [32], and hepatocellular carcinoma (HCC) [8]. Another possible explanation is that initially benign tumors reactively arise in the areas of the liver with poor perfusion, due to the lack of portal venous blood flow [33]. A recent study [34] has documented a clear path of tumoral transformation from benign FNH to malignant HCC in an adult patient with CAPV who was subsequently successfully transplanted with a whole liver graft. The proposed explanation for why very few patients with AM present with either hepatic fibrosis or cirrhosis is that, unlike the hepatotrophic cells that are affected by the disturbances in the portal blood flow, mesenchymal cells in the liver are not influenced by the lack of portal inflow [35].
In the setting of CAPV associated with tumors, a living related liver transplantation should be considered. Liver transplantation in the setting of CAPV also significantly alleviates the symptoms of intrapulmonary shunting, as well as causing a rapid withdrawal of encephalopathy.
Conclusions
Patients with Abernethy malformation presenting with benign liver tumors have an important benefit from living donor liver transplantation, but the disadvantage of APOLT is that it poses an increased risk of tumor development in the remnant liver. Moreover, in contrast with surgical resection, in the case of the living donor liver transplantation the risk of developing further tumors or of progression to malignancy of the already existing ones is virtually nonexistent.
Clearly, any recipient with CAPV is mostly benefitted by a whole liver graft in comparison with a hemiliver from a living donor. In our particular case, the extent of the unresectable liver adenomatosis was considered so important that almost any other recipient on the waiting list for a whole liver graft, even patients with hepatocarcinoma outside Milano criteria, would have qualified before her. Therefore, LDLT was chosen, not only to shorten the time on the DDLT waiting list, but also to offer her a the possibility of swift and effective treatment.
References:
- 1.Abernethy J. Account of two instances of uncommon formation, in the viscera of the human body. Phil Trans R Soc Lond. 1793;83:59–66. [Google Scholar]
- 2.Howard ER, Davenport M. Congenital extrahepatic portocaval shunts – the Abernethy malformation. J Pediatr Surg. 1997;32(3):494–97. doi: 10.1016/s0022-3468(97)90614-x. [DOI] [PubMed] [Google Scholar]
- 3.Mistinova J, Valacsai F, Varga I. Congenital absence of the portal veinecase report and a review of literature. Clin Anat. 2010;23:750e8. doi: 10.1002/ca.21007. [DOI] [PubMed] [Google Scholar]
- 4.Achiron R, Gindes L, Kivilevitch Z, et al. Prenatal diagnosis of congenital agenesis of the fetal portal venous system. Ultrasound Obstet Gynecol. 2009;34:643–52. doi: 10.1002/uog.7460. [DOI] [PubMed] [Google Scholar]
- 5.Le Borgne J, Paineau J, Hamy A, et al. Interruption of the inferior vena cava with azygos termination associated with congenital absence of portal vein. Surg Radiol Anat. 2000;22(3–4):197–202. doi: 10.1007/s00276-000-0197-x. [DOI] [PubMed] [Google Scholar]
- 6.Witters P, Maleux G, George C, et al. Congenital veno-venous malformations of the liver: widely variable clinical presentations. J Gastroenterol Hepatol. 2008;23:e390–94. doi: 10.1111/j.1440-1746.2007.05156.x. [DOI] [PubMed] [Google Scholar]
- 7.Alvarez AE, Ribeiro AF, Hessel G, et al. Abernethy malformation: One of the etiologies of hepatopulmonary syndrome. Pediatric Pulmonology. 2002;34:391–94. doi: 10.1002/ppul.10182. [DOI] [PubMed] [Google Scholar]
- 8.Hu GH, Shen LG, Yang J, et al. Insight into congenital absence of the portal vein: Is it rare? World J Gastroenterol. 2008;14(39):5969–79. doi: 10.3748/wjg.14.5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altavilla G, Cusatelli P. Ultrastructural analysis of the liver with portal vein agenesis: A case report. Ultrastruct Pathol. 1998;22:477–83. doi: 10.3109/01913129809032284. [DOI] [PubMed] [Google Scholar]
- 10.Morgan G, Superina R. Congenital absence of the portal vein: two cases and a proposed classification system for portosystemic vascular anomalies. J Pediatr Surg. 1994;29:1239–41. doi: 10.1016/0022-3468(94)90812-5. [DOI] [PubMed] [Google Scholar]
- 11.Alewine TC, Carter WR, Frew MJ. Congenital absence of the portal vein in a patient with urolithiasis. Am J Roentgenol. 2007;189:W150–52. doi: 10.2214/AJR.05.0843. [DOI] [PubMed] [Google Scholar]
- 12.Stringer MD. The clinical anatomy of congenital portosystemic venous shunts. Clinical Anatomy. 2008;21:147–57. doi: 10.1002/ca.20574. [DOI] [PubMed] [Google Scholar]
- 13.Northrup M, Mendez-Castillo A, Sethi Y, Churchill R. Congenital absence of the portal vein with an intrahepatic inferior vena cava branch showing hepatopetal flow. J Ultrasound Med. 2002;21:569–72. doi: 10.7863/jum.2002.21.5.569. [DOI] [PubMed] [Google Scholar]
- 14.Woodle ES, Thistlethwaite JR, Emond JC, et al. Successful hepatic transplantation in congenital absence of recipient portal vein. Surgery. 1990;107(4):475–79. [PubMed] [Google Scholar]
- 15.Wojcicki M, Haagsma EB, Gouw ASH, et al. Orthotopic liver transplantation for portosystemic encephalopathy in an adult with congenital absence of the portal vein. Liver Transpl. 2004;10:1203–7. doi: 10.1002/lt.20170. [DOI] [PubMed] [Google Scholar]
- 16.Iida T, Ogura Y, Doi H, et al. Successful treatment of pulmonary hypertension secondary to congenital extrahepatic portocaval shunts (Abernethy type 2) by living donor liver transplantation after surgical shunt ligation. Transpl Int. 2010;23(1):105–9. doi: 10.1111/j.1432-2277.2009.00964.x. [DOI] [PubMed] [Google Scholar]
- 17.Sanada Y, Mizuta K, Kawano Y, et al. Living donor liver transplantation for congenital absence of the portal vein. Transplant Proc. 2009;41:4214–19. doi: 10.1016/j.transproceed.2009.08.080. [DOI] [PubMed] [Google Scholar]
- 18.Soejima Y, Taguchi T, Ogita K, et al. Auxiliary partial orthotopic living donor liver transplantation for a child with congenital absence of the portal vein. Liver Transpl. 2006;12:845–49. doi: 10.1002/lt.20692. [DOI] [PubMed] [Google Scholar]
- 19.Emre S, Arnon R, Cohen E, et al. Resolution of hepatopulmonary syndrome after auxiliary partial orthotopic liver transplantation in Abernethy malformation. A Case Report. Liver Transpl. 2007;13:1662–68. doi: 10.1002/lt.21349. [DOI] [PubMed] [Google Scholar]
- 20.Hori T, Yonekawa Y, Okamoto S, et al. Pediatric orthotopic living-donor liver transplantation cures pulmonary hypertension caused by Abernethy malformation type Ib. Pediatr Transplant. 2011;15:e47–e52. doi: 10.1111/j.1399-3046.2009.01269.x. [DOI] [PubMed] [Google Scholar]
- 21.Witjes CDM, Ijzermans JNM, Noordegraaf AV, Tran TCK. Management strategy after diagnosis of Abernethy malformation: a case report. J Med Case Rep. 2012;6:167. doi: 10.1186/1752-1947-6-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakasaki H, Tanaka Y, Ohta M, et al. Congenital absence of the portal vein. Ann Surg. 1989;210(2):190–93. doi: 10.1097/00000658-198908000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pichon N, Maisonnette F, Pichon-Lefièvre F, et al. Hepatocarcinoma with congenital agenesis of the portal vein. Jpn J Clin Oncol. 2003;33(6):314–16. doi: 10.1093/jjco/hyg050. [DOI] [PubMed] [Google Scholar]
- 24.Takagaki K, Kodaira M, Kuriyama S, et al. Congenital absence of the portal vein complicating hepatic tumors. Intern Med. 2004;43(3):194–98. doi: 10.2169/internalmedicine.43.194. [DOI] [PubMed] [Google Scholar]
- 25.Sumida W, Kaneko K, Ogura Y, et al. Living donor liver transplantation for congenital absence of the portal vein in a child with cardiac failure. J Pediatr Surg. 2006;41:E9–12. doi: 10.1016/j.jpedsurg.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi N, Niwa T, Kirikoshi H, et al. Clinical classification of congenital extrahepatic portosystemic shunts. Hepatol Res. 2010;40:585–93. doi: 10.1111/j.1872-034X.2010.00667.x. [DOI] [PubMed] [Google Scholar]
- 27.Pathak A, Agarwal N, Mandliya J, et al. Abernethy malformation: a case report. BMC Pediatr. 2012;12:57. doi: 10.1186/1471-2431-12-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Starzl TE, Francavilla A, Halgrimson CG. Origin, hormonal nature and action of hepatotrophic substances in portal venous blood. Surg Gynecol Obstet. 1973;137:179–99. [PMC free article] [PubMed] [Google Scholar]
- 29.Fukushima N, Kuromatsu R, Uchiyama D, et al. Hyperplastic nodular hepatic lesions following end-to side portacaval shunting in childhood. Intern Med. 2007;46(15):1203–8. doi: 10.2169/internalmedicine.46.6419. [DOI] [PubMed] [Google Scholar]
- 30.Zhang K, Wang Q, Wang H, et al. Computed tomography and magnetic resonance imaging of multiple focal nodular hyperplasias of the liver with congenital absence of the portal vein in a Chinese girl: case report and review of the literature. Eur J Med Res. 2014;19(1):63. doi: 10.1186/s40001-014-0063-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koizumi J, Yamashita T, Dowaki S, et al. Hepatobiliary and pancreatic: Hepatic adenoma, focal nodular hyperplasia and congenital absence of the portal vein. J Gastroenterol Hepatol. 2006;21:619. doi: 10.1111/j.1440-1746.2006.04344.x. [DOI] [PubMed] [Google Scholar]
- 32.Barton JW, Keller MS. Liver transplantation for hepatoblastoma in a child with congenital absence of the portal vein. Pediatr Radiol. 1989;20:113–14. doi: 10.1007/BF02010653. [DOI] [PubMed] [Google Scholar]
- 33.Grazioli L, Alberti D, Olivetti L, et al. Congenital absence of portal vein with nodular regenerative hyperplasia of the liver. Eur Radiol. 2000;10:820–25. doi: 10.1007/s003300051012. [DOI] [PubMed] [Google Scholar]
- 34.Scheuermann U, Foltys D, Otto G. Focal nodular hyperplasia precedes hepatocellular carcinoma in an adult with congenital absence of the portal vein. Transpl Int. 2012;25(5):e67–68. doi: 10.1111/j.1432-2277.2012.01454.x. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka Y, Takayanagi M, Shiratori Y, et al. Congenital absence of portal vein with multiple hyperplastic nodular lesions in the liver. J Gastroenterol. 2003;38:288–94. doi: 10.1007/s005350300050. [DOI] [PubMed] [Google Scholar]