

SUMMARY
Proper bipolar attachment of sister kinetochores to the mitotic spindle is critical for accurate chromosome segregation in mitosis. Here we show an essential role of the formin mDia3 in achieving metaphase chromosome alignment. This function is independent of mDia3 actin nucleation activity, but is attributable to EB1-binding by mDia3. Furthermore, the microtubule binding FH2 domain of mDia3 is phosphorylated by Aurora B kinase in vitro, and cells expressing the nonphosphorylatable mDia3 mutant cannot position chromosomes at the metaphase plate. Purified recombinant mDia3 phosphorylated by Aurora B exhibits reduced ability to bind microtubules and stabilize microtubules against cold-induced disassembly in vitro. Cells expressing the phosphomimetic mDia3 mutant do not form stable kinetochore microtubule fibers; despite they are able to congress chromosomes to the metaphase plate. These findings reveal a key role for mDia3 and its regulation by Aurora B phosphorylation in achieving proper stable kinetochore microtubule attachment.
INTRODUCTION
Mammalian diaphanous-related formins (mDia) constitute a subfamily of Rho GTPase-binding formin homology (FH) proteins (Higgs and Peterson, 2005; Rivero et al., 2005). mDia formins nucleate and assemble unbranched actin structures through their FH2 domain, which forms a tethered dimer with two antiparallel actin binding domains (Xu et al., 2004). Formins are implicated in numerous actin-based cellular functions, including cytokinesis, cell morphogenesis, cell polarity, and cell migration (Goode and Eck, 2007). Several years ago, mDia formins were found to be involved in regulating microtubule-dependent processes. In migrating fibroblast, mDia stabilizes a subset of microtubules downstream of Rho signaling (Palazzo et al., 2001b), and this stabilization function is essential for cell polarization (Cook et al., 1998). Overexpression of a constitutively active mDia without the autoregulatory domains or activation of endogenous mDia with the expression of an mDia-autoinhibitory domain is sufficient to induce the formation of stable microtubules in serum-starved NIH 3T3 fibroblasts (Palazzo et al., 2001a). These stable microtubules are capped and oriented toward the wound edge (Palazzo et al., 2001a). Although the molecular mechanism of microtubule stabilization downstream of Rho-mDia signaling is still unknown, two microtubule-associated proteins, adenomatous polyposis coli (APC) and EB1, have been found to be involved in this process. mDia forms a complex with APC and EB1 and may function as a scaffold protein at cell cortex for EB1 and APC to stabilize microtubules and promote cell migration (Wen et al., 2004). Furthermore, a recent study reported that two actin nucleation mutants in a constitutively active version of mDia2 can still induce stable microtubules and bind to EB1 and APC (Bartolini et al., 2008), arguing that mDia formins are able to stabilize microtubules independent of their actin nucleation activity. Purified FH1FH2-mDia2 proteins without autoregulatory domains can directly bind to microtubules in vitro and stabilize microtubules against cold- and dilution-induced disassembly (Bartolini et al., 2008).
In mitosis, chromosomes capture microtubules through a “search and capture” process (Kirschner and Mitchison, 1986), in which proper kinetochore microtubule attachment is stabilized and improper chromosome microtubule attachment is destabilized. Numerous proteins, including motors and microtubule associated proteins, have been implicated in stable kinetochore microtubule attachment, though the precise functions of the majority of these proteins and the pathways that regulate them remain unclear (Cleveland et al., 2003; Joglekar et al., 2010; Walczak and Heald, 2008). An earlier report has suggested that formin mDia3 may also play a role in this process by acting under control of Cdc42 to regulate kinetochore microtubule attachment (Yasuda et al., 2004). HeLa cells treated with toxin B, which inactivates all Rho GTPases including Rho, Rac, and Cdc42, or depleted of endogenous mDia3 with siRNA fail to align all chromosomes at the metaphase plate (Yasuda et al., 2004). Further, immunoprecipitation analysis of mitotic cells has revealed that mDia3 binds to CENP-A at kinetochores (Yasuda et al., 2004). On the basis of these findings, the authors proposed that the Cdc42-mDia3 pathway may regulate spindle microtubule attachment and metaphase chromosome alignment. The chromosome misalignment phenotype can be caused by a number of different mechanisms, such as improper initial microtubule capture, failure to maintain biorientation, unstable and/or improper kinetochore microtubule attachment, and inability to achieve chromosome congression. Whether Cdc42-mDia3 affects any or all of these steps remains unresolved.
To test how mDia3 participates in kinetochore microtubule attachment, we now use mammalian cultured cells and purified components to establish that mDia3 microtubule binding activity and mDia3 interaction with EB1 play a key role to enhance the force generation between sister kinetochores on spindle microtubule attachment, and this function is directly regulated by Aurora B phosphorylation in response to tension between sister kinetochores.
RESULTS
mDia3, but Not Its Actin Nucleation Activity, Is Essential for Metaphase Chromosome Alignment
To understand the molecular mechanism of how mDia3 may affect metaphase chromosome alignment, we transfected a small interfering RNA (siRNA) duplex targeted to a region of human mDia3 into T98G cells, resulting in a clear reduction of mDia3 protein level (Figure 1A, lane 2) and loss of mDia3 at kinetochores by immunofluorescence microscopy (Figure 1B). To specifically test for effects of mDia3 siRNA on metaphase chromosome alignment, we treated cells with nocodazole and MG132 for 4 hr to accumulate mitotic cells and then released them into MG132 alone for 1 hr (to prevent possible premature anaphase onset) (Figure 1C). Only 32% of mDia3-depleted cells showed full chromosome alignment at the metaphase plate compared to 69% for controls with scrambled siRNA (Figures 1D and 1E). Coexpression of siRNA resistant myc-mDia3 to a level comparable to that of endogenous mDia3 (Figure 1A) was able to rescue the chromosome alignment defect (Figure 1E). These results support a role for mDia3 in metaphase chromosome alignment.
Figure 1. mDia3, but Not Its Actin Nucleation Activity, Is Essential for Metaphase Chromosome Alignment.

(A) Immunoblotting of lysates of T98G cells 48 hr posttransfection with the indicated siRNA and siRNA-resistant (siRNA-resis) myc-mDia3 expression vectors as indicated.
(B) Immunofluorescence detection of ACA, mDia3, and merged with DAPI in mitotic T98G cells 48 hr posttransfected with mock or mDia3 siRNA as indicated. ACA, magenta and mDia3, green in merged panels. Inset: colocalization of mDia3 and ACA at a pair of sister kinetochores.
(C) Schematic representation of mDia3 siRNA, rescue transfection, and chemical inhibitor treatment protocol.
(D) Knockdown of mDia3 on siRNA treatment resulted in metaphase chromosome alignment defects. Fixed cells on treatment detailed in (B) were stained with an anti-centromeric antibody (ACA, red), tubulin (green), and DNA (DAPI, blue).
(E) The actin nucleation activity is not required for mDia3 function in metaphase chromosome alignment. The percentage of mitotic cells with different degrees of chromosome alignment analyzed in cells transfected with siRNA and the myc-mDia3 constructs are indicated. Cells were characterized as fully aligned, class I unaligned (majority of the chromosomes aligned with one or two unaligned, indicated by arrow in D), and class II unaligned (with more than three unaligned chromosomes). Data represent the mean percentage of cells (mean ± standard deviation [SD], n ≥ 50 cells for each of three independent transfections).
To further document the roles of mDia3 during mitosis, unperturbed mitotic progression was followed by time-lapse analysis of chromosome behavior in HeLa cells stably expressing fluorescent histone (H2B-EYFP). Imaging mitosis in mDia3-depleted cells, but not in control cells, revealed mitotic errors, including a delay in anaphase onset and the presence of unaligned chromosomes (see Figure S1A available online), consistent with a role of mDia3 in metaphase chromosome alignment.
As mDia3 is an actin nucleator, an obvious question is whether this aspect of mDia3 function contributes to the mitotic phenotype? Alanine substitutions at Ile1431 or Lys1601 completely abolished the actin-nucleating activity of the yeast formin Bni1p (Xu et al., 2004) and corresponding mDia2 mutations (I704A and K853A) were found to be similarly defective in actin nucleation, with no effect on microtubule binding and stabilization activities (Bartolini et al., 2008). We introduced corresponding I726A and K875A mutations in the mDia3 siRNA-resistant construct and tested the ability of each mutant form to rescue the chromosome misalignment phenotype in cells depleted of endogenous mDia3. The actin nucleation-deficient constructs rescued the chromosome alignment defect as well as the wild-type mDia3 construct (Figure 1E), strongly indicating that the chromosome misalignment phenotype in mDia3-depleted cells is independent of the actin binding or nucleation activity of mDia3. Overexpression of a constitutively active form of wild-type mDia3, but not the actin nucleation-deficient mutants, was able to induce actin fibers in cultured cells, confirming the actin polymerization defect of those mutants (Figure S1B).
Stability of Kinetochore-Microtubule Connections Is Compromised in mDia3 Knockdown Cells
To determine whether the chromosome alignment defect in mDia3-depleted cells is due to lack of stable kinetochore-microtubule attachment, the presence of cold-stable kinetochore microtubule bundles was examined. In mock-depleted cells, cold treatment revealed clear stable kinetochore microtubule bundles (Figures 2A and 2B). In contrast, mDia3-depleted cells exhibited substantially fewer cold-stable microtubules with a majority of kinetochores unattached from microtubules after 10 min of cold treatment (Figures 2A and 2B). Cold-resistant stable kinetochore microtubules were restored in cells expressing siRNA-resistant wild-type mDia3 (Figures 2A and 2B). These results are consistent with a role for mDia3 in stable kinetochore-microtubule attachment.
Figure 2. The Cold Stability of Kinetochore-Microtubule Connections Is Reduced in mDia3 Knockdown Cells.

(A) T98G cells were transfected with the indicated siRNA and siRNA-resistant constructs. After 48 hr, cells were incubated at 4°C for the indicated times before they were fixed and processed for indirect immunofluorescence for tubulin (green) and ACA (magenta) antibodies.
(B) Quantification of microtubule density in (A). Average microtubule intensity (mean ± SD; n = 10 cells for each of three independent transfections, P < 0.005) was measured in each condition. Intensities are showed relative to total cellular areas and normalized against 0 min for each condition.
(C–E) mDia3 is not required for efficient kinetochore targeting of CENP-E, Hec1, and BubR1. Kinetochore localization of Hec1 (C), CENP-E (D), and BubR1 (E) in mitotic T98G cells 48 hr posttransfected with mock or mDia3 siRNA as indicated. Kinetochore proteins are shown in green in merged panels with ACA in magenta.
Kinetochore Assembly Is Not Affected in mDia3-Depleted Cells
Immunofluorescence analysis demonstrated that mDia3 only colocalized with ACA (an anticentromeric antibody used as a centromere/kinetochore marker) signals at prometaphase (Figure 1B) and metaphase cells, confirming the kinetochore localization of mDia3 (Yasuda et al., 2004). We then tested whether mDia3 is involved in recruitment or retention of other kinetochore proteins implicated in stable kinetochore-microtubule attachment and metaphase chromosome alignment. In vertebrate cells, several proteins have been implicated in this process: the Ndc80 complex (DeLuca et al., 2006; Martin-Lluesma et al., 2002), CENP-E (Kapoor et al., 2006; Wood et al., 1997), a kinetochore associated kinesin motor protein, and BubR1 (Lampson and Kapoor, 2005; Zhang et al., 2007), an essential mitotic checkpoint kinase. mDia3 siRNA efficiently and uniformly depleted mDia3 from kinetochores in cultured cells (Figure 1B). However, depletion of mDia3 by siRNA had no apparent effect on kinetochore association of Hec1 (a component of the Ndc80 complex), CENP-E, or BubR1 (Figures 2C–2E). In combination with the observation of robust mitotic arrest in mDia3-depleted cells using live cell imaging (Figure S1A), which requires an intact kinetochore to generate the checkpoint signal, these results indicate that the kinetochore structure is intact in mDia3-depleted cells and several other components of the microtubule attachment machinery are unaffected by mDia3 siRNA.
Interaction between mDia3 and EB1 Is Important for Metaphase Chromosome Alignment
Other forms of mDia have been shown to cooperate with EB1 and APC in the regulation of microtubule capture in different cell contexts. Microtubule capture at bud sites in budding yeast is regulated by the formin Bni1, the yeast ortholog of mDia formin (Kohno et al., 1996; Lee et al., 1999). The microtubule tip-binding protein Bim1 (the yeast ortholog of EB1) and Kar9 (speculated to represent a possible APC homolog) have been identified in this process (Adames and Cooper, 2000; Bloom, 2000; Schuyler and Pellman, 2001). An evolutionarily conserved pathway for microtubule capture at the leading edge of migrating fibroblasts has also been described, in which mDia2 functions as a scaffold protein for EB1 and APC at the cell cortex (Wen et al., 2004). Coimmunoprecipitation analysis revealed that mDia3 was able to associate with EB1 and APC (Figure 3A, lane 3). To directly test whether mDia3-EB1 interaction is important for metaphase chromosome alignment, we generated an mDia3 mutant, G (YEDR)-mDia3, in which Tyr735, Glu736, Asp737, and Arg739 in a conserved region adjacent to the FH2 domain were substituted with Gly with Ile738 remaining intact. A corresponding G(YEKR)-mDia2 mutant has been shown to be unable to bind to EB1, but can still bind to APC (Wen et al., 2004). G(YEDR)-mDia3 indeed lost its ability to associate with EB1 (Figure 3A, lane 6). Replacing endogenous mDia3 with this mutant resulted in an increased number of cells with misaligned chromosomes (Figures 3B and 3C), implying that the mDia3-EB1 interaction is necessary for metaphase chromosome alignment.
Figure 3. Complex Formation of mDia3-EB1-APC Is Important for Metaphase Chromosome Alignment.
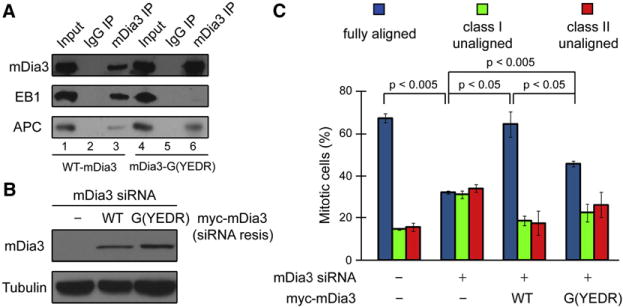
(A) mDia3 associates with EB1 and APC. mDia3 immunoprecipitates probed with mDia3 (top), EB1 (middle), or APC (bottom) antibodies as indicated.
(B and C) mDia3 and EB1 interaction is essential for metaphase chromosome alignment. (B) Immunoblotting of lysates of T98G cells 48 hr posttransfection with the indicated siRNA and siRNA-resistant (siRNA-resis) myc-mDia3 expression vectors as indicated. (C) The percentage of mitotic cells with different degrees of chromosome alignment analyzed in cells transfected with siRNA and the myc-mDia3 constructs are indicated. Cells were characterized as fully aligned, class I unaligned, and class II unaligned as described in Figure 1. Data represent the mean percentage of cells (mean ± SD, n ≥ 50 cells for each of three independent transfections).
Aurora B Kinase Phosphorylates mDia3 at Multiple Residues In Vitro
A majority of the microtubule associated proteins at the kinetochore are directly regulated by Aurora B kinase in achieving proper kinetochore microtubule attachment, such as the KMN network, a core kinetochore microtubule binding apparatus (Cheeseman et al., 2006; DeLuca et al., 2006; Welburn et al., 2010). Thus, we next tested the ability of Aurora B to phosphorylate mDia3 with a commercially available recombinant constitutively active Aurora B (Abcam, Cambridge, MA) and purified bacterial expressed recombinant mDia3 protein. Clear incorporation of (γ-32P)-Pi was observed (Figure 4A). Based on the consensus phosphorylation sequence for Aurora B (Cheeseman et al., 2002), we identified four potential phosphorylation sites in mDia3 (Figure 4B; Figure S2A). Two potential Aurora B phosphorylation sites, T66 and S196, are in the N-terminal autoinhibition regulatory domains of mDia3; whereas the other two, S820 and T882, are located in the FH2 domain. Using mass spectrometry, we detected phosphate incorporation at two of these sites, Ser196 and Thr882. Mutations of all of the four potential target residues to alanine (4A-mDia3) greatly reduced (γ-32P)-Pi incorporation in in vitro kinase assays with Aurora B (Figure 4A).
Figure 4. Aurora B Phosphorylates mDia3.

(A) Upper panel: Autoradiograph of SDS-PAGE gel of purified recombinant full-length wild-type and the nonphosphorylatable 4A-mDia3 mutant phosphorylated in the presence of (γ-32P)-ATP using purified Aurora B. Middle panel: Coomassie blue staining of purified mDia3 proteins. Bottom panel: immunoblot analysis with the pS196 phospho antibody.
(B) mDia3 protein structure showing the relative positions of GBD, DID, FH1, FH2, and DAD domains, and the Aurora B phosphorylation sites.
(C) mDia3 is phosphorylated by Aurora B in cultured cells. Interphase or mitotic cell lysates were probed with the mDia3 antibody (top panels) or the pS196 phospho antibody (bottom panels). The cell lysates were treated or untreated with λ protein phosphatase (PPase) as indicated.
(D–H) Immunofluorescence images acquired using the indicated antibodies including pS196 and mDia3 in untreated T98G cells or in cells treated with ZM447439 for 1 hr. In merged panels: ACA, magenta and mDia3, green.
(I) Expression of mDia3 nonphosphorylatable mutants resulted in metaphase chromosome misalignment. T98G cells were transfected with wild-type mDia3 or mDia3 phosphorylation mutants. Forty-eight hours posttransfection, cells were treated with nocodazole and MG132 for 4 hr and then released into MG132 alone for 1 hr. Cells were then fixed and stained with ACA, tubulin, and DNA. Cells were characterized as fully aligned, class I unaligned, and class II unaligned as described in Figure 1. Data represent the mean percentage of cells (mean ± SD, n ≥ 50 cells for each of three independent transfections).
To define the possible function of Aurora B phosphorylation of mDia3, we generated a rabbit polyclonal antibody specific for phosphorylated Ser196, which detected a strong signal of Aurora B phosphorylated wild-type mDia3, but not the 4A-mDia3 mutant, in vitro (Figure 4A). Immunoblotting of cell lysates with this phospho antibody showed a clear increase in phosphorylation of this site during mitosis (Figure 4C). Immuno-reactivity with the anti-phosphoSer196 was eliminated either by treating the lysates with λ protein phosphatase (Figure 4C), or by adsorption of the antibody with phospho peptide but not with the nonphospho peptide (Figure S2B). Immunofluorescence using this antibody in cultured cells revealed kinetochore staining that was substantially reduced by treatment with the Aurora B kinase inhibitor ZM447439 (compare Figures 4D and 4G). In contrast, an antibody that detects mDia3 regardless of its phosphorylation state did not display any obvious change in mDia3 level at the kinetochore on ZM447439 treatment (Figure 4H). Further, in cells with chromosomes aligned at the metaphase plate, we detected low mDia3 phosphorylation (Figure 4E), but normal mDia3 level (Figure 4F), at all kinetochores. Therefore, mDia3 is a substrate of Aurora B kinase in vivo, at least at S196, although we have not been able to confirm Aurora B phosphorylation of mDia3 at other three sites in vivo.
We next generated single and quadruple (4A-mDia3) non-phosphorylatable mDia3 mutants for these four potential Aurora B phosphorylation sites for phenotype analysis. These mutants were still able to associate with kinetochores (Figure S2C). Cells expressing each of the four single mutant constructs exhibited metaphase chromosome alignment defects and the misalignment phenotype became worse in cells with a multiply mutated 4A-mDia3 (Figure 4I). Replacing endogenous mDia3 with a non-phosphorylatable 4A-mDia3 mutants displayed similar chromosome misalignment phenotypes (Figure 4I). Further, live cell imaging analysis showed that cells expressing the 4A-mDia3 mutant were unable to align all chromosomes at the metaphase plate on release from monastrol treatment (Figure S2E), although they were able to form bipolar spindles (Figure S2F). These results suggest that the nonphosphorylatable mDia3 mutants can behave as dominant negative in T98G cells and affect metaphase chromosome alignment. Thus, Aurora B phosphorylation of mDia3 is essential for normal mitosis, possibly through regulation of both the activation of mDia3 (releasing from its autoinhibition) at T66 and S196 sites, and function of mDia3 FH2 domain at S820 and T882 sites.
Aurora B Phosphorylation Reduces mDia3 Ability to Stabilize Microtubules against Cold-Induced Depolymerization In Vitro
Two Aurora B phosphorylation sites, S820 and T882, are located within the FH2 domain, which has been shown to be important for mDia formin actin and microtubule activities (Bartolini and Gundersen, 2009), and possibly its interaction with EB1/APC (Wen et al., 2004). We have shown that mDia3 actin activity is not necessary for its function in mitosis (Figure 1E). To better understand how Aurora B phosphorylation affects mDia3 microtubule function and its interaction with EB1, we generated a constitutively active form of mDia3 that consists of both the FH1 and FH2 domains and lacks the regulatory GBD-DID and DAD domains (FH1FH2-mDia3) and introduced the nonphosphorylatable mutations (2A-FH1FH2-mDia3) and the phosphomimetic mutations (2E-FH1FH2-mDia3) in this fragment of mDia3 at S820 and T882 sites. A corresponding fragment of mDia2 has been shown to be able to bind directly to microtubules and stabilize microtubules against cold- and dilution-induced disassembly (Bartolini et al., 2008) and to interact with EB1/APC (Wen et al., 2004). In vitro binding assays revealed that both the nonphosphorylatable and phosphomimetic mutants can bind to EB1 as effectively as the wild-type mDia3 (Figure S3A), suggesting that Aurora B phosphorylation does not affect the ability of mDia3 to bind EB1.
Aurora B has been shown to function in destabilizing improperly attached kinetochore microtubules by phosphorylating kinetochore-associated microtubule interactors to reduce their affinity for microtubules in the absence of tension (Liu and Lampson, 2009). To test the effect of Aurora B phosphorylation on mDia3 microtubule activity, purified wild-type FH1FH2-mDia3 recombinant proteins were pre-treated with Aurora B kinase or not. Phosphorylated or nonphosphorylated mDia3 were then incubated with purified rhodamine-labeled microtubules assembled at 37°C, followed by incubation at 4°C to induce rapid microtubule depolymerization (Figure 5A, Buffer). The extent of microtubule depolymerization was visualized. When incubated with nonphosphorylated wild-type FH1FH2-mDia3 proteins, a substantial number of microtubules resisted cold depolymerization even after 10 min (Figure 5A, WT-mDia3), which is consistent with a role for mDia formins in microtubule stabilization. In contrast, incubation with the Aurora B pretreated and phosphorylated FH1FH2-mDia3 proteins resulted in a decreased number of microtubules even after 5 min of cold treatment (Figure 5A, WT-mDia3 + Aurora B). Further, purified recombinant phosphomimetic 2E-FH1FH2-mDia3 mutant proteins, but not the non-phosphorylatable 2A-FH1FH2-mDia3 mutant proteins, also exhibited reduced ability to stabilize microtubules against cold-induced disassembly (Figure 5A). The effects of these mDia3 phosphorylation mutants were not dependent on Aurora B phosphorylation (Figure S3B). We conclude that Aurora B phosphorylation of mDia3 at FH1FH2 domains affects mDia3 ability to stabilize microtubules against cold-induced depolymerization.
Figure 5. Aurora B Phosphorylation Regulates mDia3 Microtubule Binding and Stabilization Activities.
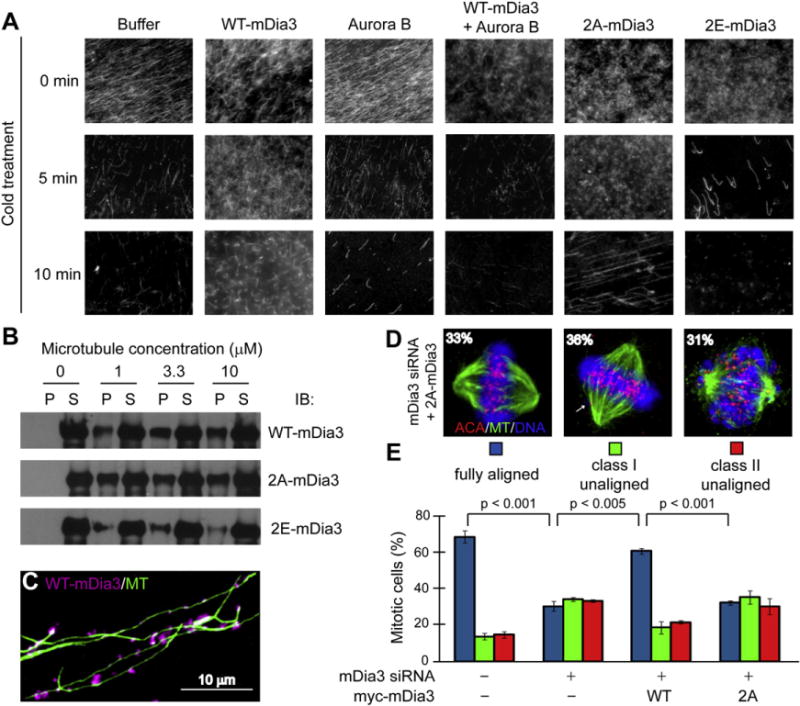
(A) Aurora B phosphorylation reduced mDia3 microtubule stabilization activity in vitro. Preassembled rhodamine-labeled microtubules were incubated with purified FH1FH2-mDia3, which is pretreated with or without an active Aurora B as indicated, or FH1FH2-mDia3 phospho mutants at 4°C and visualized at the indicated time.
(B and C) mDia3 binds directly to microtubules. (B) Immunoblotting analysis of the co-sedimentation of wild-type, 2A-, and 2E-FH1FH2-mDia3 with Taxol-assembled microtubules. (C) Immunostaining of wild-type FH1FH2-mDia3 (magenta) on Taxol-assembled microtubules (green).
(D and E) Cells expressing the nonphosphorylatable 2A-mDia3 mutant at FH2 domain cannot congress all chromosomes to the metaphase plate. Replacing endogenous mDia3 with siRNA-resistant 2A-mDia3 resulted in metaphase chromosome alignment defect. Fixed cells were stained with an anti-centromeric antibody (ACA, red), tubulin (green), and DNA (DAPI, blue). Cells were characterized as fully aligned, class I unaligned, and class II unaligned (D). In (E), data represent the mean percentage of cells (mean ± SD, n ≥ 50 cells for each of three independent transfections).
We next looked at the binding ability of purified recombinant mDia3 and phosphorylation mutants to purified microtubules. We incubated purified FH1FH2-mDia3, 2A-FH1FH2-mDia3, and 2E-FH1FH2-mDia3 with Taxol assembled microtubules, followed by high-speed centrifugation to separate microtubule pellets from soluble unbound proteins. Substantial amount of wild-type mDia3 was recovered in the pellet fraction only when microtubules were present (Figure 5B). Immunostaining also confirmed that wild-type mDia3 was localized along the length of microtubules (Figure 5C). Similar binding to microtubules was also observed for the nonphosphorylatable 2A-FH1FH2-mDia3 mutant; however, the phosphomimetic 2E-FH1FH2-mDia3 mutant had reduced ability to bind to microtubules (Figure 5B). These results are consistent with the microtubule stabilization activities against cold-induced depolymerization for mDia3 and the phosphorylation mutants.
Aurora B Phosphorylation of mDia3 FH2 Domain Is Essential for Metaphase Chromosome Alignment
To determine the role of Aurora B phosphorylation of mDia3 at FH2 domain in vivo, we generated a siRNA-resistant mutant mDia3 in which both Aurora B phosphorylation residues in the FH2 domain, S820 and T882, were mutated to alanine (myc-2A-mDia3). Chromosome alignment was analyzed by fixed cell immunofluorescence in which endogenous mDia3 was replaced with either myc-2A-mDia3 mutant or myc-wild-type mDia3 using the siRNA approach. The majority of cells (62%) expressing wild-type mDia3 contained a normal metaphase plate with fully aligned chromosomes (Figure 5E). In contrast, only 33% of scored cells expressing the 2A-mDia3 mutant had all chromosomes aligned at the metaphase plate (Figures 5D and 5E). Thirty-six percent of cells expressing the 2A-mDia3 had an incomplete metaphase plate with a few unaligned chromosomes (class I unaligned) and 31% of them did not have a visible metaphase plate (class II unaligned) (Figures 5D and 5E). These results demonstrate that the loss of Aurora B phosphorylation at mDia3 FH2 domain decreases the fidelity of metaphase chromosome alignment.
Nonphosphorylatable Form of mDia3 Is Important for Stable Kinetochore Microtubule Attachment
To better understand the function of mDia3 in kinetochore-microtubule attachment and metaphase chromosome alignment, we examined mitotic phenotypes in cells expressing the phosphomimetic 4E-mDia3 mutant using live cell microscopy. This revealed that majority of these cells were able to congress chromosomes to the metaphase plate; however, these cells stayed in a metaphase-like stage without anaphase onset much longer than cells expressing wild-type mDia3 (Figure 6A; Figure S4A). This result indicates that cells expressing the phosphomimetic 4E-mDia3 may lack proper stable kinetochore-microtubule attachment.
Figure 6. Cells Expressing the Phosphomimetic mDia3 Mutant Lack Stable Kinetochore Microtubule Attachment.

(A) Cells expressing the phosphomimetic 4E-mDia3 mutant cannot satisfy the mitotic checkpoint. Representative still frames of live cell microscopy of H2B-EYFP HeLa cells transfected with WT-mDia3 or 4E-mDia3. Arrows point to chromosome outside the smooth plate.
(B) Quantification of interkinetochore distances (mean ± SD are shown, n = 10 cells for each of three independent transfections and at least five pairs of kinetochores were measured from each cells).
(C) Transient expression of WT-mDia3 and 4E-mDia3 in cultured cells. BubR1 and ACA were observed by immunofluorescence with specific antibodies. In merge: BubR1, green; ACA, red; chromatin was visualized with DAPI, blue.
(D) Quantification of the normalized integrated intensities of the kinetochore signals of BubR1 (±SD, 30 cells per condition and at least 10 kinetochores/cell, p < 0.001).
(E) The cold stability of kinetochore microtubule connections in cells transfected with indicated mDia3 constructs (tubulin, green; ACA, magenta).
We next used three indicators to determine the possible defect in microtubule attachment in cells expressing the phosphomimetic 4E-mDia3 mutant. First, we measured inter-kinetochore spacing. We determined a mean separation of 0.74 ± 0.11 μm for unattached kinetochore pairs of control cells using ACA as a kinetochore marker. This value was increased to 1.97 ± 0.16 μm when both sister kinetochores became attached and aligned at the metaphase plate; however, inter-kinetochore spacing on fully aligned chromosomes in cells expressing 4E-mDia3 was substantially decreased (1.44 ± 0.17 μm) (Figure 6B). We next used indirect immunofluorescence to examine BubR1, a mitotic checkpoint protein whose kinetochore localization depends strongly on microtubule attachment and tension generation between sister kinetochores (Cleveland et al., 2003). In cells expressing wild-type mDia3, BubR1 became almost undetectable on attached kinetochores when chromosomes were aligned at the metaphase plate (Figures 6C and 6D). In cells expressing 4E-mDia3, however, BubR1 was detected on kinetochores of aligned chromosomes at a high level comparable to that on unattached ones (Figures 6C and 6D). BubR1 kinetochore localization in cells expressing 4E mutant was still dependent on the Aurora B kinase activity (Figure S4B). Finally, we assayed for the presence of cold-stable microtubules. In control cells, cold-stable kinetochore microtubules were present on aligned chromosomes, because kinetochore-microtubule fibers are preferentially stabilized at 4°C; however, in cells expressing the 4E-mDia3 mutant, only a few cold-stable microtubules remained after 10 min cold treatment (Figure 6E). These results collectively demonstrate that cells expressing the phosphomimetic 4E-mDia3 mutant, which has a reduced ability to bind and stabilize microtubules, can still form end-on kinetochore-microtubule attachment and are able to congress chromosomes to the metaphase plate; however, they are unable to generate normal tension to support the stability of kinetochore-microtubule attachment and to efficiently remove the checkpointprotein BubR1fromthe kinetochore.
DISCUSSION
A formin protein mDia3, but not mDia1 or mDia2, has been reported to localize to kinetochores from prometaphase to metaphase (Yasuda et al., 2004). In addition, siRNA-induced mDia3 knock down resulted in metaphase chromosome misalignment (Yasuda et al., 2004). In view of the broad range of mDia functions involving the abilities to interact with both actin filaments and microtubules (Bartolini and Gundersen, 2009; Goode and Eck, 2007), it was unclear whether the mitotic phenotypes was a direct consequence of altered kinetochore behavior. The evidence here supports a direct role for mDia3 in achieving stable kinetochore-microtubule attachment and metaphase chromosome alignment. Similar to what has been shown for the activity of mDia formin underlying microtubule stabilization in migrating cells (Bartolini et al., 2008), the mDia3 function at the kinetochore is independent of its actin nucleation activity (Figure 1). In contrast, the microtubule binding activity of mDia3 and its interaction with EB1 are important for mDia3′s kinetochore function (Figure 3). Using immunodepletion and reconstitution in cycled Xenopus egg extracts, EB1 and its binding partner, APC, have been shown to play a role in metaphase chromosome alignment (Zhang et al., 2007). APC or EB1 knock down using siRNA in mammalian cultured cells also led to less compact metaphase plates (Draviam et al., 2006; Green et al., 2005) and failure of chromosomes to reach the metaphase plate (Green et al., 2005). In metaphase cells, EB1, a microtubule tip-tracking protein, localizes only to the polymerizing plus ends of kinetochore microtubules during oscillations away from the spindle poles (Tirnauer et al., 2002). This result indicates that two distinct microtubule populations may exist for the leading and trailing sister kinetochores; however, both of them are stably attached. Thus, the interaction between mDia3 and EB1 may represent one mechanism that contributes to stable microtubule attachment at kinetochores with polymerizing microtubules.
A group of protein complexes, termed KMN network (the KNL-1, Ndc80, and Mis12 complexes), has been proposed to serve as a core microtubule binding apparatus at the kinetochore for stabilizing end-on microtubule attachment (Joglekar et al., 2010). Whether the KMN network is sufficient for this function is not clear, given the dynamic nature of attached microtubules and the weak affinity of the Ndc80 complex for microtubules in in vitro assays (Ciferri et al., 2008; Powers et al., 2009). Recent studies have demonstrated a critical role for the Ska1/RAMA complex in interacting dynamic microtubules to the kinetochore (Daum et al., 2009; Gaitanos et al., 2009; Raaijmakers et al., 2009; Welburn et al., 2009) to facilitate microtubule depolymerization-coupled motility (Welburn et al., 2009). The contribution of Ndc80 to the plus end-tracking activity of the kinetochore is unknown. The yeast Dam1 complex has been shown to confer microtubule plus end-tracking activity to Ndc80 (Lampert et al., 2010); however, no homolog of the Dam1 complex has yet been identified in mammals. In the future, it will be important to determine how mDia3-EB1-APC and the Ndc80 complex are coordinated in contributing to kinetochore microtubule attachment and microtubule dynamics, particularly at kinetochores with polymerizing microtubules.
Although we do not have an mDia3 microtubule-binding-deficient mutant to directly test the importance of mDia3 microtubule activity in stable kinetochore microtubule attachment, the requirement of mDia3 regulation by Aurora B phosphorylation for normal mitotic progress clearly supports this hypothesis. Cells expressing the phosphomimetic mDia3 mutant, which has reduced microtubule binding and stabilization activities, cannot form stable kinetochore microtubule attachments and show reduced inter-kinetochore stretching and high level of BubR1 signal even at apparently attached kinetochores (Figure 6). These cells are clearly able to form end-on kinetochore-microtubule attachments, for they can generate about half the tension compared to wild-type cells and congress to the metaphase plate. However, the reduced tension suggests that the nonphosphorylated mDia3, possibly along with EB1 and APC, plays a role in force generation between sister kinetochores, which is essential in stabilizing the attachment and dissociating mitotic checkpoint proteins, such as BubR1, from the kinetochore. Further, we have shown that Aurora B phosphorylation of mDia3 is dependent on kinetochore tension in response to microtubule attachment (Figure 4). This result clearly supports the current models that tension regulates Aurora B phosphorylation by separating the kinase residing between sister kinetochores from its outer kinetochore substrates (Liu and Lampson, 2009). This represents the mechanism for Aurora B to destabilize improperly attached kinetochore microtubules to correct attachment errors by phosphorylating kinetochore-associated microtubule interactors, such as the Ndc80 complex and mDia3, to reduce their affinity for microtubules in the absence of tension (Figure 7).
Figure 7. Aurora B Phosphorylation at FH2 Domain Regulates mDia3 Function in Kinetochore Microtubule Attachment in Response to Tension.

Model: Aurora B phosphorylation at FH2 domain regulates mDia3 function in kinetochore microtubule attachment in response to tension. Under tension: bi-oriented sister kinetochores are pulled by microtubules in opposite directions, which increases the distance between the outer kinetochore and the inner centromere, where Aurora B localizes. Thus, Aurora B cannot efficiently phosphorylate substrates at the outer kinetochores, such as the Ndc80 complex and mDia3. These microtubule interactors will contribute to stable kinetochore microtubule attachment. NOT under tension: in the misattached state (syntelic or merotelic) that is not under tension, kinetochore substrates, including the Ndc80 complex and mDia3, are phosphorylated because they are in close proximity to Aurora B at the inner centromere. The Aurora B phosphorylation leads to reduced affinity of these proteins for microtubules, resulting in destabilization of attachments. How these phosphorylation sites are subsequently regulated by kinetochore-associated phosphatase activity in response to tension remain unresolved.
Besides two phosphorylation sites in mDia3 FH2 domain, two more Aurora B phosphorylation sites, T66 and S196, are within mDia3 N-terminal GBD-DID domains, which serve in mDia formin autoinhibition through an intramolecular interaction with C-terminal DAD domain (Alberts et al., 1998). The mDia formin autoinhibition is relieved upon the binding of the small GTPase at the N-terminal GBD domain (Watanabe et al., 1999). The mDia3 kinetochore function has been reported to be regulated under upstream control by Cdc42 (Yasuda et al., 2004). However, it is unclear how membrane-bound Cdc42 is able to interact with and activate the pool of mDia3 at the kinetochore. To the best of our knowledge, there is no clear evidence to support kinetochore localization of Cdc42. Further, we found no significant increase in chromosome alignment defects relative to those produced by mDia3 siRNA using either toxin B treatment, which inactivates all Rho GTPases, or expression of a dominant-negative N17-Cdc42 mutant in T98G cells (S. Ahmad and Y. Mao, unpublished data). Thus, we speculate that two Aurora B phosphorylation sites within mDia3 GBD-DID domains, T66 and S196, could serve in regulating autoinhibition, perhaps locally at the kinetochore. There is some precedent for effects of formin phosphorylation on autoinhibition. In particular, autoinhibition of the mammalian FHOD1 has been reported to be relieved by ROCK phosphorylation at the C-terminal DAD domain (Takeya et al., 2008). It will be important to understand how mDia3 activity is tightly regulated at the kinetochore.
EXPERIMENTAL PROCEDURES
Plasmids
Human mDia3 cDNA (accession number BC117414) was cloned into Myc mammalian expressing vector (Clontech) and subcloned into pGK vector (with a GST tag) to express recombinant proteins in Escherichia coli. All mDia3 mutants were generated using the QuickChange Kit (Stratagene) and confirmed by sequencing.
Tissue Culture, Transfection, and Drug Treatment
T98G and HeLa cells were cultured in DMEM with 10% FBS and 1% penicillin-streptavidin at 37°C in 5% CO2. mDia3 siRNA were purchased from QIAGEN. Transfection was carried using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction. Transfected cells were identified with cotransfection with a Ds-Red plasmid in 1:10 ratio. In all chromosome alignment analysis, 48 hr posttransfection, cells were treated with nocodazole to accumulate mitotic cells for 4 hr and released into MG132 medium for 1 hr. Cells were then fixed and subjected to immunofluorescence analysis. Nocodazole, monastrol, and MG132 (Sigma) were added to a final concentration of 100 ng/ml, 100 μM, and 10 μM, respectively. ZM 447439 (2 μM) was a kind gift from AstraZeneca UK Ltd.
Immunofluorescence Microscopy and Live Cell Imaging
The antibodies were from Abcam except for the mDia3 antibody (Life Span Bioscience). For indirect immunofluorescence, cells grown on poly-L-lysine-coated coverslips were washed once with microtubule stabilizing buffer (MTSB: 100 mM Pipes, 1 mM EGTA, 1 mM MgSO4, and 30% of glycerol), extracted with 0.5% Triton X-100 in MTSB for 5 min, fixed with 4% formaldehyde in MTSB or methanol for 10 min, and blocked in TBS containing 0.5% Tween-20 and 1% BSA (Sigma) for 1 hr. Coverslips were subjected to primary antibodies diluted in blocking buffer for 1 hr, and to secondary antibodies (Jackson ImmunoResearch Laboratories). Image acquisition and data analysis were performed at room temperature using an inverted microscope (IX81; Olympus) with a 60× NA 1.42 plan Apo oil immersion objective lens (Olympus), a monochrome CCD camera (Sensicam QE; Cooke), and the Slidebook software package (Olympus). All images in each experiment were collected on the same day using identical exposure time. Quantitative analysis of the immunofluorescence was carried with the Slidebook software or ImageJ (NIH). For quantification of kinetochore intensities, a circular region with fixed diameter was centered on each kinetochore and normalized against ACA intensity after subtraction of background intensity measured outside the cell. Similarly, tubulin intensity was normalized against the area of the cell after subtraction of background intensity. For live-cell imaging, HeLa cells stably expressing H2B-EYFP were plated onto 35-mm glass bottom dishes (MatTek). Images were acquired using a 40× NA dry objective lens every 2 min with a 37°C chamber and were processed using Slidebook software. All statistical significance of quantification experiments was verified by Student’s t test using Microsoft Excel software.
In Vitro Kinase Assay and Mass Spectrometry
Purified GST-tagged mDia3 recombinant proteins were incubated with an active Aurora B kinase (Abcam) in the presence of 5 mM MOPS (pH7.2), 2.5 mM β-glycerophosphate, 1 mM EGTA, 0.4 mM EDTA, 5 mM MgCl2, 0.05 mM DTT, 20 μM ATP, and 5 μCi 32P-ATP for 30 min at room temperature. Reactions were terminated by addition of 2× sample buffer and subjected by 10% SDS-PAGE. Radioactive phosphate incorporated into mDia3 was visualized by autoradiography.
Aurora B phosphorylated mDia3 proteins and purified mDia3 proteins were recovered from the SDS-PAGE. The gel bands were reduced with 10 mM of DTT and alkylated with 55 mM iodoacetamide, and then digested with Sequence Grade Modified Trypsin (Promega) in ammonium bicarbonate buffer at 37°C overnight. The digestion products were extracted twice with 0.1% trifluoroacetic acid and 50% acetonitrile and 1.0% trifluoroacetic acid respectively. The extracted mixture was dried by Speed-Vac and redisolved in 10 μl 0.1% trifluoroacetic acid. Half of the extracts were injected by LC-MS/MS analysis. For LC-MS/MS analysis, each digestion product was separated by a 60-min gradient elution with the Dionex capillary/nano-HPLC system at a flow rate of 0.250 μL/min that is directly interfaced with the Thermo-Fisher LTQ-Obritrap mass spectrometer operated in data-dependent scan mode. The analytical column was a home-made fused silica capillary column (75 μm ID, 100 mm length; Upchurch) packed with C-18 resin (300 A, 5 μm, Varian). Mobile phase A consisted of 0.1% formic acid, and mobile phase B consisted of 100% acetonitrile and 0.1% formic acid. The 60-min gradients with 250 nL/min flow rate for B solvent went from 0% to 55% in 34 min and then in 4 min to 80%. The B solvent stayed at 80% for another 8 min and then decreased to 5% in 8 min. Another 6 min was used for equilibration, loading and washing. The mass acquisition method was one FT-MS scan followed by six subsequent MS/MS scan in the ion trap. The FT-MS scan was acquired at resolution 30,000 in the Orbi-trap. The six most intense peaks from the FT full scan were selected in the ion trap for MS/MS.
Microtubule Binding and Stabilization Activity Assays
For microtubule co-sedimentation assay, purified wild-type mDia3 or mDia3 phosphorylation mutants were mixed with Taxol-stabilized microtubules or unpolymerized tubulin in BRB80 and incubated in room temperature for 30 min. Binding reactions were centrifuged over 40% sucrose/BRB80 10,000 rpm for 15 min at 35°C. Recombinant proteins in the pellet and supernatant were analyzed by immunoblotting.
For microtubule stabilization activity assay, polymerization reactions were performed with Taxol-stabilized seeds and rhodamine-labeled tubulin at 37°C for 1 hr and then incubated with purified mDia3 proteins with or without pretreated with purified Aurora B. To assess microtubule stability to cold-induced depolymerization, 2 μl of each reaction was squashed under a cover-slip, and images were taken at equal exposure times on the imaging system described above.
Supplementary Material
Acknowledgments
We thank Dr. N. Ramalingam for technical support on microtubule cosedimentation analysis, Drs. G. Gundersen and R. Vallee for insight on the project, and all members of the Mao laboratory for stimulating discussion. Y. Mao is an Irma T. Hirschl Career Scientist.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and can be found with this article online at doi:10.1016/j.devcel.2011.01.008.
References
- Adames NR, Cooper JA. Microtubule interactions with the cell cortex causing nuclear movements in Saccharomyces cerevisiae. J Cell Biol. 2000;149:863–874. doi: 10.1083/jcb.149.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts AS, Bouquin N, Johnston LH, Treisman R. Analysis of RhoA-binding proteins reveals an interaction domain conserved in heterotrimeric G protein beta subunits and the yeast response regulator protein Skn7. J Biol Chem. 1998;273:8616–8622. doi: 10.1074/jbc.273.15.8616. [DOI] [PubMed] [Google Scholar]
- Bartolini F, Gundersen GG. Formins and microtubules. Biochim Biophys Acta. 2009;1803:164–173. doi: 10.1016/j.bbamcr.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolini F, Moseley JB, Schmoranzer J, Cassimeris L, Goode BL, Gundersen GG. The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J Cell Biol. 2008;181:523–536. doi: 10.1083/jcb.200709029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom K. It’s a kar9ochore to capture microtubules. Nat Cell Biol. 2000;2:E96–E98. doi: 10.1038/35014089. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Anderson S, Jwa M, Green EM, Kang J, Yates JR, 3rd, Chan CS, Drubin DG, Barnes G. Phospho-regulation of kinetochore-microtubule attachments by the Aurora kinase Ipl1p. Cell. 2002;111:163–172. doi: 10.1016/s0092-8674(02)00973-x. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Chappie JS, Wilson-Kubalek EM, Desai A. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 2006;127:983–997. doi: 10.1016/j.cell.2006.09.039. [DOI] [PubMed] [Google Scholar]
- Ciferri C, Pasqualato S, Screpanti E, Varetti G, Santaguida S, Dos Reis G, Maiolica A, Polka J, De Luca JG, De Wulf P, et al. Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell. 2008;133:427–439. doi: 10.1016/j.cell.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DW, Mao Y, Sullivan KF. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell. 2003;112:407–421. doi: 10.1016/s0092-8674(03)00115-6. [DOI] [PubMed] [Google Scholar]
- Cook TA, Nagasaki T, Gundersen GG. Rho guanosine triphos-phatase mediates the selective stabilization of microtubules induced by lysophosphatidic acid. J Cell Biol. 1998;141:175–185. doi: 10.1083/jcb.141.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum JR, Wren JD, Daniel JJ, Sivakumar S, McAvoy JN, Potapova TA, Gorbsky GJ. Ska3 is required for spindle checkpoint silencing and the maintenance of chromosome cohesion in mitosis. Curr Biol. 2009;19:1467–1472. doi: 10.1016/j.cub.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca JG, Gall WE, Ciferri C, Cimini D, Musacchio A, Salmon ED. Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell. 2006;127:969–982. doi: 10.1016/j.cell.2006.09.047. [DOI] [PubMed] [Google Scholar]
- Draviam VM, Shapiro I, Aldridge B, Sorger PK. Misorientation and reduced stretching of aligned sister kinetochores promote chromosome missegregation in EB1- or APC-depleted cells. EMBO J. 2006;25:2814–2827. doi: 10.1038/sj.emboj.7601168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaitanos TN, Santamaria A, Jeyaprakash AA, Wang B, Conti E, Nigg EA. Stable kinetochore-microtubule interactions depend on the Ska complex and its new component Ska3/C13Orf3. EMBO J. 2009;28:1442–1452. doi: 10.1038/emboj.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- Green RA, Wollman R, Kaplan KB. APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol Biol Cell. 2005;16:4609–4622. doi: 10.1091/mbc.E05-03-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs HN, Peterson KJ. Phylogenetic analysis of the formin homology 2 domain. Mol Biol Cell. 2005;16:1–13. doi: 10.1091/mbc.E04-07-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar AP, Bloom KS, Salmon ED. Mechanisms of force generation by end-on kinetochore-microtubule attachments. Curr Opin Cell Biol. 2010;22:57–67. doi: 10.1016/j.ceb.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor TM, Lampson MA, Hergert P, Cameron L, Cimini D, Salmon ED, McEwen BF, Khodjakov A. Chromosomes can congress to the metaphase plate before biorientation. Science. 2006;311:388–391. doi: 10.1126/science.1122142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner MW, Mitchison T. Microtubule dynamics. Nature. 1986;324:621. doi: 10.1038/324621a0. [DOI] [PubMed] [Google Scholar]
- Kohno H, Tanaka K, Mino A, Umikawa M, Imamura H, Fujiwara T, Fujita Y, Hotta K, Qadota H, Watanabe T, et al. Bni1p implicated in cytoskeletal control is a putative target of Rho1p small GTP binding protein in Saccharomyces cerevisiae. EMBO J. 1996;15:6060–6068. [PMC free article] [PubMed] [Google Scholar]
- Lampert F, Hornung P, Westermann S. The Dam1 complex confers microtubule plus end-tracking activity to the Ndc80 kinetochore complex. J Cell Biol. 2010;189:641–649. doi: 10.1083/jcb.200912021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson MA, Kapoor TM. The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nat Cell Biol. 2005;7:93–98. doi: 10.1038/ncb1208. [DOI] [PubMed] [Google Scholar]
- Lee L, Klee SK, Evangelista M, Boone C, Pellman D. Control of mitotic spindle position by the Saccharomyces cerevisiae formin Bni1p. J Cell Biol. 1999;144:947–961. doi: 10.1083/jcb.144.5.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Lampson MA. Regulation of kinetochore-microtubule attachments by Aurora B kinase. Biochem Soc Trans. 2009;37:976–980. doi: 10.1042/BST0370976. [DOI] [PubMed] [Google Scholar]
- Martin-Lluesma S, Stucke VM, Nigg EA. Role of hec1 in spindle checkpoint signaling and kinetochore recruitment of mad1/mad2. Science. 2002;297:2267–2270. doi: 10.1126/science.1075596. [DOI] [PubMed] [Google Scholar]
- Palazzo AF, Cook TA, Alberts AS, Gundersen GG. mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat Cell Biol. 2001a;3:723–729. doi: 10.1038/35087035. [DOI] [PubMed] [Google Scholar]
- Palazzo AF, Joseph HL, Chen YJ, Dujardin DL, Alberts AS, Pfister KK, Vallee RB, Gundersen GG. Cdc42, dynein, and dynactin regulate MTOC reorientation independent of Rho-regulated microtubule stabilization. Curr Biol. 2001b;11:1536–1541. doi: 10.1016/s0960-9822(01)00475-4. [DOI] [PubMed] [Google Scholar]
- Powers AF, Franck AD, Gestaut DR, Cooper J, Gracyzk B, Wei RR, Wordeman L, Davis TN, Asbury CL. The Ndc80 kinetochore complex forms load-bearing attachments to dynamic microtubule tips via biased diffusion. Cell. 2009;136:865–875. doi: 10.1016/j.cell.2008.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers JA, Tanenbaum ME, Maia AF, Medema RH. RAMA1 is a novel kinetochore protein involved in kinetochore-microtubule attachment. J Cell Sci. 2009;122:2436–2445. doi: 10.1242/jcs.051912. [DOI] [PubMed] [Google Scholar]
- Rivero F, Muramoto T, Meyer AK, Urushihara H, Uyeda TQ, Kitayama C. A comparative sequence analysis reveals a common GBD/FH3-FH1-FH2-DAD architecture in formins from Dictyostelium, fungi and metazoa. BMC Genomics. 2005;6:28. doi: 10.1186/1471-2164-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuyler SC, Pellman D. Search, capture and signal: games microtubules and centrosomes play. J Cell Sci. 2001;114:247–255. doi: 10.1242/jcs.114.2.247. [DOI] [PubMed] [Google Scholar]
- Takeya R, Taniguchi K, Narumiya S, Sumimoto H. The mammalian formin FHOD1 is activated through phosphorylation by ROCK and mediates thrombin-induced stress fibre formation in endothelial cells. EMBO J. 2008;27:618–628. doi: 10.1038/emboj.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirnauer JS, Canman JC, Salmon ED, Mitchison TJ. EB1 targets to kinetochores with attached, polymerizing microtubules. Mol Biol Cell. 2002;13:4308–4316. doi: 10.1091/mbc.E02-04-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak CE, Heald R. Mechanisms of mitotic spindle assembly and function. Int Rev Cytol. 2008;265:111–158. doi: 10.1016/S0074-7696(07)65003-7. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–143. doi: 10.1038/11056. [DOI] [PubMed] [Google Scholar]
- Welburn JP, Grishchuk EL, Backer CB, Wilson-Kubalek EM, Yates JR, 3rd, Cheeseman IM. The human kinetochore Ska1 complex facilitates microtubule depolymerization-coupled motility. Dev Cell. 2009;16:374–385. doi: 10.1016/j.devcel.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welburn JP, Vleugel M, Liu D, Yates JR, 3rd, Lampson MA, Fukagawa T, Cheeseman IM. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol Cell. 2010;38:383–392. doi: 10.1016/j.molcel.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Eng CH, Schmoranzer J, Cabrera-Poch N, Morris EJ, Chen M, Wallar BJ, Alberts AS, Gundersen GG. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat Cell Biol. 2004;6:820–830. doi: 10.1038/ncb1160. [DOI] [PubMed] [Google Scholar]
- Wood KW, Sakowicz R, Goldstein LS, Cleveland DW. CENP-E is a plus end-directed kinetochore motor required for metaphase chromosome alignment. Cell. 1997;91:357–366. doi: 10.1016/s0092-8674(00)80419-5. [DOI] [PubMed] [Google Scholar]
- Xu Y, Moseley JB, Sagot I, Poy F, Pellman D, Goode BL, Eck MJ. Crystal structures of a Formin Homology-2 domain reveal a tethered dimer architecture. Cell. 2004;116:711–723. doi: 10.1016/s0092-8674(04)00210-7. [DOI] [PubMed] [Google Scholar]
- Yasuda S, Oceguera-Yanez F, Kato T, Okamoto M, Yonemura S, Terada Y, Ishizaki T, Narumiya S. Cdc42 and mDia3 regulate microtubule attachment to kinetochores. Nature. 2004;428:767–771. doi: 10.1038/nature02452. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ahmad S, Mao Y. BubR1 and APC/EB1 cooperate to maintain metaphase chromosome alignment. J Cell Biol. 2007;178:773–784. doi: 10.1083/jcb.200702138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.