

Abstract
Pheochromocytomas and paragangliomas are rare tumors with high morbidity rates caused by excessive catecholamine secretion, even though the majority of tumors are benign. The use of perioperative blockade regimens, together with improved surgical techniques, has greatly impacted the perioperative morbidity associated with these tumors. The old dogma of the “tumor of tens” no longer holds true. For example, at least one third of all pheochromocytomas and paragangliomas are hereditary, with mutations in 1 of 10 well‐characterized susceptibility genes, and one quarter of all tumors are malignant. This review focuses on the perioperative management of pheochromocytoma and paragangliomas and the clinical implications of the associated genetic mutations.
Pheochromocytomas (PCCs) and paragangliomas (PGLs) are rare but unique tumors. They are associated with excessive catecholamine secretion that leads to high morbidity rates even though the majority of tumors are benign, and they are associated with a wider range of susceptibility genes than any other solid tumor type. PCCs are derived from the adrenal medulla, while PGLs are histologically identical tumors derived from ganglia outside the adrenal gland. PGLs can be further subdivided into those occurring in the head and neck (HNPGL), derived from parasympathetic ganglia and often nonsecretory, and those outside the head and neck, termed extra‐adrenal PGL, most often derived from sympathetic ganglia that hypersecrete catecholamines. Interestingly, despite excessive catecholamines in the circulation, some patients do not experience any symptoms, and this can complicate diagnosis.1 Many patients do develop symptoms, including the classic triad of diaphoresis, palpitations, and headache, or even life‐threatening cardiovascular emergencies such as myocardial infarctions, cardiomyopathy, and stroke. These effects of catecholamine oversecretion can cause significant perioperative morbidity and mortality. Early reports suggest surgical mortality rates of 30% to 45%; however, with current medical management and new surgical techniques, the surgical mortality rate is significantly improved at 0% to 2.9%.2 This review describes the perioperative management of PCC/PGL and the association of PCC/PGL with 10 well‐characterized genetic mutations.
Epidemiology
PCCs/PGLs have an estimated incidence of 2 to 8 per million.3 PCCs/PGLs are the cause of hypertension in 0.2% to 0.6% of patients and are present in 4% of adrenal incidentalomas.4 PCCs/PGLs may be underdiagnosed, as shown by one autopsy study that found only 24 of 54 PCCs were diagnosed pre‐mortum.5 Although most PCCs/PGLs are benign, 10% to 15% of PCCs and 20% to 50% of PGLs are malignant.6, 7, 8 The true rate of malignancy is difficult to determine given the variable definition in the literature. The World Health Organization (WHO) definition of malignant PCC/PGL is the presence of distant metastases at sites where chromaffin tissue is not normally present.3 The most common sites of metastatic disease are lymph nodes, bones, liver, and lung. There are no reliable markers for malignant potential, although studies have found that increased size (>5 cm), extra‐adrenal location (regardless of tumor size), and the succinate dehydrogenase subunit B (SDHB) mutation carry a higher risk of malignancy.6, 7 The Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) is a histologic scoring system from 0 to 20 that was developed in 2002 to predict malignant potential.9 A score <4 denotes tumors that act clinically benign, while scores of ≥4 carry an increased risk of malignant potential. However, the PASS is not necessarily reliable as it is prone to great interobserver and intraobserver variability and should be used with caution.10
Genetics
Population‐based studies, mostly from European countries, suggest that up to 32% of PCCs/PGLs have a germline mutation in known susceptibility genes.11 At our US‐based PCC/PGL referral center, the practice is to send all patients with PCC/PGL for genetic testing, and our mutation detection rate is 41%.12 In patients with a positive family history of nonsyndromic PCC/PGL, mutation rates can be as high as 79%,13 and in patients with HNPGL, the mutation rate is 54%.14 There are currently 10 well‐characterized PCC/PGL susceptibility genes (Table 1). Three genes cause well known cancer susceptibility syndromes: NF1 (Neurofibromatosis type 1) is a GTPase that regulates Ras in the MAPK pathway, VHL (von Hippel‐Lindau disease) has ubiquitin ligase activity and regulates HIFα in the hypoxia pathway, and RET (Multiple Endocrine Neoplasia Type 2) is a transmembrane tyrosine kinase receptor that signals through the PI3K pathway.11 In addition, mutations in any of the succinate dehydrogenase (SDH) complex (complex II of the mitochrondrial respiratory chain) subunits can increase susceptibility of PCC/PGL including SDH subunits (SDHA, 15 SDHB, 16 SDHC, 17 SDHD 18) and the SDH cofactor gene, SDHAF2, also known as SDH5.19 Since 2010, two more susceptibility genes have been identified. TMEM127 encodes a transmembrane protein in the early endosome and is involved in the mTOR pathway,20 and MAX encodes a transcription factor that heterodimerizes with Myc to control downstream gene transcription.21
Table 1.
Pheochromocytoma and Paraganglioma Susceptibility Genes
| Gene | Function | PCC/PGL Location | Risk of Malignancy | Biochemical Association |
|---|---|---|---|---|
| NF1 | GTPase | Adrenal PCC | Intermediate | NE, NM, E, M |
| RET | Tyrosine kinase receptor | Adrenal PCC | Low | E, M |
| VHL | Ubiquitin ligase activity |
Adrenal PCC (often bilateral) |
Low | NM, NE |
| SDHA | Succinate dehydrogenase subunit | Unknown | Unknown | Unknown |
| SDHB | Succinate dehydrogenase subunit |
Extra‐adrenal PGL Adrenal PCC HNPGL |
High | DA/MT, NM, M |
| SDHC | Succinate dehydrogenase subunit | HNPGL | Low | Nonsecretory |
| SDHD | Succinate dehydrogenase subunit |
HNPGL Extra‐adrenal PGL Adrenal PCC |
Low | Nonsecretory, DA/MT, NM, M |
| SDHAF2 | Co‐factor for succinate dehydrogenase complex |
HNPGL (multiple) |
Low | Nonsecretory |
| TMEM127 | Transmembrane protein |
Adrenal PCC HNPGL Extra‐adrenal PGL |
Low | Unknown |
| MAX | Transcription factor | Adrenal PCC (often bilateral) |
Unknown (possibly intermediate) |
Unknown |
Abbreviations: DA, dopamine; E, epinephrine; HNPGL, head and neck paraganglioma; M, metanephrine; MT, methoxytyramine; NE, norepinephrine; NM, normetanephrine; PCC, pheochromocytoma; PGL, paraganglioma.
There are phenotype/genotype correlations between each susceptibility gene and the location of PCC/PGL (Table 1). Mutations in NF1, VHL, and RET usually cause adrenal‐based PCC, with rare PGL reported in some cases.11 Mutations in SDHC, SDHD, and SDHAF2 lead to HNPGLs.19, 22, 23, 24 Mutations in SDHB lead most commonly to extra‐adrenal PGL; however, SDHB (and SDHD) mutation carriers can develop multiple primary PCC/PGL in any location, including within the adrenal gland.22 TMEM127 mutations were first believed to be associated with adrenal PCC only but have since been found in tumors in all locations.20, 25, 26 MAX‐associated tumors seem to be more common in the adrenal gland and tend to be bilateral.21 Early work suggests tumors with MAX mutations also have a higher rate of metastatic potential (~10%) than most other susceptibility genes (<5%) aside from SDHB.21 The risk of malignancy with SDHB mutations varies in the literature from 31% to 71%,22, 27 but a recent meta‐analysis suggests that the risk may be less than previously appreciated (~13%–23%) when accounting for incident and prevalent cases.28 Clinical genetic testing for patients with PCC/PGL is of utmost importance given the different risks associated with the development of multiple primary tumors, recurrence, or metastastic disease. No formal guidelines exist for surveillance of patients with PCC/PGL or for screening in unaffected mutation carriers, but we11 and others29, 30 have published expert opinion recommendations.
Catecholamines
Catecholamines are derived from the amino acid tyrosine, and the synthesis pathway is well defined (Figure). The catecholamines epinephrine and norepinephrine have a short half‐life of only a few minutes when circulating in the blood. Epinephrine and norepinephrine are degraded by methylation through catechol‐O‐methyltransferase to metanephrine and normetanephrine, respectively, or by deamination through monoamine oxidase to vanillymandelic acid (Figure 1).
Figure 1.
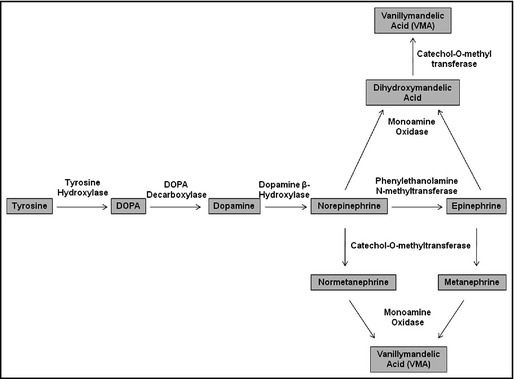
Synthesis and metabolism of catecholamines.
Screening for PCC/PGL relies on biochemical testing for plasma‐free metanephrines and/or urinary fractionated metanephrines and catecholamines. Plasma‐free metanephrines have at least equal sensitivity and specificity (98% and 92%, respectively) as 24‐hour urine collections and are more convenient for patients.31 It is important to note that catecholamine measurements (and, to a lesser degree, metanephrines) are likely to be falsely elevated as a result of interfering medications.32 In addition, urinary dopamine is not a reliable measurement because much of the excreted dopamine is made locally in the kidney. Interestingly, different heritable mutations are associated with different patterns of catecholamine production. VHL‐associated PCCs/PGLs have low expression of phenylethanolamine‐N‐methyltransferase (PNMT) and, therefore, these tumors have excessive production of the precursor molecules normetanephrine and norepinephrine, rather than the metabolites metanephrine and epinephrine.33 On the other hand, tumors with RET mutations often overexpress PNMT and, therefore, have high levels of epinephrine.33 NF1‐associated PCC have elevation of both epinephrine and metanephrine, while SDHB‐associated tumors have a normetanephrine and dopamine predominance.33 If patients are not tested for plasma dopamine (or methoxytyramine, a metabolite of dopamine), oversecretion can be missed. Methyoxytyramine levels are not yet commercially available. Tumors with SDHC, SDHD, and SDHAF2 are often nonsecretory since they are most commonly derived from parasympathetic ganglia in the head and neck. In fact, if biochemical testing results show high levels of catecholamines in patients with known HNPGL, then the presence of another primary tumor in the abdomen or pelvis should be considered and investigated. The biochemical profiles of tumors associated with mutations in TMEM127, MAX, and SDHA have not been well‐defined.
There are no established biomarkers for metastatic PCC/PGL. Plasma‐free metanephrines and urinary fractionated metanephrines and catecholamines are followed for disease recurrence (either a new primary tumor, local recurrence, or metastatic disease). In addition, although nonspecific and not always sensitive, chromogranin A has been shown in some studies to correlate with tumor size and malignancy.34 Initial work on the dopamine metabolite methoxytyramine suggests that it may correlate with the presence of metastases in patients with or without SDHB mutations.35 More research is needed to discover reliable biomarkers to monitor patients with metastatic disease.
Perioperative Management
Surgical resection is the treatment of choice for PCC/PGL, but the hypersecretion of catecholamines can be life‐threatening perioperatively. The physical stress of intubation, surgical incision, and the manipulation of the tumor mass can precipitate a hypertensive crisis, and profound hypotension with shock can occur after tumor resection because of the acute removal of catecholamines.36 Anesthesiologists need to be prepared for hemodynamic swings intraoperatively with close arterial blood pressure monitoring, availability of rapid‐acting vasodilators, and aggressive volume resuscitation. In addition, improved outcomes have been associated with laprascopic surgery for PCC compared with open procedures.37, 38
Risk factors for hemodynamic instability include large tumors >3 to 4 cm, higher catecholamine levels, uncontrolled blood pressure, or orthostatic hypotension preoperatively.2 Therefore, medical management, including perioperative blockade, is believed to be important to lower morbidity and mortality of PCC/PGL resection. The data on perioperative blockade are not robust, and, in fact, no standardized guidelines exist. Many different medical regimens are used to control the effects of catecholamine hypersecretion including the use of α‐blockers, calcium channel blockers, and tyrosine hydroxylase inhibitors (Table 2). Since no randomized trial to determine safety, efficacy, or adverse events currently exists, mostly small retrospective analyses or case reports in the literature serve as data to support decision making for perioperative management in patients with PCC/PGL.
Table 2.
Common Medications for Perioperative Blockade of Patients With Pheochromocytoma and Paraganglioma
| Drug | Action | Characteristics | Common Dosing | Common Side Effectsa |
|---|---|---|---|---|
| Phenoxybenzamine | Nonselective α1‐ and alpha2‐blocker | Noncompetitive antagonist | 10 mg 2–3×daily (maximium 60 mg per day) | Orthostasis, nasal congestion |
| Doxazosin | Selective α1‐blocker | Competitive antagonist | 2–4 mg 2–3×daily | Orthostasis, dizziness |
| Prazosin | Selective α1‐blocker | Competitive antagonist | 1–2 mg twice daily | Orthostasis, dizziness |
| Terazosin | Selective α1‐blocker | Competitive antagonist | 1–4 mg once daily | Orthostasis, dizziness |
| Nicardipine | Calcium channel blocker | Dihydropyridine long‐acting | 30 mg twice daily | Headache, edema, vasodilatation |
| Amlodipine | Calcium channel blocker | Dihydropyridine long‐acting | 5–10 mg daily | Headache, edema, palpitations |
| Metyrosine | Tyrosine hydroxylase inhibitor | Decreases catecholamine production | 250–500 mg 4× daily (dose‐escalated every 2 days) | Severe lethargy, extrapyramidal neurological side effects and gastrointestinal upset |
| Metoprolol | Selective β1‐blocker | Used to treat reflex tachycardia only after full α‐blockade achieved | 25–50 mg 1–2× daily | Fatigue, dizziness |
Many common side effects are expected and are suggestive of complete perioperative blockade. If possible and when appropriate, these side effects should be managed without dose reduction.
α‐Blockers are the mainstay of perioperative management in patients with PCC/PGL. In response to excess catecholamine secretion, α receptor activation leads to severe vasoconstriction, which can cause hypertension, arrhythmia, or even myocardial infarction. Competitive and noncompetitive α‐blockers are used in perioperative management. Phenoxybenzamine is a noncompetitive inhibitor that covalently binds to α1 and α2 receptors. Therefore, the inhibition of receptors by phenoxybenzamine is not easily overcome by the extra release of catecholamines intraoperatively with manipulation of the tumor mass as de novo synthesis of α receptors is required. This helps to lower the risk of intraoperative hypertensive crisis; however, this irreversible binding can lead to hypotension after the tumor is resected, thereby removing the source of catecholamine production. Vassopressor support and intravenous fluids may be required for 24 to 48 hours postoperatively to maintain blood pressure. Side effects of phenoxybenzamine include orthostasis and nasal congestion.
Other α‐blockers used include the selective α1 receptor blockers doxazosin, terazosin, and prazosin. These are competitive inhibitors with relatively shorter durations of action and therefore can be overcome by the extra catecholamine release intraoperatively, which can lead to hypertensive crisis. However, the shorter half‐life makes these inhibitors less likely to result in hypotension after tumor removal.
Other antihypertensive medications are used occasionally in the perioperative blockade. Calcium channel blockers, such as nicardipine or amlodipine, work by inhibiting norepinephrine‐mediated transmembrane calcium influx into smooth muscle. Some physicians prefer the use of calcium channel blockers given the cardiac and renal protective effects. β‐Blockers should never be used alone in patients with PCC/PGL because the unopposed α‐adrenergic effect can cause severe vasoconstriction leading to hypertensive crisis. β‐Blockers, such as the selective β1 antagonist metoprolol, do have a role in perioperative blockade after full α‐blocking has been achieved as a known side effect of a full α‐blockade is reflex tachycardia. The tachycardia induced by the alpha blockade is a desired side effect indicating that complete α‐blockade has been achieved. At this point, β‐blockers can be added to reduce the tachycardia.
A tyrosine hydroxylase inhibitor called metyrosine is sometimes used to control excessive catecholamine production. Tyrosine hydroxylase is involved in the first step of the synthesis of catecholamines, and inhibition of this enzyme leads to decreased production of all catecholamine precursors and metabolites. The medication is expensive but can offer significant hemodynamic stability in patients perioperatively, as the lack of excess catecholamine production will prevent the potential intraoperative hypertension and hypotension experienced before and after tumor mass resection.39, 40 There are also major side effects associated with metyrosine, including severe lethargy, extra‐pyramidal neurological side effects, and gastrointestinal upset.
Several retrospective studies examine the different types of perioperative blockade with varying results. The largest study is a retrospective series comparing the perioperative management algorithm used at the Mayo Clinic vs the one used at the Cleveland Clinic.41 Patients with PCC/PGL at the Mayo Clinic were treated with phenoxybenzamine for 1 to 4 weeks prior to surgery and were dosed to orthostatic hypotension to ensure full α‐blockade. β‐Blockers were added if the heart rate was >80 beats per minute and a calcium channel blocker was used if the patient was still hypertensive. In addition, metyrosine was added in a few patients with very large tumors. Patients with PCC/PGL at the Cleveland Clinic were treated with doxazosin often with the addition of a calcium channel blocker. β‐Blockade was added if tachycardia occurred. The retrospective analysis showed that the phenoxybenzamine regimen resulted in a trend for shorter duration of severe intraoperative hypertension but was associated with more postoperative hypotension. In fact, 56% of patients treated with pheyoxybenzamine required phenylephrine support vs 27% of patients treated with doxazosin. Despite this, no differences between treatment regimens were found in terms of postoperative surgical outcomes or in the length of hospital stay.41 Of course, there are many confounding variables in this retrospective analysis including comparing techniques at two different institutions with different surgeons, patient populations, and intraoperative care.
Other single‐center studies found no differences in perioperative regimens. One study retrospectively compared the efficacy of phenoxybenzamine (n=21) with doxazosin (n=17) and prazosin (n=11) without any identified differences in intraoperative hypertension, postoperative blood pressure, or volume‐replacement needs.42 Another small retrospective study examined 8 patients treated with phenoxybenzamine compared with 27 patients treated with doxazosin and also found no difference between preoperative and intraoperative hemodyamic parameters.43
At our own institution, our practice is to manage all preoperative blockade as an outpatient and use phenoxybenzamine for at least 2 weeks to control blood pressure perioperatively. The goal blood pressure is in the high normal range with systolic BP 120 to 140 mm Hg and diastolic BP 70 to 90 mm Hg. We expect to see a reflex tachycardia when full α‐blockade is achieved, and, if the heart rate is consistently above 100 beats per minute, we add a β‐blocker. We also add metyrosine for a period of 8 to 14 days to decrease catecholamine production to help control intraoperative hemodynamic swings. Dosing and duration of metyrosine depends on the level of the catecholamines with a tendency to use higher dosing and longer duration with tumors associated with very high catecholamine levels. Postoperative hypotension is often observed and can be an indication of complete and appropriate preoperative blockade. In this setting, we recommend giving intravenous fluids and vasopressor support with α agonists such as levophed as needed. In our experience, postoperative hypotension requiring vasopressor support and intravenous fluids rarely lasts more than 24 hours.
In a small retrospective study at our institution, we evaluated the perioperative management of 65 consecutive patients (43 women and 22 men) undergoing surgery for histologically proven PCC/PGL.44 The mean age of patients was 51±12 years, with 51 cases of adrenal PCC and 14 cases of extra‐adrenal PGL. Patients were prescribed phenoxybenzamine for 29±16 days and metyrosine for 14±2 days. Thirty of 65 patients received a β‐blocker preoperatively. Fifty‐six patients were admitted on the day of surgery, and 2 patients were admitted 1 to 2 days early. One patient had severe hypertension that had normalized by arrival in the emergency department and a second patient had symptomatic hypotension. Seven patients were admitted the night before for logistical reasons. The mean postoperative length of stay was 4.3±2.1 days and only 21 of 65 patients required postoperative pressor support. Of those 21, only 3 required more than 24 hours of pressor support. No patients experienced intraoperative or postoperative cardiovascular complications such as stroke or myocardial infarction.
Hypertensive crisis can occur in patients with known PCC/PGL or as the presenting sign in patients with no prior diagnosis. In these circumstances, we recommend controlling blood pressure using intravenous α‐blockade with a phentolamine drip. Other intravenous vasodilators such as sodium nitroprusside or nicardipine can also be used if needed.
Cardiovascular Effects of Medical Treatment for Malignant Pheochromocytoma
Metastatic disease occurs in about 25% of patients, and those with an SDHB mutation carry a substantially higher risk of malignancy compared with other known susceptibility genes.11 The presence of metastasis can occur at diagnosis of the primary tumor or even 20 years later, and patients with metastatic PCC/PGL have an overall 5‐year survival rate of 50%. The mainstays of treatment include surgical debulking, 131I‐MIBG, and systemic chemotherapy regimens including cytoxan, vincristine, and decarbazine.6 New treatment options with targeted therapies including tyrosine kinase inhibitors, mTOR inhibitors, and other receptor blockers are being actively explored.6 All of these treatment options have associated side effects, many of which are cardiovascular, including severe hypertension, and can make management of these cases very challenging.
Conclusions
Although rare, PCCs/PGLs are more commonly associated with germline mutations than any other solid tumor type. As a result of increased availability of genetic testing, more patients and families are being identified as mutation carriers and require lifelong screening for these potentially life‐threatening tumors. Once diagnosis has been established, there are various regimens to manage perioperative blockade, which, in combination with improved surgical techniques, appear to reduce morbidity and mortality in the perioperative period. Patients need lifelong surveillance because of risk of recurrence and malignancy. Patients who develop metastatic disease may require chronic hypertension management that can be complicated by the cardiovascular side effects associated with the treatments for metastatic disease.
Disclosure
The authors have no conflicts of interest to disclose. Funding support to L.F. was received from the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through grant No. KL2TR000139.
J Clin Hypertens (Greenwich). 2013;15:428–434©2013 Wiley Periodicals, Inc.23730992
References
- 1. Cohen DL, Fraker D, Townsend RR. Lack of symptoms in patients with histologic evidence of pheochromocytoma: a diagnostic challenge. Ann N Y Acad Sci. 2006;1073:47–51. [DOI] [PubMed] [Google Scholar]
- 2. Bruynzeel H, Feelders RA, Groenland TH, et al. Risk factors for hemodynamic instability during surgery for pheochromocytoma. J Clin Endocrinol Metab. 2010;95:678–85. [DOI] [PubMed] [Google Scholar]
- 3. DeLellis RA, Lloyd RV, Heitz PU, et al. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Endocrine Organs. Lyon, France: IARC Press; 2004. [Google Scholar]
- 4. Kasperlik‐Zaluska AA, Roslonowska E, Slowinska‐Srzednicka J, et al. 1,111 patients with adrenal incidentalomas observed at a single endocrinological center: incidence of chromaffin tumors. Ann N Y Acad Sci. 2006;1073:38–46. [DOI] [PubMed] [Google Scholar]
- 5. Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50‐year autopsy series. Mayo Clin Proc. 1981;56:354–60. [PubMed] [Google Scholar]
- 6. Parenti G, Zampetti B, Rapizzi E, et al. Updated and new perspectives on diagnosis, prognosis, and therapy of malignant pheochromocytoma/paraganglioma. J Oncol. 2012;2012:872713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ayala‐Ramirez M, Feng L, Johnson MM, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. 2011;96:717–25. [DOI] [PubMed] [Google Scholar]
- 8. Goffredo P, Sosa JA, Roman SA. Malignant pheochromocytoma and paraganglioma: a population level analysis of long‐term survival over two decades. J Surg Oncol. 2012; Dec 11. [DOI] [PubMed] [Google Scholar]
- 9. Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551–66. [DOI] [PubMed] [Google Scholar]
- 10. Wu D, Tischler AS, Lloyd RV, et al. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol. 2009;33:599–608. [DOI] [PubMed] [Google Scholar]
- 11. Fishbein L, Nathanson KL. Pheochromocytoma and paraganglioma: understanding the complexities of the genetic background. Cancer Genet. 2012;205:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fishbein L, Merrill S, Fraker D, et al. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol. 2012. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cascon A, Pita G, Burnichon N, et al. Genetics of pheochromocytoma and paraganglioma in Spanish patients. J Clin Endocrinol Metab. 2009;94:1701–5. [DOI] [PubMed] [Google Scholar]
- 14. Burnichon N, Rohmer V, Amar L, et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab. 2009;94:2817–27. [DOI] [PubMed] [Google Scholar]
- 15. Burnichon N, Briere JJ, Libe R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–70. [DOI] [PubMed] [Google Scholar]
- 18. Baysal BE, Ferrell RE, Willett‐Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–51. [DOI] [PubMed] [Google Scholar]
- 19. Bayley JP, Kunst HP, Cascon A, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010;11:366–72. [DOI] [PubMed] [Google Scholar]
- 20. Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Comino‐Mendez I, Gracia‐Aznarez FJ, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43:663–7. [DOI] [PubMed] [Google Scholar]
- 22. Ricketts CJ, Forman JR, Rattenberry E, et al. Tumor risks and genotype‐phenotype‐proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat. 2010;31:41–51. [DOI] [PubMed] [Google Scholar]
- 23. Schiavi F, Savvoukidis T, Trabalzini F, et al. Paraganglioma syndrome: SDHB, SDHC, and SDHD mutations in head and neck paragangliomas. Ann N Y Acad Sci. 2006;1073:190–7. [DOI] [PubMed] [Google Scholar]
- 24. Kunst HP, Rutten MH, de Monnink JP, et al. SDHAF2 (PGL2‐SDH5) and hereditary head and neck paraganglioma. Clin Cancer Res. 2011;17:247–54. [DOI] [PubMed] [Google Scholar]
- 25. Neumann HP, Sullivan M, Winter A, et al. Germline Mutations of the TMEM127 Gene in Patients with Paraganglioma of Head and Neck and Extraadrenal Abdominal Sites. J Clin Endocrinol Metab. 2011;96:E1279–E1282. [DOI] [PubMed] [Google Scholar]
- 26. Yao L, Schiavi F, Cascon A, et al. Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA. 2010;304:2611–9. [DOI] [PubMed] [Google Scholar]
- 27. Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812–8. [DOI] [PubMed] [Google Scholar]
- 28. van Hulsteijn LT, Dekkers OM, Hes FJ, et al. Risk of malignant paraganglioma in SDHB‐mutation and SDHD‐mutation carriers: a systematic review and meta‐analysis. J Med Genet. 2012;49:768–76. [DOI] [PubMed] [Google Scholar]
- 29. Amar L, Fassnacht M, Gimenez‐Roqueplo AP, et al. Long‐term postoperative follow‐up in patients with apparently benign pheochromocytoma and paraganglioma. Horm Metab Res. 2012;44:385–9. [DOI] [PubMed] [Google Scholar]
- 30. Gimenez‐Roqueplo AP, Caumont‐Prim A, Houzard C, et al. Imaging Work‐Up for Screening of Paraganglioma and Pheochromocytoma in SDHx Mutation Carriers: a Multicenter Prospective Study from the PGL.EVA Investigators. J Clin Endocrinol Metab. 2013;98:E162–73. [DOI] [PubMed] [Google Scholar]
- 31. Eisenhofer G, Siegert G, Kotzerke J, et al. Current progress and future challenges in the biochemical diagnosis and treatment of pheochromocytomas and paragangliomas. Horm Metab Res. 2008;40:329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eisenhofer G, Goldstein DS, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true‐ from false‐positive test results. J Clin Endocrinol Metab. 2003;88:2656–66. [DOI] [PubMed] [Google Scholar]
- 33. Eisenhofer G, Lenders JW, Timmers H, et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem. 2011;57:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bilek R, Safarik L, Ciprova V, et al. Chromogranin A, a member of neuroendocrine secretory proteins as a selective marker for laboratory diagnosis of pheochromocytoma. Physiol Res. 2008;57(Suppl 1):S171–9. [DOI] [PubMed] [Google Scholar]
- 35. Eisenhofer G, Lenders JW, Siegert G, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2012;48:1739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mannelli M, Dralle H, Lenders JW. Perioperative management of pheochromocytoma/paraganglioma: is there a state of the art? Horm Metab Res. 2012;44:373–8. [DOI] [PubMed] [Google Scholar]
- 37. Agarwal G, Sadacharan D, Aggarwal V, et al. Surgical management of organ‐contained unilateral pheochromocytoma: comparative outcomes of laparoscopic and conventional open surgical procedures in a large single‐institution series. Langenbecks Arch Surg. 2012;397:1109–16. [DOI] [PubMed] [Google Scholar]
- 38. Dickson PV, Alex GC, Grubbs EG, et al. Posterior retroperitoneoscopic adrenalectomy is a safe and effective alternative to transabdominal laparoscopic adrenalectomy for pheochromocytoma. Surgery. 2011;150:452–8. [DOI] [PubMed] [Google Scholar]
- 39. Perry RR, Keiser HR, Norton JA, et al. Surgical management of pheochromocytoma with the use of metyrosine. Ann Surg. 1990;212:621–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Steinsapir J, Carr AA, Prisant LM, Bransome ED Jr. Metyrosine and pheochromocytoma. Arch Intern Med. 1997;157:901–6. [PubMed] [Google Scholar]
- 41. Weingarten TN, Cata JP, O'Hara JF, et al. Comparison of two preoperative medical management strategies for laparoscopic resection of pheochromocytoma. Urology. 2010;76:508–e6. [DOI] [PubMed] [Google Scholar]
- 42. Kocak S, Aydintug S, Canakci N. Alpha blockade in preoperative preparation of patients with pheochromocytomas. Int Surg. 2002;87:191–4. [PubMed] [Google Scholar]
- 43. Prys‐Roberts C, Farndon JR. Efficacy and safety of doxazosin for perioperative management of patients with pheochromocytoma. World J Surg. 2002;26:1037–42. [DOI] [PubMed] [Google Scholar]
- 44. Richardson KM, Cohen DL. Safety and efficacy of outpatient preoperative alpha blockade of pheochromocytoma. Endocr Rev. 2010;31(3 suppl 1):12–56. [Google Scholar]