

Abstract
Hepatocyte apoptosis and energy metabolism in mitochondria have an important role in the mechanism of acute liver failure (ALF). However, data on the association between apoptosis and the energy metabolism of hepatocytes are lacking. The current study assessed the activity of several key enzymes in mitochondria during ALF, including citrate synthase (CS), carnitine palmitoyltransferase-1 (CPT-1) and cytochrome c oxidase (COX), which are involved in hepatocyte energy metabolism. A total of 40 male Sprague-Dawley rats were divided into five groups and administered D-galactosamine and lipopolysaccharide to induce ALF. Hepatic pathology and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling examinations indicated that hepatocyte apoptosis was observed at 4 h and increased 8 h after ALF. Hepatocyte necrosis appeared at 12 h and was significantly higher at 24 h with inflammatory cell invasion. The results measured by electron microscopy indicated that ultrastructural changes in mitochondria began at 4 h and the mitochondrial outer membrane was completely disrupted at 24 h resulting in mitochondrial collapse. The expression of CS, CPT-1 and COX was measured and analyzed using assay kits. The activity and protein expression of CS, CPT-1 and COX began to increase at 4 h, reached a peak at 8 h and decreased at 12 h during ALF. The activities of CS, CPT-1 and COX were enhanced during hepatocyte apoptosis suggesting that these enzymes are involved in the initiation and development of ALF. Therefore, these results demonstrated that energy metabolism is important in hepatocyte apoptosis during ALF and hepatocyte apoptosis is an active and energy-consuming procedure. The current study on how hepatocyte energy metabolism affects the transmission of death signals may provide a basis for the early diagnosis and development of an improved therapeutic strategy for ALF.
Keywords: mitochondria, carnitine palmitoyltransferase-1, citrate synthase, cytochrome c oxidase, liver failure, acute
Introduction
Acute liver failure (ALF) is defined as severe liver damage induced by multiple factors and has a mortality rate of 80–90% (1). Current studies have verified that hepatocyte apoptosis is important in the pathology of ALF (1–5). The basic function of the mitochondrion is energy metabolism, which provides all of the energy necessary for life. A variety of studies have indicated that, in addition to energy metabolism, modulation of cellular apoptosis is the second main function of mitochondria (2,6,7). For example, the release of cytochrome c and pro-apoptotic proteins into the cytoplasm, calcium mobility and the generation of reactive oxygen species (ROS) result in an alteration in mitochondrial permeability and ATP depletion (3).
Hepatocytes are enriched with mitochondria that comprise 13–20% of the liver volume. The liver is the chemical center of the human body, consuming 20% of oxygen in the entire body and is important in the metabolism of sugar, fat, protein, water, salt and vitamins. The energy supply to the liver predominantly originates from the oxidization of fatty acids. The liver has a central position in lipid metabolism and is the location of fatty acid β-oxidization (4). Therefore, investigating the association between apoptosis and energy metabolism in hepatocyte mitochondria during ALF has important practical value for understanding the mechanisms underlying ALF, providing a basis for the early diagnosis of ALF and developing a reasonable therapy for ALF.
Metabolic pathways in the body consist of a series of chemical reactions catalyzed by enzymes, of which the speed and direction are determined by one or several key enzymes. The modulation of energy metabolism, however, is primarily achieved by modulating the activities of key enzymes (8). Citrate synthase (CS) is the key enzyme and the first rate-limiting enzyme in the tricarboxylic acid cycle (TCA). The CS of eukaryotes is coded by the nuclear genome, synthesized in cytoplasmic ribosomes and exerts its function in the mitochondrial matrix (9). The CS is the rate-limiting enzyme of the TCA cycle and its activity can modulate the cycle (10–12). Carnitine palmitoyltransferase-1 (CPT-1) is located in the outer membrane of mitochondria and catalyzes long-chain fatty acyl-CoA and carnitine to synthesize fatty acyl carnitine, which is the first rate-limiting reaction of the oxidation procedure of fatty acids in mitochondria (13). Cytochrome c oxidase (COX) is the final complex of electron transmission in the respiratory chain and the key enzyme in oxidative phosphorylation in mitochondria (14), and also plays an important role in energy production (15). These three enzymes are rate-limiting and are the key enzymes in mitochondrial energy metabolism. Their activities can reflect the mitochondrial energy metabolic function. Measuring the alterations in the activities of these three enzymes can indirectly reflect alterations in mitochondrial function. Previous studies have indicated that decreases in the activities of CS, CPT-1 and COX induced oxidative stress. This created excessive ROS (2), which increased the Ca2+ concentration in the cytoplasm (16,17), initiated caspase reactions (5,18), modulated the expression of the apoptosis-modulation proteins p53 and Bcl-2 (19,20) and produced cell apoptosis through lipid peroxidation, amino acid oxidation/glycosylation of intracellular biomembranes, including the mitochondrial membrane, endoplasmic reticulum membrane and nuclear membrane, and base oxidation of DNA (21). In addition, oxidative stress produced an accumulation of excessive acidic substances, activated lysosomal pathways leading to cellular death, increased cellular sensitivity to tumor necrosis factor (TNF)-α (22) and also initiated apoptosis (23). Therefore, determining the association between apoptosis and mitochondrial energy metabolism warrants further investigation, particularly during the course of ALF.
The present study aimed to observed the metabolic characteristics of hepatocyte death during ALF and verify that the change in energy metabolism is involved in ALF, particularly the enhanced activities of CS, CPT-1 and COX during hepato-cyte apoptosis.
Materials and methods
Animals and treatment
Male Sprague-Dawley (SD) rats weighing 200±20 g were provided by the Experimental Animal Center of Beijing Youan Hospital, Capital Medical University (Beijing, China). The animals were housed at a room temperature of 22±2°C, with a light cycle between 07:00 and 19:00 and free access to food and water. All experimental procedures were approved by the Ethics Committee of Animal Care and Usage of Capital Medical University. A total of 40 rats were randomly divided into five groups (n=8): Group A, control group; Group B, 4 h after administration of D-galactosamine (D-GalN) + lipopolysaccharide (LPS; ALF 4 h group); Group C, 8 h after administration of D-GalN + LPS (ALF 8 h group); Group D, 12 h after administration of D-GalN + LPS (ALF 12 h group); and Group E, 24 h after administration of D-GalN + LPS (ALF 24 h group). The rats in Groups B–E were injected intraperitoneally (i.p.) with D-GalN (800 mg/kg; Sigma-Aldrich, St. Louis, MO, USA) and LPS (100 µg/kg; Sigma-Aldrich). The rats in Group A (control) were injected i.p. with 1 ml of 0.9% saline. At selected time points following D-GalN/LPS treatment, rats were anesthetized and blood was collected. The liver was isolated and used immediately to prepare mRNA. The mRNA and liver tissues were stored at −75°C for later analysis.
Assays of biochemical parameters
Serum alanine amino-transferase (ALT), aspartate aminotransferase (AST) and total bilirubin (TBIL) were examined using an automatic biochemical analyzer (Olympus AU5400; Olympus, Tokyo, Japan); the prothrombin time (PT) and prothrombin activity (PTA) were determined using an automatic coagulation analyzer (SYSMEX CA-7000; Siemens Healthcare, Erlangen, Germany).
Histopathological analysis
Liver tissues were removed immediately and fixed in 10% neutral formalin solution for at least 24 h, then embedded in paraffin wax and sectioned (4 µm thickness) for histopathological evaluation. Liver sections were stained with hematoxylin and eosin (H&E) using the Hematoxylin and Eosin Staining kit (C0105; Beyotime Institute of Biotechnology, Haimen, China), then the sections were analyzed by light microscopy (CKX41 Optical Microscope; Olympus).
Mitochondrial extraction
Mitochondrial extraction was immediately performed on 400 mg of liver tissue from the middle portion of the right lobe using a mitochondria extraction kit (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) according to the manufacturer's instructions. Half of the mitochondria were kept in the 50 µl storage solution from the kit, from which 10 µl of solution was used to measure the activities of CS, CPT-1 and COX using spectrophotometry (UV-2600 Ultraviolet-Visible Spectrometer; Unico (Shanghai) Instrument Co., Ltd., Shanghai, China) following protein quantification. The other half of the mitochondria was fixed with glutaraldehyde for electron microscopic observation of mitochondrial ultrastructure.
In situ apoptosis detection by TUNEL
Fresh liver tissue from the right lobe was fixed with 4% paraformaldehyde for 24 h, and following paraffin-embedding, was cut into 4 µm sections for TUNEL (Boehringer Mannheim, Ingelheim am Rhein, Germany). Under the mediation of terminal deoxynucleotidyl transferase, biotinylated deoxyuridine triphosphate was labeled at the 3′-OH terminal formed following the DNA break. Using specific binding of biotin and avidin, peroxidase was linked to the DNA breakpoint. Substrate was finally added to observe apoptotic cells with brown nuclei labeled by 3,3′-diaminobenzidine staining under light microscopy. In total, 10 fields of view were randomly selected from sections of every sample and the apoptotic cells were counted under a lens (magnification, ×200) to calculate the apoptotic index (AI, expressed as a percentage) in every 100 cells. Apoptotic cells fit the following criteria: Single, no surrounding inflammatory cells, membrane shrinkage and nuclei densely stained with brown particles or debris.
Measurement of CS, CPT-1 and COX
The activities of CS, CPT-1 and COX were examined using a CS assay kit (GMS50130.1; Genmed Scientifics, Inc., Shanghai, China), a CPT-1 assay kit (GMS50118.1.2; Genmed Scientifics, Inc.) and a COX assay kit (GMS10014.2; Genmed Scientifics, Inc.), respectively, according to the manufacturers' instructions.
Western blot analysis
Protein was extracted from liver tissue with RIPA buffer [20 mM Tris, pH 7.4, 137 mM NaCl, 0.5% sodium dodecyl sulfate (SDS), 10% glycerol and 1% Triton X-100] with phosphatase inhibitor 1 (100X), phosphatase inhibitor 2 (100X) and a protease inhibitor cocktail (100X). Protein concentrations were determined using a Bio-Rad detergent compatible Protein Assay kit (microtiter plate; Bio-Rad Laboratories, Inc., Hercules, CA, USA). Proteins (40 mg sample) in SDS-loading buffer [50 mM Tris (pH 7.6), 10% glycerol, 1% SDS] were subjected to SDS-12% polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane (Bio-Rad Laboratories, Inc.). The membrane was blocked with 5% dry milk and 0.1% Tween 20. Antibodies against COX1 (cat. no. sc-23982; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), CPT1 (cat. no. ab83862), CS (cat. no. ab129088) and β-actin (cat. no. ab6276) (Abcam, San Francisco, CA, USA) were used for western blot analysis. Membranes were probed with the primary antibodies (1:1,000) in 10 ml blocking buffer overnight at 4°C. Following washing, membranes were further probed with the appropriate secondary antibody (1:2,000; cat. nos. cat. 115-035-003 and 111-035-003; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) in 10 ml of blocking buffer for 1 h at room temperature. SuperSignal® West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Rockford, IL, USA) was used for chemiluminescence development.
Statistical analysis
The results are presented as the mean ± standard deviation. Statistical comparisons among groups were performed by analysis of variance followed by the t-test for independent samples between two groups. The bilateral 95% confidence interval was calculated using SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 (two tailed) was considered to indicate a statistically significant difference.
Results
Characteristic alterations of ALF induced by D-GalN/LPS administration
The effects of D-GalN/LPS administration on the liver of rats were initially assessed. Serum ALT, AST, TBIL, PT and PTA and histological changes in the liver of rats were investigated following D-GalN/LPS treatment. The results indicated that serum ALT, AST, TBIL and PT were significantly elevated, whereas PTA was significantly reduced, in a time-dependent manner following D-GalN/LPS exposure (Table I). ALT and AST began to increase at 4 h, reached a peak at 12 h and decreased at 24 h after ALF. TBIL increased at 8 h and peaked at 24 h and PT began to increase while PTA decreased at 4 h after ALF.
Table I.
Alterations in serum ALT, AST, TBIL, PT and PTA in the liver of D-GalN/LPS-treated rats.
| Group | ALT (U/l) | AST (U/l) | TBIL (µmol/l) | PT (sec) | PTA (%) |
|---|---|---|---|---|---|
| Control | 40.05±3.93 | 132.89±19.62 | 1.41±0.77 | 9.68±0.83 | 112.69±13.06 |
| ALF 4 h | 121.25±52.33a | 315.84±106.46a | 1.54±0.87 | 16.28±4.93a | 66.44±24.23a |
| ALF 8 h | 827.18±359.48a | 2489.06±1181.33a | 6.76±3.63a | 19.55±6.93a | 58.88±20.12a |
| ALF 12 h | 7778.60±4026.25a | 9461.90±5238.06a | 52.71±10.43a | 41.35±12.61a | 27.96±9.99a |
| ALF 24 h | 73.28±31.92b | 116.08±12.18 | 64.23±5.59a | 71.99±21.55a | 15.60±4.44a |
P<0.01, compared with the control group;
P<0.05, compared with the control group. ALT, alanine aminotransferase; AST, aspartate amino-transferase; TBIL, total bilirubin; PT, prothrombin time; PTA, prothrombin activity; D-GalN, D-galactosamine; LPS, lipopolysaccharide; ALF, acute liver failure.
Histological examination revealed that the livers from control group animals demonstrated complete hepatic lobules without degeneration or necrosis by H&E staining. At ALF 4 h, hepatic tissue underwent ballooning degeneration without clear inflammatory invasion in the sinusoids. At ALF 8 h, there was apparent hepatocyte apoptosis, partial hyperemia in the sinusoids, piecemeal necrosis and inflammatory invasion. At ALF 12 h, a large quantity of hepatocytes underwent piecemeal necrosis and there was rupture of hepatic cords. At ALF 24 h, there was marked necrosis of hepatocytes and severe hyperemia in the sinusoids, the boundary of hepatocytes became obscure, the fiber mesh scaffold collapsed and there was clear inflammatory cell invasion (Fig. 1).
Figure 1.
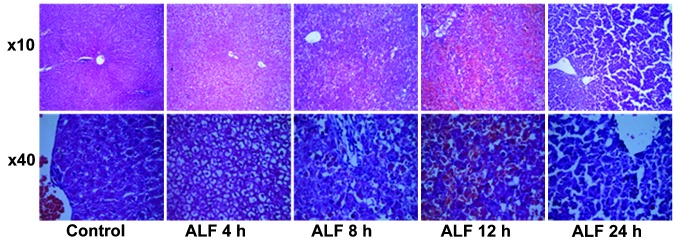
Histological examination of acute liver damage induced by D-galactosamine/lipopolysaccharide treatment in rats. Liver sections were stained with hematoxylin and eosin. ALF 4 h, hepatic tissue underwent ballooning degeneration without inflammatory invasion in the sinusoids; ALF 8 h, hepatocyte apoptosis and partial hyperemia was visible in the sinusoids and piecemeal necrosis and inflammatory invasion was observed; ALF 12 h, a large quantity of hepatocytes underwent piecemeal necrosis and there was rupture of hepatic cords; ALF 24 h, marked necrosis of hepatocytes and severe hyperemia was observed in the sinusoids, the boundary of hepatocytes became obscure, the fiber mesh scaffold collapsed and there was clear inflammatory invasion. ALF, acute liver failure.
Hepatocyte apoptosis during the early stages of ALF
TUNEL staining demonstrated that cellular injury occurred predominantly in an apoptotic pattern during the early stages of ALF. As shown in (Fig. 2A–C), apoptotic cells were predominantly located around the central vein and were characterized by their shrunken, round shape, small cell body, concentrated cytoplasm, increased density of nuclear chromatin, which congregated near the nuclear membrane, and their invaginated cellular membrane, which separated the cell into membrane-encapsulated apoptotic bodies varying in size. The AI was significantly different between the control group (10.60±2.61), ALF 4 h (48.90±10.76) and ALF 8 h groups (74.60±14.85; Fig. 2D).
Figure 2.

Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling detection of apoptosis in rat livers during the early stages of ALF. (A–C) Apoptotic cells were visible 4 and 8 h after D-galactosamine/lipopolysaccharide treatment in rats and were located predominantly around the central vein, as indicated by arrows; (D) 10 fields of view were randomly selected from sections of every sample and the apoptotic cells were counted under a lens (magnification, ×200) to calculate the number of apoptotic cells in every 100 cells. ALF, acute liver failure.
Morphological alterations in liver mitochondria during ALF
The hepatocyte mitochondria from the control group were round or oval with complete membrane structures and concentric circles or longitudinally arranged ridges that were tidy and compact. In the ALF 4 h group, the mitochondria were reduced in number, fairly structured, fairly complete and had a complete structure of ridges that had a fairly tidy arrangement but with concentrated stroma. In the ALF 8 h group, the mitochondria were swollen and clearly damaged but had somewhat complete outer membranes and disorganized ridge structures. In the ALF 12 h group, the mitochondria were swollen and their inner structures were not easily visible. In the ALF 24 h group, the outer membrane was disrupted and the mitochondria had collapsed (Fig. 3).
Figure 3.

Morphological alterations in liver mitochondria during ALF. Control group, the mitochondria were round or oval with complete membrane structures and concentric circles or longitudinally arranged ridges that were tidy and compact; ALF 4 h, the mitochondria were reduced in number, fairly structured, relatively complete and had a complete structure of ridges that had a relatively tidy arrangement but with concentrated stroma; ALF 8 h, the mitochondria were swollen and clearly damaged but had somewhat complete outer membranes and disorganized ridge structures; ALF 12 h, the mitochondria were clearly swollen and their inner structures were not easily visible; ALF 24 h, the outer membrane was disrupted and the mitochondria had collapsed. ALF, acute liver failure.
Activities of CS, CPT-1 and COX in liver mitochondria and their expression in liver tissue
To assess the dynamic alterations of CS, CPT-1 and COX during the course of ALF, the present study examined the activities of CS, CPT-1 and COX in liver mitochondria and the expression level of CS, CPT-1 and COX in liver tissue. As shown in (Fig. 4A and B) the activities of CS, CPT-1 and COX in liver mitochondria began to increase at 4 h (Fig. 4C) peaked at 8 h, began to decrease at 12 h and decreased markedly at 24 h. The present study also determined the expression level of CS, CPT-1 and COX in liver tissue during D-GalN/LPS-induced hepatic failure. As shown in Fig. 4D, compared with the control group, the expression levels of CS, CPT-1 and COX were upregulated at 4 h and then decreased after 8 h in liver tissue.
Figure 4.

Activities of CS, CPT-1 and COX in liver mitochondria and their protein expression level in liver tissue. (A–C) The activity of CS, CPT-1 and COX in liver mitochondria began to increase at 4 h, peaked at 8 h and began to decrease at 12 h. (D) Western blotting demonstrated that the protein expression levels of CS, CPT-1 and COX were upregulated at 4 h and then decreased after 12 h in liver tissue. ALF, acute liver failure; CS, citrate synthase; CPT-1, carnitine palmitoyltransferase-1; COX, cytochrome c oxidase; D-GalN, D-galactosamine; LPS, lipopolysaccharide.
Discussion
Mitochondria are critical during hepatocyte death and the dysfunction of mitochondria is an obligatory process in hepa-tocyte death. The increase in mitochondrial permeability and dysfunction can induce hepatocyte apoptosis and necrosis. Mitochondria undertake the metabolic function of producing energy through the respiratory chain and the mitochondrial inner and outer membranes can simultaneously quarantine pro-apoptotic proteins in the periplasmic space. However, the metabolic function and characteristics of mitochondria and their association with hepatocyte apoptosis remain to be elucidated.
The combined administration of D-GalN and LPS is a common method of establishing an ALF model in rodent animals (24). LPS can directly stimulate Kupffer cells to produce TNF-α and other inflammatory factors, producing hepatocyte injury (25). D-GalN is a specific hepatic toxicant that can deplete hepatic uridine and induce hepatic injury (26). Combined administration of D-GalN and LPS can enhance sensitivity to LPS and result in ALF (26). The present study successfully duplicated this animal model.
ALF is characterized by broad hepatocyte death or acute disruption of liver function and is the common outcome of all severe liver diseases. Regardless of the pathogenesis, the final manifestation is large-scale death of hepatocytes and other liver cells. Currently, it is known that there are two types of hepatocyte death: Apoptosis and necrosis. Apoptosis is a physiological cellular suicide process that is induced by in vivo and/or in vitro factors, is active and energy-consuming, highly ordered, genetically controlled and involves a series of enzymes (27). Necrosis, however, is an acute, irreversible, and passive process that is characterized by metabolic loss and destruction of cellular integrity (28). Previous studies indicated that energy metabolism is critical in determining patterns of cell death (29–32). Although there have been numerous studies investigating ALF (13,16–18,28,33), the mechanism underlying hepatocyte death during ALF remains to be elucidated. Particularly, studies regarding the energy metabolic characteristics and the association between key enzymes in energy metabolism and hepatocyte apoptosis during ALF are controversial. For example, previous studies testified that fatty acid metabolism altered during ALF, however, this was predominantly determined by measuring the plasma ketone level to reflect the oxidation level of fatty acids in the liver (33,34); and the plasma ketone level is affected by numerous factors, including glucose supplementation and starvation.
The present study indicated that apoptosis and necrosis occurred in hepatocytes during ALF, but with different patterns at different stages. During the early stages of ALF, hepatocyte apoptosis was predominant. Under light microscopy, apoptosis was apparent at ALF 8 h, while necrosis was visible during and after ALF 12 h. TUNEL staining also indicated that apoptosis was most significant at ALF 8 h. The increase in serum ALT and AST was due to the release of intracellular enzymes into the blood following necrosis. The present study indicated that serum ALT and AST peaked at ALF 12 h, while the activities of mitochondrial COX, CS and CPT-1 peaked at ALF 8 h, earlier than the ALT and AST peaks, indicating that the activities of COX, CS and CPT-1 were enhanced during apoptosis and decreased during necrosis. This implies that apoptosis is an active and energy-consuming process (35,36). However, this result disagreed with previous studies suggesting that decreased activities of COX, CS and CPT-1 induced apoptosis, the production of ROS and the release of cytochrome c and pro-apoptosis factors (37–40). The reason for this discrepancy may be that apoptosis is an active and energy-consuming process that requires a large quantity of ATP supplementation at this stage, and this signal stimulates the synthesis of enzymes to ensure the smooth progression of apoptosis, as reflected in the enhancement of enzymatic activities. In addition, cell apoptosis is a complex process and numerous important factors are involved, including toxins, cytokines and hormones. Therefore, hepatocyte apoptosis during the early stage of ALF, may be mediated by other factors, while the energy metabolism of mitochondria provides the necessary energy for apoptosis. By contrast, enhanced enzymatic activities may also be a compensatory mechanism.
The present study only observed the association between the activities of CS, CPT-1 and COX and hepatocyte apoptosis; it did not investigate the association between enzyme activities and other factors, including cytokines, pro-apoptotic proteins and inhibitors of apoptosis proteins. In addition, the association between the enzymatic activity and signaling pathways of hepatocyte apoptosis and necrosis is not clear. The present study could not explain why the enzymatic activities in different mitochondrial locations were enhanced, while the mitochondrial structure altered. The presumption is that the enzymatic activity is closely associated with the structure and permeability of the mitochondrial outer membranes, but is less associated with the structure of the mitochondrial inner membrane and ridge. Therefore, the roles of key enzymes in different locations and alterations in mitochondrial structures, in particular the permeability of the mitochondrial outer membranes, should be investigated further to determine the mechanism underlying changes in enzymatic activity during ALF. Further studies investigating how hepatocyte energy metabolism affects the transmission of death signals will provide a basis for the early diagnosis and development of an improved therapeutic strategy for ALF.
Acknowledgments
This study is supported by the Chinese State key Projects for Basic Research (grant no. 2007CB512801), the Chinese National Research Grant of the Eleventh Five Year Plan for the Key Projects in Infectious Diseases (grant no. 2008ZX100002-005-03) and the Technology Projects of Fengtai Distinct, Beijing (grant no. xm101210).
Abbreviations
- ALF
acute liver failure
- CS
citrate synthase
- CPT-1
carnitine palmitoyltransferase-1
- COX
cytochrome c oxidase
- ROS
reactive oxygen species
- TCA
tricarboxylic acid cycle
- D-GalN
D-galactosamine
- LPS
lipopolysaccharide
- TNF-α
tumor necrosis factor-α
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- TBIL
total bilirubin
- PT
prothrombin time
- PTA
prothrombin activity
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay
References
- 1.Liver Failure and Artificial Liver Group. Chinese Society of Infectious Diseases. Chinese Medical Association. Severe Liver Diseases and Artificial Liver Group Chinese Society of Hepatology, Chinese Medical Association: Diagnostic and treatment guidelines for liver failure. Zhonghua Gan Zang Bing Za Zhi. 2006;14:643–646. In Chinese. [PubMed] [Google Scholar]
- 2.Orrenius S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev. 2007;39:443–455. doi: 10.1080/03602530701468516. [DOI] [PubMed] [Google Scholar]
- 3.Kushnareva Y, Newmeyer DD. Bioenergetics and cell death. Ann NY Acad Sci. 2010;1201:50–57. doi: 10.1111/j.1749-6632.2010.05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryu SY, Peixoto PM, Teijido O, Dejean LM, Kinnally KW. Role of mitochondrial ion channels in cell death. Biofactors. 2010;36:255–263. doi: 10.1002/biof.101. [DOI] [PubMed] [Google Scholar]
- 5.Ohta S. A multi-functional organelle mitochondrion is involved in cell death, proliferation and disease. Curr Med Chem. 2003;10:2485–2494. doi: 10.2174/0929867033456440. [DOI] [PubMed] [Google Scholar]
- 6.Zhao BC. Role of liver in lipid metabolism. In: Zhou AR, editor. Biochemistry (Chin) 5th edition. Vol. 18. People's Medical Publishing House; Beijing: 2003. p. 365. [Google Scholar]
- 7.Zhao BC. Regulation of activities of enzymes. In: Zhou AR, editor. Biochemistry (Chin) 5th edition. Vol. 4. People's Medical Publishing House; Beijing: 2003. pp. 63–64. [Google Scholar]
- 8.Wiegand G, Remington S, Deisenhofer J, Huber R. Crystal structure analysis and molecular model of a complex of citrate synthase with oxaloacetate and S-acetonyl-coenzyme. A J Mol Biol. 1984;174:205–219. doi: 10.1016/0022-2836(84)90373-5. [DOI] [PubMed] [Google Scholar]
- 9.Cheng TL, Liao CC, Tsai WH, Lin CC, Yeh CW, Teng CF, Chang WT. Identification and characterization of the mitochondrial targeting sequence and mechanism in human citrate synthase. J Cell Biochem. 2009;107:1002–1015. doi: 10.1002/jcb.22200. [DOI] [PubMed] [Google Scholar]
- 10.Donald LJ, Duckworth HW. Molecular cloning of the structural gene for Acinetobacter citrate synthase. Biochem Biophys Res Commun. 1986;141:797–803. doi: 10.1016/S0006-291X(86)80243-1. [DOI] [PubMed] [Google Scholar]
- 11.Bloxham DP, Parmelee DC, Kumar S, Wade RD, Ericsson LH, Neurath H, Walsh KA, Titani K. Primary structure of porcine heart citrate synthase. Proc Natl Acad Sci USA. 1981;78:5381–5385. doi: 10.1073/pnas.78.9.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerner J, Hoppel C. Fatty acid import into mitochondria. Biochim Biophys Acta. 2000;1486:1–17. doi: 10.1016/S1388-1981(00)00044-5. [DOI] [PubMed] [Google Scholar]
- 13.Ostermeier C, Iwata S, Michel H. Cytochrome c oxidase. Curr Opin Struct Biol. 1996;6:460–466. doi: 10.1016/S0959-440X(96)80110-2. [DOI] [PubMed] [Google Scholar]
- 14.Kadenbach B, Barth J, Akgün R, Freund R, Linder D, Possekel S. Regulation of mitochondrial energy generation in health and disease. Biochim Biophys Acta. 1995;1271:103–109. doi: 10.1016/0925-4439(95)00016-W. [DOI] [PubMed] [Google Scholar]
- 15.Kadenbach B, Hüttemann M, Arnold S, Lee I, Bender E. Mitochondrial energy metabolism is regulated via nuclear-coded subunits of cytochrome c oxidase. Free Radic Biol Med. 2000;29:211–221. doi: 10.1016/S0891-5849(00)00305-1. [DOI] [PubMed] [Google Scholar]
- 16.Jezek P, Hlavatá L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues and organism. Int J Biochem Cell Biol. 2005;37:2478–2503. doi: 10.1016/j.biocel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 17.Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185–194. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 18.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 19.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh CL, Huang CN, Lin YC, Peng RY. Molecular action mechanism against apoptosis by aqueous extract from guava budding leaves elucidated with human umbilical vein endothelial cell (HUVEC) model. J Agric Food Chem. 2007;55:8523–8533. doi: 10.1021/jf071858b. [DOI] [PubMed] [Google Scholar]
- 21.Rana SV. Metals and apoptosis: Recent developments. J Trace Elem Med Biol. 2008;22:262–284. doi: 10.1016/j.jtemb.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 22.Giordano A, Calvani M, Petillo O, Grippo P, Tuccillo F, Melone MA, Bonelli P, Calarco A, Peluso G. tBid induces alterations of mitochondrial fatty acid oxidation flux by malonyl-CoA-independent inhibition of carnitine palmitoyl-transferase-1. Cell Death Differ. 2005;12:603–613. doi: 10.1038/sj.cdd.4401636. [DOI] [PubMed] [Google Scholar]
- 23.Harada H, Grant S. Apoptosis regulators. Rev Clin Exp Hematol. 2003;7:117–138. [PubMed] [Google Scholar]
- 24.Deutschman CS, Haber BA, Andrejko K, Cressman DE, Harrison R, Elenko E, Taub R. Increased expression of cytokine-induced neutrophil chemoattractant in septic rat liver. Am J Physiol. 1996;271:R593–R600. doi: 10.1152/ajpregu.1996.271.3.R593. [DOI] [PubMed] [Google Scholar]
- 25.Decker K, Keppler D. Galactosamine hepatitis: Key role of the nucleotide deficiency period in the pathogenesis of cell injury and cell death. Rev Physiol Biochem Pharmacol. 1974;71:77–106. doi: 10.1007/BFb0027661. [DOI] [PubMed] [Google Scholar]
- 26.Qiu Z, Kwon AH, Tsuji K, Kamiyama Y, Okumura T, Hirao Y. Fibronectin prevents D-galactosamine/lipopolysac-charide-induced lethal hepatic failure in mice. Shock. 2006;25:80–87. doi: 10.1097/01.shk.0000185797.04589.5c. [DOI] [PubMed] [Google Scholar]
- 27.Canbay A, Friedman S, Gores GJ. Apoptosis: The nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 28.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: A tale of two deaths? Hepatology. 2006;43(2 Suppl 1):S31–S44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 29.Vaquero J, Blei AT. Etiology and management of fulminant hepatic failure. Curr Gastroenterol Rep. 2003;5:39–47. doi: 10.1007/s11894-003-0008-8. [DOI] [PubMed] [Google Scholar]
- 30.Van Herreweghe F, Festjens N, Declercq W, Vandenabeele P. Tumor necrosis factor-mediated cell death: To break or to burst, that's the question. Cell Mol Life Sci. 2010;67:1567–1579. doi: 10.1007/s00018-010-0283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis CW, Hawkins BJ, Ramasamy S, Irrinki KM, Cameron BA, Islam K, Daswani VP, Doonan PJ, Manevich Y, Madesh M. Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic Biol Med. 2010;48:306–317. doi: 10.1016/j.freeradbiomed.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 33.Clemmesen O. Splanchnic circulation and metabolism in patients with acute liver failure. Dan Med Bull. 2002;49:177–193. [PubMed] [Google Scholar]
- 34.Saibara T, Maeda T, Onishi S, Yamamoto Y. Plasma exchange and the arterial blood ketone body ratio in patients with acute hepatic failure. J Hepatol. 1994;20:617–622. doi: 10.1016/S0168-8278(05)80349-6. [DOI] [PubMed] [Google Scholar]
- 35.Lee SJ, Kwon CH, Kim YK. Alterations in membrane transport function and cell viability induced by ATP depletion in primary cultured rabbit renal proximal tubular cells. Korean J Physiol Pharmacol. 2009;13:15–22. doi: 10.4196/kjpp.2009.13.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zischka H, Larochette N, Hoffmann F, Hamöller D, Jägemann N, Lichtmannegger J, Jennen L, Müller-Höcker J, Roggel F, Göttlicher M, et al. Electrophoretic analysis of the mitochondrial outer membrane rupture induced by permeability transition. Anal Chem. 2008;80:5051–5058. doi: 10.1021/ac800173r. [DOI] [PubMed] [Google Scholar]
- 37.Marín-García J, Goldenthal MJ, Damle S, Pi Y, Moe GW. Regional distribution of mitochondrial dysfunction and apoptotic remodeling in pacing-induced heart failure. J Card Fail. 2009;15:700–708. doi: 10.1016/j.cardfail.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Chinta SJ, Rane A, Yadava N, Andersen JK, Nicholls DG, Polster BM. Reactive oxygen species regulation by AIF- and complex I-depleted brain mitochondria. Free Radic Biol Med. 2009;46:939–947. doi: 10.1016/j.freeradbiomed.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puig T, Relat J, Marrero PF, Haro D, Brunet J, Colomer R. Green tea catechin inhibits fatty acid synthase without stimulating carnitine palmitoyltransferase-1 or inducing weight loss in experimental animals. Anticancer Res. 2008;28:3671–3676. [PubMed] [Google Scholar]
- 40.Mazzarelli P, Pucci S, Bonanno E, Sesti F, Calvani M, Spagnoli LG. Carnitine palmitoyltransferase I in human carcinomas: A novel role in histone deacetylation? Cancer Biol Ther. 2007;6:1606–1613. doi: 10.4161/cbt.6.10.4742. [DOI] [PubMed] [Google Scholar]