

Abstract
Due to an increasing problem of drug resistance among almost all parasites species ranging from protists to worms, there is an urgent need to explore new drug targets and their inhibitors to provide new and effective parasitic therapeutics. In this regard, there is growing interest in exploring known drug leads of human epigenetic enzymes as potential starting points to develop novel treatments for parasitic diseases. This approach of repurposing (starting with validated targets and inhibitors) is quite attractive since it has the potential to reduce the expense of drug development and accelerate the process of developing novel drug candidates for parasite control. Lysine deacetylases (KDACs) are among the most studied epigenetic drug targets of humans, and a broad range of small-molecule inhibitors for these enzymes have been reported. In this work, we identify the KDAC protein families in representative species across important classes of parasites, screen a compound library of 23 hydroxamate- or benzamide-based small molecules KDAC inhibitors, and report their activities against a range of parasitic species, including the pathogen of malaria (Plasmodium falciparum), kinetoplastids (Trypanosoma brucei and Leishmania donovani), and nematodes (Brugia malayi, Dirofilaria immitis and Haemonchus contortus). Compound activity against parasites is compared to that observed against the mammalian cell line (L929 mouse fibroblast) in order to determine potential parasite-versus-host selectivity). The compounds showed nanomolar to sub-nanomolar potency against various parasites, and some selectivity was observed within the small panel of compounds tested. The possible binding modes of the active compounds at the different protein target sites within different species were explored by docking to homology models to help guide the discovery of more selective, parasite-specific inhibitors. This current work supports previous studies that explored the use of KDAC inhibitors in targeting Plasmodium to develop new anti-malarial treatments, and also pioneers experiments with these KDAC inhibitors as potential new anthelminthics. The selectivity observed begins to address the challenges of targeting specific parasitic diseases while limiting host toxicity.
Author Summary
Due to pandemic drug resistance in the treatment of parasitic infections, there is an urgent need to identify novel drug targets and their associated drug compounds. Although “drug repurposing”, i.e. the application of known drugs and compounds to new indications such as infectious diseases, provides a cost effective approach in the development of novel therapeutics, selectivity is one of the major obstacles to overcome in getting such compounds into clinical trials as anti-parasitic drugs. Using the lysine deacetylases (KDACs) as an example, we explored the activities of a panel of known inhibitors against the KDAC targets in a range of parasitic organisms. The computational study of their binding modes to the targets (by docking the compounds to the homology models within different organisms in comparison with the human proteins) helps to rationalize the different activities observed and provide insight on the optimization of lead compounds to improve selectivity. Our work provides support of “drug repurposing” in the treatment of parasitic diseases, and demonstrates the necessity of optimizing these leads for the ultimate goal of preparing them for clinical use.
Introduction
Neglected tropical diseases are the most common infections of the poorest populations around the globe, causing massive burdens on the countries’ general population and inhibiting economic development [1]. Treatments for these diseases usually rely on a single drug, or limited options of drugs. Most drugs used for treating neglected diseases are quite old, have unknown mechanisms of action, and often have limited effectiveness with poor safety profiles. Furthermore, drug resistance has been observed following the treatment of almost all parasitic pathogens, including protists, helminthes (roundworms) and Platyhelminthes (flatworms) [2]. Parasitic genome sequencing is now being exploited to help accelerate the development of much-needed compounds with novel mechanisms of action.
Lysine deacetylase (KDACs) is a more specific term for enzymes that remove the e-acetyl group from lysine side chains, and have emerged as an important class of drug targets with the potential to treat a variety of diseases in human, ranging from psychiatric diseases and neurodegenerative diseases to cancer [3, 4]. Given the significance of KDACs in epigenetic modulation, numerous small compounds have been developed to inhibit their activity, originally directed at altering chromatin structure and thus modulating gene expression [5]. In humans, KDACs belong to a large family with 18 members [6] divided into the Zn-dependent (Class I and Class II) and NAD-dependent (Class III) enzymes. The Zn-dependent enzymes have been the focus of intense research since they compose the majority of the KDAC family members and are the primary targets of the known inhibitors [7]. In light of “drug repurposing” for the treatment of parasitic diseases [8, 9], KDACs have been identified as an emerging drug target in all the major human parasitic pathogens [10], but no systematic characterization has been conducted to date, except in Plasmodium [11]. Considerable efforts were also made to utilize the collected information of the known KDAC inhibitors to explore the potential of targeting orthologs in the parasitic pathogens, ranging from P. falciparum [12–15] to Schistosoma mansoni [16, 17]. Andrews et al. have pursued KDAC inhibitors as antimalarial drugs [12, 18, 19]; One of their studies presents comparative gene expression profiling of P. falciparum in response to exposure to three different KDAC inhibitors [20]. Despite structural similarity between the three inhibitors, diverse transcriptional effects were observed in the study, and were attributed to possibly subtle differences in their inhibition of KDAC isoforms, or cellular distribution. Marek et al. [16] recently reported that lysine deacetylase 8 from S. mansoni (smKDAC8), the most expressed class I KDAC isoform in this organism, was a functional acetyl-L-lysine deacetylase with an essential role in parasite infectivity. Crystal structures were obtained for different inhibitors bound to smKDAC8, and their binding modes were compared for the optimization of the lead inhibitor, in order to achieve better potency and selectivity for smHDAC8 [17]. In a more recent study, many KDAC inhibitors that are currently in clinical trials for oncology were evaluated as therapeutic leads to target the kinetoplastid Trypanosoma brucei for human African trypanosomiasis (HAT) [21]. These inhibitors were found to be moderately to strongly potent in blocking proliferation of blood-stage T. brucei in culture; however, none of these drugs were lethal to cultured parasites when tested at human-tolerated doses. This again confirms that parasitic selectivity is the major issue to address before repurposing these drugs as anti-parasitic therapeutics [8, 10]. Similar observations were also made in other work [22]. Most of the studies to date have focused on a single enzyme isoform (or at the cellular level for the target parasite), and have neglected to explore the potential interaction of inhibitors acting on different KDAC orthologs and isoforms, in particular, the human ortholog/isoforms. In pursuing parasitic-specific or selective KDAC inhibitors, a systematic evaluation of the KDAC targets as well as their interactions with small molecular inhibitors is warranted to gain better insights at the molecular level for the improvement of inhibitor selectivity.
It should be noted that lysine deacetylases are generally referred to as histone deacetylases (HDACs; a historical imperative, as epigenetic modification of histones was described in 1964 by Allfrey et al. [23]), but it has been shown by proteomics that over 1700 proteins in cells besides histones undergo dynamic acetylation [3]. Thus, here, we use the more accurate terminology of lysine deacetylase (KDAC), acetylase (KAT) and methylase (KMT).
In this work, we took advantage of the genomes (deduced proteomes) of many parasite species (resulting from recently-emerging sequencing technologies) to characterize the KDAC family in parasites, including the less frequently-studied nematode species. KDAC family members within these species were characterized using an orthology-based approach. Most importantly, known human KDAC inhibitors were screened for activity against a few representative parasite species including the pathogen of malaria (Plasmodium falciparum), kinetoplastids (T. brucei and Leishmania donovani), and nematodes (Brugia malayi, Dirofilaria immitis and Haemonchus contortus). Activity observed in vitro against these parasites was compared with that observed for toxicity with a mammalian cell line to determine the potential for host-versus-parasite selectivity. We also examined potential molecular mechanisms by which active compounds could be acting on the targets. This work provides some perspective on the prospect of targeting KDACs in parasites and paves the way for developing more selective KDAC ligands as novel drugs to control parasitic infections of humans and animals.
Methods
Data Collection
Whole proteome data from 26 eukaryotic species were collected. The datasets were comprised of 11 species of nematodes, 4 species of Platyhelminthes, 5 species of protists (kinetoplastids and pathogen of malaria) and 6 species of hosts/outgroups. Data were downloaded as follows: for the outgroups, Homo sapiens and Mus musculus were from Ensembl [24] release 67; and Bos taurus, Canis lupus familiaris, Sus scrofa and Ovis aries were from Genbank [25] release 102, 102, 103 and 100 respectively. For the nematodes, Caenorhabditis elegans and Brugia malayi were from Wormbase [26] WS230; Trichinella spiralis, Dirofilaria immitis, Ascaris suum, Haemonchus contortus and Necator americanus were from published data [27–31]. Trichuris muris was from the Sanger Institute release (ftp://ftp.sanger.ac.uk/pub/pathogens/Trichuris/muris/). Loa was from Broad Institute release (http://www.broadinstitute.org). The other 2 nematode species, Ancylostoma ceylanicum and Trichuris suis were from our in-house sequencing datasets. For the Platyhelminthes, Schistosoma japonicum was from Chinese National Human Genome Center at Shanghai (http://lifecenter.sgst.cn/schistosoma/en/schistosomaCnIndexPage.do#Download); Schistosoma mansoni was from the Sanger Institute release (ftp://ftp.sanger.ac.uk/pub/pathogens/Schistosoma/mansoni/genome/gene_predictions/GeneDB_Smansoni_Proteins.v4.0g.gz, retrieved on 02/29/2009); Schistosoma haematobium was downloaded from SchistoDB (http://SchistoDB.net) [32] on 02/01/2012; and Clonorchis sinensis was downloaded from NCBI (NCBI bioproject 72781 [33]). All the kinetoplastids (Trypanosoma brucei, Trypanosoma cruzi, Leishmania major and Leishmania donovani) were downloaded from TriTrypDB (http://tritrypdb.org) [34] on 01/07/2014 (release 6.0). Plasmodium falciparum was downloaded from NCBI (ftp://ftp.ncbi.nih.gov/genomes/Protozoa/Plasmodium_falciparum/) on 01/07/2014. Isoforms of these downloaded sequences were examined against the coding genes, and only the longest ones were kept when applicable.
Protein Family Definition and Identification of KDAC Protein Families
Protein families (orthologous groups) were defined utilizing the Markov cluster algorithm [35] of the OrthoMCL package [36, 37] with an inflation factor 1.5 based on the proteomes. Each protein family consists of at least two proteins from one or more species. The gene annotations of KDAC proteins for human in Ensembl, as well as those reported in literature for the pathogens malaria, toxoplasmosis, trypanosomiasis, schistosomiasis and leishmaniasis [10] were used to identify and manually curate the KDAC protein families. The number of proteins in each of these protein families was used to cluster the 26 species, using Manhattan clustering with average linkage using the software package GENE-E (http://www.broadinstitute.org/cancer/software/GENE-E/). A heatmap based on orthologous protein data was generated in MS Excel Version 2010.
Experimental Compound Screening in Parasitic Species and Mammalian Cell Line
A representative selection of 20 compounds (Fig 1) from a library of several hundred KDAC inhibitors that were synthesized in the Mai laboratory at the Sapienza Universita in Rome was screened. Additionally, largazole and two analogs were supplied by Prof. Robert Williams’ laboratory at Colorado State University. The compounds were selected based on the following criteria: 1). Known KDAC inhibitors which have been well studied and characterized in human studies, usually used as controls, e.g. GRM1 (SAHA), GRM2 (Tubastatin), and GRM3 (Entinostat); 2). Cyclic depsipeptide based, class I-selective KDAC inhibitors and their analogs, e.g. SD-L-148 (Largazole), SD-L-256, JMF-1080; 3). Other hydroxamate- or benzamide-based small molecules which have been shown to be human KDAC inhibitors in purified enzyme-based assays.
Fig 1. Chemical structures of compounds used in parasite screening assays.

Compounds were tested against 3 parasite groups (2 roundworms, 2 protists and malaria, Table 1) and a mammalian cell line (L929 mouse fibroblast; NCTC clone 929 [L cell, L-929, derivative of Strain L] (ATCC CCL-1) was obtained from ATCC). Compound screening against roundworms was conducted using three organisms with very different modes of parasitism: the blood feeding and gut dwelling H. contortus, and the animal and human tissue-dwelling filarial nematodes D. immitis and B. malayi. Experimental procedures were described in previous studies [38, 39]. The kinetoplastids (T. brucei strain S427 and L. donovani strain MHOM/SD/00/LS) viability assays were conducted with exponentially growing trypomastigotes, oraxenic amastigotes, for each species respectively in 96-well plates using automated liquid-handling equipment. Test compounds in DMSO were added to each well at 2–5 μM for T. brucei and 5–10 μM for L. donovani followed by incubation with the parasite for 72 hours at 37°C with 5% CO2. Known anti-trypanosomal compounds, i.e. pentamidine and suramin, were included in each plate to serve as positive controls. Parasite viability was determined by addition of resazurin and evaluation of plates using a fluorescent plate reader. Compounds showing ≥75% inhibition in primary assays were selected and titrated to confirm their activity and to generate IC50 values. Activity/base protocols were used to calculate IC50 values and generate quality control parameters for each plate. Compounds with IC50 ≤ 1 μM for T. brucei and IC50s ≤ 5 μM for L. donovani were tested versus mammalian cells to determine parasite versus host-cell selectivity. A P. falciparum viability assay [40] was conducted with the 3D7 strain of P. falciparum known to be sensitive to all antimalarial drugs. Assays were performed in 96-well microtiter plates, and each well contained 100 μl of parasite culture maintained in media supplemented with human red blood cells (0.5% parasitemia, 2.5% hematocrit) in a humidified atmosphere at 37°C, 5% O2 and 5% CO2. Test compounds in DMSO were added to each well at 2–5 μM. After incubation, 85% of the supernatant was removed and cells were washed with PBS. A DNA-specific dye (SYBR Green or DAPI) was added in the presence of lysis agents, saponin and Triton X-100. Plates were incubated for 15 min and then read in a fluorescent microplate reader. Compounds showing ≥75% inhibition in primary assays were cherry-picked and titrated to confirm activity and generate IC50 values. Activity/Base protocols were used to calculate IC50 values and generate quality control parameters for each plate. Compounds with IC50 ≤ 1 μM were tested against mammalian cells (to determine parasite versus host-cell selectivity) and also against a selection of drug-resistant strains of P. falciparum.
Table 1. Compound screening in host cells and parasites.
Only measured activities were reported in the table, in the units of nM. The vertical line in the table separates host and parasites. Abbreviation used: CYT vt: cytotoxicity viability assay; Endoparasites_DR: endoparasites dose response assay; HAT vt: Human African trypanosoma viability assay; LEI axe: Leishmania axenic amastigote assay; MAL vt: Malaria viability assay. L929: L929 mouse fibroblast; TbbS427: T. brucei strain S427; Ld1S: L. donovani strain MHOM/SD/00/LS; PfDd2: P. falciparum 3D7 strain.
| Assay | CYT vt | Endoparasites_DR | HAT vt | LEI axe | MAL vt | ||
|---|---|---|---|---|---|---|---|
| Cell line / Species | L929 | B. malayi | D. immitis | H. contortus | TbbS427 | Ld1S | PfDd2 |
| Timepoint | 5 day | 72 hour | 96 hour | 72 hour | 72 hour | 15 minute | |
| MC2984 | |||||||
| SDM141 | |||||||
| SDM146 | |||||||
| MC2727a* | 2.35 | 0.96 | |||||
| MC2726* | 1.92 | 0.189 | |||||
| MC2625* | 0.311 | 1.18 | >5 | 0.022 | |||
| MC2664 | |||||||
| MC2780* | 4.81 | 2.53 | 8.14 | 0.623 | 0.473 | 0.056 | |
| MC2776* | >10 | 4.39 | >10 | ||||
| MC3031* | 0.555 | 0.267 | >5 | ||||
| MC3004 | |||||||
| MC3079 | |||||||
| MC3050 | |||||||
| MC1742* | 1.51 | <0.01 | |||||
| MC1862* | 7.12 | 1.15 | |||||
| MC2126* | 1.76 | 0.441 | >5 | 1.92 | |||
| MC2129 | |||||||
| JMF-1080 | |||||||
| SD-L-256 | 0.333 | >10 | >10 | 0.9 | |||
| SD-L-148 | 0.101 | 7.1 | |||||
| GRM1 | 0.155 | 1.81 | >5 | 0.152 | |||
| GRM2* | 6.22 | 2.7 | >5 | ||||
| GRM3 | |||||||
* compounds with lower activity in at least one parasite species compared to L929.
Protein Structural Modeling and Ligand Docking
For those KDAC isotypes in parasitic species targeted by active compounds, homology models were built by using the X-ray structure of the human ortholog as template, using the ROSETTA3.4 macromolecular modeling package [41]. The catalytic zinc ion at the active site was modeled explicitly following the approach of Wang et al to mimic the square-based pyramidal geometry as observed in crystal structures [42][43]. After the initial comparative modeling and loop building, each protein model was relaxed with the following constraints to achieve the desired geometry: the zinc ion was constrained to have the axial position coordinated to the conserved histidine residue (HIS, deprotonated Nε), two equatorial positions coordinated to the conserved aspartic acid residues (ASP, deprotonated hydroxyl oxygen) and the remaining two equatorial positions coordinated to solvent water molecules. 100 models for each target were generated using the constrained relaxation procedure, and the one with the lowest total energy was chosen as the final protein model for subsequent docking studies. For each small ligand to be docked, OMEGA [44] was used to generate a conformer library; OpenEye's AM1-BCC implementation [45] was used to calculate partial charges. The hydroxamate group was deprotonated in the modeling process, as suggested by previous docking and virtual screening reports [46]. The ligands were docked to the models in ROSETTA using the ligand_dock application by specifying a constraint of the hydroxamate group to be coordinated to the zinc, replacing the two water molecules used in modeling the zinc geometry. One hundred poses were generated for each compound at each target, the 5 best-scoring poses were selected for manual inspection, and a representative pose was finally chosen for interpretation.
Identification of Active Site Variances
For each KDAC protein isotype, a representative X-ray structure from its human ortholog was chosen as the structural template. Any residue with an atom within a distance cutoff (10 Å) to the catalytic zinc ion was defined as an active-site residue. Sequence alignments of other parasite orthologs with the human protein (built by MUSCLE [47] for each KDAC family) were used to identify residues that were different in the parasite, and these residues were identified as variants at the active site.
Results
The KDAC Protein Families in the Parasites
Protein families were constructed from all longest isoform sequences in the proteomes of 26 eukaryotic species, which included 20 parasitic species and 6 host species (OrthoMCL clusters with multiple sequences were defined as protein families). The dataset includes 399,592 proteins in total, from which 44,531 protein families were derived. We identified all of the KDAC protein families within the parasitic species, based on the annotations of the human orthologs and the annotations available for a few parasitic species. The annotations of C. elegans KDAC proteins from WormBase [48] were also used for inference and manual curation.
As shown in Fig 2, all 11 Zn-dependent KDAC protein isoforms were not present within the parasites. From the human ortholog annotations, all the human KDACs were clustered into 6 separate families. KDAC1 and KDAC2 were clustered into one family (note A), and the class IIA isotypes (4, 5, 7, and 9) and IIB isotypes (6 and 10) were each clustered into their own families (notes B and C). Isotype KDAC3 from most species was clustered into one family (as was KDAC11), while KDAC8s from all the hosts were clustered in one family.
Fig 2. KDAC proteins inferred for the parasitic species within protein families.

Those with the same superscripts (A, B, C) are clustered within the same family. Color codes provide the number of total proteins from each species within an orthologous protein family.
In the class IIA family, almost all of the parasites had only one member in the family containing human orthologs, except P. falciparum, which had no member present. Based on the C. elegans annotation, there should be orthologs of KDAC4 for the roundworms. Some roundworms (A. ceylanicum, A. suum, N. americanus, H. contortus, L. loa and D. immitis) also have the KDAC5 ortholog, which clustered with the C. elegans KDAC5 into a new protein family. Primary sequence similarity results suggested that the kinetoplastids share higher homology to the human KDAC5 protein, so these were annotated as KDAC5. Two flatworm species (S. mansoni and S. haematobium) also had a KDAC9 ortholog, both of which were clustered into a separate protein family. From the current analysis, no KDAC7 ortholog was present in any parasitic species examined.
Although there are only two isotypes in the class IIB family, two human protein members (KDAC6/10) showed considerable expansion/deletion among the parasites. The kinetoplastids only contained one member, while many of the roundworms and one of flatworms (S. japonicum) have three or more members. Some of the roundworms had only one ortholog in this family, with one of them (D. immitis) having no member present.
For the other isotypes, a single KDAC3 homolog was uniquely shared among Leishmania species (and another among Trypanosoma species), with all of the hosts and flatworms and most of the roundworms sharing orthology to the human KDAC3 protein, suggesting divergence of this protein among the kinetoplastids. Orthologs to the human KDAC8 were only found among host species, but a separate S. mansoni KDAC8 orthologous group was found to be conserved in (and uniquely found in) flatworms. Finally, KDAC11 was conserved only among host and roundworm species.
Based on the number of protein members within the protein families, the different species were clustered according to their phylogenetic distances to each other (Fig 2). Kinetoplastids, flatworms and hosts were all clustered with their own species groups, but the roundworms clustered into two groups, largely due to the expansion in the number of KDAC6 and KDAC10 orthologs among the roundworms in the top cluster. P. falciparum clustered separately from all of the other species, as it contained only one protein (124507060) with detected orthology to any KDACs identified in other species. This protein was orthologous to KDAC1/2 family members, previously characterized as a KDAC1 ortholog [49]. Two of the three other KDAC homologs previously identified in P. falciparum [10] are orthologous to each other, but share no orthology to any other species examined here. There are far fewer proteins in the P. falciparum proteome compared to the other species (just 5,337 proteins compared to more than 8,000 in every other species), but a smaller number of proteins did not necessarily limit the representation of KDAC orthologs, as seen in the roundworm species in Fig 2.
In Vitro Screening of Compounds against Various Parasite Species
For compound screening, we adopted cell-based approaches, since these compounds have been previously reported to have activities in isolated enzyme assays on human KDAC proteins. A total of 13 compounds out of the 23 screened showed efficacy in at least one parasite, with all of them also showing some kind of activity on the mammalian cell lines (Table 1). Most of the active compounds showed extremely high (nM to sub-nM IC50) potency in the inhibition of P. falciparum growth. This was consistent with reports for the hydroxamate-based KDAC inhibitors acting on the malaria pathogen [12]. Approximately 10 compounds had IC50/E50 lower (ratio < 0.5) in at least one parasite species compared to the host-cell line. One of the compounds (MC2776), a pyrrole-based hydroxyamate derivative, shows considerable potency (EC50 = 4.39 μM) on the nematode B. malayi, without detectable activity in the host-cell line (> 10 μM), making it a candidate for further optimization and in-depth study. The activities of MC2776 and MC2780 observed in our work were in accord with those reported recently (8b and 10c therein) [50], with higher than 10 μM IC50 on human cancer cell lines for MC2776 (8b) and more potent IC50 for MC2780 (10c). None of the benzamide analogs showed activities on these cell lines (> 20 μM), although in vitro activities of these compounds was reported on human KDACs [51]. This suggested a possible role for the hydroxyamate/benzamide group on cell permeability/transport.
Homology Models and Ligand Binding to the KDAC Proteins
The active compounds from in vitro screening were used to study ligand binding to the KDAC proteins, using computational methods. Two of the representative compounds, MC2776 and MC2780, were docked to the KDAC1 isotypes of the host (human) and each of the parasitic species (B. malayi, L. donovani and P. falciparum). The KDAC1 isotype was chosen because it is ubiquitously expressed in all tissues within all the organisms studied, as reported recently in a tissue-specific expression profiling for 10 A. suum tissues (Fig 3; [52]). In addition, the recently reported X-ray structure of the human protein (pdb code: 4BKX [53]) facilitates structural modeling of the parasitic orthologs with side-by-side comparison of ligand binding.
Fig 3. Expression level of A. suum KDAC1 gene (GS_10652) in different tissues.

Gene expression values are in depth of coverage per million reads (DCPMs). The expression values are averaged across male and female samples.
Homology models were built for the KDAC1 orthologs from three parasitic species (B. malayi, L. donovani and P. falciparum) respectively using the human crystal structure as template. The sequence identity and similarity between each target and the template are high, especially for the nematode B. malayi (S1 Table), suggesting the models should have adequate resolution for the subsequent docking study. The RMSD (root-mean-square-deviation) values for each model after each step in the modeling process remain stable at below 2Å, indicating that the models show high similarities to the human structure, and that there are only subtle differences in the loop regions and side chain conformations which may lead to differences in binding modes.
To validate our docking procedure, a benchmark docking study was also performed for the crystal structure, using the bound ligand (acetate ion). The experimental pose was successfully obtained for acetate (RMSD between lowest energy ligand conformation and crystal structure: 0.74 Å). This validates the potential utility of the docking procedure. The subsequent docking of the two ligands from the screening suggests that both ligands could bind relatively well with the orthologs, but shows some differences at the different ortholog binding sites, especially for the roundworm-selective ligand MC2776 (Fig 4). The models showed that when viewed from above, the ligand the pyrrole ring was almost perfectly in the plane of the hydroxymate in human KDAC1 with the hydroxymate group chelated with catalytic zinc. However, in the B. malayi ortholog, the pyrrole ring rotated counter-clockwise in order to accommodate the tyrosine residue (Y296) at the opening at the binding channel (Fig 4a). The tyrosine residue is conserved across all the KDAC isotypes among almost all organisms, and has been implicated to play a critical role in the selective ligand binding to KDAC8 of S. mansoni [16]. The different orientation of the Y296 in B. malayi could be attributed to a nearby point mutation (C254N). The small hydrophobic residue in other species is tightly packed beneath the binding pocket; while in B. malayi, the bulkier, more hydrophilic asparagine led to a propagation of rearrangements of the two strands nearby, resulting in a misaligned tyrosine residue at the protein surface. In contrast, because of the lack of the pyrrole ring, MC2780 showed very similar binding modes in the KDAC1 proteins of human and B. malayi (Fig 5). The tert-butylcarbamate group at position 3 of the terminal phenyl ring extended toward the outer portion of the binding gorge, contacting one of the loops lining the rim of the catalytic tunnel (residues G677–G686), while in the protist proteins, the same group tilted away to the other side of the channel (S1 Fig). The different binding modes of MC2776 and MC2780 at KDAC1 may partially explain the different affinities among different organisms. Although quantitative binding energies cannot be obtained from simple docking simulations, the distances of the catalytic zinc atom to its coordinating atoms from the protein and ligand were measured in all the models for comparison (Table 2). In general, the ortholog protein with higher binding affinity with the ligand shows shorter distance (both mean and standard deviation) to the zinc atom for most of the coordinating atoms, to maintain the optimal square-based pyramidal geometry. Docking results showed that both MC2776 and MC2780 could bind the KDAC1 of the protists (L. donovani and P. falciparum); However, MC2776 did not show any efficacy in any of the protists, while MC2780 demonstrated pan-parasite potential. This may be due to its inability to reach its target under assay conditions due to metabolism, transport, or other issues [54].
Fig 4. Compound MC2776 docked to the KDAC1 protein.

(A) in the B. malayi protein and (B) in the H. sapiens protein. MC2776 is shown as yellow stick model along with important residues for ligand binding. (C) shows a close-up view of the zinc-centered square based pyramid, Distances for these are shown in Table 2. (D) and (E) show the rendered surface models of the cartoon representations from (A) and (B).
Fig 5. Compound MC2780 docked to the KDAC1 protein.
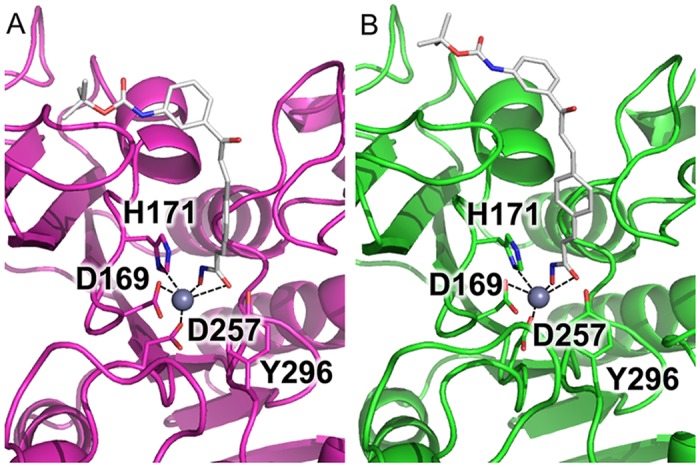
A) in the B. malayi protein and B) in the H. sapiens protein. MC2780 is shown as grey stick model along with important residues for ligand binding.
Table 2. Distances of the catalytic zinc atom to the binding residue and ligand chelating atoms in the models of MC2776 and MC2780 docked to KDAC1 protein of different species.
The distances shown are in the unit of Angstroms. The chemical formulae for both ligands are depicted in Fig 1. O1 and O2 are from hydroxamate group of the ligands. Other atoms are from the protein, as shown in Figs 4 and 5. The coordinates of the models as pdb file are available in S1 Supporting Materials.
| Compound | MC2776 | MC2780 | ||||||
|---|---|---|---|---|---|---|---|---|
| Species | H. sapiens | B. malayi | L. donovani | P. falciparum | H. sapiens | B. malayi | L. donovani | P. falciparum |
| ZN-ND1 (His171) (a) | 3.18 ± 0.47 | 2.80 ± 0.04 | 2.51 ± 0.18 | 2.64 ± 0.12 | 2.76 ± 0.09 | 2.75 ± 0.09 | 2.30 ± 0.05 | 2.66 ± 0.15 |
| ZN-O (Asp169) (b) | 2.26 ± 0.13 | 2.22 ± 0.05 | 2.43 ± 0.03 | 2.39 ± 0.07 | 2.12 ± 0.04 | 2.17 ± 0.03 | 2.41 ± 0.02 | 2.46 ± 0.11 |
| ZN-O (Asp257) (c) | 2.68 ± 0.31 | 2.18 ± 0.08 | 2.17 ± 0.08 | 2.18 ± 0.06 | 2.44 ± 0.03 | 2.16 ± 0.06 | 2.09 ± 0.09 | 2.20 ± 0.04 |
| ZN-O1 (Ligand) (d) | 2.24 ± 0.29 | 2.08 ± 0.02 | 2.06 ± 0.06 | 2.09 ± 0.05 | 2.04 ± 0.01 | 2.11 ± 0.04 | 2.05 ± 0.04 | 2.13 ± 0.08 |
| ZN-O2 (Ligand) (e) | 2.59 ± 0.41 | 2.26 ± 0.05 | 2.83 ± 0.41 | 2.48 ± 0.26 | 2.62 ± 0.15 | 3.09 ± 0.08 | 3.10 ± 0.21 | 2.21 ± 0.11 |
| EC50(IC50) (mM) | >10 | 4.39 | - | - | 4.81 | 2.53 | 0.47 | 0.06 |
Active-Site Variance of the KDAC Proteins for Selective Ligand Design
To facilitate selective ligand design, we systematically examined all of orthologous proteins within the parasite species and identified the variant residues near the active site for comparison with human protein structures. X-ray crystal structures of the catalytic domain have been reported for 6 of the 11 zinc-containing KDAC isotypes in human, i.e. KDAC1, 2, 3, 8 (class I) and 4, 7 (class II). Among them, KDAC7 lacks a clear ortholog in any of the parasite species, so active-site variants were reported for KDAC1, 2, 3, and 8 together with the only class II isotype KDAC4 (Table 3), showing that active-site residues were well conserved in most of parasite orthologs (especially for KDAC1). The kinetoplastid orthologs have slightly more variance, with 8 out of the 50 residues being different from the human protein. All the other orthologs of KDAC1 in the parasite proteins had 5 or fewer residues different from the human protein, with the exception of B. malayi, which had 6 different residues. Interestingly, the one uniquely different residue (C254N) is the also a contributing factor of the different binding mode of MC2776, as suggested by docking. This demonstrates that variation at active sites could play an important role in the pursuit of selective ligands. It is not surprising to see a higher variances for KDAC3 in the kinetoplastids, since each of the two genera has been grouped into its own orthologous protein cluster (Leishmania and Trypanosoma), suggesting their divergent distance from the human ortholog. KDAC8s of trematodes also clustered into a separate cluster than the mammalian orthologs, but still showed quite high conservation at the active sites, with only 8 out of the 48 residues different. In the class IIA family cluster, only one member was found for the parasites except for P. falciparum. Annotated KDAC4 proteins (roundworms and flatworms) were compared with the human KDAC4 structure and were well-conserved except for A. ceylanicum, with 22 different residues out of a total of 49. A closer examination revealed that this was due to the fragmented sequence within that region, since at positions 21, there are gaps instead of amino acid residues. This is likely an artifact resulted from the draft nature of the genome (proteome) data.
Table 3. Sequence variations of KDAC proteins of parasitic species in comparison with the host (H. sapiens) orthologs at the active sites.
Active-site residues were defined as any residue with a distance less than 10 Å to the catalytic zinc in the crystal structure. Abbreviations were used for the names of all the species following the rule of “the first letter of genus + first three letter of species”.
| Class | I | IIA | ||||
|---|---|---|---|---|---|---|
| Target Protein | KDAC1 | KDAC2 | KDAC3 | KDAC8 | KDAC4 | |
| Human gene (H. sapiens; ENSG00000XXXXXX) | 116478 | 196591 | 171720 | 147099 | 68024 | |
| PDB code | 4BKX | 4LXZ | 4A69 | 1T67 | 4CBY | |
| Total defined residues | 50 | 49 | 50 | 48 | 49 | |
| Nematode | Acey | 1 | - | 3 | - | 22 |
| Asuu | 1 | 0 | 3 | - | 5 | |
| Bmal | 6 | 0 | 2 | - | 13 | |
| Cele | 5 | 3 | 3 | - | 5 | |
| Dimm | 2 | 0 | 4 | - | 6 | |
| Hcon | 1 | 1 | 3 | - | 5 | |
| Lloa | 2 | 0 | 4 | - | 6 | |
| Name | 1 | 12 | 3 | - | 5 | |
| Tmur | 4 | - | 4 | - | 14 | |
| Tspi | 3 | - | 4 | - | 9 | |
| Tsui | 3 | - | 3 | - | 13 | |
| Kinetoplastid | Tcru | 8 | - | 21 | - | - |
| Tbru | 8 | - | 26 | - | - | |
| Lmaj | 8 | - | 14 | - | - | |
| Ldon | 8 | - | 14 | - | - | |
| Malaria | Pfal | 3 | - | - | - | - |
| Trematode | Sman | 4 | - | 3 | 8 | 14 |
| Sjap | 1 | - | 3 | 8 | 7 | |
| Shem | 1 | - | 3 | 8 | 9 | |
| Csin | 4 | - | 6 | 8 | 7 | |
Discussion
We presented the first systematic examinations of all zinc-dependent KDAC proteins in representative parasites from nematode, trematode, kinetoplastid and malaria pathogens, and showed that some human KDAC isotypes lack clear orthologs in the parasites, with the only conserved isotype across all the species studied being KDAC1. KDAC1 enzymes are primarily localized in the nucleus and are expressed in all tissues almost ubiquitously. As a classical KDAC protein, it has been studied extensively both in human and in the parasites such as P. falciparum as a novel drug target [6, 12]. KDAC1 shares high sequence similarities among all the species studied. It has been reported that KDAC1 of P. falciparum shares over 55% sequence identity to yeast, human, chicken, and frog KDAC orthologs [55]. This also consistent for the nematode/trematode KDAC1 in comparison with host orthologs, but the homology was lower in the kinetoplastids, which had ~40% sequence identity with the remaining species (S2 Fig). It was also reflected in active-site residues, with most of the variants coming from the kinetoplastid orthologs. As shown in S2 Fig, out of the 12 active-site variants from all the parasite proteins, 3 are specifically present in the kinetoplastids, indicating a higher level of divergence. It has been found that the KDACs of kinetoplastids branched very early from the eukaryotic lineages, especially for the class I isotopes [56]. This suggests that it would be relatively easier to design selective compounds against the kinetoplastid KDAC1. Achieving species selectivity within the other parasite species would still be difficult, due to the higher level of conservations of protein sequence especially at the active site. In this regard, the molecular ligand binding modeling of B. malayi KDAC1 offered potential insight on improving parasitic selectivity. In fact, a BLAST search of the protein sequence against other nematode proteomes suggests that the C254N mutation was present within KDAC1 of other Brugia species (B. timori and B. pahangi), as well as the Onchocerca species (O. ochengi, O. flexuosa, and O. volvulus).
The other members in the class I isotype were tentatively labeled as KDAC3 for the kinetoplastids, although they were each clustered into their own families (Leishmania/Trypanosoma). They showed even greater divergence in comparison with other species, raising the possibility that they were completely new isotypes instead of KDAC3 orthologs. Nevertheless, given their divergence from any other KDACs, it might be worthwhile to pursue their roles as selective drug targets. Much work has been done on the trematode KDAC8 (specifically in S. mansoni) as a novel drug target for the control of schistosomaisis, since it is the most highly expressed class I KDAC isotype in this organism [16]. Our results indicate that KDAC8 is in a separate family of KDACs within the flatworms, with some divergence from host orthologs. Structural characterization of S. mansoni KDAC8 confirmed our findings and supported the observation that selective ligands can be designed to explore subtle conformational differences at the active site [16].
Interestingly, among the class II families, IIA and IIB proteins showed different gene expansion/deletion patterns within the parasite species compared to hosts. Four members have been identified for the class IIA proteins in the hosts (4, 5, 7, and 9), while only 1 or 2 members were present in the parasites. In contrast, in the class IIB families, some parasites (especially the roundworm species) showed considerable expansions. This has been observed before in the non-parasitic nematode C. elegans [57] and was confirmed in many of the parasitic roundworms analyzed in this study. However, some of roundworms (like all the protists) also showed a loss of genes, none or just one protein among the two class IIB protein families (6, 10). The highly variable gene expansion/loss pattern implied different functional roles for the class IIB isotypes in nematodes, which is a topic worth further exploration.
The much less-studied KDAC11, as the sole member of the class IV isotype, was only present in hosts and nematodes. KDAC11 has been found to be differentially expressed among different tissues and was suggested to be a novel drug target in human carcinomas [58]. Further characterization of its roles in nematodes could decipher its specificities and reveal any potential for targeted therapeutics. It was surprising that two of the class II KDACs in P. falciparum were clustered into a completely new protein family, with none of the known KDAC proteins from other species present. This protein family contained the most members (1271 proteins) among all the families generated, including 1244 P. falciparum proteins and 27 proteins from the other 12 species. Besides the two KDACs, the 1244 P. falciparum proteins within this protein family were assigned annotations ranging from protein enzymes (kinase, polymerase, protease, transferase, and etc.) to transporters, ion channels and many others, with over half of them annotated as unknown functions. This indicates that the protein family was a promiscuous assembly with no consensus functions identified, and also showed that the two P. falciparum KDAC proteins have the largest evolutionary distances from other species (consistent with previous observations [10, 12]).
In this work, we report some preliminary results of compound screening against a panel of parasitic species, and explored the mechanisms of their activities at the molecular level for certain KDAC isotypes. Docking calculations were performed on the homology models for all the parasitic protein orthologs. Caution should be exercised when interpreting the observations made for protein-ligand interactions, since homology models have limited representation of different loop conformations between the target structure and the template. In our case, the sequence identity between template and target was relatively high (ranging from 45% for L. donovani and 62% for P. falciparum, to 72% for B. malayi for the full length, and over 80% for all the species at the active site as shown in Table 3 and S1 Table and S2 Fig), so the docking result in homology models should be reliable at this level [59]. However, it is still premature to draw conclusions based on the docking results on a single isotype, since the "inactivity" of compounds in a full-organism assay may be attributed to a number of possible explanations including: (i) the compound not penetrating the cell (inability to reach the target), (ii) the compound being metabolized in the organism, active excretion from the cell, or (iii) the target of the compound not being essential. In addition, although KDAC1 is the most important target across all the organisms (since it is ubiquitous), the compounds’ molecular interactions on other KDAC isotypes or even other proteins were not examined in detail. The work reported is "preliminary" evidence of KDACs as drug targets, nevertheless it should inspire both biochemical experimental and computational studies, for more detailed characterization of KDAC targets within the parasite species and help generate new and improved selective compounds to target parasitic disease.
Conclusions
A systematic study of all KDAC proteins within parasitic species from protists to nematodes as novel drug targets is reported. Although much work is still required to elucidate their functions and essential nature of these KDACs in parasites, preliminary compound screening suggests that they are inhibited by known host KDAC ligands; such compounds also showed different degrees of selectivity within the parasitic panel. Molecular modeling in combination with genomic profiling offered insight in the mechanism of selectivity and suggested future directions for selectively targeting parasitic lysine deactylases.
Supporting Information
MC2780 is shown as grey stick model along with important residues for ligand binding as Fig 5.
(TIFF)
Active-site residues (within 10 Å distance to catalytic zinc atom) in the crystal structure (4BKX, chain B) are marked as “+” underneath. Any variant residues within the parasitic species are marked as “*” at the bottom.
(TIF)
(XLSX)
(ZIP)
Acknowledgments
We acknowledge members of the Mitreva and Marshall groups for helpful discussions. We also thank the anonymous reviewers and editor for excellent inputs and constructive suggestions for the improvement of the manuscript.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the National Institute of Health (NIH) National Institute of Allergy and Infectious Diseases (NIAID) Grant #AI081803 to MM. Part of the study was also supported by Scynexis, Inc. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hotez PJ, Pecoul B. "Manifesto" for advancing the control and elimination of neglected tropical diseases. PLoS neglected tropical diseases. 2010;4(5):e718 10.1371/journal.pntd.0000718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morens DM, Folkers GK, Fauci AS. The challenge of emerging and re-emerging infectious diseases. Nature. 2004;430(6996):242–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–40. Epub 2009/07/18. 10.1126/science.1175371 [DOI] [PubMed] [Google Scholar]
- 4. Nebbioso A, Carafa V, Benedetti R, Altucci L. Trials with 'epigenetic' drugs: an update. Molecular oncology. 2012;6(6):657–82. 10.1016/j.molonc.2012.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gregory PD, Wagner K, Horz W. Histone acetylation and chromatin remodeling. Experimental cell research. 2001;265(2):195–202. [DOI] [PubMed] [Google Scholar]
- 6. Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Molecular cancer research : MCR. 2007;5(10):981–9. [DOI] [PubMed] [Google Scholar]
- 7. Ficner R. Novel structural insights into class I and II histone deacetylases. Current topics in medicinal chemistry. 2009;9(3):235–40. [DOI] [PubMed] [Google Scholar]
- 8. Andrews KT, Fisher G, Skinner-Adams TS. Drug repurposing and human parasitic protozoan diseases. International journal for parasitology Drugs and drug resistance. 2014;4(2):95–111. 10.1016/j.ijpddr.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Panic G, Duthaler U, Speich B, Keiser J. Repurposing drugs for the treatment and control of helminth infections. International journal for parasitology Drugs and drug resistance. 2014;4(3):185–200. 10.1016/j.ijpddr.2014.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andrews KT, Haque A, Jones MK. HDAC inhibitors in parasitic diseases. Immunology and cell biology. 2012;90(1):66–77. 10.1038/icb.2011.97 [DOI] [PubMed] [Google Scholar]
- 11. Deshmukh AS, Srivastava S, Dhar SK. Plasmodium falciparum: epigenetic control of var gene regulation and disease. Sub-cellular biochemistry. 2013;61:659–82. 10.1007/978-94-007-4525-4_28 [DOI] [PubMed] [Google Scholar]
- 12. Andrews KT, Tran TN, Fairlie DP. Towards histone deacetylase inhibitors as new antimalarial drugs. Current pharmaceutical design. 2012;18(24):3467–79. [PubMed] [Google Scholar]
- 13. Giannini G, Battistuzzi G, Vignola D. Hydroxamic acid based histone deacetylase inhibitors with confirmed activity against the malaria parasite. Bioorganic & medicinal chemistry letters. 2015;25(3):459–61. [DOI] [PubMed] [Google Scholar]
- 14. Hansen FK, Skinner-Adams TS, Duffy S, Marek L, Sumanadasa SD, Kuna K, et al. Synthesis, antimalarial properties, and SAR studies of alkoxyurea-based HDAC inhibitors. ChemMedChem. 2014;9(3):665–70. 10.1002/cmdc.201300469 [DOI] [PubMed] [Google Scholar]
- 15. Miao J, Lawrence M, Jeffers V, Zhao F, Parker D, Ge Y, et al. Extensive lysine acetylation occurs in evolutionarily conserved metabolic pathways and parasite-specific functions during Plasmodium falciparum intraerythrocytic development. Molecular microbiology. 2013;89(4):660–75. 10.1111/mmi.12303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marek M, Kannan S, Hauser AT, Moraes Mourao M, Caby S, Cura V, et al. Structural basis for the inhibition of histone deacetylase 8 (HDAC8), a key epigenetic player in the blood fluke Schistosoma mansoni. PLoS pathogens. 2013;9(9):e1003645 10.1371/journal.ppat.1003645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stolfa DA, Marek M, Lancelot J, Hauser AT, Walter A, Leproult E, et al. Molecular basis for the antiparasitic activity of a mercaptoacetamide derivative that inhibits histone deacetylase 8 (HDAC8) from the human pathogen schistosoma mansoni. Journal of molecular biology. 2014;426(20):3442–53. 10.1016/j.jmb.2014.03.007 [DOI] [PubMed] [Google Scholar]
- 18. Trenholme K, Marek L, Duffy S, Pradel G, Fisher G, Hansen FK, et al. Lysine acetylation in sexual stage malaria parasites is a target for antimalarial small molecules. Antimicrobial agents and chemotherapy. 2014;58(7):3666–78. 10.1128/AAC.02721-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hansen FK, Sumanadasa SD, Stenzel K, Duffy S, Meister S, Marek L, et al. Discovery of HDAC inhibitors with potent activity against multiple malaria parasite life cycle stages. European journal of medicinal chemistry. 2014;82:204–13. 10.1016/j.ejmech.2014.05.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andrews KT, Gupta AP, Tran TN, Fairlie DP, Gobert GN, Bozdech Z. Comparative gene expression profiling of P. falciparum malaria parasites exposed to three different histone deacetylase inhibitors. PloS one. 2012;7(2):e31847 10.1371/journal.pone.0031847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carrillo AK, Guiguemde WA, Guy RK. Evaluation of histone deacetylase inhibitors (HDACi) as therapeutic leads for human African trypanosomiasis (HAT). Bioorganic & medicinal chemistry. 2015. [DOI] [PubMed] [Google Scholar]
- 22. Kelly JM, Taylor MC, Horn D, Loza E, Kalvinsh I, Bjorkling F. Inhibitors of human histone deacetylase with potent activity against the African trypanosome Trypanosoma brucei. Bioorganic & medicinal chemistry letters. 2012;22(5):1886–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 1964;51:786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kinsella RJ, Kahari A, Haider S, Zamora J, Proctor G, Spudich G, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database: the journal of biological databases and curation. 2011;2011:bar030 10.1093/database/bar030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benson DA, Karsch-Mizrachi I, Clark K, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Research. 2012;40(Database issue):D48–D53. 10.1093/nar/gkr1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harris TW, Baran J, Bieri T, Cabunoc A, Chan J, Chen WJ, et al. WormBase 2014: new views of curated biology. Nucleic acids research. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li BW, Rush AC, Jiang DJ, Mitreva M, Abubucker S, Weil GJ. Gender-associated genes in filarial nematodes are important for reproduction and potential intervention targets. PLoS Negl Trop Dis. 2011;5(1):e947 Epub 2011/02/02. 10.1371/journal.pntd.0000947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang YT, Gao X, Rosa BA, Abubucker S, Hallsworth-Pepin K, Martin J, et al. Genome of the human hookworm Necator americanus. Nature genetics. 2014;46(3):261–9. 10.1038/ng.2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cantacessi C, Gasser RB, Strube C, Schnieder T, Jex AR, Hall RS, et al. Deep insights into Dictyocaulus viviparus transcriptomes provides unique prospects for new drug targets and disease intervention. Biotechnol Adv. 2011;29(3):261–71. Epub 2010/12/25. 10.1016/j.biotechadv.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laing R, Kikuchi T, Martinelli A, Tsai IJ, Beech RN, Redman E, et al. The genome and transcriptome of Haemonchus contortus, a key model parasite for drug and vaccine discovery. Genome Biol. 2013;14(8):R88 Epub 2013/08/30. 10.1186/gb-2013-14-8-r88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Godel C, Kumar S, Koutsovoulos G, Ludin P, Nilsson D, Comandatore F, et al. The genome of the heartworm, Dirofilaria immitis, reveals drug and vaccine targets. FASEB J. 2012;26(11):4650–61. 10.1096/fj.12-205096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zerlotini A, Heiges M, Wang H, Moraes RL, Dominitini AJ, Ruiz JC, et al. SchistoDB: a Schistosoma mansoni genome resource. Nucleic Acids Res. 2009;37(Database issue):D579–82. Epub 2008/10/10. 10.1093/nar/gkn681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang X, Chen W, Huang Y, Sun J, Men J, Liu H, et al. The draft genome of the carcinogenic human liver fluke Clonorchis sinensis. Genome biology. 2011;12(10):R107 10.1186/gb-2011-12-10-r107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic acids research. 2010;38(Database issue):D457–62. 10.1093/nar/gkp851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30(7):1575–84. Epub 2002/03/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, et al. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Current protocols in bioinformatics / editoral board, Andreas D Baxevanis [et al]. 2011;Chapter 6:Unit 6 12 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li L, Stoeckert CJ Jr., Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome research. 2003;13(9):2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Taylor CM, Wang Q, Rosa BA, Huang SC, Powell K, Schedl T, et al. Discovery of anthelmintic drug targets and drugs using chokepoints in nematode metabolic pathways. PLoS Pathog. 2013;9(8):e1003505 Epub 2013/08/13. 10.1371/journal.ppat.1003505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Taylor CM, Martin J, Rao RU, Powell K, Abubucker S, Mitreva M. Using existing drugs as leads for broad spectrum anthelmintics targeting protein kinases. PLoS pathogens. 2013;9(2):e1003149 10.1371/journal.ppat.1003149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson JD, Dennull RA, Gerena L, Lopez-Sanchez M, Roncal NE, Waters NC. Assessment and continued validation of the malaria SYBR green I-based fluorescence assay for use in malaria drug screening. Antimicrobial agents and chemotherapy. 2007;51(6):1926–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Das R, Baker D. Macromolecular modeling with rosetta. Annual review of biochemistry. 2008;77:363–82. [DOI] [PubMed] [Google Scholar]
- 42. Wang C, Vernon R, Lange O, Tyka M, Baker D. Prediction of structures of zinc-binding proteins through explicit modeling of metal coordination geometry. Protein science: a publication of the Protein Society. 2010;19(3):494–506. Epub 2010/01/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, et al. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem. 2008;283(39):26694–704. Epub 2008/07/11. 10.1074/jbc.M803514200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. Journal of chemical information and modeling. 2010;50(4):572–84. 10.1021/ci100031x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. Journal of computational chemistry. 2002;23(16):1623–41. Epub 2002/10/24. [DOI] [PubMed] [Google Scholar]
- 46. Irwin JJ, Raushel FM, Shoichet BK. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry. 2005;44(37):12316–28. [DOI] [PubMed] [Google Scholar]
- 47. Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC bioinformatics. 2004;5:113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harris TW, Baran J, Bieri T, Cabunoc A, Chan J, Chen WJ, et al. WormBase 2014: new views of curated biology. Nucleic acids research. 2014;42(Database issue):D789–93. 10.1093/nar/gkt1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Joshi MB, Lin DT, Chiang PH, Goldman ND, Fujioka H, Aikawa M, et al. Molecular cloning and nuclear localization of a histone deacetylase homologue in Plasmodium falciparum. Molecular and biochemical parasitology. 1999;99(1):11–9. [DOI] [PubMed] [Google Scholar]
- 50. Valente S, Trisciuoglio D, Tardugno M, Benedetti R, Labella D, Secci D, et al. tert-Butylcarbamate-containing histone deacetylase inhibitors: apoptosis induction, cytodifferentiation, and antiproliferative activities in cancer cells. ChemMedChem. 2013;8(5):800–11. 10.1002/cmdc.201300005 [DOI] [PubMed] [Google Scholar]
- 51. Patel V, Mazitschek R, Coleman B, Nguyen C, Urgaonkar S, Cortese J, et al. Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. Journal of medicinal chemistry. 2009;52(8):2185–7. 10.1021/jm801654y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rosa BA, Jasmer DP, Mitreva M. Genome-wide tissue-specific gene expression, co-expression and regulation of co-expressed genes in adult nematode Ascaris suum. PLoS neglected tropical diseases. 2014;8(2):e2678 10.1371/journal.pntd.0002678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, et al. Class I HDACs share a common mechanism of regulation by inositol phosphates. Molecular cell. 2013;51(1):57–67. 10.1016/j.molcel.2013.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. An WF, Tolliday N. Cell-based assays for high-throughput screening. Molecular biotechnology. 2010;45(2):180–6. 10.1007/s12033-010-9251-z [DOI] [PubMed] [Google Scholar]
- 55. Darkin-Rattray SJ, Gurnett AM, Myers RW, Dulski PM, Crumley TM, Allocco JJ, et al. Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc Natl Acad Sci USA. 1996;93(23):13143–7. Epub 1996/11/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Horn D. Histone deacetylases. Advances in experimental medicine and biology. 2008;625:81–6. 10.1007/978-0-387-77570-8_7 [DOI] [PubMed] [Google Scholar]
- 57. Yang XJ, Gregoire S. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Molecular and cellular biology. 2005;25(8):2873–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Deubzer HE, Schier MC, Oehme I, Lodrini M, Haendler B, Sommer A, et al. HDAC11 is a novel drug target in carcinomas. International journal of cancer Journal international du cancer. 2013;132(9):2200–8. 10.1002/ijc.27876 [DOI] [PubMed] [Google Scholar]
- 59. Rodrigues JP, Melquiond AS, Karaca E, Trellet M, van Dijk M, van Zundert GC, et al. Defining the limits of homology modeling in information-driven protein docking. Proteins. 2013;81(12):2119–28. 10.1002/prot.24382 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MC2780 is shown as grey stick model along with important residues for ligand binding as Fig 5.
(TIFF)
Active-site residues (within 10 Å distance to catalytic zinc atom) in the crystal structure (4BKX, chain B) are marked as “+” underneath. Any variant residues within the parasitic species are marked as “*” at the bottom.
(TIF)
(XLSX)
(ZIP)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.