

Abstract
Purpose.
Retinal pigment epithelial (RPE) cell death is an important feature of the advanced forms of AMD. Complement alternative pathway (AP) activation is associated with RPE cell death in AMD. In this study, we developed a new model to initiate AP activation on RPE cells and investigated the cellular mechanisms modulating AP activation–mediated RPE cell death.
Methods.
An anti-RPE antibody was developed. A spontaneously arising human RPE cell line (ARPE-19) and donor RPE cells were primed with this antibody followed by stimulation with 6% C1q-depleted human serum (C1q-Dep) to activate AP. Complement activation was evaluated by flow cytometry and immunofluorescent staining. Cellular response to complement activation was examined by measurement of intracellular calcium and adenosine triphosphate (ATP) release. Cell viability was assessed by Sytox orange, tetrazolium salt, and lactate dehydrogenase release assays.
Results.
Alternative pathway complement–mediated RPE cell death was associated with membrane attack complex formation and a rapid rise in intracellular calcium followed by release of ATP. Downregulation of membrane complement regulatory proteins and protein kinase C (PKC) inhibition increased cell susceptibility to complement attack. Pretreatment of RPE cells with either hydrogen peroxide or hydroquinone enhanced cell death. Chronic repetitive treatment of RPE cells with low levels of oxidants also enhanced complement-mediated cell death.
Conclusions.
Activation of complement through the alternative pathway induces sublytic and lytic phases of complement attack on RPE cells, leading to cell death modulated by extracellular calcium, membrane complement regulatory proteins, and intracellular signaling mechanisms. Single-dose oxidant exposure and low-dose repetitive oxidant exposure rendered RPE cells more susceptible to complement-mediated death.
Keywords: AMD, RPE, complement, oxidative stress, calcium, PKC
A new model was developed to initiate complement attack on retinal pigment epithelial (RPE) cells through the alternative complement pathway. This new model elucidates mechanisms of complement-mediated RPE injury and is useful to study the functional effects of complement and complement/oxidant interactions.
Introduction
Activation and deposition of complement in the sub retinal pigment epithelial (RPE) space and choroid is one of the early pathological findings in tissues from eyes with AMD.1–4 The identification of a genetic susceptibility locus within complement factor H5–8 provides an additional link between complement activation and disease susceptibility. These two key observations have led to a significant interest in complement both as a predictor of disease occurrence as well as a therapeutic target. The latter is highlighted by the entry of the complement inhibitor POT-4 into clinical trials for AMD in 2007 and the entry of at least three additional complement-directed therapeutics (Eculizumab, FCFD4514S, ARC1905),9 and recent favorable results of factor D inhibitor (FCFD4514S) to treat geographic atrophy (GA) secondary to nonneovascular AMD.10 Complement activation is clearly involved in the pathology of AMD; however, the impact of complement on ocular function and the mechanisms by which it contributes to disease susceptibility are still not well understood.
Retinal pigment epithelial cell death is an important feature of the advanced forms of AMD. Complement activation products C3a, C5a, and membrane attack complex (MAC) regulate RPE cell expression of cytokines, pro-inflammatory molecules, and growth factors.11–14 They also modulate disease progression in the laser choroidal neovascularization (CNV) model of wet AMD.15,16 Localized accumulations of complement activation products adjacent to the RPE point to the RPE as a site of complement activation, either by a bystander effect through formation of MAC or through receptor-mediated activation by C3a and C5a.
The activation of the complement cascade can occur through the classical, alternative, and lectin pathways. C1q is an initiator protein of classical pathway (CP) activation and factor B (FB) is a component of alternative pathway (AP) activation.17 Classical pathway function requires both calcium and magnesium ions, whereas the AP requires only magnesium ions.18–20
Nucleated cells are protected against bystander-mediated complement attack by multiple cell surface membrane complement regulatory proteins (mCRPs) including CD46, CD55, CD59, and CR1.21 These proteins regulate complement activity by increasing the rate of C5 convertase turnover through the following mechanisms: dissociation of the convertase complex by decay accelerating factor (DAF/CD55), active proteolysis of the complex by membrane cofactor protein (MCP/CD46) in conjunction with factor I, and assembly inhibition of the final MAC by CD59.21 Additionally, cells have a secondary mechanism that involves calcium-dependent removal of MAC protein from the cell surface.22,23
To better understand the effect of complement activation and MAC deposition on RPE cell death, we developed a model that induces sublytic and lytic phases of complement attack through AP using an anti-RPE antibody followed by the addition of C1q-depleted human serum (C1q-Dep). In the current study, we used this model to investigate the cellular mechanisms modulating RPE cell death in response to AP activation, to determine whether calcium and protein kinase C (PKC) protect RPE cells from complement-mediated cell lysis, to assess the relative contribution of mCRPs on RPE cell death, and to determine functional effects of complement/oxidant interactions.
Materials and Methods
Materials
Fluo-4AM, TrypLE, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA), Sytox Orange were purchased from Invitrogen (Carlsbad, CA, USA). Collagen type I, tert-butyl hydroperoxide (t-BH), and hydroquinone (HQ) were purchased from Sigma (St. Louis, MO, USA). Hydrogen peroxide (H2O2) was purchased from VWR (West Chester, PA, USA). Normal human serum (NHS), C1q-Dep, C5 depleted serum (C5-Dep), FB depleted serum (FB-Dep), and anti-FB antibody were purchased from Quidel Corp. (San Diego, CA, USA). Protein G purified normal sheep immunoglobulin G (IgG) was purchased from Molecular Innovations, Inc. (Novi, MI, USA). SpectraMax M2 with SoftMax Pro software, MetaMorph image analysis software and FLIPR Tetra device were purchased from Molecular Devices (Sunnyvale, CA, USA). ON-Targetplus SMARTpool small interfering RNA (siRNA) targeted to human MCP, DAF, CD59 and non-target “control” was purchased from Thermo Scientific (Chicago, IL, USA). A spontaneously arising human RPE cell line (ARPE-19) cells were obtained from ATCC (Manassas, VA, USA). Adenosine triphosphate (ATP) Bioluminescence Assay Kit HS II, lactate dehydrogenase (LDH) kit, and tetrazolium salt WST-1 (4-[3-(4-lodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1.3-benzene disulfonate) were purchased from Roche Applied Science (Indianapolis, IN, USA). Murine monoclonal anti-human C5b-9 clone aE11 was purchased from Dako (Carpinteria, CA, USA). Fluorescein isothiocyanate goat anti-mouse IgG + IgM and anti-human CD59 clone p282 (H19), CD46 clone E4.3, CD55 clone IA10, and CD35/CR1 clone E11 antibodies were purchased from BD Biosciences (San Jose, CA, USA). Rabbit polyclonal anti-ZO-1 (cat# 40-2300) and Alexa Fluor 488 donkey anti-sheep IgG were purchased from Invitrogen. Mouse monoclonal anti-cytokeratin peptide 18 (clone CY-90) was purchased from Sigma. Horseradish peroxidase conjugated donkey anti-sheep IgG was purchased from Jackson ImunoResearch Laboratories, Inc. (West Grove, PA, USA).
RPE Cell Culture
ARPE-19 cells (the donor with CFHYH402 variant) were passaged upon reaching 80% to 90% confluence. Human donor eyes were obtained within 24 hours after death from the North Carolina Organ Donor and Eye Bank, Inc. (Winston-Salem, NC, USA) in accordance with the provisions of the Declaration of Helsinki for research involving human tissue. Retinal pigment epithelial cells for primary culture studies were harvested from eyes obtained from three donors (a 61-year-old donor with CFHYH402 variant, a 51-year-old donor with CFHHH402 variant, and a 62-year-old donor with CFHYY402 variant) as we have previously described.24 The identity of RPE cells was confirmed by cytokeratin-18 and ZO-1 stain. All plates were coated with 0.1 mg/mL collagen type I for 1 hour at room temperature (RT), rinsed with distilled H2O and air-dried for 1 hour prior to plating cells. Cells were grown in MEM containing 10% FBS, 1× penicillin/streptomycin (Pen/Strep) at 37°C in a humidified environment containing 5% CO2.
Generation of Anti-RPE Antibody
An anti-RPE polyclonal antibody denoted S-58 (named to indicate the origin from sheep, “S,” animal number “58”) was generated by inoculating sheep with paraformaldehyde (PFA)-fixed ARPE-19 cells (1×106 cells in complete Freund's adjuvant) followed by repeated monthly immunizations in incomplete Freund's adjuvant over a 5-month period. Immunoreactivity was monitored by florescence-activated cell sorting (FACS) analysis using test bleeds obtained 8 days following each boost. At the end of the immunization protocol, production bleeds were obtained and immunoglobulins were purified on an affinity Protein G column (Lampire Biological Laboratories, Pipersville, PA, USA).
FACS Analysis
For detection of mCRPs, ARPE-19 cells were trypsinized, resuspended in FACS buffer (Dulbecco's phosphate-buffered saline [DPBS], 0.05% sodium azide, 1% BSA) and then incubated with primary antibodies at 1:150 dilutions. After incubation with primary antibodies in FACS buffer for 1 hour at 4°C, cells were washed then incubated using the appropriate secondary antibodies at 1:200 dilutions for 30 minutes at 4°C, washed, and analyzed.
For S-58 binding assay, ARPE-19 cells were primed with S-58 (1.2 mg/mL) for 30 minutes at 37°C and detached with 1× TrypLE. S-58 binding to ARPE-19 cells was measured as previously described.25 Alexa 488-conjungated donkey anti-sheep antibody was used at a dilution of 1:1000.
For MAC deposition, ARPE-19 cells were primed with S-58 (1.2 mg/mL) for 30 minutes at 37°C, washed, and incubated with either 6% C1q-Dep or 6% HiC1q-Dep (as negative control) at 37°C for 20 minutes. C1q-Dep was used as a source of complement for all experiments. Cells were detached with 1× TrypLE and fixed in 0.5% PFA for 10 minutes at 37°C followed by chilling on ice for 1 minute. Cell surface MAC deposition (aE11, 1:25 in 1% BSA/PBS) was measured as previously described.25 Alexa 488-conjungated goat anti-mouse antibody was used at a dilution of 1:600.
Western Blot and Immunofluorescent Staining
ARPE-19 and donor RPE cells were incubated with S-58 at various concentrations for 30 minutes at 37°C. Cell extracts were prepared and Western blot was performed as we have previously described.25 To identify bound S-58, cells were fixed in 4% PFA for 15 minutes, blocked in 5% normal donkey serum for 30 minutes at RT, then incubated with Alexa 488 anti-sheep IgG (1:500 in 1% BSA/PBS) for 1 hour at RT. To detect cellular MAC deposition, cells from a 62-year-old donor were incubated with S-58 antibody (1.2 mg/mL) for 30 minutes at 37°C, washed, and then incubated for 30 minutes with either 6% C1q-Dep or heat inactivated C1q-Dep (HiC1q-Dep, 30 minutes at 56°C, as a negative control). The cells were then fixed in 4% PFA for 15 minutes, blocked with 5% normal goat serum for 30 minutes at RT, followed by incubation with mouse anti-human C5b-9 antibody (aE11, 1:25 diluted in 1% BSA/PBS) overnight at 4°C, and then with goat anti-mouse Alexa 568 antibody (1:500 in 1% BSA/PBS) for 1 hour at RT. Fluorescent staining was observed with a confocal microscope (Nikon C90i; Nikon, Garden City, NY, USA).
Determination of Free Intracellular Calcium
ARPE-19 cells were plated in a 96-well plate and grown to confluence. S-58 was serially diluted into H/BSA (Hank's balanced salt solution, 1% BSA, 1% HEPES, pH 7.4). Samples (100 μL/well) were added in triplicate and incubated for 1 hour at 37°C in the presence of 2 μM Fluo-4AM Cells were washed twice with H/BSA. A baseline fluorescence reading was obtained prior to stimulation with 6% C1q-Dep or C5-Dep in H/BSA. Six percent serum was chosen based on serum dose-response experiments to identify a serum concentration that gave the maximal dynamic range in response to changing levels of priming antibody. We held the serum concentration constant and varied the antibody concentration so that in subsequent experiments, we could alter complement attack levels while controlling for serum. Calcium binding was monitored using an excitation wavelength of 470 to 495 nm and an emission wavelength 515 to 575 nm using a FLIPR system (Molecular Devices).
Determination of Extracellular Adenosine triphosphate (ATP) Release
ARPE-19 cells were plated in a 96-well plate and grown to confluence. S-58 was serially diluted into H/BSA and incubated for 1 hour at 37°C. Cells were washed twice with H/BSA, and then incubated for 3 hours at 37°C with either 6% C1q-Dep or C5-Dep in H/BSA. Following complement attack, 100 μL of supernatant was removed from each well, placed in a separate plate and centrifuged at 200× g. Fifty microliters of supernatant were removed from the centrifuged plate and ATP levels were quantified with an ATP Bioluminescence Assay Kit HS II along with a SpectraMax M2 containing SoftMax Pro Software.
Sytox Orange Nucleic Acid Stain
Cells were plated in a 96-well plate and grown to confluence. S-58 was serially diluted into H/BSA and incubated for 30 minutes at 37°C. Cells were washed twice with H/BSA and complement attack was initiated by adding either 6% C1q-Dep, FB-Dep, or C5-Dep. Six percent NHS was used for positive control of lysis. The relative numbers of lysed cells were determined by incubating cells in the presence of membrane impermeant dye Sytox Orange. The number of positively stained nuclei, or membrane-compromised “dead” cells, was quantified using a ×20 objective on a Nikon TE300 inverted microscope fitted with an ASI MS-2000 automated stage (Applied Scientific Instruments, Eugene, OR, USA) and Co-olsnap CCD camera (Photometrics Scientific, Tucson, AZ, USA) clarity for with excitation at 547 nm and emission at 570 nm. Image capture automation was conducted using MetaMorph Image capture software in which three nonoverlapping fields were captured per well. The average of the three nonoverlapping fields was used as the number of stained nuclei per well.
For the calcium depletion experiments, cells were briefly washed in Ca2+-free HBSA containing EGTA (HBSS –Ca2+/–Mg2+, 0.02% MgCl2, 1% BSA, 0.5 mM EGTA), and then washed in Ca2+-free H/BSA with no EGTA to obtain extracellular calcium-free conditions. Media supplemented with 6% serum in Ca2+-free HBSA (without EGTA) were then added to the cells in the absence or presence of calcium at the indicated levels. Inhibition of the alterative pathway with anti-FB antibody was achieved by pre-incubation of serum on ice with 20 μg/mL anti-FB antibody prior to addition of antibody/serum mixture to primed cells.
For experiments evaluating the effect of inhibitors (PKC and MEK1), cells were initially primed with S-58 (1.2 mg/mL) and then incubated with chelerythrine and PD98059 at the indicated concentrations for 10 minutes. At the end of the incubation period, the media were removed and replaced with fresh media containing both inhibitors and 6% C1q-Dep.
SiRNA Transfection
ARPE-19 cells were plated at subconfluent densities in 10-cm dishes. Twenty-four hours after plating, cells were transfected for 6 hours with 2 nM siRNA using Lipofectamine and then cell culture media were replaced with media lacking Pen/Strep. After transfection, cells were cultured for 72 hours and then replated in a 96-well plate for analysis of complement-mediated lysis or in 10 cm dishes for FACS analysis, performed 24 hours later. This time point was chosen based on preliminary studies in which the maximal cell surface mCRP-downregulation occurred at 24 hours. Fold changes in death (Sytox Orange stained nuclei/×20 field measured as described above) over control cells were used to normalize responses over multiple experiments.
Determination of Reactive Oxygen Species (ROS)
Retinal pigment epithelial cells from a 62-year-old donor were plated on collagen-coated 96-well black plates with clear bottoms. On day 6 after plating, the cells were washed with serum-free and phenol-free (SPF) medium, loaded with 20 μM CM-H2DCFDA in SPF medium for 30 minutes at 37°C and then washed twice. Baseline fluorescence was measured using a fluorescence plate reader (490-nm excitation, 522-nm emission) with bottom reading. Cells were then treated with or without various oxidant concentrations in SPF medium, and fluorescence measurements were obtained at the indicated times. The oxidants were then removed; cells were washed once and incubated with SPF medium for another 2 hours. Fluorescence was then measured followed by cell viability determination via WST assay described below.
LDH Assay
Human donor and ARPE-19 cells were plated on collagen-coated 96-well plates. Cells reached 80% to 100% confluence 24 hours after plating and were then grown for an additional 5 days. On day 6 after plating, the cells were washed once with SPF-MEM (donor RPE) or SPF-DMEM-F12 (ARPE-19) and then stimulated for 1.5 hours at 37°C with various oxidant concentrations in the corresponding media. Cells were washed once and then subjected to complement attack as described above. Lactate dehydrogenase release was measured using a “cytotoxicity detection kit (LDH)” as we have previously described.26
WST and Cell Count Assay
Cell plating and treatment with oxidants and complement were performed as described above. Cell viability was measured using a “cell proliferation reagent,” WST-1 as we have previously described.26 After colorimetric measurement, the cells were trypsinized and viable cell number was determined with a trypan blue exclusion assay using a hemocytometer (Hausser Scientific, Horsham, PA, USA).
Repetitive Oxidant Exposure Model and Nonproliferating Cell Model
Cells were plated as described above. For the repetitive exposure model, cells were stimulated every other day for 1 week (four exposures) with various oxidant concentrations in phenol-free MEM containing 1% FBS. At the completion of the repetitive oxidant exposure, cells were challenged by complement activation. Cell death was assessed with the WST assay and cell count assay as described above. For the nonproliferating cell model, cells were cultured for 3 weeks and then counted to confirm that they were no longer proliferating. Oxidative stress (single exposure) and complement attack were performed as described above.
Statistical Analysis
Data are expressed as the mean ± SD. Student's t-test was used to determine whether there were statistically significant differences between treatment groups as determined by LDH release assay, WST assay, cell count assay, FACS, ROS, and calcium depletion assay. Nonparametric Kruskal-Wallis multiple comparisons were used to determine the difference among the targeted siRNA treatments, and a pair-wise t-test was used to determine the difference between control siRNA and targeted siRNA treatments. A P value less than 0.05 was considered statistically significant. All assays were replicated in at least three independent experiments with similar results.
Results
Identification of Cultured Human RPE Cells
To confirm the epithelial nature of cultured human RPE cells, cells were stained for cytokeratin and ZO-1 (Supplementary Fig. S1). Cytoplasmic cytokeratin-18, an epithelial cell marker and cell membrane associated ZO-1, a junctional marker, were detected in donor RPE cells and ARPE-19 cells.
Generation of an Anti-RPE Antibody and Elicitation of Complement Attack Through AP Activation
A “complement-fixing” polyclonal antibody recognizing RPE cells was developed by immunizing sheep with PFA-fixed ARPE-19 cells. Bleeds were screened by FACS for reactivity. From the three immunized sheep, one IgG from sheep numbered 58 was identified that recognized ARPE-19 cells and denoted “S-58” (Supplementary Fig. S2A). To determine whether S-58–primed RPE cells fixed complement, we measured cell surface MAC formation in the presence of serum. S-58 priming efficiently led to membrane MAC deposition (Supplementary Fig. S2B)
To confirm that S-58 recognized donor RPE cells, Western blot and immunofluorescent microscopy were performed under identical conditions on ARPE-19 cells and donor RPE cells. S-58 bound to both ARPE-19 and donor RPE cells in a dose-dependent manner (Supplementary Fig. S3A) and the ARPE-19 cell-membrane staining pattern was similar to that observed in donor RPE cells (Supplementary Fig. S3B). Membrane attack complex deposition on the cell surface was confirmed in donor RPE cells by immunofluorescence microscopy (Supplementary Fig. S3C). To examine the specificity of S-58 to induce cell lysis in the presence of human serum, we used protein G purified normal sheep IgG as negative control. In the presence of C1q-Dep, S-58 priming caused donor RPE cell death in a dose-dependent manner (Supplementary Figs. S4A, S4B).
The ability of S-58 to induce cell swelling, raise intracellular Ca2+, cause plasma membrane breakdown, and produce ATP release was examined to assess the functionality of the formed MAC in the presence of human serum. As shown in Figure 1A, S-58–primed ARPE-19 cells did not swell in the presence of C5-Dep; however, pronounced cell swelling occurred when primed cells were incubated with C1q-Dep. Coincident with the observed swelling was a rapid S-58 concentration-dependent increase in intracellular calcium that did not occur in C5-Dep human serum (Fig. 1B). Adenosine triphosphate release mirrored the observed dose response with calcium and was associated with plasma membrane disruption as indicated by the penetrance of a membrane impermeant nuclear dye Sytox orange (Fig. 1C).
Figure 1.

S-58 primed ARPE-19 cells in the presence of C1q-Dep form functional MAC, which kills cells through the AP but not the CP. (A) ARPE-19 cells were primed with S-58 (1.2 mg/mL) and then incubated with either 6% C1q-Dep or 6% C5-Dep. Complement attack on primed ARPE-19 cells leads to cell swelling (right panel) as compared with primed cells treated with C5-Dep (left panel). (B) Cells were primed with varying S-58 concentrations (a = 2.8 mg/mL, b = 1.8 mg/mL, c = 1.2 mg/mL, d = 0.8 mg/mL, e = 0.6 mg/mL, and f = 2.8 mg/mL in the absence of serum) or g = serum alone, with Fluo-4AM (2 μM) washed twice, and then incubated with 6% C1q-Dep or 6% C5-Dep. Intracellular calcium was determined by Fluo-4AM fluorescence intensity measurement. (C) Cells were primed with S-58 at the indicated concentrations and then incubated for 3 hours with either 6% C1q-Dep or 6% C5-Dep. The number of Sytox Orange-stained nuclei of three nonoverlapping fields per well using a ×20 objective was quantified by MetaMorph image analysis software, and is an indicator of cell death. Adenosine triphosphate release was quantified with an ATP Bioluminescence Assay Kit HS II. (D) Cells primed with S-58 (1.2 mg/mL) were washed with Ca2+-free HBSA containing EGTA and then washed in Ca2+-free HBSA without EGTA and then challenged with 6% NHS, 6% C1q-Dep, or 6% FB-Dep, in the presence or absence of exogenous Ca2+, or in the presence of an inhibitory anti-FB monoclonal antibody (20 μg/mL). Cell death was determined as described in (C). Negative control: absence of S-58.
The effect of S-58 on CP and AP activation was assessed with NHS and with C1q-Dep or FB-Dep, respectively. The CP was also examined for its contribution by calcium depletion, and the AP was examined using an anti-FB inhibitory antibody (Fig. 1D). Depletion of C1q had no effect on complement-mediated ARPE-19 cell death compared to that observed in complete NHS, indicating that the complement-mediated death was not primarily dependent on the CP FB depletion resulted in nearly complete inhibition of cell death, suggesting the dependence of cell lysis on a functional AP. Depletion of extracellular calcium in either NHS or C1q-Dep enhanced cell lysis, but calcium depletion did not reverse cell death inhibition in FB-Dep. These data suggest that cell lysis, in response to S-58, is dependent on the AP.
Protection by CD46, CD59, and Calcium Against AP-Mediated RPE Cell Death
Cells express mCRPs on their surface to prevent bystander injury when complement is activated on adjacent tissue. Consistent with previous reports,25,27 CD46, CD55, and CD59 were present on the surface of ARPE-19 cells; CR1 was not detected (Supplementary Fig. S5). To determine the relative contribution of each regulatory molecule in RPE cells, siRNA was used to knock down mCRP expression individually or in combination. Membrane complement regulatory proteins levels were measured by FACS analysis on the day of complement-mediated attack. The percentage of mCRP knockdown was calculated based on mean fluorescence intensity corresponding to histographs as shown in Supplementary Figure S6. In targeted siRNA-treated cells, the CD46, CD55, and CD59 were knocked down by 75% to 87%, 33% to 70%, and 35% to 65%, respectively, relative to control siRNA-treated cells. As shown in Figure 2A, exposure of S-58 primed cells to serum with either of the individual CD46 or CD59 knockdowns resulted in a 2-fold enhancement in complement-mediated cell death. CD55 knockdown had little effect on complement-mediated cell death. The greatest cell-mediated death was observed when either CD46 or CD55 was knocked down in combination with CD59 or when all three regulators were simultaneously targeted. The variability in percentage of mCRP knockdown corresponding to fold change in cell death was shown in Fig. 2B.
Figure 2.

Effects on cell survival of siRNA-mediated knockdown of ARPE-19 cell mCRPs. (A) ARPE-19 cells were transfected for 6 hours with either nontargeting control siRNA or the indicated siRNAs (2 nM) followed by media replacement. Transfected cells were cultured for 72 hours, replated in 96-well plates and cultured for an additional 24 hours. Cells were then incubated with S-58 (1.2 mg/mL) and then treated with 6% C1q-Dep. Cell death was determined by penetration of membrane impermeant dye Sytox Orange as described in the Methods. *P < 0.05 versus control siRNA-treated cells. (B) ARPE-19 cells were transfected, replated, and cultured as described in (A). Cells were then harvested and processed for FACS analysis. Individual mCRP knockdown experiments were repeated three or more times (CD46: n = 3, CD55: n = 5, CD59: n = 3) and combination mCRP knockdowns (excluding CD46/CD59) were repeated twice. The average knockdown percentage was calculated based on mean fluorescence intensity as described in figure S6. Error bars correspond to SDs of replicate experiments.
Nucleated cells internalize or actively shed formed cell surface MAC to further decrease their susceptibility to complement attack.28,29 Activation of these protective mechanisms has been correlated with a rise in intracellular calcium from the extracellular space.22,30 To examine if extracellular calcium levels also played a role in complement-mediated ARPE-19 cell lysis, S-58–primed RPE cells were challenged with complement under extracellular calcium-free conditions as described in methods. As shown in Figure 3, removal of extracellular Ca2+ from the incubation buffer–enhanced RPE susceptibility to complement-mediated cell lysis by 5- to 6-fold at levels of activation above 1.2 mg/mL S-58 when compared with that observed in cells maintained with 1.5 mM extracellular calcium in the incubation buffer (Fig. 3A). The protective effect of extracellular calcium was dose-dependent and pronounced effects on cell death were seen when extracellular calcium concentrations were below 0.015 mM (Fig. 3B). These data suggest that while mCRPs are important protective molecules, a rise in intracellular calcium also protects RPE cells from complement-mediated lysis.
Figure 3.

Calcium depletion and PKC or MEK1 inhibition modulate the response of ARPE-19 cells to complement attack. (A) To obtain extracellular calcium-free conditions, S-58-primed cells were washed with Ca2+-free HBSA containing EGTA (HBSS –Ca2+/–Mg2+ containing 0.02% MgCl2, 1% BSA, 0.5 mM EGTA) and then washed in Ca2+-free HBSA with no EGTA. Cells were then incubated with media supplemented with 6% C1q-Dep in Ca2+-free HBSA without EGTA in the presence of 1.5 mM Ca2+or in the absence of Ca2+. (B) Extracellular calcium-free conditions were obtained in S-58 primed cells as described in (A). Media supplemented with 6% C1q-Dep in Ca2+-free HBSA without EGTA were then added to the cells in the presence or absence of calcium at the indicated levels. The 0 mM calcium refers to the conditions before serum was added. (C, D). Cells were primed for 30 minutes with S-58 (1.2 mg/mL), washed and incubated for 10 minutes with chelerythrine (C) and PD98059 (D) at the indicated concentrations. Pre-incubation solution was removed, and then 6% C1q-Dep diluted in HBSS with the corresponding inhibitors was added. Cell death was determined as described in Figure 1C.
Protein kinase C–induced cell surface MAC reduction and PKC-dependent activation of MEK have been associated with cell protection against complement-mediated lysis.31–33 We found that cell death was enhanced in a dose dependent manner when chelerythrine was used to inhibit PKC. However, in contrast to previous reports, cell death was reduced when cells were treated with the MEK inhibitor PD98059 (Figs. 3C, 3D).
Enhancement of Cell Death in Response to Oxidative Stress
To determine if oxidative stress enhances complement-mediated cell death, ARPE-19 cells were stimulated with sub-lethal H2O2 followed by an induction of complement attack using S-58 in the presence of C1q-Dep. Pretreatment of RPE cells with H2O2 enhanced the level of cell death (Fig. 4A). We next examined whether the response to H2O2-enhanced complement-mediated RPE cell death differed among RPE cells from various donors. Retina pigment epithelial cells obtained from three donors were treated with low doses of H2O2 followed by complement attack. Similar responses to oxidant and complement were observed among these three donors (Fig. 4B). The results shown in Fig. 4B were confirmed by trypan blue exclusion assay (not shown). The enhanced effects of complement on RPE have been correlated with the modulation of cell surface mCRPs.27 However, in our experimental setting, levels of the mCRPs were affected minimally by H2O2 despite the fact that H2O2 significantly enhanced complement-mediated death (Fig. 4C).
Figure 4.

Oxidative stress synergizes with complement to enhance cell death. (A) The ARPE-19 cells were pretreated with the indicated concentrations of H2O2 for 1.5 hours, primed with S-58 (1.2 mg/mL), and then incubated with 6% C1q-Dep. Cell viability was determined by WST-1 assay. *(P < 0.05) versus medium alone or oxidant alone. **(P < 0.05) versus complement alone or oxidant alone. (B) Retinal pigment epithelial cells from three donors were stimulated with H2O2 (200 μM) for 1.5 hours, primed with S-58 (1.2 mg/mL), and then incubated with 6% C1q-Dep. Cell viability was examined by WST-1 assay. *(P < 0.05) versus MEM alone or oxidant alone. **(P < 0.05) versus complement alone or oxidant alone. (C) The ARPE-19 cells were treated for 1.5 hours with H2O2 (0.5 mM). Cell surface mCRP levels were determined by FACS.
Cigarette smoking is an important risk factor for AMD; HQ is a major oxidant in cigarette smoke and atmospheric pollutants.34 To determine whether HQ enhances complement-mediated cell death, RPE cells from a donor (51-year old) were treated with HQ and complement as described above. Consistent with enhancement of complement-mediated cell death induced by H2O2, pretreatment of RPE cells with HQ enhanced the level of cell death by increasing cell permeability (Fig. 5A), decreasing cell viability (Fig. 5B) and decreasing viable cell numbers (not shown). Similar responses were observed in RPE cells from three donors and ARPE-19 cells (not shown), suggesting the observed responses were independent of CFH genotype. The results obtained from these three assays described above were also confirmed in nonproliferating cells (not shown).
Figure 5.
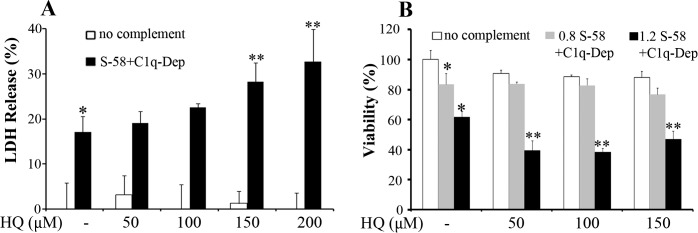
Hydroquinone sensitizes donor RPE cells to complement-mediated cell injury. (A, B). Retinal pigment epithelial cells were pretreated with the indicated concentrations of HQ for 1.5 hours, incubated with S-58 Ab (1.2 mg/mL in [A], 0.8 mg/mL and 1.2 mg/mL denoted “0.8” and “1.2” in [B]), and then incubated with 6% C1q-Dep. Cell permeability and cell viability were determined by LDH assay (A) and WST-1 assay (B), respectively, in RPE cells from a 51-year-old donor. *(P < 0.05) versus MEM alone and **(P < 0.05) versus complement alone or oxidant alone.
To investigate whether ROS generation contributes to the sensitization to complement attack, intracellular ROS was measured at various time points induced by H2O2, HQ, all transretinal (atRal, 24% sheep anti-RPE antibody equal 1.2 mg/mL in the publication),34 and t-BH, which also sensitized RPE cells to AP attack (not shown). During incubation with oxidants, H2O2 and t-BH significantly increased ROS level when compared with HQ and atRal at doses tested (Fig. 6A, in a donor with CFHYY402 variant). Two hours after removing oxidants, the ROS level was higher in cells treated with atRal when compared with those treated with HQ despite the similar cell viability. ROS level was significant lower in cells treated with H2O2 as to t-BH–treated cells (Fig. 6B). Similar responses were observed in RPE cells from a donor with CFHHH402 variant and ARPE-19 cells, the donor with CFHYH402 variant (not shown).
Figure 6.

Different oxidants generate different ROS levels. Retinal pigment epithelial cells from a 62-year-old donor were loaded with 20 μM CM-H2DCFDA for 30 minutes and washed twice. (A) Cells were treated with or without various oxidant concentrations, and ROS production was measured by relative fluorescence units (RFU) at the indicated times using a fluorescence plate reader (490-nm excitation, 522-nm emission). *P < 0.05 versus DMEM, HQ and atRal. (B) Cells assayed in (A) were washed and replaced with serum-free medium for 2 hours. Reactive oxygen species was measured followed by cell viability determination by WST assay. *P < 0.05 versus medium. **P < 0.05 versus HQ.
Donor RPE cells were stimulated repetitively with HQ or H2O2 followed by complement challenge to mimic chronic in vivo sublethal RPE cell oxidative stress. Similar to acute oxidative stress, chronic repetitive treatment of RPE cells with low levels of oxidants also enhanced complement-mediated cell death (Figs. 7A, 7B). Similar responses were observed in RPE cells from three donors and ARPE-19 cells (not shown), suggesting the observed responses were independent of CFH genotype. Hydroquinone at 50 μM is normally a sublethal dose when given to RPE cells as a single treatment; however, cell number was decreased when it was given in repetitive exposures, an effect that was further enhanced by complement. In contrast, single treatment, or repeated treatments with 200 μM H2O2 was not toxic to RPE cells, but this dose still enhanced complement-mediated cell death, when given as a single or repeated exposure (Figs. 7A, 7B).
Figure 7.

Repetitive HQ and H2O2 exposures sensitize donor RPE cells to complement-mediated cell injury. (A) and (B) Retinal pigment epithelial cells were treated with the indicated concentrations of HQ and H2O2 in phenol free MEM containing 1% FBS every other day for 1 week (four exposures), primed with S-58 (1.2 mg/mL) for 30 minutes, washed once, and then incubated with 6% C1q-Dep for determination of cell viability (A) and viable cell number (B) in RPE cells from a 62-year-old donor. *(P < 0.05) versus MEM alone and **(P < 0.05) versus complement alone or oxidant alone.
Discussion
In the present study, an RPE cell-priming antibody has been shown to activate complement attack in the presence of C1q-Dep on cultured, human, RPE cell surfaces through the alternate complement pathway. Using this model, factors that protect against and sensitize cells to complement-mediated cell death were identified. Protective factors include calcium-dependent activation of PKC as well as mCRPs, which, when downregulated, increase susceptibility to complement-mediated death. Conversely, exposure to sublethal oxidative stress increases RPE cell susceptibility to the cytotoxic effects of complement activation.
Several approaches have been taken to study the functional effects of complement on RPE cells.12,13,15 It has been challenging to evaluate the effects of MAC on RPE function because of the abundant mCRPs on the cell surface that block C5b-9 complex formation. Choroidal flat mounts and a priming monoclonal antibody showed promise in a mouse ex vivo assay.36–38 However, the results may not be applied to complement regulators in human cells due to differences between human and mouse complement regulatory mechanisms.36–38 A cell-based assay using oxidative stress at the time of complement addition to sensitize human RPE cells and activate complement has provided insight into complement effects on RPE behavior; however, the assay neither activated complement through the AP nor did it allow the examination of complement from the sublytic to lytic phase.27 Recently, complement-mediated lysis was induced by incubating ARPE-19 cells in a polyclonal anti-human CD59 antiserum followed by stimulation with NHS to investigate sublytic MAC-mediated functional changes in ARPE-19 cells; however, the assay neither activated complement AP nor excluded the possibility of CD59 blockage due to application of anti-CD59 antiserum.39 In the present studies, a priming antibody against human RPE cells was developed that elicits MAC formation through the AP on RPE cells in the presence of C1q-Dep, resulting in both sublytic and lytic phases of complement attack. This approach has been useful to study the functional effects of complement and complement/oxidant interactions.
The relative contribution to AMD of locally produced, or systemic complement components is not fully understood. Retinal pigment epithelial cells in vivo are thought to not synthesize all complement components sufficient to initiate complement attack. The complement components synthesized by extra-ocular sources such as the liver are deposited in the serum and are likely important contributors to the effect of complement on RPE cells. Accordingly, to more closely simulate in vivo pathophysiology, we sought to mimic those extra-ocular complement components by using human serum as the source of complement and adding to our MAC assay, to capture the contribution of systemic complement components.
Activation of complement attack on RPE cells induced a response with several measurable endpoints. While the lectin pathway may contribute to these responses, this pathway does not appear to be required since use of C2 depleted serum had no effect on the ability to induce complement-mediated cell death (not shown). Furthermore, assays performed with C1q-Dep showed responses independent of the CP, which requires C1q to initiate complement activation.
Retinal pigment epithelial cells express mCRPs. Using siRNA we reduced individual mCRPs levels, but did not completely abolish expression on the cell surface. This failure to obtain complete knockdown of protein expression may reflect both efficacy of the siRNA and residency time of the individual proteins on the cell surface. Although the contribution of CD46 was expected based on earlier studies25,40,41 the current study identifies CD59 as an important protector of ARPE-19 cells from complement attack. Furthermore, while knockdown of CD55 alone had a modest effect, inhibition of CD55 in combination with either CD46 or CD59 enhanced the level of cell death. CD55 accelerates the decay of the C3/C5 convertase. The minimal effect observed following selective CD55 knockdown in the presence of functional CD46 and CD59 suggests that by itself, accelerated C3/C5 convertase decay is not a critical RPE cell complement protective mechanism. More robust complement-induced cell death occurred when levels of CD46 and CD59 are attenuated, indicating that these pathways play a more central role in the protection of RPE cells.
The modest increases in cell death observed in the individual mCRP-knockdowns indicate that a significant decrease in mCRP activity is needed to see an impact on complement-mediated cell death. Additionally, when extracellular calcium was depleted or when cells were challenged with sublethal H2O2 and HQ doses, complement-mediated cell death was enhanced. The observation that extracellular calcium depletion enhances cell death is consistent with previous observations in which calcium signaling induces cellular protective mechanisms.29,42,43 However, acute and massive calcium influx has also been associated with complement-mediated cell death associated with mitochondrial toxicity.44 Thus, the activity of calcium in association with complement-mediated attack on nucleated cells is complex with calcium involvement in both sublytic and lytic events.
Protein kinase C and MEK1 have been shown to protect cells from a lytic response.31–33,45 Similar to that observed in K562 and human carcinoma cells,32,45 PKC inhibition resulted in enhanced complement-mediated RPE cell death. In contrast to those cells,32,45 MEK1 inhibition protected RPE cells from complement-mediated death. These findings suggest that the signaling pathways that regulate RPE cell survival and death following complement attack may differ from those used by other nucleated cells.
The high level of cell death observed when RPE cells were pretreated with sublethal doses of H2O2 or HQ followed by complement attack emphasizes that oxidative stress predisposes cells to complement-mediated death. In these assays, RPE cells displayed a relatively greater sensitivity to HQ toxicity when compared to H2O2, suggesting that cells may respond to different oxidants by different mechanisms. We confirmed that ROS levels varied in cells treated with different oxidants. Hydroquinone is an exogenous oxidant and H2O2 is a byproduct of cell metabolism. Consequently, RPE cells may be better equipped to prevent damage from this oxidant that can be endogenously generated.
The effect of different oxidant concentrations on RPE cell toxicity varies among publications.46–48 In general, we observed lower toxicity in response to higher HQ concentrations, than that which has been previously reported.49 It is possible that this lesser toxicity observed in our system reflects the relatively short oxidant exposure time (1.5 hours), when compared with other studies.49
CFH variant phenotypes are associated with greater or lesser AMD risk. Accordingly, it was of interest to determine whether different RPE cell phenotypes responded differently to oxidant and complement attack. Similar responses were observed in ARPE-19 cells, which have the CFHYH402 variant and RPE cells from three donors each with different CFH variants. The results indicate that the observed responses were independent of the CFH genotype.
Previous studies examining complement and H2O2-mediated oxidative stress have suggested that oxidant-mediated downregulation of cell surface mCRPs is one factor responsible for enhanced sensitivity to complement attack.27 However, in our system, cell surface mCRP levels after challenge with oxidants did not correlate with observed cell death levels. These data suggest that additional mechanisms may operate to increase susceptibility to complement attack by oxidative stress. Our data indicated that intracellular ROS production might be one of these additional mechanisms. Experiments are currently underway in our laboratory to further explore how calcium influences cell death, the pathways involved in the enhanced sensitivity of RPE cells to H2O2- and HQ-induced oxidative stress, and whether these oxidants employ similar or different mechanisms to enhance RPE cell sensitivity to complement attack.
In the present report, we conducted in vitro experiments to examine the effect of a human source of complement on cultured human cells. Clearly, a tissue culture model does not replicate all aspects of in vivo pathophysiology. Nonetheless, in our system that used human cells and human tissue–derived reagents it was possible to design rigorously controlled experiments that simulate specific aspects of human physiology and pathophysiology, and to generate a hypothesis that can be tested in in vivo models. Data from animal models have supported a role for complement and oxidative stress in AMD, and help to validate the in vitro model described in the present report. Of particular relevance, in a recent study, it was shown that AP activation is required for complement factor-B−/− mice to develop AMD-like pathology, when exposed to oxidant stress in the form of cigarette smoke.50
Very recently reported data from a randomized phase II clinical trial further support our in vitro experiments, and provide additional evidence that complement plays an important role in RPE cell survival and is involved in GA progression in advanced AMD. In the MAHALO study, it was found that lampalizumab (antifactor D) can slow the progression of GA in patients with advanced dry AMD and the treatment effect was specifically observed in patients positive for the factor I genetic biomarker.10 These data, which are the first to show a beneficial treatment effect with a complement inhibitor in GA, are consistent with our in vitro experiments, and highlight the importance of in vitro and in vivo models to examine the mechanisms of complement-mediated RPE injury.
Acknowledgements
This work was supported, in part, by National Institutes of Health (Bethesda, MD, USA) 5P30EY005722 (Core grant) and Howard Hughes Medical Institute Medical Research Fellows Program (Chevy Chase, MD, USA; [JEB]).
Disclosure: P. Yang, None; P. Baciu, Allergan, Inc. (E); B.C. Parker Kerrigan, Allergan, Inc. (E); M. Etheridge, Allergan, Inc. (E); E. Sung, Allergan, Inc. (E); B.A. Toimil, None; J.E. Berchuck, None; G.J. Jaffe, None
References
- 1. Van der Schaft TL, Mooy CM, de Bruijn WC, de Jong PT. Early stages of age-related macular degeneration: an immunofluorescence and electron microscopy study. Br J Ophthalmol. 1993; 77: 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001; 73: 887– 896. [DOI] [PubMed] [Google Scholar]
- 3. Miller RA, Bookstein F, Van der Meulen J, et al. Candidate biomarkers of aging: age-sensitive indices of immune and muscle function covary in genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 1997; 52: B39– B47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000; 14: 835– 846. [PubMed] [Google Scholar]
- 5. Edwards AO, Ritter R III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005; 308: 421– 424. [DOI] [PubMed] [Google Scholar]
- 6. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005; 308: 385– 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005; 308: 419– 421. [DOI] [PubMed] [Google Scholar]
- 8. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005; 102: 7227– 7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yehoshua Z, Rosenfeld PJ, Albini TA. Current Clinical Trials in Dry AMD and the Definition of Appropriate Clinical Outcome Measures. Semin Ophthalmol. 2011; 26: 167– 180. [DOI] [PubMed] [Google Scholar]
- 10. Williams D. MAHALO phase II study: safety, tolerability and activity of lampalizumab (anti-factor D) in patients with geographic atrophy. Paper presented at the 31st Annual Meeting of the American Society of Retina Specialists; August 27, 2013; Toronto, Ontario, Canada. [Google Scholar]
- 11. Cortright DN, Meade R, Waters SM, Chenard BL, Krause JE. C5a, but not C3a, increases VEGF secretion in ARPE-19 human retinal pigment epithelial cells. Curr Eye Res. 2009; 34: 57– 61. [DOI] [PubMed] [Google Scholar]
- 12. Fukuoka Y, Strainic M, Medof ME. Differential cytokine expression of human retinal pigment epithelial cells in response to stimulation by C5a. Clin Exp Immunol. 2003; 131: 248– 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fukuoka Y, Medof EM. C5a receptor-mediated production of IL-8 by the human retinal pigment epithelial cell line, ARPE-19. Curr Eye Res. 2001; 23: 320– 325. [DOI] [PubMed] [Google Scholar]
- 14. Kunchithapautham K, Rohrer B. Sublytic membrane-attack-complex (MAC) activation alters regulated rather than constitutive vascular endothelial growth factor (VEGF) secretion in retinal pigment epithelium monolayers. J Biol Chem. 2011; 286: 23717– 23724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006; 103: 2328– 2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bora PS, Sohn JH, Cruz JM, et al. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J Immunol. 2005; 174: 491– 497. [DOI] [PubMed] [Google Scholar]
- 17. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010; 20: 34– 50. [DOI] [PubMed] [Google Scholar]
- 18. Arlaud GJ, Gaboriaud C, Thielens NM, Rossi V. Structural biology of C1. Biochem Soc Trans. 2002; 30: 1001– 1006. [DOI] [PubMed] [Google Scholar]
- 19. James K. Complement: activation, consequences, and control. Am J Med Technol. 1982; 48: 735– 742. [PubMed] [Google Scholar]
- 20. Major B, Kardos J, Kekesi KA, Lorincz Z, Zavodszky P, Gal P. Calcium-dependent conformational flexibility of a CUB domain controls activation of the complement serine protease C1r. J Biol Chem. 2010; 285: 11863– 11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009; 9: 729– 740. [DOI] [PubMed] [Google Scholar]
- 22. Carney DF, Hammer CH, Shin ML. Elimination of terminal complement complexes in the plasma membrane of nucleated cells: influence of extracellular Ca2+ and association with cellular Ca2+. J Immunol. 1986; 137: 263– 270. [PubMed] [Google Scholar]
- 23. Carney DF, Lang TJ, Shin ML. Multiple signal messengers generated by terminal complement complexes and their role in terminal complement complex elimination. J Immunol. 1990; 145: 623– 629. [PubMed] [Google Scholar]
- 24. Jaffe GJ, Earnest K, Fulcher S, Lui GM, Houston LL. Antitransferrin receptor immunotoxin inhibits proliferating human retinal pigment epithelial cells. Arch Ophthalmol. 1990; 108: 1163– 1168. [DOI] [PubMed] [Google Scholar]
- 25. Yang P, Tyrrell J, Han I, Jaffe GJ. Expression and modulation of RPE cell membrane complement regulatory proteins. Invest Ophthalmol Vis Sci. 2009; 50: 3473– 3481. [DOI] [PubMed] [Google Scholar]
- 26. Yang P, Peairs JJ, Tano R, Jaffe GJ. Oxidant-mediated Akt activation in human RPE cells. Invest Ophthalmol Vis Sci. 2006; 47: 4598– 4606. [DOI] [PubMed] [Google Scholar]
- 27. Thurman JM, Renner B, Kunchithapautham K, et al. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J Biol Chem. 2009; 284: 16939– 16947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cole DS, Morgan BP. Beyond lysis: how complement influences cell fate. Clin Sci (Lond). 2003; 104: 455– 466. [DOI] [PubMed] [Google Scholar]
- 29. Pilzer D, Gasser O, Moskovich O, Schifferli JA, Fishelson Z. Emission of membrane vesicles: roles in complement resistance, immunity and cancer. Springer Semin Immunopathol. 2005; 27: 375– 387. [DOI] [PubMed] [Google Scholar]
- 30. Morgan BP, Dankert JR, Esser AF. Recovery of human neutrophils from complement attack: removal of the membrane attack complex by endocytosis and exocytosis. J Immunol. 1987; 138: 246– 253. [PubMed] [Google Scholar]
- 31. Kraus S, Fishelson Z. Cell desensitization by sublytic C5b-9 complexes and calcium ionophores depends on activation of protein kinase C. Eur J Immunol. 2000; 30: 1272– 1280. [DOI] [PubMed] [Google Scholar]
- 32. Kraus S, Seger R, Fishelson Z. Involvement of the ERK mitogen-activated protein kinase in cell resistance to complement-mediated lysis. Clin Exp Immunol. 2001; 123: 366– 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jurianz K, Ziegler S, Donin N, Reiter Y, Fishelson Z, Kirschfink M. K562 erythroleukemic cells are equipped with multiple mechanisms of resistance to lysis by complement. Int J Cancer. 2001; 93: 848– 854. [DOI] [PubMed] [Google Scholar]
- 34. Pons M, Cousins SW, Csaky KG, Striker G, Marin-Castano ME. Cigarette smoke-related hydroquinone induces filamentous actin reorganization and heat shock protein 27 phosphorylation through p38 and extracellular signal-regulated kinase 1/2 in retinal pigment epithelium: implications for age-related macular degeneration. Am J Pathol. 2010; 177: 1198– 1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berchuck JE, Yang P, Toimil BA, Ma Z, Baciu P, Jaffe GJ. All-trans-retinal sensitizes human RPE cells to alternative complement pathway-induced cell death. Invest Ophthalmol Vis Sci. 2013; 54: 2669– 2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ramo K, Cashman SM, Kumar-Singh R. Evaluation of adenovirus-delivered human CD59 as a potential therapy for AMD in a model of human membrane attack complex formation on murine RPE. Invest Ophthalmol Vis Sci. 2008; 49: 4126– 4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ma KN, Cashman SM, Sweigard JH, Kumar-Singh R. Decay accelerating factor (CD55)-mediated attenuation of complement: therapeutic implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010; 51: 6776– 6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sweigard JH, Cashman SM, Kumar-Singh R. Adenovirus-mediated delivery of CD46 attenuates the alternative complement pathway on RPE: implications for age-related macular degeneration. Gene Ther. 2011; 18: 613– 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lueck K, Wasmuth S, Williams J, et al. Sub-lytic C5b-9 induces functional changes in retinal pigment epithelial cells consistent with age-related macular degeneration. Eye (Lond). 25: 1074– 1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vogt SD, Curcio CA, Wang L, et al. Retinal pigment epithelial expression of complement regulator CD46 is altered early in the course of geographic atrophy. Exp Eye Res. 2011; 93: 413– 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fett AL, Hermann MM, Muether PS, Kirchhof B, Fauser S. Immunohistochemical localization of complement regulatory proteins in the human retina. Histol Histopathol. 2012; 27: 357– 364. [DOI] [PubMed] [Google Scholar]
- 42. Wood A, Wing MG, Benham CD, Compston DA. Specific induction of intracellular calcium oscillations by complement membrane attack on oligodendroglia. J Neurosci. 1993; 13: 3319– 3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reiter Y, Ciobotariu A, Jones J, Morgan BP, Fishelson Z. Complement membrane attack complex, perforin, and bacterial exotoxins induce in K562 cells calcium-dependent cross-protection from lysis. J Immunol. 1995; 155: 2203– 2210. [PubMed] [Google Scholar]
- 44. Papadimitriou JC, Phelps PC, Shin ML, Smith MW, Trump BF. Effects of Ca2+ deregulation on mitochondrial membrane potential and cell viability in nucleated cells following lytic complement attack. Cell Calcium. 1994; 15: 217– 227. [DOI] [PubMed] [Google Scholar]
- 45. Donin N, Jurianz K, Ziporen L, Schultz S, Kirschfink M, Fishelson Z. Complement resistance of human carcinoma cells depends on membrane regulatory proteins, protein kinases and sialic acid. Clin Exp Immunol. 2003; 131: 254– 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu L, Hackett SF, Mincey A, Lai H, Campochiaro PA. Effects of different types of oxidative stress in RPE cells. J Cell Physiol. 2006; 206: 119– 125. [DOI] [PubMed] [Google Scholar]
- 47. Marin-Castano ME, Csaky KG, Cousins SW. Nonlethal oxidant injury to human retinal pigment epithelium cells causes cell membrane blebbing but decreased MMP-2 activity. Invest Ophthalmol Vis Sci. 2005; 46: 3331– 3340. [DOI] [PubMed] [Google Scholar]
- 48. Bailey TA, Kanuga N, Romero IA, Greenwood J, Luthert PJ, Cheetham ME. Oxidative stress affects the junctional integrity of retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2004; 45: 675– 684. [DOI] [PubMed] [Google Scholar]
- 49. Strunnikova N, Zhang C, Teichberg D, et al. Survival of retinal pigment epithelium after exposure to prolonged oxidative injury: a detailed gene expression and cellular analysis. Invest Ophthalmol Vis Sci. 2004; 45: 3767– 3777. [DOI] [PubMed] [Google Scholar]
- 50. Woodell A, Coughlin B, Kunchithapautham K, et al. Alternative complement pathway deficiency ameliorates chronic smoke-induced functional and morphological ocular injury. PLoS One. 2013; 8: e67894. [DOI] [PMC free article] [PubMed] [Google Scholar]