

Abstract
Following the first CNS Anticancer Drug Discovery and Development Conference, the speakers from the first 4 sessions and organizers of the conference created this White Paper hoping to stimulate more and better CNS anticancer drug discovery and development. The first part of the White Paper reviews, comments, and, in some cases, expands on the 4 session areas critical to new drug development: pharmacological challenges, recent drug approaches, drug targets and discovery, and clinical paths. Following this concise review of the science and clinical aspects of new CNS anticancer drug discovery and development, we discuss, under the rubric “Accelerating Drug Discovery and Development for Brain Tumors,” further reasons why the pharmaceutical industry and academia have failed to develop new anticancer drugs for CNS malignancies and what it will take to change the current status quo and develop the drugs so desperately needed by our patients with malignant CNS tumors. While this White Paper is not a formal roadmap to that end, it should be an educational guide to clinicians and scientists to help move a stagnant field forward.
Keywords: brain metastasis, chemotherapy, glioma, medulloblastoma, pharmacokinetics, pharmacology
Introduction and the Challenge of Drug Discovery and Development for Brain Tumors
The first CNS Anticancer Drug Discovery and Development Conference (ADDDC) was organized and convened in 2014 by scientists and physicians frustrated by the dearth of available and efficacious anticancer drugs for the treatment of primary infiltrative and secondary metastatic tumors of the central nervous system. The Organizing and Scientific Program Committees were tasked with designing the conference using educational lectures covering many, but not all, areas relevant to anticancer drug discovery/development. By all indications, they were successful in that goal. By nearly universal agreement, committee members and speakers felt that a White Paper, summarizing approaches and concerns voiced at the conference, should be written and made available to the academic and pharmaceutical industry communities.
For those who were unable to attend the 2014 ADDDC, the speakers in attendance agreed to summarize salient points from their talks. It was hoped that, following a review of the topics covered, the authors of this White Paper would offer suggestions that might move the field of CNS anticancer drug discovery/development forward in the coming years.
Magnitude of The Cancer Problem
In the United States, it is expected that there will be ∼69 850 new cases of primary malignant and “nonmalignant” brain and CNS tumors in 2015.1 It is projected that ∼21 800 people will develop malignant brain and CNS tumors in 2015 and that 13 800 will die of these tumors. The most common malignant primary CNS tumor is glioblastoma (World Health Organization [WHO] grade IV), with medulloblastoma (WHO grade IV) being the most common malignant CNS tumor in children. Primary malignant CNS tumors account for ∼1.4% of all cancers, 22% of all childhood cancers, and 2.4% of all cancer deaths.
In 2015, nonmalignant primary tumors were predicted to afflict ∼45 300 people.1 Unfortunately, many of the nonmalignant tumors, such as meningioma and nerve sheath tumors, can grow and transform into more malignant phenotypes, producing significant morbidity and mortality.
In addition, a large number of tumors will metastasize to the CNS. Because such cases are not tracked by national cancer registries, we can only estimate their totals based on the number of new cancer cases and expectations for frequency of metastases to the CNS. Some sources have cited a frequency of 20% to 40% of all cancers.2 Thus, one can assume an occurrence of 98 000 to 170 000 new cases of cancers that will metastasize to the CNS.3,4 The most common primary cancers metastasizing to the brain are lung cancer (50%), breast cancer (15%–20%), unknown primary cancer (10%–15%), melanoma (10%), and colon cancer (5%).2,4
Outcome (Survival) Expectations for Patients With Primary Malignant Gliomas
During a period of 36 years (1978–2014), the increase in overall survival (OS) for patients diagnosed with glioblastoma has increased from 7 months to ∼14 months (Fig. 1). Actual OS may not even be that high given the change in WHO criteria for diagnosing glioblastoma that occurred in about 1993, which allowed high-grade gliomas with vascular proliferation, but not necrosis, previously called anaplastic astrocytoma (AA), to be called glioblastoma.5 These changes in WHO criteria were not immediately adopted and many studies published in the early 2000s adhered to earlier criteria, including pseudopalisading necrosis for glioblastoma multiforme (GBM). For AA (WHO grade III) and anaplastic oligoastrocytoma tumors, the gains in OS between 1990 and 2009 have been associated with increases of ∼2 years (Fig. 2). This is primarily because of the good results from 2 studies of post-irradiation adjuvant chemotherapy with a 6-thioguanine plus BCNU (carmustine) combination and PCV (procarbazine, CCNU [lomustine], and vincristine) combined with eflornithine.6–8 These studies used the older, pre-1993 criteria for AA and GBM and may have been the last large studies to do so.
Fig. 1.

Plot of median overall survival (mOS) for patients treated with adjuvant chemotherapy in phase III clinical trials for GBM over the years 1978–2014.6,177–187
Fig. 2.

Plot of median overall survival (mOS) for patients with anaplastic astrocytoma (and a small number of patients with anaplastic oligoastrocytoma) treated with adjuvant chemotherapy in clinical trials during the years 1990–2009.6,8,179,188–193
For recurrent/progressive high-grade gliomas, the outcomes are also quite poor. The results shown in Table 1 were analyzed for the case number–weighted mean objective response rate (ORR) and progression-free survival at 6 months (PFS-6), which were 6% and 17%, respectively. A similar analysis for noncytotoxic drugs (Table 2) found an ORR of 4% and PFS-6 of 9%. The results for noncytotoxic anti-angiogenic drugs was a little better, with an ORR of 14% and PFS-6 of 19%, but are still not encouraging for cancer trials of chemotherapy.
Table 1.
Compilation of phase II trials of cytotoxic drugs in GBMa
| Agent | N Patients | Response Criteria | ORR, % | PFS-6, % |
|---|---|---|---|---|
| NABTC119 | 437 | Macdonald | 7 | 16 |
| Carboplatin + thymidine120 | 45 | Macdonald | 2.2 | Not stated |
| BCNU121 | 40 | Macdonald | 15 | 17.5 |
| BCNU + TMZ122 | 36 | Macdonald | 5.5 | 21 |
| Carboplatin + erlotinib123 | 43 | Macdonald | 2.3 | 14 |
| CCNU124,125 | 157 | Levin or RANO | 4–9 | 19–25 |
| CCNU + cloretazine125 | 32 | Macdonald | 0 | 6 |
| Hydroxyurea +/− imatinib126 | 120 | Macdonald | 0.8 | 5 |
| Irinotecan127,128 | 128 | Macdonald | 0–17 | NR |
| PCV129,130 | 146 | Macdonald | 4–11 | 32–38 |
| Procarbazine131 | 113 | Macdonald | 5.3 | 8 |
| TMZ (5/28)131,132 | 240 | Macdonald | 5–8 | 18–21 |
| TMZ (7/7)133 | 45 | Macdonald | 15.5 | 43.8 |
| TMZ (21/28)134,135 | 91 | Macdonald | 9–13 | 11–30 |
| TMZ (28/28)–PD off TMZ136 | 29 | Macdonald | 11.1 | 35.7 |
| TMZ + thalidomide137 | 43 | Macdonald | 7 | 24 |
Abbreviations: TMZ, temozolomide; NABTC, North American brain tumor consortium.
aThe case number–weighted mean ORR and PFS-6 for these studies is 6% and 17%, respectively.
Table 2.
Compilation of phase II trials of noncytotoxic and non–anti-angiogenic drugs in GBM
| Agent | N Patients | Response Criteria | ORR, % | PFS-6, % |
|---|---|---|---|---|
| AMG102138 | 61 | Macdonald | 0 | 15.0–17.9 |
| Cilengitide139 | 81 | Macdonald | 9 | 10%–15% |
| Cis-retinoic acid + celecoxib140 | 25 | Macdonald | 0 | 19 |
| Enzastaurin124 | 174 | Levin | 2.9 | 11 |
| Erlotinib141,142 | 102 | Macdonald | 3.7–6.3 | 11.4–18.3 |
| Erlotinib + sirolimus143 | 32 | Macdonald | 0 | 3.1 |
| Fenretinide144 | 23 | Not stated | 0 | 0 |
| Gefitinib145 | 57 | Macdonald | 0 | 13 |
| Imatinib146 | 34 | Macdonald | 5.9 | 3 |
| Imatinib147 | 51 | Macdonald | 5.9 | 16 |
| Imatinib + hydroxyurea126,148,149 | 384 | Macdonald | 1.7–9 | 7–27 |
| Lapatinib150 | 17 | Levin | 0 | Not stated |
| Temsirolimus151 | 65 | Macdonald | 0 | 7.8 |
| Tipifarnib152 | 67 | Macdonald | 7.5 | 16.7 |
| Vorinostat153 | 66 | Macdonald | 3.0 | 15.2 |
The case number–weighted mean ORR and PFS-6 for these studies is 4% and 9%, respectively.
Improvements in OS over the years have been noted not so much for advances in therapy as for parsing patients on the basis of molecular/genetic characterization of their tumors. These observations have fallen into 2 categories. The first relates to genetic characteristics that may better define a tumor phenotype, such as codeletion of chromosomes 1p and 19q,9–12 isocitrate dehydrogenase 1/2 mutation,13,14 epidermal growth factor receptor (EGFR) amplification or mutation,15–17 and phosphatase and tensin homolog (PTEN) deletion.15,18 The second set of observations relate to tumor response to external beam radiation therapy and chemotherapy with alkylating drugs, for which it has been shown that reduction in DNA repair (eg, O6-DNA methylguanine-methyltransferase) favors OS.19,20 To date, none of these molecular/genetic observations have led to efficacious new drugs for the treatment of gliomas.
Outcome (Survival) Expectations for Patients With Medulloblastoma
During the period 1953–1999, the increase in OS for diagnoses of medulloblastoma was marked and reflected improvements in neurosurgery and anesthesia, neuroimaging, radiation therapy, and chemotherapy. To put these advances in perspective, the 5-year OS following surgery and craniospinal irradiation was 41% in 1953,21 but by 1994, 5-year PFS was 90% for localized disease and 67% for medulloblastoma with metastatic spread (M4).22 In addition to the spread of tumor outside the confines of the cerebellum, studies confirmed risk factors of age (≥3 year), residual tumor burden after surgery (≤1.5 cm2),23,24 and the impact of adjuvant chemotherapy.24 Thus, by 1999, it was shown that external beam radiation therapy with adjuvant vincristine, lomustine, and procarbazine chemotherapy produced a 5-year PFS of 78% for residual tumor ≤1.5 cm2, and 54% if residual tumor was >1.5 cm2; 5-year PFS was 78% for children over 3 years with only local disease and <1.5 cm2 residual tumor.
The treatment of progressive/recurrent medulloblastoma with cytotoxic drugs occurred, for the most part, before 2000. The drugs used were, generally, the same drugs used for the treatment of gliomas. Table 3 is a compilation of some of the small studies that formed the literature at the time. Those studies typically used what today is called the clinical benefit rate (CBR), which includes response and stable disease based on neuroimaging methods available at the time. For single drugs, the mean CBR was 46% and for drug combinations 62%, with the better median time to tumor progression being 10 to 19 months, an outcome considerably better than for GBM, another WHO grade IV CNS malignancy.
Table 3.
Cytotoxic chemotherapies used for progressive/recurrent medulloblastoma compiled from literature prior to 2001154a
| Single Drugs | CBR, % |
| Diaziquone (AZQ)155,156 | 28 |
| Carboplatin157,158 | 35 |
| i.v. Methotrexate159–161 | 38 |
| Cisplatin162,163 | 40 |
| i.v. Melphalan164 | 50 |
| Dibromodulcitol165 | 51 |
| Vincristine166–168 | 73 |
| Lomustine169–171 | 80 |
| Weighted mean CBR (n = 166) | 46 |
| Drug Combinations | CBR |
| Vincristine, prednisone, procarbazine172 | 25 |
| TPDCV154,173b | 60 |
| PCV174 | 62 |
| Nitrogen mustard, vincristine, procarbazine175 | 73 |
| Carboplatin + tenoposide176 | 72 |
| Weighted mean CBR (n = 82) | 62 |
aThe range of median time-to-progression values for the better treatments is 10 to 19 months. CBR is the clinical benefit rate and includes patients with complete response, partial response, and stable disease.
bTPDCV, thioguanine, procarbazine, dibromodulcitol, CCNU, vincristine.
Like glioma therapies, molecular/genetic studies showed patients having variable outcomes, from very good to very bad. An excellent summary paper by Taylor and colleagues defines 4 subgroups.25 This paper reflected the clinical conclusions of a consensus conference in Boston in the fall of 2010. The 4 main subgroups of medulloblastoma were Wnt, Sonic hedgehog, Group 3, and Group 4 (Fig. 3). The participants outlined the demographic, transcriptional, genetic, and clinical differences among the 4 subgroups and determined that the molecular classification of medulloblastoma will evolve in the future as larger cohorts are studied at greater depth. To date, no molecular/genetic observations have led to new drugs for the treatment of medulloblastoma, and therefore this molecular grouping has some limitation in its value to the clinician until new drugs are developed.
Fig. 3.

Comparison of the subgroups of medulloblastoma and their association with published papers on molecular subgrouping.25,194–199 SHH, Sonic hedgehog. Reprinted with permission from Michael Taylor.25
Premise for Why Drugs Failed and The Dearth of New Drugs
The conference addressed many of the reasons why some anticancer drugs were not more effective against CNS tumors, why so few new ones have been developed, and why none are considered sufficiently efficacious to change medical practice. While many colleagues other than conference attendees have expressed the opinion that the main reason drugs have failed is that we have insufficient information on the target drivers for gliomas and other CNS tumors, many participants at the conference felt that target identification was only one part of the problem. The following list conveys the conference consensus:
Most anticancer drugs were not developed specifically for infiltrative CNS tumors.
Most therapeutic targets were not unique to CNS tumors and, therefore, were likely to produce a poor therapeutic ratio. The advent of public genetic databases, such as The Cancer Genome Atlas, provides opportunities to identify unique drug targets.
Not all “target-specific” drugs have proven to be truly specific and are, therefore, likely to produce unforeseen host toxicity and a poor therapeutic ratio and are difficult to use in combination with other drugs.
Drugs may not reach infiltrative tumor cells in the CNS because of physicochemical constraints that limit their endothelial and tumor cell permeability and/or because they are substrates for active endothelial and tumor cell efflux pumps that lower intracellular drug levels.
Free drug concentrations (free fraction) were not considered when evaluating preclinical data, which thus gave false indication of drug concentration capable of exerting pharmacological effect.
Evaluation of drugs in models that did not recapitulate real CNS tumors gave false indication of expected efficacy.
Drug-to-target binding (drug target residence time) may not have been optimal for the expected target effect. Currently, most covalent bond (irreversible) targeted drugs are older cytotoxic drugs, whereas many of the newer targeted drugs are reversible binding drugs that require constant dosing, are likely to produce a poor therapeutic ratio, and are difficult to use in combination with other drugs.
Quantitative Structure–Activity Relationship Studies and Brain Penetrant Anticancer Drugs
The general concept that hydrophilic drugs are more brain penetrant than hydrophobic drugs has been part of the pharmaceutical chemistry culture for many decades. During the past 60 years, this understanding has become increasingly elucidated for anticancer drugs and CNS tumors. The 1960s gave voice to the initial quantitative studies of brain capillary permeability and structure–activity relationships based on parameters such as partition coefficient (log P), or the logarithm of the ratio of the concentrations of the un-ionized solute in solvents (octanol/buffered water), molecular weight, charge, and other relevant measures. The early studies of Corwin Hansch, the father of computer-assisted molecule design, and colleagues provided the basis for illuminating the quantitative structure–activity relationship (QSAR) and some of the first studies using anticancer drugs.26–39 QSAR studies of nitrosoureas defined tumor cell kill and increased survival as a function of an optimal log P, with different values depending on where the tumor grew. For example, in murine L120 leukemia, optimal log P was about—0.6,31 in murine Lewis lung carcinoma log P was 0.8,40 and against an intracerebral rat brain 9 L tumor model log P was ∼0.4, with a close second being a log P of 1.5.41 While this approach provided reasonable data for a complex alkylating agent, it was appreciated that additional factors would influence antitumor activity as well. Other QSAR studies in the 1970s defined the optimal log P as between 1.5 and 3.2 for imidazole carboxamides in intracerebral murine G26 tumors.28 These and other experimental studies supported clinical trial outcomes in infiltrative gliomas, leading to FDA approval of carmustine (BCNU; log P = 1.5), lomustine (CCNU; log P = 2.8), and temozolomide (log P = 1.4) for the treatment of high-grade gliomas.
CNS Drug Penetration and Drug Modeling
Studies of brain capillary permeability of standard compounds and anticancer drugs provided more precise information regarding lipophilicity (log P) and molecular size and formed a rationale for quantitative predictive modeling that is used by the pharmaceutical industry and medicinal chemists today to guide drug discovery and development of CNS penetrant drugs and to, conversely, protect the brain from drugs that need to be excluded from the CNS. One example, used by the pharmaceutical industry, is the CNS multiparameter optimization score, which can be calculated using the formula proposed by Wager and colleagues.42 The calculation is based on values such as the logarithm of the acid dissociation constant of the most basic center or creatinine phosphokinase-MB, logarithm of the partition coefficient, logarithm of the distribution coefficient at pH 7.4, molecular mass, topological polar surface area, and number of hydrogen bond donors (HBDs). This approach was used by Genentech to develop a new and specific brain penetrant inhibitor of phosphoinositide 3-kinase (PI3K) specifically for the treatment of high-grade gliomas.43
In addition to understanding rules-based approaches to drug penetration from blood to brain, it is important to understand how CNS tumors grow and what impact tumor cell location might have on drug action. First, we should briefly consider the differences between the intact blood–brain barrier (BBB) and the CNS blood–tumor barrier (BTB). This is schematized in Fig. 4. In the normal BBB, inter-endothelial tight junctions lead to passive permeability dependent on molecular size and lipophilicity, whereas active efflux pumps further restrict CNS extracellular accumulation of specific heterocyclic drugs and chemicals (Figs 4 and 5). Brain tumors, whether primary or secondary, are capable of changing these relationships such that BTB manifests new vessels produced by the tumor, as well as the release of ligands such as vascular endothelial growth factor (VEGF), resulting in a reduced number of inter-endothelial tight junctions and, with them, less restriction on the molecular size of penetrant drugs with fluid movement driven by hydrostatic force from blood into tumor extracelluar space and with the elaboration of a protein-rich edema.
Fig. 4.

These illustrations compare and contrast the differences between the BBB and the more leaky BTB “barrier.”
Fig. 5.

Depiction of some of the efflux pumps responsible for endothelial brain barrier phenomena. The size of the circle is a relative approximation of efflux pump activity in brain endothelia. Abbreviations: PGP, P-gp; OATP, organic anion-transporting polypeptide; OAT3, organic anion transporter.
Fig. 6 shows the expected relationship between blood-to-tissue permeation, depicted as ki, for radiolabeled standard compounds and drugs diffusible across the BBB and the BTB. From this plot, it is apparent that higher-molecular-weight compounds that are diffusion limited distribute more quickly and to a greater extent into tumors than into the CNS.
Fig. 6.

This plot depicts the the relationship of the transfer constant ki, permeability × surface area, for i.c. rat 9 L tumor vs normal mouse brain200,201 and temozolomide from a human study.201 The dashed line has slope of unity and is not fit of that data. 1, 3HOH; 2, NaCl; 3, urea; 4, glycerol; 5, creatinine; 6, 5-fluorouracil; 7, dianhydrogalactitol; 8, galactitol; 9, misonidazole; 10, procarbazine; 11, α-difluoromethylornithine; 12, dibromodulcitol; 13, sucrose; 14, epipodophyllotoxin; 15, bleomycin; 16, inulin; and 17, temozolomide.
Given the infiltrative and invasive nature of many brain tumors, it is critical to develop drugs that can permeate across the BBB.44 Many new molecularly targeted agents that are able to inhibit signaling pathways critical for tumor growth and proliferation have failed to elicit any clinical benefit in the treatment of CNS tumors, either primary or metastatic.45 This is in spite of many of these molecules having favorable physicochemical properties for passive diffusion across an intact BBB. Compared with treatment of other types of tumors, targeting tumors of the CNS is particularly challenging due to the location of the tumor in a pharmacological and immunological sanctuary within the CNS. The BBB presents a major obstacle to systemic chemotherapy and is capable of significantly limiting drug response.46 Many talks at the conference showed how drug efflux transporters at the BBB restrict the passage of drugs into the brain and thus shield the tumor cells from exposure to targeted agents and cytotoxic chemotherapy. In addition to the BBB, the presence of similar drug efflux pumps within tumor cells further protects them from chemotherapy.
Many studies have tried to correlate brain penetration and CNS activity of a drug or chemical to their physicochemical properties. These studies vary in their approach and methodology for predicting BBB permeability. In general, it has been shown that compounds with high activity within the CNS tend to have high lipophilicity, few hydrogen bond donors, low polar surface area, and low molecular weight.47–50 Based on accumulated knowledge, it is not surprising that these drug properties impart high membrane permeability to the drug molecule and enhanced transport to the brain.50 However, several molecules with these favorable physical properties have been found to have lower permeability into the brain, as a result of active efflux transporters that make the BBB impermeable to these transporter-substrate compounds.47,51 The BBB is fortified by numerous drug transport proteins (Fig. 5), many of which pump (transport) drugs out of the brain. For instance, ATP-dependent transporters can markedly restrict brain penetration of some therapeutic agents, even though these molecules have favorable physicochemical properties that would predict unrestricted permeability across the BBB.47,51 A majority of these efflux transporters belong to 2 superfamilies: the ATP-binding cassette (ABC) and the solute carrier. P-glycoprotein (P-gp, ABCB1), breast cancer resistance protein (BCRP, ABCG2), and multidrug resistance-associated proteins (MRPs, ABCC) are important members of efflux proteins found in the BBB. These transporters have been shown to have high expression patterns in the brain capillary endothelium in both animal models and humans.52,53
The advent of transporter knockout mice, especially P-gp (Mdr1) and BCRP, has greatly enhanced the capability to examine transporter liability of specific drug candidates using in vivo preclinical models. These models have been characterized for transporter expression and tight junction function in the capillary endothelium and have been useful in determining whether active efflux transport is an important mechanism in limiting BBB permeability and hence effective delivery to infiltrative tumor cells in the brain. Interestingly, it has been shown that both transporters work in concert to efflux several dual substrate molecules and can compensate for each other in efficiently limiting drug BBB permeability.54
The downregulation of transporters in the BBB may improve efficacy of some of the failed molecularly targeted agents through improved targeted delivery to the invasive tumor cells that reside behind an intact BBB. Preclinical studies have highlighted the feasibility and proof-of-concept of this possible treatment strategy.55 Several preclinical examples of improved delivery using this strategy, with the resulting enhanced efficacy, were presented at the conference. The necessity of finding the mechanistic causes of decreased permeability in regions of tumor burden that have an intact BBB, resplendent with a full complement of efflux transporters, is highlighted by clinical imaging studies that use multiple imaging modalities to examine tumor location with respect to the integrity of the BBB. Overlay images using T1-weighted MRI, with contrast, and 18F-DOPA ([18F]-L-dihydroxyphenylalanine) PET show that there are frequently significant regions (volumes) of brain that contain tumor, but these are not contrast enhanced (Fig. 7). This clearly indicates that the old adage of the BBB being “broken down” where there is tumor is not only incorrect but highly misleading when considering the reasons for clinical failure of promising agents.56,57
Fig. 7.

Current diagnostic imaging modalities fail to adequately describe entire tumor location. (A) T2-FLAIR image of GBM patient with tumor area outlined in blue. (B) T1-gadolinium (GAD) image of the same patient tumor (outlined in red). Contour includes postoperative cavity (not just enhancement). (C) PET image from the same patient with active tumor outlined in yellow. Tumor volume comparison of T1-GAD, T2-FLAIR, and PET indicates a large volume of active tumor (PET) outside of the area where the BBB is leaky (T1-GAD). (From Parrish et al57).
Examining drug delivery to specific regions of the brain tumor and surrounding brain is critical to understanding the reasons for failure or success of therapeutic options. Rational choice and testing of therapeutic agents for brain tumors must take into account the genetic heterogeneity of the tumor and the heterogeneity of delivery across the BBB. Given the overall reasons for failure to effect a cure in primary and secondary brain tumor, we must be prepared to treat residual tumor after surgical resection, that is, effectively treat “those left behind.”58
Building on our understanding of how drugs distribute from blood into the CNS and tumors and additional information gleaned from years of experimental studies, we created a scheme (Fig. 8) to depict the relationship of tumor cell burden to capillary permeability based on physiological and pathological observations.59
Fig. 8.

Depiction of the relationship of tumor cell burden to capillary permeability based on physiological and pathological observations.59,200
Tumor Heterogeneity
Conceptually, it is important to consider that different tumor cells will—depending on their location in the tumor or whether they infiltrate the adjacent brain and the regional and plasma pharmacokinetics (PK) of the drug being administered—have disparate vulnerability to the drug. This variable tumor cell vulnerability is very difficult to study and quantitate, yet importantly, it is a crucial factor to take into account when considering tumor heterogeneity. Tumor heterogeneity was discussed broadly at the conference. It was appreciated that tumor cells by location in the tumor and adjacent brain will differ in their proximity to capillaries, nutrients, and therapeutic levels of drugs. Tumor cells will differ in their cell cycle location, exposure to hypoxia, genetic profile, methylation status, protein levels, and protein activity. Much of this information cannot be easily gleaned from an individual tumor and certainly not in situ with today's technology; therefore, it is difficult in many cases to predict the impact of drug effects on their target(s).
The only example cited at the conference was that of the target protein ornithine decarboxylase (ODC), the target for eflornithine (α-difluoromethylornithine, DFMO). For this and other drug target enzymes, the heterogeneity of enzyme activity and drug binding to targets will be critical elements for antitumor efficacy. Studies of ODC level (a surrogate for ODC activity) in WHO grades III and IV glioma tumors found that median ODC levels could vary ∼2-fold within individual tumors and ∼5-fold between grade III and grade IV gliomas.60 While ODC can increase with tumor malignancy,60–62 it was interesting that even a modest 2-fold change in ODC activity could result in a survival gain of a much greater magnitude of ∼10-fold when glioma treatment included eflornithine, a specific ODC inhibitor.63
In addition to drug target variability, tumor drug distribution can impact drug response. Unfortunately, even in preclinical models, it is difficult to obtain regional PK/pharmacodynamic (PD) values. Using a serial brain tumor sectioning protocol, Sharma and colleagues64,65 developed a sensitive and robust method to characterize the intratumoral PK using liquid chromatography–tandem mass spectrometry and PD using phosphorylated extracellular signal-regulated kinase (ERK) in antibody-based detection of gefitinib in small amounts of glioblastoma tumor samples obtained from mice bearing intracerebral tumors treated with gefitinib. Concentrations of gefitinib, a reversible EGFR tyrosine kinase inhibitor, showed up to 2.4-fold intratumoral variability in PK and 1.5-fold variability in PD in a standard U87 GBM model. The tumor sectioning protocol facilitated binning of the PK/PD data to biological and physiological characteristics of the tumor. The methods are sufficiently accessible and could be applied to other anticancer drugs and tumor models to obtain greater resolution of intratumoral PK and PD.
Metastatic Tumor Therapy
It could be argued that patients with systemic non-CNS tumors will have fewer PK problems than those associated with infiltrative gliomas and would be expected to have greater options for treatment with anticancer therapies. Over the years, CNS anticancer drug therapy has changed from a BTB-centric approach that emphasized BTB permeability as a predictor of drug delivery—a PK characteristic—to an integrated PK/PD approach. In the newer PK/PD paradigm, the linkage between local drug concentrations—and manipulating drug delivery, transport, and metabolism to selectively enhance tumor delivery—and molecular activity (receptor and enzyme), cell signaling and downstream biomarkers, and mechanistic PK/PD modeling has become a new mantra.
In the limited discussion of metastatic tumor therapeutic strategies at the conference, emphasis was placed on understanding active efflux transporter expression and relevance to tumor heterogeneity as well as the following PK considerations.
BTB passive permeability, often significantly elevated in brain metastases, varies widely within and among tumors. Even though tumors that metastasize to the CNS are frequently less infiltrative than gliomas, the consensus was that achievement of active drug concentrations should be based on penetration through an intact BBB to maximize regional tumor PK/PD.
Delivery of the major drugs used in the treatment of metastatic breast cancer (eg, taxanes, anthracyclines, vincas, lapatinib) is currently about 2% to 10% of that used for extracranial tumors.
Therapeutic drug concentrations—low end of the dose-response curve—are likely reached in only a subset of CNS metastases, and thus, unlike non-CNS tumors, PK failures contribute to ineffective drug therapy.
Discussed at the conference were various strategies to improve drug delivery and action for CNS metastases. These strategies include:
Selective enhancement of BTB passive permeability (eg, phosphodiesterase type 5 inhibitors) or facilitated drug influx (organic anion-transport polypeptide [OATP], equilibrative nucleoside transporter)
Inhibition of BTB/BBB active efflux of P-gp, BCRP, and MRP pumps
Enhancement of receptor-mediated BBB/BTB transcytosis using vector-drug conjugates
Preferential enzymatic prodrug activation at site of action within CNS metastases and beyond the BBB
Active drug species catabolism at site of action within CNS metastases
Selective up- or downregulation of metabolism or drug transport
Downregulation of cytoprotective factors (eg, glutathione) or upregulation of cytotoxic pathways (ie, caspase)
Current-Generation Brain Penetrant Drugs and Their Targets
At the conference, current BBB penetrant alkylating agents such as BCNU, CCNU, procarbazine, and temozolomide were not discussed, as the focus of the conference was on the future of CNS anticancer drug development. Attendees heard first from Russell Petter about the impact and importance of irreversible covalent binding kinase inhibitors using Bruton's tyrosine kinase (BTK) inhibitors such as CC-292 from Celgene Avilomics Research. Also presented were 3 new brain penetrant anticancer drugs: (i) a reversible PI3 K inhibitor (GNE317) from Genentech, (ii) an irreversible pan erbB (EGFR, EGFR variant III, human epidermal growth factor receptor [HER]4, HER2) inhibitor (NT113) from NewGen Therapeutics, and (iii) a reversible anaplastic lymphoma kinase/ROS1 kinase inhibitor (PF-06463922) from Pfizer. Each of these drugs demonstrates rapid brain penetration, free drug levels in brain of 30%–100% of free plasma levels, and low efflux in cell lines overexpressing P-gp and other cancer-resistant pump proteins. Discussed during this session was the increased understanding of unbound brain concentrations and the relationship with transporter-mediated efflux (eg, P-gp, BCRP). In vitro assays are available to determine whether potential drugs are substrates of transporters that would prevent efficacious exposure in the brain. Additionally, drug discovery programs can utilize in silico calculations to predict whether or not molecules would be transporter substrates. Other newer drugs in development and hypothesized were also discussed, including peptide drug conjugates.
Another topic of interest concerned large-molecular-weight biologics that can, under special circumstances, achieve activity against brain metastases and glial neoplasms. While monoclonal antibodies (mAbs) directed against tumor-specific proteins such as HER2 have been demonstrated to reduce tumor size and increase survival, large-molecular-weight biologics have a limited ability to cross the BBB and, therefore, a low impact on CNS tumors. Xenobiotics are restricted by the BBB, but nutrients, hormones, and other required molecules enter the brain by processes such as receptor-mediated transcytosis. Low-density lipoprotein receptor–related protein 1 (LRP1) is known to perform this transport function in BBB endothelial cells, and accordingly, scientists at Angiochem have created a family of peptides (angiopeps) designed for LRP1 recognition. Conjugation of angiopep-2 (An2) to confer brain permeability has been demonstrated for small molecules, peptides, and enzymes. The approach has been accomplished with ANG1005, an An2-paclitaxel conjugate that has been shown to achieve high concentrations in human brain tumors that were surgically removed 4–6 h after dosing. These and other promising antitumor effects in phase I and phase IIa clinical studies support continued development of ANG1005, which is currently in phase IIb trials for breast cancer brain metastases and phase IIa trials for primary brain tumors. Other studies using a conjugate of An2 and a mAb to HER2 (ANG4043) showed brain i.c. tumor penetration and activity against i.c. rodent breast tumors.66
Impact of Drug-Target Binding Kinetics on Drug Action: Principles and Practice
Peter Tonge's presentation at the conference focused on the importance to the target of drug residence time and how this can be used to improve drug discovery/development. Despite enormous investments in time and resources, ∼90% of drug candidates fail in human clinical trials. A common reason for this attrition is the inability to achieve the drug exposure predicted to be required for efficacy in phase I trials without unacceptable side effects (safety). Thus, there is a knowledge gap between how in vitro vs preclinical data are used to predict efficacy in humans. Lead compounds are normally selected and advanced based on in vitro measurements performed at constant drug concentrations. Thermodynamic metrics, such as half-maximal inhibitory concentration (IC50), dissociation constant (Kd), and Ki (inhibition constant), are used to quantitate the affinity of compounds for the target, and strenuous efforts are made to increase target affinity while decreasing the affinity for any known off-target proteins. Thus, (thermodynamic) selectivity is a key driver in drug discovery. In addition, measurements of drug activity in live cell assays are used for compound prioritization, and, again, these studies are usually performed at constant drug concentrations. However, this approach neglects a critical fact: that in vivo drug concentrations vary with time. If drug-target binding kinetics are slow relative to drug PK, then drug and target may not be at equilibrium, and in such situations thermodynamic parameters will not be able to fully predict target engagement in the human body.67 In an ideal world, the preferred drug penetrates from blood to tumor cells rapidly, binds to the cellular target rapidly, dissociates slowly from the target, but clears rapidly from surrounding tissue. This should improve the therapeutic index through kinetic selectivity (assuming that dissociation from off-target proteins is rapid by comparison). A caveat to our assumptions is that often we do not know the drug concentration at the target site (inside a tumor cell), so presuming that it is constant is probably a large approximation.
We thus propose that knowledge of drug-target kinetic data will improve lead optimization by selecting and advancing compounds that have improved target engagement under nonequilibrium conditions. In particular, compounds with increased residence time should improve target engagement at lower drug concentrations, reducing the drug exposure required for efficacy and improving safety.
The kinetic lifetime of the drug-target complex can be quantified by residence time (tR = 1/koff), where koff is the rate at which the complex dissociates to form unoccupied (active) target.68 To exemplify the critical nature of residence time and its intimate relationship with drug PK, in Fig. 9 we have calculated target occupancy as a function of time for 3 targets with the same thermodynamic affinity for the drug (Kd = 14 nM), but different residence times (tR).67 Target 1 is assumed to be the therapeutic target (tR = 104 h), while targets 2 and 3 are off-target proteins, binding to which leads to toxicity (tR = 11.5 h and 1 s). In addition, drug concentration is assumed to reach an initial concentration (Cmax) of 500 nM and clear with a half-life of 1 h.
Fig. 9.

Variation in drug-target occupancy as a function of time for three targets. It is assumed that drug concentration (Cmax) is 500 nM after dosing and clears with t1/2 of 1 h and that drug binds to 3 different targets with the same Kd (14 nM). Target occupancy (%) is calculated for targets 1 and 2 assuming no rebinding and residence times (tR = 1/koff) of 104 and 11.5 h, respectively. For target 3, Kd is used to calculate occupancy, that is, kon and koff are fast relative to the change in [drug]: here tR = 1 s based on diffusion-controlled kon.67
Based on thermodynamic measurements of drug “potency” (affinity), the drug shows no selectivity—because the Kd values for the 3 binding partners are the same. At this stage in a drug discovery program, the compound could be abandoned. However, selectivity is time dependent: after 12 h there is a clear discrimination between the 3 targets: target 3 is basically free of drug, whereas target 1 is still 87% occupied. The key point is that kinetic selectivity and thus therapeutic index are time dependent and vary based on drug concentration (PK) and the residence time of the drug on target and off-target proteins. Thus, a strategy in which PK and dosing algorithms are coupled with knowledge of residence-time effects should lead to improved therapeutic indices (ie, safer, better drugs). In pursuit of this goal, we are formulating mechanistic PK/PD models that implicitly include the kinetics of drug-target interactions, and recently we used this approach to predict the efficacy of an LpxC inhibitor in an animal model of Pseudomonas aeruginosa infection.69 There are numerous molecular design strategies that might afford agents that achieve kinetic selectivity: focus on targets that engage ligands in slow-off states, select for slow koff inhibitors during the optimization stage, or pursue inhibitors that are irreversible by virtue of covalent modification.
Fig. 10 summarizes some of the key factors that modulate the interplay among drug concentration, target engagement, and drug PD and that are required to fully parameterize PK/PD models. These include target vulnerability and the percent of target that must be occupied to cause the desired PD effect.
Fig. 10.

Some factors that affect target engagement. Many time-dependent inhibitors bind through an induced-fit 2-step mechanism where interconversion between an initial (EI) and final (EI*) enzyme inhibitor complex is slow. Here koff ≈ k6 so tR = 1/k6.
To implement the use of drug-target kinetics in lead compound selection and optimization, structure-kinetics relationships are needed to inform the design of inhibitors with altered residence times. Examples are now starting to appear in the literature where such information is being obtained and applied. This includes work on antibacterial agents.70 These efforts are being expanded to kinases, which are important targets in cancer drug discovery. A recent emphasis in kinase inhibitor discovery includes the design and synthesis of irreversible inhibitors, which have very long (infinite) residence times on their targets.71 In the talk at the conference, several examples were given in which drug-target kinetics were being applied to understand the efficacy of kinase inhibitors, including those that target EGFR72 and BTK.73,74 Celgene Avilomics Research has developed CC-292, a small molecule irreversible inhibitor of BTK, and a chemical probe derived from CC-292 has been used to quantitate target engagement in vivo.74
In addition to quantifying target engagement, knowledge of drug concentration at the target site is of crucial importance to fully deploy mechanistic PK/PD modeling. To provide this information, we are developing radiolabeled drugs to noninvasively image drug biodistribution in animals and humans using PET.75 Because PET is an approved method for quantifying drug concentration in the human body, its use is expected to improve the ability to translate knowledge of in vitro drug action into predictions of drug efficacy in humans.
Drug Targets and Discovery
One of the age-old questions remains: what do we want a drug to do? We discussed at length which characteristics are needed for a drug to reach its tumor target in the CNS. We also addressed the question about how long a drug should be resident on a target for maximal biological effect. We have not discussed what the targets might be, and for good reason. We do not have a sure answer for this issue. Uncertainty breeds multiple approaches and follows the adage that if you do not know where you are going, any road will get you there. Scientists have used genetic information along with focused mechanistic studies related to cell cycle control, DNA function, and protein signaling to guide anticancer drug discovery, with many drug discovery efforts focusing upon protein tyrosine kinases. Speakers at the conference addressed some of these areas and had various viewpoints about how current knowledge in these areas might help future drug development issues. Examples from their presentations follow.
Master Regulators and Drivers of Oncogenesis of Glioblastoma
Antonio Iavarone presented his research that focuses on targets of gliomagenesis through his studies of regulators and drivers of glioblastoma. He believes that the genome era is an exciting time in scientific discovery. His group is trying to contextualize the alterations of each genetic network in the natural environment of a specific tumor and identify the key driving modules (mutations, epigenetic changes, and others) on which specific tumor subgroups rely for growth, survival, and progression. With this information in hand, he believes it will be possible to target the critical alterations with specific drugs, often already available for other types of diseases. By focusing on one of the most lethal forms of human tumors, GBM, this group has been able to make incredible progress along this line in the last 2 years. They discovered 2 transcription factors—STAT3 (signal transducer and activator of transcription 3) and C/EBP (cytidine-cytidine-adenosine-adenosine-thymidine [CCAAT]–enhancer binding homologous protein)—which are responsible for activation and maintenance of the most aggressive gene expression signature of high-grade glioma, the mesenchymal signature. The therapeutic implication of this work has arisen from our ability to efficiently target the 2 transcription factors in preclinical mouse models with consequent collapse of the mesenchymal signature and extended survival.76
More recently, the lavarone group has identified the first examples of highly oncogenic and recurrent gene fusions in GBM, targeting their dependency in a particular tumor subtype and observing dramatic antitumor effects. Recurrent gene fusions in GBM result in the constitutive activation of receptor tyrosine kinase genes (fibroblast growth factor receptor [FGFR], EGFR, and neurotrophic tyrosine kinase receptor type 1) that render tumors addicted to the driver events.77,78 Among them, the FGFR–transforming acidic coiled-coil (TACC) gene fusion is the addicting oncogenic event with the highest therapeutic value. First, this is a recurrent oncogenic event in several types of human cancer besides GBM (head/neck, lung, bladder, and others). Second, FGFR-TACC fusions are potent oncogenes that transform normal cells by activation of noncanonical substrates and precipitation of aneuploidy. Finally, human tumors harboring FGFR-TACC fusions acquire marked sensitivity to FGFR inhibitory compounds. It is not surprising that this line of investigation has matured toward clinical trials.79 The strongly addicting oncogenic activity of FGFR-TACC fusions is endowed in the unique, dual oncogenic mechanism implemented by these genetic events, which is summarized in Fig. 11. The FGFR-TACC fusion proteins aberrantly localize on top of the spindle pole of mitotic cells. This results in mislocalization at a crucial site for the organization of mitosis of a constitutively active tyrosine kinase that ultimately perturbs mitotic progression and promotes chromosome missegregation and aneuploidy, a hallmark of malignant neoplasms. However, chromosome missegregation and aneuploidy per se would be associated with loss of cellular fitness, but they are exploited for neoplastic transformation and maintenance by the strong noncanonical signaling events triggered by FGFR-TACC.
Fig. 11.
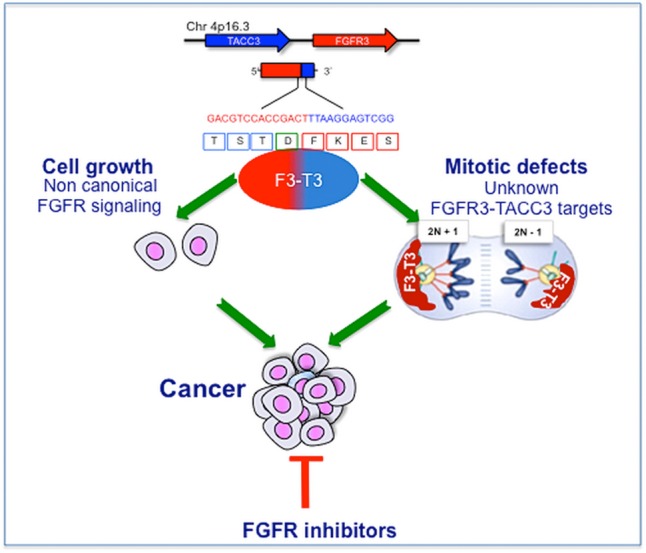
The mechanism of action of FGFR-TACC fusions in glioblastoma.
The integrated computational-experimental pipeline that we developed plus our ability to functionalize any genetic brain tumor module was recently applied to the entire landscape of copy number variations (CNVs), somatic mutations, and gene fusions of human GBM. This information is quickly advancing our ability to translate each new genetic finding into the personalized context of the clinical setting.
Targeting Proteins for Drug Discovery
Forest White discussed the need for better understanding protein targets in order to develop next-generation drugs. Clearly, an increased understanding of cancer genomics has revolutionized our knowledge of the genomic alterations and their consequent effects on transcriptional regulation in brain cancer. Through analysis of hundreds of tumor specimens, selected pathways have been highlighted by common mutations in a significant proportion of patient tumors. However, it is not obvious that mutated or amplified genes are necessarily present at the protein level, and genomic and transcriptomic data do not directly identify signaling network activity.
To identify activated networks and thereby determine potential therapeutic targets, White's group at MIT has applied functional proteomics to quantify tyrosine phosphorylation signaling networks in a panel of human GBM tumor tissues. The results of this analysis highlight a high degree of interpatient heterogeneity, as each tumor features a dramatically different set of tyrosine phosphorylated proteins. In fact, despite clustering into subtypes based on transcriptional data, these tumors no longer cluster due to either protein expression or tyrosine phosphorylation data. Even with the high degree of complexity embedded in phosphorylation data across patient tumors, correlation analysis recapitulates known activated signaling networks present in selected tumors, potentially indicating patient-specific targets defined by highly interconnected nodes within these networks. Finally, to identify the phosphorylation sites driving tumor cell proliferation and diffuse invasion, MR images from these patients were analyzed by Kristin Swanson's lab at Northwestern University to extract quantitative phenotypic information, net dispersal rates, and proliferation rates for each tumor. A correlation analysis of phenotypic and phosphorylation datasets was performed, yielding multiple significantly correlated phosphorylation sites for each phenotype. Intriguingly, this unsupervised analysis highlighted several novel phosphorylation sites in addition to well-characterized phosphorylation sites known to drive tumor cell proliferation and migration/invasion. Further functional studies of these novel phosphorylation sites are warranted.
One of the other main issues in glioma treatment is the apparent lack of efficacy of targeted therapeutics, based on minimal improvement in PFS or OS. In evaluating the efficacy of targeted therapeutics, one of the primary questions is whether the therapy effectively hit the target. To directly address this question, we have altered our functional phosphoproteomics platform to provide absolute quantification of selected phosphorylation sites on therapeutic targets and their downstream pathways. Using this system, we have quantified the effect of treating GBM patient-derived xenograft flank tumors with several different EGFR small-molecule inhibitors. In each case, the inhibitor effectively hit the target, resulting in ∼90% decrease in EGFR tyrosine phosphorylation. Erlotinib, dacomitinib, and NT-113 each effectively decreased phosphorylation on EGFR scaffold proteins and the ERK1/2 mitogen-activated protein kinase cascade, with NT-113 having a slightly greater effect. Going forward, this methodology can be used for longitudinal studies, to track the efficacy of target inhibition at multiple time points during tumor treatment to assess resistance mechanisms as the tumor progresses on therapy. This platform is also being used to investigate therapeutic efficacy in orthotopic tumors, to assess the impact of brain penetration of selected targeted therapeutics.
Kinase Chemical Genetics and Cancer Drug Discovery
Arvin Dar described an approach combining kinase-focused chemistry with genetic screens in Drosophila to develop polypharmacological compounds with extremely high therapeutic indices that are concomitantly safe and effective. In the approach used by the Dar group, the chemical design aspect of the method is based on generating focused libraries of small-molecule compounds that allow inhibition of one or more drivers of tumor progression. Candidate compounds that meet this criterion are then tested in Drosophila screens to empirically determine optimal target profiles. The disease model presented was based on a species of Drosophila that expresses a mutant form of Ret (Rearranged during Transfection), a known driver of medullary thyroid carcinomas. The screens determine the ability of the compounds to rescue flies from oncogene-induced lethality during their development life cycle. The screens allow lead identification that is tumor selective and possesses a relatively wide therapeutic index. Leads that have been identified in flies have performed well in other animals, including mice80 and humans.81 Another advantage of conducting screens in flies is that one can quickly determine the mechanism of action through functional assays and genetic perturbations.
Dar reported that flies are exquisitely sensitive to small structural changes in lead compounds in a manner that is not captured by traditional cell culture or biochemical assays.82 For example, in RetMEN2B screens, there were significant differences in the therapeutic indices among the initial hit, AD57, and several close analogs (Fig. 12). Removing a single –CF3 group (AD58) resulted in a highly toxic compound. Adding a single methylene group (AD36) removed most drug efficacy. It is noteworthy that in cell culture and in in vitro Ret kinase assays, all 3 compounds acted similarly.
Fig. 12.
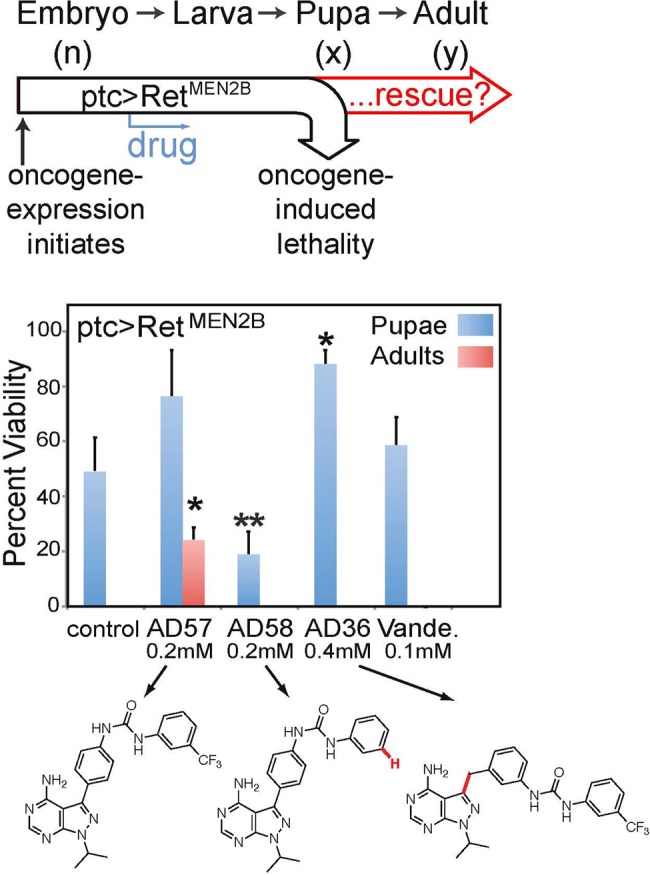
Drosophila whole body assays are extremely sensitive to small changes in chemical structure. (Top) An oncogenic mutant of Ret kinase (RetMEN2B) was expressed under the patched (ptc) promoter in the fly, leading to early lethality. (Middle) Compounds were assayed for pharmacological rescue from Ret-induced lethality. Pupae and fully enclosed adults were counted (n = 200, 75, 98, 54). Vande. = Vandetinib. (Bottom) AD57, AD58, and AD36 are chemically similar yet produced vastly different biological responses. Cross comparison of these compounds led to the discovery of mechanistic basis of efficacy (targets) and dose-limiting toxicity (anti-targets). This figure is modified from Dar et al.80
Comparisons of AD57, AD58, and AD36 across several different assay platforms (biochemistry, cell culture, and fly phenotyping) led to the concept of the kinase “anti-target.” For example, the fly and biochemical data indicated that the toxicity of AD58 was due to its low activity against Raf (target) coupled with its high activity against mammalian target of rapamycin (mTOR; anti-target), triggering an “incoherent feed-forward loop.”82 Dar and colleagues were able to use this information to “dial out” activity against the anti-target mTOR, and the optimized molecules performed extremely well in both fly and mouse xenograft models.
Historically, Drosophila species have proven to be powerful models for understanding the mechanisms that direct development, signal transduction, and cell biology. Further, flies have clear orthologs for ∼75% of human disease–related genes83,84 and are increasingly used to explore diseases, including cancer.82,85,86 Accordingly, Dar and his collaborators in the Cagan Lab at Mount Sinai Hospital, and other labs elsewhere, continue to develop fly-based models of various cancers, including brain cancers. The Read Lab at Emory University has been instrumental in this regard.87
Drug Discovery Using Functional Genetics, Patient Isolates, and BBB-Traversing “optide” Libraries
Patrick Paddison presented the efforts of his group to identify new candidates for drug targets for GBM functional genetic approaches in patient tumor stemlike cell isolates, as well as a new therapeutics program at Fred Hutchinson Cancer Research Center using “optimized peptides,” or “optides.” His talk covered work from his laboratory and that of a collaborator, James Olson.
GBM is the most aggressive and common form of CNS cancer in adults, and the inability to develop new, more effective therapies may arise from preclinical models that inadequately predict a therapeutic window and from the fact that many new anticancer drugs being used for GBM are “hand-me-downs” from other cancers and were not specifically developed for treating CNS tumors. To identify patient-tailored drug targets for GBM, Paddison and colleagues have performed a series of functional genetic screens in patient-derived GBM stemlike cells (GSCs) as well as nontransformed human neural stem cells (NSCs). GSCs retain tumor-initiating potential and tumor-specific genetic and epigenetic signatures, even during extended outgrowth in serum-free culture. NSCs represent nontransformed candidate cell of origin controls, which share similar gene expression signatures and identical in vitro growth conditions. Using these systems along with RNA interference or CRISPR (clustered regularly interspaced short palindromic repeat)/Cas9 (CRISPR-associated nuclease 9) platforms, they identified multiple molecular vulnerabilities specific to GSCs, which appear to be largely driven by oncogenic transformation, in processes ranging from kinetochore regulation to 3′ pre-mRNA splice site recognition.88–91 They are currently performing small-molecule screens for lead compounds that exacerbate 3′ splice recognition defects in GBM isolates at the National Institutes of Health Chemical Genomics Center (in collaboration with Dr Marc Ferrer) and are developing several other screening pipelines for other novel targets.
Paddison and colleagues have also created an optimized peptides/optides novel drug development pipeline for brain tumors (www.fredhutch.org/en/treatment/treatment-research/optides.html) (Fig. 13). The main optide class they recently focused on is a natural product called knottin. Naturally occurring knottin drugs protect plants from insects, help spiders and scorpions immobilize prey, and serve a host of other predatory or protective purposes.92–94 Knottins are 20–60 amino acid peptides with 3-4 disulfide bridges that tether the protein in a knot configuration.95 In multiple instances, knottins have been engineered or have evolved to have picomolar or nanomolar affinity for targets to which the original scaffold knottin bound poorly or not at all.96–101 Paddison and collaborators created a knottin-derived bioconjugate (Tumor Paint) that delivers fluorescence to pediatric brain tumors and other cancers, with the goal of guiding surgeons to optimize surgical resection of the cancer.102,103 The FDA recently allowed human clinical testing of the clinical candidate (BLZ-100) in brain tumor patients following a successful phase I study in skin cancer patients in Australia. The research team are currently assessing the BBB penetration of 88 knottins from scorpions and spiders.
Fig. 13.
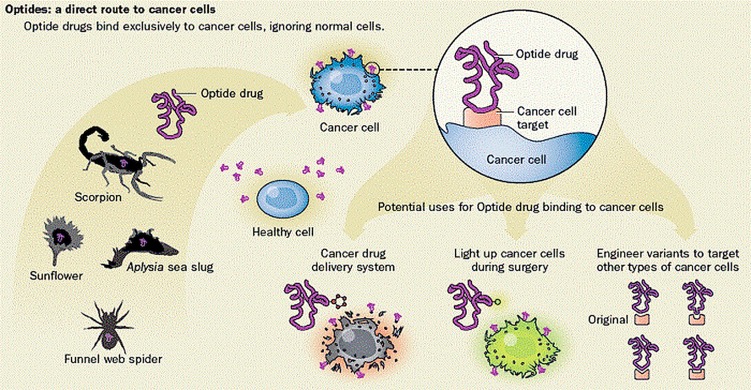
Multiple sources of optide starting scaffolds exist in nature, from the venom of spiders and scorpions to plants and flowers. Some of these have been found to bind to cancer cells, and mutations can be made to enhance their specificity. This allows them to be used as targeting agents for fluorescent surgical aids or cytotoxic warheads.
Similar to many natural product drugs, knottins are quite complicated to synthesize. People have known for decades that these would be good starting points for drug discovery, but the difficulty in making them has hampered drug research, especially when it comes to making variants. A major question from this research was whether these molecules could be synthesized in sufficient quantities and with proper folding to conduct in vivo experiments. Many knottins are insoluble when produced in the laboratory or conjugated to other molecules. To that end, Paddison and colleagues have been able to create reliable amino acid substitution algorithms that sharply improve the fraction of optides that are successfully produced. Further, since Escherichia coli and yeast fail to properly fold and excrete most knottin peptides, the Paddison team created a mammalian expression system that enabled production of as many as 20 000 pooled knottins or 50 individual candidates per month, with further scaling potential.104,105 With these problems solved, a toolbox of bioconjugates was created that could be added to knottins to induce equilibrium with albumin or otherwise extend serum half-life to more than 8 h without adversely affecting target engagement.103,106 Importantly, the group recently established a Molecular Design and Therapeutics Core at the Fred Hutchinson Cancer Research Center, for the cost- and time-effective production of optides for in vitro and in vivo work. They eventually hope to have several BBB-traversing optide variants in clinical trials within the next 5–10 years for both pediatric and adult CNS tumors.
Systems Pharmacology Approaches
While different groups of scientists within a pharmaceutical company will look at various aspects of drug pharmaceutics, pharmacology, antitumor activity, toxicity, and clinical efficacy, increasingly efforts in academia are focusing on a systems approach that was first adopted by some medical school teaching programs in the mid- to late 1990s. Marc Birtwistle discussed the need for a broader and more nuanced systems pharmacology approach for studying potential drugs and how they react against cellular targets and the host to exert the desired effect and, when the drug fails to work as expected (or as hoped for), to define the reason for the failure.
This approach is rooted in describing the biochemical and biophysical processes underlying GBM progression and growth through physicochemical formalisms such as chemical kinetics and reaction diffusion. The resulting computational models are typically systems of coupled ordinary, partial, and/or hybrid stochastic differential equations that, much like engineering analysis of weather patterns, can be simulated to predict response to therapy. Such models have as adjustable parameters measurable quantities such as CNV, gene expression level, and well-studied mutations. Importantly, they have the ability to account for the quantitative and dynamic aspects of drug action that are particularly relevant not only in GBM, but in many tumor types, since most PK models of drug dynamics and distribution are also based on differential equations. This has given rise to the idea that such models may be described as next-generation or “enhanced” PD models, which are becoming a hallmark of systems pharmacology. Encapsulated within this drug action aspect is the inherent polypharmacology of most of the “targeted” therapeutics, which, although specifically designed to inhibit a particular target kinase, usually show a broad spectrum of inhibition across kinases to varying degrees.
Researchers see such models as providing 2 potentially transformative capabilities. First, they can be used to generate hypotheses for potentially effective new drug combinations and treatment regimens, including dose and timing, tailored to individual patients. A simulation example of how one might perform such a task is illustrated by a recent paper by the group107 in which they propose how to take a “canonical” computational model of the disease biochemistry, adjust it to represent (to the best of one's ability) what is known about the particular patient, analyze the model to understand points of fragility in the biochemical network, which correspond to good drug targets, and then simulate millions of drug combinations, doses, and timings to predict the best regimens.
Second, model analysis can suggest which patients might respond or not respond to a drug. Such information could be used to, for example, include and exclude patients for clinical trials, evaluate the likelihood of a drug's efficacy (or, with the right model, toxicity) early in the pharmaceutical development pipeline, or suggest indications and contraindications for approved drugs. The Birtwistle and Gallo groups have performed a preliminary analysis of this sort using gene expression data from 16 GBM patients in The Cancer Genome Atlas, to “personalize” a kinetic model of GBM-relevant signal transduction using the measured gene expression profiles (Fig. 14). The response of each of these 16 different patients was simulated after what would be a 14-day regimen of an investigational drug, ON123300, a kinase inhibitor with known affinities for mTOR, cyclin-dependent kinase 4, platelet derived growth factor receptor, and FGFR. By simulating the 14-day drug regimen using a traditional PK model and looking at key signaling endpoints, active Akt and ERK and a marker of apoptosis, cleaved poly(ADP-ribose) polymerase (PARP), a range of dynamic responses was predicted to drug administration across this simulated 16-patient population. The drug was predicted to uniformly inhibit Akt signaling but paradoxically activate ERK signaling to differing degrees in different simulated patients (different colors). The prediction of increased ERK signaling was independently validated in cell line studies.108 Cell death responses (cleaved PARP) were predicted for most patients but at markedly different times during the simulated regimen. Interestingly, the simulated patients with the largest ERK activation responses were predicted to not undergo a cell death response within the regimen time window, suggesting that they would not be suited for treatment with ON123300. These results demonstrate how systems pharmacology models can take as input diverse patient variables and then make specific predictions about response to therapy. With the ability to simulate a patient's response assuming a single genomic context, extension to multiple genomic contexts to account for intratumoral heterogeneity is straightforward. When coupled with considerations of drug distribution and dynamics, such mechanistic modeling approaches are potentially suited for contributing to improved ability to treat GBM patients clinically, in terms of both using existing drugs more intelligently and developing new drugs.
Fig. 14.
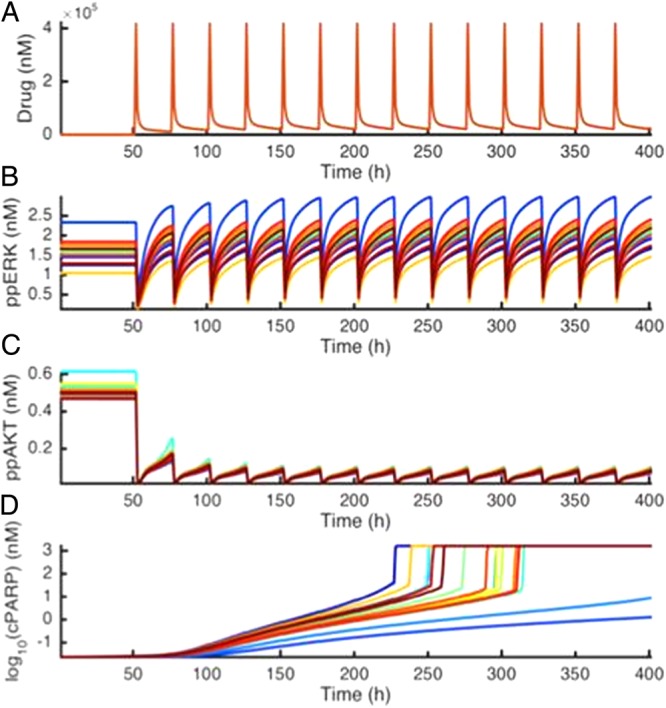
Simulation results from The Cancer Genome Atlas (TCGA) patient-tailored GBM cell signaling model. The mRNA expression for 16 patients from TCGA was used to set initial protein concentrations. Response to an investigational drug, ON123300, with multiple cellular targets mTOR, FGFR, platelet derived growth factor receptor, cyclin-dependent kinase 4 was simulated. (A) Drug concentration in plasma over time. (B) Phosphorylated pancreatic (pp)ERK levels over time. Each color represents a different patient. (C) ppAkt levels over time. (D) Cleaved PARP levels over time. Note the log scale.
Of course, much of the data used to develop and constrain such models are based on in vitro cell culture work, and such results do not always translate to mouse models, much less to the clinical arena. Thus, one can expect the earliest implementations of such systems pharmacology approaches in clinical settings to be tenuous at best. Yet, as with any computational modeling exercise, the model is ever improved by challenging it further with new datasets and situations, to understand where the model is inadequate. The lack of viable alternatives for dealing with the complexity and heterogeneity of brain tumors (and for that matter most cancers) to propose effective treatment strategies makes the investment in and development of such systems pharmacology approaches worthwhile. It is understood that models, at some point, require actual clinical validation data.
Clinical Paths for Drug Study and Regulatory Approval
To many in pharmaceutical and biotech industries, the path available for FDA drug approval and the market size for an anticancer drug define whether they will spend funds and research and development time on a new drug. This has been especially problematic for primary CNS tumors that represent about 1% of cancer. Metastatic CNS tumors are interrelated with the primary cancer, so are a mixed bag for the companies that may already be developing drug therapies for non-small-cell lung carcinoma or breast cancer. In any case, the impetus to develop CNS anticancer therapies is not great, although once CNS target genes shared with other tumors are identified, the process may be intensified.
Speakers at the conference addressed consensus clinical and radiographic endpoints, current FDA guidance for drug approval, and newer statistical approaches that may have relevance to new targeted therapies, especially those coupled to biomarkers. From the outset it was apparent that there is no singular approach to clinical study endpoints other than to improve OS. For some tumors, improvement in tumor vessel leakiness can be imaged with gadolinium contrast for MRI and iodinated contrast CT scans and quantified by 2-dimensional measurements in a manner akin to the use of Response Evaluation Criteria In Solid Tumors criteria and the methodologies used for extracranial cancers.109,110 For tumors that do not contrast-enhance, it can be more difficult to image tumor progression and response to therapy using traditional imaging modalities. In addition, many tumors that grow in the CNS do not respond quickly or dramatically to therapy, and/or dead tumor cell removal is so slow that it can be difficult to see response to treatment over periods of months. It is uncommon to see what is called a complete response for primary CNS tumors, yet intracranial tumors that populate the Virchow Robin spaces, like primary CNS lymphoma, can frequently show complete responses. Slow dead-cell removal of primary infiltrative gliomas has been shown to occur after nitrosourea chemotherapy in rodent models and is consistent with clinical experience and the infrequency of gliomas responding to chemotherapy with complete or even partial responses.111 In addition, postsurgical changes, radiation damage, and even chemotherapy damage can further complicate the assessment of response and progression by MRI and CT scan. These observations led to methodologies that evolved with technical advances from radionuclide brain scans to CT and MRI scans over the years.112–116
Endpoints for Clinical Trials
Patrick Wen discussed the evolving process of evaluating patients to ensure that treatment effects can be defined and quantified by their efficacy and toxicity. The measures used, taken from the start of a treatment, are OS, PFS, clinical response, and, to a lesser extent, clinical response, performance status, and use of supportive treatments like corticosteroids. Aside from death, certainty regarding progression and response requires reliable and reproducible radiographic imaging. To this end, the uniqueness of the CNS complicates imaging studies and the definition of progression and response. The intactness of the BBB can be compromised by ligands such as VEGF that will modulate the inter-endothelial tight junctions, cause edema, and initiate new non-CNS capillaries that leak contrast dyes. These events can occur weeks to months after radiation therapy and lead to subacute radiation effect, or what is today called “pseudoprogression,” a term used to distinguish it from true tumor progression. Fortunately the process responds partially to corticosteroids and bevacizumab, a humanized antibody that sequesters circulating VEGF, thereby reducing CNS edema and new leaky small vessels. Methodology and criteria established over the years allow patients with pseudoprogression to remain on study until it is certain that the radiographic changes are due to pseudoprogression rather than tumor growth.
Another complication of therapy directed against VEGF or its endothelial receptors by drugs like bevacizumab is to improve the MRI without causing significant tumor cell kill. For studies with these agents, special imaging techniques are sometimes needed to evaluate the efficacy of drugs used in combination with bevacizumab or VEGF-receptor drugs.
Wen and colleagues are developing guiding principles for clinical trials using consensus committee approaches of the Response Assessment in Neuro-Oncology (RANO) Working Group. For high-grade infiltrative tumors, the basic criteria follow:
Product of the maximal cross-sectional enhancing diameters will be used to determine the size of the contrast-enhancing lesions.
Enhancing and nonenhancing tumor areas are evaluated (at present it is easier to measure the contrast lesion than the nonenhancing region).
Response (complete, partial) requires confirmatory scan at least 4 weeks after the first response seen.
Steroid dose and clinical status must be recorded.
RANO criteria for clinical trials further stipulate and define what constitutes measurable and nonmeasurable disease, provide minimal criteria for entry into clinical trials, exclude patients for studies of recurrent disease unless the study is longer than 12 weeks after chemoradiation to minimize pseudoprogression, define durability of response, and provide guidance for patients with equivocal changes by allowing them to continue treatment and repeating MRI in 4 weeks. These are all sensible criteria for contrast-enhancing tumors. Additionally, these criteria are acceptable to the FDA for contrast-enhancing CNS tumors.
For non-contrast-enhancing tumors, size remains important, as does the appearance of new contrast enhancement. RANO criteria are in development for low-grade infiltrative gliomas117 and will be developed for mid-grade infiltrative gliomas in the future. There is a perception among neuro-oncologists, neuroradiologists, and neurosurgeons that the field will need new and more sophisticated (automated) neuroimaging algorithms to better quantitate response and progression for tumors whose growth is defined by T2 hyperintensity rather than contrast enhancement. Some approaches under investigation are contrast-enhanced T1-weighted digital subtraction maps with volumetric analyses, T2 and/or T2 fluid attenuated inversion recovery (FLAIR) volumetric analyses, diffusion, dynamic susceptibility contrast and dynamic contrast enhanced MRI, and maybe even amino acid PET. In addition, for large clinical trials it will be helpful to standardize imaging protocols across multicenter trials, employ central readers, and use automated/semi-automated segmentation and volumetrics.
FDA Regulatory Framework and Guidance
Joohee Sul discussed the charges of FDA's Center for Drug Evaluation and Research, Office of Hematology and Oncology Products, to define appropriate clinical trial endpoints and approval pathways for oncology drugs, addressing the specific challenges of each disease, in this case, neuro-oncology.
Regular marketing approval for oncology drugs requires substantial evidence of effectiveness from adequate and well-controlled clinical trials. This is accomplished by providing direct evidence that a drug or biologic confers clinical benefit to patients, which is defined as improvement in how the patient “functions, feels, or survives.” In 1992, subpart H was added to the New Drug Application regulations to allow accelerated approval of therapeutic agents for diseases that are serious or life-threatening and in cases for which the new drug appears to provide benefit over standard therapy. Accelerated approval can be granted on the basis of a surrogate endpoint that is reasonably likely to predict clinical benefit. Postmarketing confirmatory trials are required to demonstrate that treatment with the drug is in fact associated with clinical benefit, and such trials are generally expected to be under way at the time that accelerated approval is granted.
The FDA Safety and Innovation Act of 2012 introduced the Breakthrough Therapy Designation (BTD) pathway, intended to facilitate and expedite development and review of new drugs to address an unmet medical need in the treatment of a serious or life-threatening condition. In order to be granted BTD, there must be preliminary clinical evidence that the drug may demonstrate substantial improvement over existing therapies for one or more clinically significant endpoints. Since its inception in 2012, the number of BTD applications has risen dramatically from 2 in 2012 to 97 in 2014.
Oncology Clinical Trial Endpoints
OS is the “gold standard” for measurement of direct clinical benefit in oncology clinical trials. While OS as an outcome measure is not subject to bias, it often requires a large sample size and a long follow-up period. PFS is defined as the time from randomization to progressive disease or death and is able to capture treatment effect, as it is not obscured by subsequent therapies administered after disease progression and before death. Measurement of ORR includes partial and complete responses and should also capture duration of response. Finally, clinical outcomes assessments (COAs) measuring patients' symptoms or function are valuable as they emphasize clinical benefit from the patient's perspective.
Neuro-oncology Endpoints
Drug approvals in neuro-oncology have been few and far between. Nitrosoureas were approved based on ORR in the 1970s for primary and metastatic brain tumors. Temozolomide received accelerated approval for treatment of refractory AA in 1999, based on durable ORR, and for first-line treatment of GBM in 2005, based on improved OS demonstrated in confirmatory postmarketing trials. More recently, bevacizumab has received approval for recurrent GBM based on durable ORR, although the recent results of 2 large randomized trials did not confirm benefit in the newly diagnosed population.
Determining the most appropriate clinical outcome measures for clinical trials in neuro-oncology has been challenging, more so now in the era of immunotherapies and molecularly targeted agents that may confound assessment of ORR and PFS as determined by neuroimaging. The FDA has participated in the efforts of the neuro-oncology community to modify imaging response criteria and standardize imaging, as well as discussions on the use of COA in clinical trials. Regardless of the endpoints selected and how they are measured, it is paramount to keep in mind that ultimately, treatments for brain tumors must demonstrate a direct clinical benefit in patients, as measured by how, as stated, the patient functions, feels, or survives.
Statistical Considerations
Donald Berry discussed innovative statistical approaches being used for the study of breast cancer, associated biomarkers, and newer agents as an example of how statistical nuances of these approaches might be incorporated into future neuro-oncology trials. The discussion that follows also reflects a recent publication of his.118 Clinical trials for CNS tumors have been limited in scope for a number of reasons, not the least of which is a dearth of agents to evaluate. Most studies today seek to use resources to answer minor questions with regard to the timing of very few therapies—radiation, temozolomide, CCNU, the PCV combination, and bevacizumab. These clinical trials address scientific questions that do not help to unravel the nature of complex diseases such as gliomas and medulloblastoma. In addition, some studies represent backward steps because they occupy resources that would better serve if allocated to create new therapies. In general in medical oncology, where there are more drugs to contemplate, many clinical trials are attempting to use molecular features to parse treatments and narrow treatment populations to those with the highest likelihood of potential treatment benefit. To an extent, this has been advocated in glioma and medulloblastoma therapy, but sadly there are no alternative agents to use if a patient has biomarkers that indicate a poor prognosis using current available treatments. It is difficult to place an objective value on giving patients hope, albeit not necessarily evidence based.
As we continue to learn more about causation and drivers of CNS cancers as well as non-CNS cancers, we expect a large number of opportunities for creating novel drug combinations, yet we are stymied because the majority of anticancer drugs available today are not ideal for use against infiltrating CNS tumors, and the potential combinations make for an essentially infinite number of therapies.
Possibly because of clinical naiveté, we take the same clinical trial approach to evaluate all experimental therapies and frequently fail to experiment with the design of the clinical trial itself. This approach may be detrimental especially given the rarity of many of the glioma and medulloblastoma tumors that are likely to require unique drug and treatment approaches and that have a short survival. Focusing on large and long-duration (5–10 year) clinical trials makes therapies more expensive and often unfeasible because of survival considerations, and will ultimately delay their availability to patients. As we learn more about the molecular, genetic, and protein biology of CNS cancers, we expect it will lead to even smaller subsets of patients that might benefit and will require more nuanced clinical trial and statistical approaches and, possibly, utilization of staged regulatory approaches to encourage pharmaceutical participation. A stumbling block to progress has long been the small numbers of patients who choose to enroll or qualify for enrollment in clinical trials because of criteria for inclusion and exclusion in such trials. One of those exclusions has been presence of brain metastases, although some trials now allow participation by patients whose metastases are controlled. Some early-stage clinical trials have dropped brain metastasis as an exclusion criterion.
Hypothesis testing is the focus of traditional clinical trials, including control of type I error rate and statistical power. Suppose investigators propose a clinical trial comparing an experimental arm with a control and they want to be able to detect a 25% reduction in the death hazard with 90% power and a 5% 2-sided type I error rate. For a median OS of 3 years, accrual of 4 patients/month, and minimum follow-up of 3 years, 650 patients would be required and, maybe a decade later, final results might be available. No pharmaceutical company will support such a trial in glioma or medulloblastoma. Even if PFS is the endpoint rather than OS, most regulatory agencies would require powering the study to OS anyway so that study size and duration would change little. Imagine that scenario using 2 new drugs and then understand why it is so difficult to develop new drugs for CNS cancers.
Future trials in CNS cancers must explicitly consider disease prevalence. They must also consider the rapidity of advances in biology and the rate at which alternative therapies are being developed. This will mean smaller, shorter, and more focused trials. The new clinical trial paradigm should have the goal of delivering good medicine to patients who have or will have the narrowly defined disease in question. Randomization will continue to play a role, albeit one that is more refined. It is likely that the role of hypothesis testing will at most be ancillary. No one knows what that future will look like and what drug development and drug regulation will be like, but change is, as always, inevitable.
Newer clinical-trial designs that may have value going forward are platform trials and basket trials. Both approaches attempt to merge research with clinical practice. It is hoped that with change and new insights, clinical trials will become smaller, as large clinical trials in narrowly defined diseases are impractical. This is especially true in glioma and medulloblastoma, where total subsets in the US per year could be as small as 50 to 1000 patients. In addition, unlike many cancers, infiltrative glioma and, to a lesser extent, medulloblastoma tumor patients do not easily or frequently achieve complete or partial response. Part of this situation is caused by the slow pace of dead-cell removal in the brain. MRI artifacts of surgery and radiation therapy further complicate the true evaluation of response. For these reasons, clinicians rely a great deal on the failure of tumor to progress (stable disease or no change) and tumor progression (progressive disease). Sometimes response can take 6 months or longer to appreciate by CT or MRI.
The expectation is that the business model for pharmaceutical companies and the statistical design of clinical trials will be different. Donald Berry expects that type I and type II error probabilities will fade out of existence for many cancer clinical trials and be replaced by the delivery of effective therapy for patients who have specific cancer characteristics.
Accelerating Drug Discovery and Development for Brain Tumors
This section of the White Paper is based on the discussion among conference speakers and attendees in response to the question, What could be done to accelerate CNS anticancer drug discovery and development for primary and secondary CNS tumors?.
The conference focused on 4 key areas which, when adequately addressed, will help bring effective treatments to patients with primary and secondary CNS tumors.
Identification of robust drug targets within specific patient populations for which investment in clinical trials will not be a barrier
Appreciation by pharmaceutical and medicinal chemists that methods are available to enhance the transport of CNS anticancer agents into brain to reach infiltrating tumor in the CNS
Appreciation of and methods to overcome patient tumor heterogeneity and resistance to treatment within primary tumors and metastases so that drug therapy is effective in all tumor regions
Creation of a specific lexicon and drug development pipeline focused on drugs specifically intended for use in brain tumors, thereby establishing a consistent foundation spanning academia and industry. The hope is that this will lead to more effective and safer drugs with increased therapeutic indices resulting in higher free brain drug concentrations with fewer off-target effects and better chances for achieving antitumor efficacy
As was made abundantly clear in the first few sessions of this symposium, the lack of effective CNS anticancer drugs is due primarily to the inability of drugs to penetrate into brain tumors with concentrations of drug sufficient to achieve an adequate duration of action. Most contemporary clinical trials in neuro-oncology have systematically neglected to perform PK and/or PD studies in intracranial tumors, either in rodents or in humans. That is not to say that some have not attempted to measure drug levels and their effect(s) on target molecules in human gliomas and metastatic tumors at the time of surgery. The problem is that single measurements provide limited information, and without carefully obtained and curated drug effects within tumor cells at varying distances from leaky and intact capillary regions, little really can be learned from a PK/PD perspective. Thus, in most cases, we really do not know whether the negative outcomes of the studies can be attributed, at least in part, to defective penetration of the drug through the BTB or to target binding. Many attendees felt such studies should always be performed in normal brain, and when possible, relevant intracranial orthotopic models before clinical studies are undertaken. Similarly, any clinical-study testing of new drugs in neuro-oncology patients should always be preceded and/or accompanied by phase 0 studies to determine whether that particular drug is able to reach therapeutic concentrations in human CNS tumors.
Numerous examples were presented in which a drug showed promise in a preclinical xenograft or orthotopic cell line model, but because such models did not recapitulate the inherent BBB biology in real CNS tumors, they tended to overestimate the effectiveness of the studied drugs. Moreover, it would seem that at least some chemotherapeutic agents that are currently effective against non-CNS tumor types might have been selected specifically for lack of brain penetration to prevent neurotoxicity. Thus, major efforts are needed to learn how to engineer, from a medicinal chemistry standpoint, current and new drugs to improve brain uptake. In addition, when new brain penetrant agents are in development, thought must be given to their potential for creating neurotoxicity.
At odds with other areas of clinical oncology, thus far, the targeted therapies tested against malignant brain tumors have resulted in failure. There are likely many reasons for this outcome. However, a major shortcoming in neuro-oncology clinical studies is that they are usually performed without a full understanding of the value of the targeted molecular alterations for tumor maintenance. Furthermore, tumor heterogeneity (hypoxia, cell cycle status, metabolic factors, enzyme activation, phosphorylation state, treatment resistance, and other reasons) can be an inconvenient limitation for any targeted agent, even assuming that an addicting role of a driver/genomic alteration exists in a subpopulation of tumor cells harboring a particular alteration/mutation. Along these lines, the screening and personalized validation paradigm presented by the Iavarone group, focused upon the FGFR-TACC fusion protein in patients with isocitrate dehydrogenase–wild-type glioma, provides a guide for how to combine the comprehensive characterization of strength of evidence of oncogenic activity and size of anticipated targeting effect for the selection of a “druggable” genetic alteration in GBM patients.79 This paradigm may or may not lead to a drug of value to treat infiltrative gliomas, and only time will tell the value of this approach.
A number of contributors believe that an approach to drug discovery for CNS cancers would likely be best served by incorporating newer studies with an appreciation for drug-target kinetics (a case of residence time) used to inform the development of novel CNS anticancer drugs. Because this aspect of drug discovery is relatively unexplored, it would be helpful to gather data on lead molecules that have come out of anticancer drug discovery programs, determine their kinetic profiles, and correlate the target binding kinetics of these compounds with their cellular and in vivo anticancer activity. These studies can and probably should be extended to related analogs in the compound series that resulted in identifying the drug lead for further clarification of the following:
Knowledge of the structural factors that control the on and off rates for binding of the drug to the target. The resulting structure-kinetic relationship could ultimately be used to design compounds with improved drug-target residence times.
The development of correlations between the thermodynamics and kinetics of drug-target interactions and the drug PD. To flesh out these correlations, cellular activity data will need to be obtained under nonequilibrium conditions (ie, drug wash-out experiments). In principle, this may result in reprioritization of the top leads selected for in vivo efficacy studies.
A number of contributors also opined that a universal academic and industrial failing over the years has been the absence of informative regional PK studies of potential CNS anticancer agents that can be used to define biodistribution and binding as well as impact of tumor heterogeneity on drug PD. Most attendees believe that it remains important to study promising radiolabeled compounds to determine their biodistribution in animal tumor models. Although more difficult, it would also be helpful to study drug biodistribution in humans using the noninvasive modalities of PET and MRI. These studies would, for example, provide valuable information for correlating drug PK and PD by determining drug concentration at the target site. Approaches such as these have been attempted to correlate the extent of target engagement with drug PD of the BTK inhibitor CC-292 (Celgene Avilomics Research). The data would also be used to populate mechanistic PK/PD models to predict efficacious drug doses in humans.
It is universally accepted that tumors are highly heterogeneous in anatomical and morphological features as well as genomic driver mutations, protein expression, and protein activity. From a PK perspective, tumor cells at varying distances from capillaries will receive variable amounts of free drug and hence will experience heterogeneity in drug distribution and, therefore, drug activity. Depending on where in the tumor one looks, one finds different genomic driver mutations—for example, PTEN loss in biopsy #1, but PTEN wild type and EGFR amplification in biopsy #2 taken from a different location. Moreover, it would be naïve to assume that such genomic parameters are static; rather, they are more likely to dynamically evolve and adapt. Thus, the notion that a patient's tumor can be characterized by a single set of driver mutations and that such genomic data can inform therapy is too simplistic. “Liquid biopsies,” such as characterizing circulating tumor DNA, may 1 day be used in place of tissue biopsies and are being used in addition to them to inform such tumoral dynamics. One needs to consider that a wide range of mutations is present throughout the tumor, and thus drug sensitivity is a highly heterogeneous and dynamic reaction of tumors to treatment. Thus, even if one observes a potentially “sensitizing mutation,” for example, EGFR copy number amplification for an EGFR kinase inhibitor, trials may well fail due to the presence of cells with different genomic drivers and, thus, diverse drug sensitivities. When one couples the likelihood that not all cells in the tumor are sensitive to the same drugs with the difficultly of delivering the drug to the brain in appreciable concentrations, the challenges are steep. Optimistically, it would seem that the genomic landscape of tumors is far from infinite, as has been revealed by the now impressive amount of genomic data (eg, The Cancer Genome Atlas) that have been collected not only from primary and secondary CNS tumors, but from all tumor types. A handful of major driver mutations recurrently show up in certain tumor types. A major task, therefore, is first to understand which combinations of drugs effectively “cordon” the tumor in the genomic sense, meaning that biologically the tumor has “nowhere to run,” and every possible tumor cell is sensitive to the combination of drugs given. This is a challenge for systems biology and systems pharmacology. Then, we must design combinations of drugs that can penetrate the BBB effectively, all likely within a narrow therapeutic window. This is a challenge for traditional medicinal chemistry and clinical pharmacology.
When it comes to target identification, the adage referred to earlier, “if you do not know where you are going, any road will get you there,” rings true. To develop new classes of drugs for brain cancers, some scientists prefer samples from patient tumors, and others would like to see more work using whole animal approaches as well as fly-based approaches. Phenotypic screens provide different chemical leads than those arising from biochemical or single-target approaches, and these early starting points are intriguing to pursue in the development of small-molecular-weight drugs.
Also offered was a philosophy that oncology and biologics departments and CNS departments should work together more closely and question the perception that it is best to avoid brain targets because biologic agents cannot enter the brain. This belief has been countered by progress made in developing modifications to biologic agents, such as mAbs that confer brain permeability.
One suggestion voiced by participants is that pharmaceutical companies could develop most anticancer agents to penetrate the BBB and avoid efflux pumps. The major rebuttal to that belief is not that these drugs would not work against other non-CNS cancers, but rather that this approach would lead to unintended neurotoxicity. In some cases, this might be a possibility, but many behavioral animal models could be used to determine whether individual drugs would adversely affect neurological function. MRI and pathology tissue studies could predict whether these drugs could produce white matter or other effects on brain vessels, neurons, and glia.
A number of academic investigators proposed an ambitious agenda focusing on National Cancer Institute (NCI)–based funding for a CNS cancer consortium grant program. The concept would be to bring together teams of clinicians and basic scientists to tackle clinical translation of existing data and new candidate therapeutic approaches (including new cytotoxic agents, precision targets, and immunotherapy). On the other hand, academic/industry partnerships to create new anticancer drugs and exploit new cellular targets are well accepted, with a long history of programs to do just that. Examples of this are the NCI synthetic drug contracts of the 1960s and 1970s, as well as the NCI-sponsored National Cooperative Drug Discovery Group program, which was initiated in 1982, and of which one of us (V.A.L.) was the first grant recipient to create inhibitors of the protein tyrosine kinase in c-Src. Without doubt, the future of CNS anticancer drug discovery and development will certainly require a greater number of financial incentives than presently exist, since CNS cancer accounts for only 1% of cancers and these rare CNS cancers are plagued by some of the most difficult PK/PD conundrums and the most elusive druggable targets. It will certainly require academic/industry partnerships to offset research and development costs and to stimulate new thinking and approaches to drug development.
From the Clinical Paths Discussion, some ideas and concerns were presented that may be relevant to this section. The majority of clinical trials today are for patients with rapidly growing GBM, and few, if any, are designed for more slowly growing low- and mid-grade gliomas (astrocytoma, oligodendroglioma, oligoastrocytoma, ependymoma, and other variants). The only caveat to this is that the first drugs approved for anaplastic gliomas were alkylating agents (BCNU, CCNU) that were approved on the basis of ORR. Parts of the discussion also focused on the problem of designing single and drug combination studies for tumors that grow more slowly than GBM and might primarily prevent tumor growth rather than leading to short duration tumor cell kill. For the mid- and lower-grade gliomas, this problem is compounded by the financial risk undertaken by pharmaceutical companies that require financial incentives to develop new drugs and bring them to market. Since studies in low- and mid-grade gliomas can take 5–10 years to attain OS data, the financial incentive is low based on remaining patent protection or orphan drug exclusivity.
Some opined that without previous regulatory approvals and a paved regulatory path to follow, it is too hard (risky) to develop a drug for CNS tumors, whereas others believe that this approach is incorrect. In addition, regulatory agencies will need to recognize that stable disease and clinical response rates for nonenhancing tumors represent disease control as well as valid endpoints, an important concept in the brain, where space is limited and tumor growth can easily compromise healthy tissue—so stopping tumor progression becomes an important clinical outcome, especially for the patient with a CNS tumor.
Furthermore, as many participants expect that it will require 2 to 4 selective target inhibiting drugs working in concert to truly control CNS tumor growth and extend OS, the structure of clinical studies and acceptable regulatory endpoints that will, on the one hand, convince clinicians and the regulatory agencies that the drug(s) are worthy of approval and, on the other hand, justify the large financial cost to pharmaceutical companies to develop new drugs to treat rare tumors are daunting and unresolved issues at this time. In many ways, these problems are also societal in nature, as those with money to invest are loath to do so if the investing horizon is many years away, with no assurance of profit. Improving quality and length of life for CNS cancer patients has been and will likely continue to be less important than making a financial profit in a short period of time. Until that perspective changes and new incentives make it economically feasible to develop these drugs, we are liable to see very little movement in new CNS anticancer drug discovery/development.
Funding
This manuscript was the product of the first CNS Anticancer Drug Discovery and Development Conference, which was supported by Genentech (a member of the Roche Group), the American Brain Tumor Association, the Society for Neuro-Oncology, Bristol-Myers Squibb, Celgene, and Novartis.
Acknowledgments
The authors would like to thank Joann Aaron for her contribution and thoughtful editing of this White Paper.
Conflict of interest statement. None of the authors were paid to write this article. Many of the authors are supported by the companies and universities they work for, which might be engaged in anticancer drug research. Because of this employment and potential conflicts of interest, some of the coauthors did not contribute to the Accelerating Drug Discovery and Development for Brain Tumors sections of the paper that focus on directions that the pharmaceutical industry, academia, and the FDA might need to take in the future to move the field covered by the conference forward.
References
- 1.Ostrom QT, Gittleman H, Farah P, et al. CBTRUS Statistical Report: primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013;15(suppl 2):ii1–ii56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patchell RA. The management of brain metastases. Cancer Treat Rev. 2003;29(6):533–540. [DOI] [PubMed] [Google Scholar]
- 3.Hutter A, Schwetye KE, Bierhals AJ, McKinstry RC. Brain neoplasms: epidemiology, diagnosis, and prospects for cost-effective imaging. Neuroimaging Clin N Am. 2003;13(2):237–250, x-xi. [DOI] [PubMed] [Google Scholar]
- 4.Mehta M, Vogelbaum MA, Chang S, et al. Neoplasms of the central nervous system. In: DeVita VTJ, Lawrence TS, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2011:1700–1749. [Google Scholar]
- 5.Kleihues P, Burger PC, Scheithauer BW. The new WHO classification of brain tumours. Brain Pathol. 1993;3(3):255–268. [DOI] [PubMed] [Google Scholar]
- 6.Prados MD, Larson DA, Lamborn K, et al. Radiation therapy and hydroxyurea followed by the combination of 6-thioguanine and BCNU for the treatment of primary malignant brain tumors. Int J Radiat Oncol Biol Phys. 1998;40(1):57–63. [DOI] [PubMed] [Google Scholar]
- 7.Levin VA, Hess KR, Choucair A, et al. Phase III randomized study of postradiotherapy chemotherapy with combination alpha-difluoromethylornithine-PCV versus PCV for anaplastic gliomas. Clin Cancer Res. 2003;9(3):981–990. [PubMed] [Google Scholar]
- 8.Levin VA, Hess KR, Choucair AK, et al. Final report for evaluable patients treated on DM92–035, phase III randomized study of post-irradiation PCV versus DFMO-PCV, for anaplastic gliomas (AG). Neuro Oncol. 2012;14(Supplement 6):vi74. [Google Scholar]
- 9.Bello MJ, Leone PE, Vaquero J, et al. Allelic loss at 1p and 19q frequently occurs in association and may represent early oncogenic events in oligodendroglial tumors. Int J Cancer. 1995;64(3):207–210. [DOI] [PubMed] [Google Scholar]
- 10.Kraus JA, Koopmann J, Kaskel P, et al. Shared allelic losses on chromosomes 1p and 19q suggest a common origin of oligodendroglioma and oligoastrocytoma. J Neuropathol Exp Neurol. 1995;54(1):91–95. [DOI] [PubMed] [Google Scholar]
- 11.Smith JS, Alderete B, Minn Y, et al. Localization of common deletion regions on 1p and 19q in human gliomas and their association with histological subtype. Oncogene. 1999;18(28):4144–4152. [DOI] [PubMed] [Google Scholar]
- 12.Jenkins RB, Curran WB, Scott C, Cairncross G. Pilot evaluation of 1p and 19q deletions in anaplastic oligodendrogliomas collected by a national cooperative cancer treatment group. Am J Clin Oncol. 2001;24(5):506–508. [DOI] [PubMed] [Google Scholar]
- 13.Lewandowska MA, Furtak J, Szylberg T, et al. An analysis of the prognostic value of IDH1 (isocitrate dehydrogenase 1) mutation in polish glioma patients. Mol Diagn Ther. 2014;18(1):45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabha N, Knobbe CB, Maganti M, et al. Analysis of IDH mutation, 1p/19q deletion, and PTEN loss delineates prognosis in clinical low-grade diffuse gliomas. Neuro Oncol. 2014;167:914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith JS, Tachibana I, Passe SM, et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst. 2001;93(16):1246–1256. [DOI] [PubMed] [Google Scholar]
- 16.Jin W, Xu X, Yang T, Hua Z. p53 mutation, EGFR gene amplification and loss of heterozygosity on chromosome 10, 17 p in human gliomas. Chin Med J (Engl). 2000;113(7):662–666. [PubMed] [Google Scholar]
- 17.Gil-Benso R, Lopez-Gines C, Benito R, et al. Concurrent EGFR amplification and TP-53 mutation in glioblastomas. Clin Neuropathol. 2007;26(5):224–231. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki H, Zlatescu MC, Betensky RA, et al. PTEN is a target of chromosome 10q loss in anaplastic oligodendrogliomas and PTEN alterations are associated with poor prognosis. Am J Pathol. 2001;1591:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–1354. [DOI] [PubMed] [Google Scholar]
- 20.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. [DOI] [PubMed] [Google Scholar]
- 21.Paterson E, Farr RF. Cerebellar medulloblastoma: treatment by irradiation of the whole central nervous system. Acta Radiol. 1953;39(4):323–336. [DOI] [PubMed] [Google Scholar]
- 22.Packer RJ, Sutton LN, Elterman R, et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg. 1994;81(5):690–698. [DOI] [PubMed] [Google Scholar]
- 23.Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: a Children's Cancer Group study. J Clin Oncol. 1999;17(7):2127–2136. [DOI] [PubMed] [Google Scholar]
- 24.Zeltzer PM, Boyett JM, Finlay JL, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children's Cancer Group 921 randomized phase III study. J Clin Oncol. 1999;17(3):832. [DOI] [PubMed] [Google Scholar]
- 25.Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leo A, Hansch C, Church C. Comparison of parameters currently used in the study of structure-activity relationships. J Med Chem. 1969;12(5):766–771. [DOI] [PubMed] [Google Scholar]
- 27.Leo A, Jow PY, Silipo C, Hansch C. Calculation of hydrophobic constant (log P) from pi and f constants. J Med Chem. 1975;18(9):865–868. [DOI] [PubMed] [Google Scholar]
- 28.Levin VA, Crafts D, Wilson CB, et al. Imidazole carboxamides: relationship of lipophilicity to activity against intracerebral murine glioma 26 and preliminary phase II clinical trial of 5-[3,3-bis(2-chloroethyl)-1-triazeno]imidazole-4-carboxamide (NSC-82196) in primary and secondary brain tumors. Cancer Chemother Rep. 1975;59(2 Pt 1):327–331. [PubMed] [Google Scholar]
- 29.Hansch C, Rockwell SD, Jow PY, et al. Substituent constants for correlation analysis. J Med Chem. 1977;20(2):304–306. [DOI] [PubMed] [Google Scholar]
- 30.Fink SI, Leo A, Yamakawa M, et al. The quantitative structure-selectivity relationship of anthracycline antitumor activity and cardiac toxicity. Farmaco [Sci]. 1980;35(12):965–979. [PubMed] [Google Scholar]
- 31.Hansch C, Leo A, Schmidt C, et al. Antitumor structure-activity relations. Nitrosoureas vs. L-1210 leukemia. J Med Chem. 1980;23(10):1095–1101. [DOI] [PubMed] [Google Scholar]
- 32.Denny WA, Cain BF, Atwell GJ, et al. Potential antitumor agents. 36. Quantitative relationships between experimental antitumor activity, toxicity, and structure for the general class of 9-anilinoacridine antitumor agents. J Med Chem. 1982;25(3):276–315. [DOI] [PubMed] [Google Scholar]
- 33.Hansch C, Bjorkroth JP, Leo A. Hydrophobicity and central nervous system agents: on the principle of minimal hydrophobicity in drug design. J Pharm Sci. 1987;76(9):663–687. [DOI] [PubMed] [Google Scholar]
- 34.Lober G, Geller K, Hanschmann H, et al. Cationic anticancer drugs and their modes of action. Physiologie. 1989;26(4):305–316. [PubMed] [Google Scholar]
- 35.Hansch C, Hoekman D, Leo A, et al. The expanding role of quantitative structure-activity relationships (QSAR) in toxicology. Toxicol Lett. 1995;79(1–3):45–53. [DOI] [PubMed] [Google Scholar]
- 36.Kurup A, Garg R, Hansch C. Comparative QSAR study of tyrosine kinase inhibitors. Chem Rev. 2001;101(8):2573–2600. [DOI] [PubMed] [Google Scholar]
- 37.Hansch C, Hoekman D, Leo A, et al. Chem-bioinformatics: comparative QSAR at the interface between chemistry and biology. Chem Rev. 2002;102(3):783–812. [DOI] [PubMed] [Google Scholar]
- 38.Hansch C, Steinmetz WE, Leo AJ, et al. On the role of polarizability in chemical-biological interactions. J Chem Inf Comput Sci. 2003;43(1):120–125. [DOI] [PubMed] [Google Scholar]
- 39.Hansch C, Leo A, Mekapati SB, Kurup A. QSAR and ADME. Bioorg Med Chem. 2004;12(12):3391–3400. [DOI] [PubMed] [Google Scholar]
- 40.Montgomery JA, Mayo JG. Quantitative structure-activity relationships in anticancer agents. Activity of selected nitrosoureas against a solid tumor, the Lewis lung carcinoma. J Med Chem. 1974;17(5):477–480. [DOI] [PubMed] [Google Scholar]
- 41.Levin VA, Kabra P. Effectiveness of the nitrosoureas as a function of their lipid solubility in the chemotherapy of experimental rat brain tumors. Cancer Chemother Rep. 1974;58(6):787–792. [PubMed] [Google Scholar]
- 42.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chemical Neuroscience. 2010;1(6):435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salphati L, Heffron TP, Alicke B, et al. Targeting the PI3K pathway in the brain–efficacy of a PI3 K inhibitor optimized to cross the blood-brain barrier. Clin Cancer Res. 2012;18(22):6239–6248. [DOI] [PubMed] [Google Scholar]
- 44.Agarwal S, Sane R, Oberoi R, et al. Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med. 2011;13:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen PY, Brandes AA. Treatment of recurrent high-grade gliomas. Curr Opin Neurol. 2009;22(6):657–664. [DOI] [PubMed] [Google Scholar]
- 46.Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2(1):3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pardridge WM. CNS drug design based on principles of blood-brain barrier transport. J Neurochem. 1998;70(5):1781–1792. [DOI] [PubMed] [Google Scholar]
- 48.Ajay, Bemis GW, Murcko MA. Designing libraries with CNS activity. J Med Chem. 1999;42(24):4942–4951. [DOI] [PubMed] [Google Scholar]
- 49.Vilar S, Chakrabarti M, Costanzi S. Prediction of passive blood-brain partitioning: straightforward and effective classification models based on in silico derived physicochemical descriptors. J Mol Graph Model. 2010;28(8):899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahar Doan KM, Humphreys JE, Webster LO, et al. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002;303(3):1029–1037. [DOI] [PubMed] [Google Scholar]
- 51.Golden PL, Pollack GM. Blood-brain barrier efflux transport. J Pharm Sci. 2003;92(9):1739–1753. [DOI] [PubMed] [Google Scholar]
- 52.Agarwal S, Uchida Y, Mittapalli RK, et al. Quantitative proteomics of transporter expression in brain capillary endothelial cells isolated from P-glycoprotein (P-gp), breast cancer resistance protein (Bcrp), and P-gp/Bcrp knockout mice. Drug Metab Dispos. 2012;40(6):1164–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uchida Y, Ohtsuki S, Katsukura Y, et al. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J Neurochem. 2011;117(2):333–345. [DOI] [PubMed] [Google Scholar]
- 54.Agarwal S, Hartz AM, Elmquist WF, Bauer B. Breast cancer resistance protein and P-glycoprotein in brain cancer: two gatekeepers team up. Curr Pharm Des. 2011;17(26):2793–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Agarwal S, Mittapalli RK, Zellmer DM, et al. Active efflux of Dasatinib from the brain limits efficacy against murine glioblastoma: broad implications for the clinical use of molecularly targeted agents. Mol Cancer Ther. 2012;11(10):2183–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pafundi DH, Laack NN, Youland RS, et al. Biopsy validation of 18F-DOPA PET and biodistribution in gliomas for neurosurgical planning and radiotherapy target delineation: results of a prospective pilot study. Neuro Oncol. 2013;15(8):1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parrish KE, Sarkaria JN, Elmquist WF. Improving drug delivery to primary and metastatic brain tumors: Strategies to overcome the blood-brain barrier. Clin Pharmacol Ther. 2015;97(4):336–346. [DOI] [PubMed] [Google Scholar]
- 58.Berens ME, Giese A. “… those left behind.” Biology and oncology of invasive glioma cells. Neoplasia. 1999;1(3):208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levin VA, Freeman-Dove M, Landahl HD. Permeability characteristics of brain adjacent to tumors in rats. Arch Neurol. 1975;32(12):785–791. [DOI] [PubMed] [Google Scholar]
- 60.Levin VA, Jochec JL, Shantz LM, et al. Tissue-based assay for ornithine decarboxylase to identify patients likely to respond to difluoromethylornithine. J Histochem Cytochem. 2004;52(11):1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ernestus RI, Rohn G, Schroder R, et al. Polyamine metabolism in brain tumours: diagnostic relevance of quantitative biochemistry. J Neurol Neurosurg Psychiatry. 2001;71(1):88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ernestus RI, Rohn G, Schroder R, et al. Polyamine metabolism in gliomas. J Neurooncol. 1996;29(2):167–174. [DOI] [PubMed] [Google Scholar]
- 63.Levin VA, Jochec JL, Shantz LM, Aldape KD. Relationship between ornithine decarboxylase levels in anaplastic gliomas and progression-free survival in patients treated with DFMO-PCV chemotherapy. Int J Cancer. 2007;121(10):2279–2283. [DOI] [PubMed] [Google Scholar]
- 64.Sharma J, Lv H, Gallo JM. Analytical approach to characterize the intratumoral pharmacokinetics and pharmacodynamics of gefitinib in a glioblastoma model. J Pharm Sci. 2012;101(11):4100–4106. [DOI] [PubMed] [Google Scholar]
- 65.Sharma J, Lv H, Gallo JM. Intratumoral modeling of gefitinib pharmacokinetics and pharmacodynamics in an orthotopic mouse model of glioblastoma. Cancer Res. 2013;73(16):5242–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Regina A, Demeule M, Tripathy S, et al. ANG4043, a novel brain-penetrant peptide-mab conjugate, is efficacious against HER2-positive intracranial tumors in mice. Mol Cancer Ther. 2014;141:129–140. [DOI] [PubMed] [Google Scholar]
- 67.Lu H, Tonge PJ. Drug-target residence time: critical information for lead optimization. Curr Opin Chem Biol. 2010;14(4):467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5(9):730–739. [DOI] [PubMed] [Google Scholar]
- 69.Walkup GK, You Z, Ross PL, et al. Translating slow-binding inhibition kinetics into cellular and in vivo effects. Nat Chem Biol. 2015;116:416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu H, England K, am Ende C, et al. Slow-onset inhibition of the FabI enoyl reductase from Francisella tularensis: residence time and in vivo activity. ACS Chem Biol. 2009;4(3):221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nature Reviews. 2011;10(4):307–317. [DOI] [PubMed] [Google Scholar]
- 72.Barkovich KJ, Hariono S, Garske AL, et al. Kinetics of inhibitor cycling underlie therapeutic disparities between EGFR-driven lung and brain cancers. Cancer Discov. 2012;2(5):450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu L, Di Paolo J, Barbosa J, et al. Antiarthritis effect of a novel Bruton's tyrosine kinase (BTK) inhibitor in rat collagen-induced arthritis and mechanism-based pharmacokinetic/pharmacodynamic modeling: relationships between inhibition of BTK phosphorylation and efficacy. J Pharmacol Exp Ther. 2011;338(1):154–163. [DOI] [PubMed] [Google Scholar]
- 74.Evans EK, Tester R, Aslanian S, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346(2):219–228. [DOI] [PubMed] [Google Scholar]
- 75.Weinstein EA, Liu L, Ordonez AA, et al. Noninvasive determination of 2-[18F]-fluoroisonicotinic acid hydrazide pharmacokinetics by positron emission tomography in Mycobacterium tuberculosis-infected mice. Antimicrob Agents Chemother. 2012;56(12):6284–6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463(7279):318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frattini V, Trifonov V, Chan JM, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45(10):1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337(6099):1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Di Stefano AL, Fucci A, Frattini V, et al. Detection, characterization and inhibition of FGFR-TACC fusions in IDH wild type glioma. Clin Cancer Res. 2015;2114:3307–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature. 2012;486(7401):80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wells SA, Jr., Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clinical Oncol. 2012;30(2):134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miles WO, Dyson NJ, Walker JA. Modeling tumor invasion and metastasis in Drosophila. Dis Model Mech. 2011;4(6):753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chien S, Reiter LT, Bier E, Gribskov M. Homophila: human disease gene cognates in Drosophila. Nucleic Acids Res. 2002;30(1):149–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adams MD, Celniker SE, Holt RA, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287(5461):2185–2195. [DOI] [PubMed] [Google Scholar]
- 85.Pandey UB, Nichols CD. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev. 2011;63(2):411–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rudrapatna VA, Cagan RL, Das TK. Drosophila cancer models. Dev Dynamics. 2012;241(1):107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Read RD. Drosophila melanogaster as a model system for human brain cancers. Glia. 2011;59(9):1364–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ding Y, Hubert CG, Herman J, et al. Cancer-Specific requirement for BUB1B/BUBR1 in human brain tumor isolates and genetically transformed cells. Cancer Discovery. 2013;3(2):198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hubert CG, Bradley RK, Ding Y, et al. Genome-wide RNAi screens in human brain tumor isolates reveal a novel viability requirement for PHF5A. Genes Dev. 2013;27(9):1032–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Toledo CM, Herman JA, Olsen JB, et al. BuGZ is required for Bub3 stability, Bub1 kinetochore function, and chromosome alignment. Dev Cell. 2014;28(3):282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Herman JA, Toledo CM, Olson JM, et al. Molecular pathways: regulation and targeting of kinetochore-microtubule attachment in cancer. Clin Cancer Res. 2015;212:233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Undheim EA, Sunagar K, Herzig V, et al. A proteomics and transcriptomics investigation of the venom from the barychelid spider Trittame loki (brush-foot trapdoor). Toxins (Basel). 2013;5(12):2488–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chouabe C, Eyraud V, Da Silva P, et al. New mode of action for a knottin protein bioinsecticide: pea albumin 1 subunit b (PA1b) is the first peptidic inhibitor of V-ATPase. J Biol Chem. 2011;286(42):36291–36296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Louis S, Delobel B, Gressent F, et al. Broad screening of the legume family for variability in seed insecticidal activities and for the occurrence of the A1b-like knottin peptide entomotoxins. Phytochemistry. 2007;68(4):521–535. [DOI] [PubMed] [Google Scholar]
- 95.Le Nguyen D, Heitz A, Chiche L, et al. Molecular recognition between serine proteases and new bioactive microproteins with a knotted structure. Biochimie. 1990;72(6–7):431–435. [DOI] [PubMed] [Google Scholar]
- 96.Moore SJ, Hayden Gephart MG, Bergen JM, et al. Engineered knottin peptide enables noninvasive optical imaging of intracranial medulloblastoma. Proc Natl Acad Sci USA. 2013;110(36):14598–14603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moore SJ, Leung CL, Norton HK, Cochran JR. Engineering agatoxin, a cystine-knot peptide from spider venom, as a molecular probe for in vivo tumor imaging. PLoS One. 2013;8(4):e60498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moore SJ, Cochran JR. Engineering knottins as novel binding agents. Methods Enzymol. 2012;503:223–251. [DOI] [PubMed] [Google Scholar]
- 99.Kimura RH, Jones DS, Jiang L, et al. Functional mutation of multiple solvent-exposed loops in the Ecballium elaterium trypsin inhibitor-II cystine knot miniprotein. PLoS One. 2011;6(2):e16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lahti JL, Silverman AP, Cochran JR. Interrogating and predicting tolerated sequence diversity in protein folds: application to E. elaterium trypsin inhibitor-II cystine-knot miniprotein. PLoS Comput Biol. 2009;5(9):e1000499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kimura RH, Levin AM, Cochran FV, Cochran JR. Engineered cystine knot peptides that bind alphavbeta3, alphavbeta5, and alpha5beta1 integrins with low-nanomolar affinity. Proteins. 2009;77(2):359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stroud MR, Hansen SJ, Olson JM. In vivo bio-imaging using chlorotoxin-based conjugates. Curr Pharm Des. 2011;17(38):4362–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Veiseh M, Gabikian P, Bahrami SB, et al. Tumor paint: a chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 2007;67(14):6882–6888. [DOI] [PubMed] [Google Scholar]
- 104.Correnti C, Richardson V, Sia AK, et al. Siderocalin/Lcn2/NGAL/24p3 does not drive apoptosis through gentisic acid mediated iron withdrawal in hematopoietic cell lines. PLoS One. 2012;7(8):e43696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bandaranayake AD, Correnti C, Ryu BY, et al. Daedalus: a robust, turnkey platform for rapid production of decigram quantities of active recombinant proteins in human cell lines using novel lentiviral vectors. Nucleic Acids Res. 2011;39(21):e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee MJ, Veiseh O, Bhattarai N, et al. Rapid pharmacokinetic and biodistribution studies using cholorotoxin-conjugated iron oxide nanoparticles: a novel non-radioactive method. PLoS One. 2010;5(3):e9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang XY, Birtwistle MR, Gallo JM. A general network pharmacodynamic model-based design pipeline for customized cancer therapy applied to the VEGFR pathway. CPT Pharmacometrics Syst Pharmacol. 2014;3:e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang X, Lv H, Zhou Q, et al. Preclinical pharmacological evaluation of a novel multiple kinase inhibitor, ON123300, in brain tumor models. Mol Cancer Ther. 2014;13(5):1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. [DOI] [PubMed] [Google Scholar]
- 110.Villaruz LC, Socinski MA. The clinical viewpoint: definitions, limitations of RECIST, practical considerations of measurement. Clin Cancer Res. 2013;19(10):2629–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rosenblum MK, Knebel KD, Vasquez DA, Wilson CB. Brain-tumor therapy. Quantitative analysis using a model system. J Neurosurg. 1977;46(2):145–154. [DOI] [PubMed] [Google Scholar]
- 112.Levin VA, Crafts DC, Norman DM, et al. Criteria for evaluating patients undergoing chemotherapy for malignant brain tumors. J Neurosurg. 1977;47(3):329–335. [DOI] [PubMed] [Google Scholar]
- 113.Macdonald DR, Cascino TL, Schold SC, Jr., Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8(7):1277–1280. [DOI] [PubMed] [Google Scholar]
- 114.Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: Response Assessment in Neuro-Oncology Working Group. J Clin Oncol. 2010;28(11):1963–1972. [DOI] [PubMed] [Google Scholar]
- 115.Vogelbaum MA, Jost S, Aghi MK, et al. Application of novel response/progression measures for surgically delivered therapies for gliomas: Response Assessment in Neuro-Oncology (RANO) Working Group. Neurosurgery. 2012;70(1):234–243. discussion 243–234. [DOI] [PubMed] [Google Scholar]
- 116.Reardon DA, Galanis E, DeGroot JF, et al. Clinical trial end points for high-grade glioma: the evolving landscape. Neuro Oncol. 2011;13(3):353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van den Bent MJ, Wefel JS, Schiff D, et al. Response assessment in neuro-oncology (a report of the RANO group): assessment of outcome in trials of diffuse low-grade gliomas. Lancet Oncol. 2011;12(6):583–593. [DOI] [PubMed] [Google Scholar]
- 118.Berry D. The brave new world of clinical cancer research: adaptive biomarker-driven trials integrating clinical practice with clinical research. Molecular Oncology. 2015;95:951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lamborn KR, Yung WKA, Chang SM, et al. Progression-free survival: An important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol. 2008;10(2):162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Robins HI, Chang SM, Prados MD, et al. A phase II trial of thymidine and carboplatin for recurrent malignant glioma: a North American Brain Tumor Consortium Study. Neuro Oncol. 2002;4(2):109–114. [PMC free article] [PubMed] [Google Scholar]
- 121.Brandes AA, Tosoni A, Amista P, et al. How effective is BCNU in recurrent glioblastoma in the modern era? A phase II trial. Neurology. 2004;63(7):1281–1284. [DOI] [PubMed] [Google Scholar]
- 122.Prados MD, Yung WK, Fine HA, et al. Phase 2 study of BCNU and temozolomide for recurrent glioblastoma multiforme: North American Brain Tumor Consortium study. Neuro Oncol. 2004;6(1):33–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.de Groot JF, Gilbert MR, Aldape K, et al. Phase II study of carboplatin and erlotinib (Tarceva, OSI-774) in patients with recurrent glioblastoma. J Neurooncol. 2008;90(1):89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wick W, Puduvalli VK, Chamberlain MC, et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol. 2010;28(7):1168–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Batchelor TT, Mulholland P, Neyns B, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31(26):3212–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dresemann G, Weller M, Rosenthal MA, et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J Neurooncol. 2010;96(3):393–402. [DOI] [PubMed] [Google Scholar]
- 127.Santisteban M, Buckner JC, Reid JM, et al. Phase II trial of two different irinotecan schedules with pharmacokinetic analysis in patients with recurrent glioma: North Central Cancer Treatment Group results. J Neurooncol. 2009;92(2):165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Friedman HS, Petros WP, Friedman AH, et al. Irinotecan therapy in adults with recurrent or progressive malignant glioma. J Clin Oncol. 1999;17(5):1516–1525. [DOI] [PubMed] [Google Scholar]
- 129.Kappelle AC, Postma TJ, Taphoorn MJ, et al. PCV chemotherapy for recurrent glioblastoma multiforme. Neurology. 2001;56(1):118–120. [DOI] [PubMed] [Google Scholar]
- 130.Schmidt F, Fischer J, Herrlinger U, Dietz K, Dichgans J, Weller M. PCV chemotherapy for recurrent glioblastoma. Neurology. 2006;66(4):587–589. [DOI] [PubMed] [Google Scholar]
- 131.Yung WK, Albright RE, Olson J, et al. A phase II study of temozolomide vs.: procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000;83(5):588–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brada M, Hoang-Xuan K, Rampling R, et al. Multicenter phase II trial of temozolomide in patients with glioblastoma multiforme at first relapse. Ann Oncol. 2001;12(2):259–266. [DOI] [PubMed] [Google Scholar]
- 133.Wick A, Felsberg J, Steinbach JP, et al. Efficacy and tolerability of temozolomide in an alternating weekly regimen in patients with recurrent glioma. J Clin Oncol. 2007;25(22):3357–3361. [DOI] [PubMed] [Google Scholar]
- 134.Brandes AA, Tosoni A, Cavallo G, et al. Temozolomide 3 weeks on and 1 week off as first-line therapy for recurrent glioblastoma: phase II study from gruppo italiano cooperativo di neuro-oncologia (GICNO). Br J Cancer. 2006;95(9):1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Norden AD, Lesser GJ, Drappatz J, et al. Phase 2 study of dose-intense temozolomide in recurrent glioblastoma. Neuro Oncol. 2013;15(7):930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Perry JR, Belanger K, Mason WP, et al. Phase II trial of continuous dose-intense temozolomide in recurrent malignant glioma: RESCUE study. J Clin Oncol. 2010;28(12):2051–2057. [DOI] [PubMed] [Google Scholar]
- 137.Groves MD, Puduvalli VK, Chang SM, et al. A North American Brain Tumor Consortium (NABTC 99–04) phase II trial of temozolomide plus thalidomide for recurrent glioblastoma multiforme. J Neurooncol. 2007;81(3):271–277. [DOI] [PubMed] [Google Scholar]
- 138.Wen PY, Schiff D, Cloughesy TF, et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol. 2011;13(4):437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reardon DA, Fink KL, Mikkelsen T, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008;26(34):5610–5617. [DOI] [PubMed] [Google Scholar]
- 140.Levin VA, Giglio P, Puduvalli VK, et al. Combination chemotherapy with 13-cis-retinoic acid and celecoxib in the treatment of glioblastoma multiforme. J Neurooncol. 2006;78(1):85–90. [DOI] [PubMed] [Google Scholar]
- 141.van den Bent MJ, Brandes AA, Rampling R, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27(8):1268–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yung WK, Vredenburgh JJ, Cloughesy TF, et al. Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro Oncol. 2010;12(10):1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Reardon DA, Desjardins A, Vredenburgh JJ, et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol. 2010;96(2):219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Puduvalli VK, Yung WK, Hess KR, et al. Phase II study of fenretinide (NSC 374551) in adults with recurrent malignant gliomas: A North American Brain Tumor Consortium study. J Clin Oncol. 2004;22(21):4282–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rich JN, Reardon DA, Peery T, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22(1):133–142. [DOI] [PubMed] [Google Scholar]
- 146.Wen PY, Yung WK, Lamborn KR, et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99–08. Clin Cancer Res. 2006;12(16):4899–4907. [DOI] [PubMed] [Google Scholar]
- 147.Raymond E, Brandes AA, Dittrich C, et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: A European Organisation for Research and Treatment of Cancer Brain Tumor Group study. J Clin Oncol. 2008;26(28):4659–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Reardon DA, Egorin MJ, Quinn JA, et al. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol. 2005;23(36):9359–9368. [DOI] [PubMed] [Google Scholar]
- 149.Reardon DA, Egorin MJ, Desjardins A, et al. Phase I pharmacokinetic study of the vascular endothelial growth factor receptor tyrosine kinase inhibitor vatalanib (PTK787) plus imatinib and hydroxyurea for malignant glioma. Cancer. 2009;115(10):2188–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Thiessen B, Stewart C, Tsao M, et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol. 2010;65(2):353–361. [DOI] [PubMed] [Google Scholar]
- 151.Galanis E, Buckner JC, Maurer MJ, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23(23):5294–5304. [DOI] [PubMed] [Google Scholar]
- 152.Cloughesy TF, Wen PY, Robins HI, et al. Phase II trial of tipifarnib in patients with recurrent malignant glioma either receiving or not receiving enzyme-inducing antiepileptic drugs: a North American Brain Tumor Consortium Study. J Clin Oncol. 2006;24(22):3651–3656. [DOI] [PubMed] [Google Scholar]
- 153.Galanis E, Jaeckle KA, Maurer MJ, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27(12):2052–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Levin VA, Leibel SA, Gutin PH. Neoplasms of the central nervous system. In: DeVita VTJ, Hellman S, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. 6th edn. Philadelphia: Lippincott-Raven; 2001:2100–2160. [Google Scholar]
- 155.Schold SC, Jr., Friedman HS, Bjornsson TD, Bigner DD. Treatment of human glioma and medulloblastoma tumor lines in athymic mice with diaziquone and diaziquone-based drug combinations. Cancer Res. 1984;44(6):2352–2357. [PubMed] [Google Scholar]
- 156.Allen JC, Bloom J, Ertel I, et al. Brain tumors in children: current cooperative and institutional chemotherapy trials in newly diagnosed and recurrent disease. [review]. Semin Oncol. 1986;13(1):110–122. [PubMed] [Google Scholar]
- 157.Gaynon PS, Ettinger LJ, Baum ES, et al. Carboplatin in childhood brain tumors. A Children's Cancer Study Group phase II trial. Cancer. 1990;66(12):2465–2469. [DOI] [PubMed] [Google Scholar]
- 158.Allen JC, Walker R, Luks E, et al. Carboplatin and recurrent childhood brain tumors. J Clin Oncol. 1987;5(3):459–463. [DOI] [PubMed] [Google Scholar]
- 159.Rosen G, Ghavimi F, Nirenberg A, et al. High-dose methotrexate with citrovorum factor rescue for the treatment of central nervous system tumors in children. Cancer Treat Rep. 1977;61(4):681–690. [PubMed] [Google Scholar]
- 160.Djerassi I, Kim JS, Shulman K. High-dose methotrexate-citrovorum factor rescue in the management of brain tumors. Cancer Treat Rep. 1977;61:691. [PubMed] [Google Scholar]
- 161.Mooney C, Souhami R, Pritchard J. Recurrent medulloblastoma: lack of response to high-dose methotrexate. Cancer Chemother Pharmacol. 1983;10:135. [DOI] [PubMed] [Google Scholar]
- 162.Walker R, JC A. Treatment of recurrent primary intracranial childhood tumors with cis-diamine-dichloroplatinum. Ann Neurol. 1983;14:371. [Google Scholar]
- 163.Sexauer CL, Khan A, Burger PC, et al. Cisplatin in recurrent pediatric brain tumors. A POG Phase II study. A Pediatric Oncology Group Study. Cancer. 1985;56(7):1497–1501. [DOI] [PubMed] [Google Scholar]
- 164.Friedman HS, Schold SC, Jr., Mahaley MS, Jr., et al. Phase II treatment of medulloblastoma and pineoblastoma with melphalan: clinical therapy based on experimental models of human medulloblastoma. J Clin Oncol. 1989;7(7):904–911. [DOI] [PubMed] [Google Scholar]
- 165.Levin VA, Edwards MS, Gutin PH, et al. Phase II evaluation of dibromodulcitol in the treatment of recurrent medulloblastoma, ependymoma, and malignant astrocytoma. J Neurosurg. 1984;61(6):1063–1068. [DOI] [PubMed] [Google Scholar]
- 166.Rosenstock JG, Evans AE, Schut L. Response to vincristine of recurrent brain tumors in children. J Neurosurg. 1976;45:135. [DOI] [PubMed] [Google Scholar]
- 167.Smart CR, Ottoman RE, Rochlin DB, et al. Clinical experience with vincristine in tumors of the central nervous system and other malignant diseases. Cancer Chemother Rep. 1968;52:733. [PubMed] [Google Scholar]
- 168.Haddy TB, Ferbach DJ, Watkins WL, et al. Vincristine in uncommon malignant disease in children. Cancer Chemother Rep. 1964;41:41. [PubMed] [Google Scholar]
- 169.Wilson CB, Gutin P, Boldrey EB, et al. Single-agent chemotherapy of brain tumors. A five-year review. Arch Neurol. 1976;33(11):739–744. [DOI] [PubMed] [Google Scholar]
- 170.Ward HWC. CCNU in the treatment of recurrent medulloblastoma. Br Med J. 1974;1:642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fewer D, Wilson CB, Boldrey EB, et al. Phase II study of 1-(2-choloroethyl)-3-cyclohexyl-1-nitrosourea (CCNU) in the treatment of brain tumors. Cancer Chemother Rep. 1972;56:421. [PubMed] [Google Scholar]
- 172.Cangir A, Ragab AH, Steubner P, et al. Combination chemotherapy with vincristine, procarbazine, prednisone with or without nitrogen mustard (MOOP vs OPP) in children with recurrent brain tumors. Med Pediatr Oncol. 1984;12:1. [DOI] [PubMed] [Google Scholar]
- 173.Edwards MS, Levin VA, Wilson CB. Chemotherapy of recurrent pediatric posterior fossa tumors. Clin Neurosurg. 1983;30:209–225. [DOI] [PubMed] [Google Scholar]
- 174.Crafts DC, Levin VA, Edwards MS, et al. Chemotherapy of recurrent medulloblastoma with combined procarbazine, CCNU, and vincristine. J Neurosurg. 1978;49(4):589–592. [DOI] [PubMed] [Google Scholar]
- 175.van Eys J, Baram TZ, Cangir A, et al. Salvage chemotherapy for recurrent primary brain tumors in children. J Pediatr. 1988;113:601. [DOI] [PubMed] [Google Scholar]
- 176.Gentet JC, Doz F, Bouffet E, et al. Carboplatin and VP 16 in medulloblastoma: a phase II Study of the French Society of Pediatric Oncology (SFOP). Med Pediatr Oncol. 1994;23(5):422–427. [DOI] [PubMed] [Google Scholar]
- 177.Walker MD, Alexander E, Jr., Hunt WE, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg. 1978;49(3):333–343. [DOI] [PubMed] [Google Scholar]
- 178.Levin VA, Wara WM, Davis RL, et al. Northern California Oncology Group protocol 6G91: response to treatment with radiation therapy and seven-drug chemotherapy in patients with glioblastoma multiforme. Cancer Treat Rep. 1986;70(6):739–743. [PubMed] [Google Scholar]
- 179.Levin VA, Silver P, Hannigan J, et al. Superiority of post-radiotherapy adjuvant chemotherapy with CCNU, procarbazine, and vincristine (PCV) over BCNU for anaplastic gliomas: NCOG 6G61 final report. Int J Radiat Oncol Biol Phys. 1990;18(2):321–324. [DOI] [PubMed] [Google Scholar]
- 180.Phillips TL, Levin VA, Ahn DK, et al. Evaluation of bromodeoxyuridine in glioblastoma multiforme: a Northern California Cancer Center Phase II study. Int J Radiat Oncol Biol Phys. 1991;21(3):709–714. [DOI] [PubMed] [Google Scholar]
- 181.Dinapoli RP, Brown LD, Arusell RM, et al. Phase III comparative evaluation of PCNU and carmustine combined with radiation therapy for high-grade glioma. J Clin Oncol. 1993;11(7):1316–1321. [DOI] [PubMed] [Google Scholar]
- 182.Levin VA, Maor MH, Thall PF, et al. Phase II study of accelerated fractionation radiation therapy with carboplatin followed by vincristine chemotherapy for the treatment of glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 1995;33(2):357–364. [DOI] [PubMed] [Google Scholar]
- 183.Groves MD, Maor MH, Meyers C, et al. A phase II trial of high-dose bromodeoxyuridine with accelerated fractionation radiotherapy followed by procarbazine, lomustine, and vincristine for glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 1999;45(1):127–135. [DOI] [PubMed] [Google Scholar]
- 184.Levin VA, Uhm JH, Jaeckle KA, et al. Phase III randomized study of postradiotherapy chemotherapy with alpha-difluoromethylornithine-procarbazine, N-(2-chloroethyl)-N'-cyclohexyl-N-nitrosurea, vincristine (DFMO-PCV) versus PCV for glioblastoma multiforme. Clin Cancer Res. 2000;6(10):3878–3884. [PubMed] [Google Scholar]
- 185.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 186.Souhami L, Seiferheld W, Brachman D, et al. Randomized comparison of stereotactic radiosurgery followed by conventional radiotherapy with carmustine to conventional radiotherapy with carmustine for patients with glioblastoma multiforme: report of Radiation Therapy Oncology Group 93–05 protocol. Int J Radiat Oncol Biol Phys. 2004;60(3):853–860. [DOI] [PubMed] [Google Scholar]
- 187.Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Jeremic B, Jovanovic D, Djuric LJ, et al. Advantage of post-radiotherapy chemotherapy with CCNU, procarbazine, and vincristine (mPCV) over chemotherapy with VM-26 and CCNU for malignant gliomas. J Chemother. 1992;4(2):123–126. [DOI] [PubMed] [Google Scholar]
- 189.Levin VA, Prados MR, Wara WM, et al. Radiation therapy and bromodeoxyuridine chemotherapy followed by procarbazine, lomustine, and vincristine for the treatment of anaplastic gliomas. Int J Radiat Oncol Biol Phys. 1995;32(1):75–83. [DOI] [PubMed] [Google Scholar]
- 190.Prados MD, Scott C, Curran WJ, Jr, et al. Procarbazine, lomustine, and vincristine (PCV) chemotherapy for anaplastic astrocytoma: A retrospective review of radiation therapy oncology group protocols comparing survival with carmustine or PCV adjuvant chemotherapy. J Clin Oncol. 1999;17(11):3389–3395. [DOI] [PubMed] [Google Scholar]
- 191.Prados MD, Scott C, Sandler H, et al. A phase 3 randomized study of radiotherapy plus procarbazine, CCNU, and vincristine (PCV) with or without BUdR for the treatment of anaplastic astrocytoma: A preliminary report of RTOG 9404. Int J Radiat Oncol Biol Phys. 1999;45(5):1109–1115. [DOI] [PubMed] [Google Scholar]
- 192.Levin VA, Yung WK, Bruner J, et al. Phase II study of accelerated fractionation radiation therapy with carboplatin followed by PCV chemotherapy for the treatment of anaplastic gliomas. Int J Radiat Oncol Biol Phys. 2002;53(1):58–66. [DOI] [PubMed] [Google Scholar]
- 193.Prados MD, Seiferheld W, Sandler HM, et al. Phase III randomized study of radiotherapy plus procarbazine, lomustine, and vincristine with or without BUdR for treatment of anaplastic astrocytoma: final report of RTOG 9404. Int J Radiat Oncol Biol Phys. 2004;58(4):1147–1152. [DOI] [PubMed] [Google Scholar]
- 194.Cho YJ, Tsherniak A, Tamayo P, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol. 2011;29(11):1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Northcott PA, Dubuc AM, Pfister S, Taylor MD. Molecular subgroups of medulloblastoma. Expert Rev Neurother. 2012;12(7):871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kool M. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3:e3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kool M, Korshunov A, Remke M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123(4):473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24(12):1924–1931. [DOI] [PubMed] [Google Scholar]
- 200.Levin VA. Pharmacokinetics and central nervous system chemotherapy. In: Hellmann K, Carter SK, eds. Fundamentals of Cancer Chemotherapy. New York: McGraw-Hill; 1987:28–40. [Google Scholar]
- 201.Rosso L, Brock CS, Gallo JM, et al. A new model for prediction of drug distribution in tumor and normal tissues: pharmacokinetics of temozolomide in glioma patients. Cancer Res. 2009;69(1):120–127. [DOI] [PubMed] [Google Scholar]