

Abstract
Common variable immunodeficiency (CVID) is a late-onset humoral deficiency characterized by B lymphocyte dysfunction or loss, decreased immunoglobulin production, and recurrent bacterial infections. CVID is the most frequent human primary immunodeficiency but still presents challenges in the understanding of its etiology and treatment. CVID in equine patients manifests with a natural impairment of B lymphocyte differentiation, and is a unique model to identify genetic and epigenetic mechanisms of disease. Bone marrow transcriptome analyses revealed decreased expression of genes indicative of the pro-B cell differentiation stage, importantly PAX5 (p≤0.023). We hypothesized that aberrant epigenetic regulation caused PAX5 gene silencing, resulting in the late-onset and non-familial manifestation of CVID. A significant increase in PAX5 enhancer region methylation was identified in equine CVID patients by genome-wide reduced-representation bisulfite sequencing and bisulfite PCR sequencing (p=0.000). Thus, we demonstrate that integrating transcriptomics and epigenetics in CVID enlightens potential mechanisms of dysfunctional B lymphopoiesis or function.
Keywords: CVID, bone marrow, B cell differentiation, PAX5, transcriptome, equine
INTRODUCTION
Common variable immunodeficiency (CVID) is characterized by late-onset impaired immunoglobulin production and, consequently, recurrent bacterial infections. Although CVID is the most common human primary immunodeficiency, variability in phenotype, genetic background, and age of onset have precluded rapid definitive diagnosis and understanding of its etiology [1, 2]. Most patients are diagnosed between the ages of 20 and 40 years, some of them presenting with B lymphopenia, whereas others with normal B lymphocyte numbers fail to produce immunoglobulins [2]. The heterogeneity of CVID cases has complicated classification into categories, though attempts have been made based on clinical phenotypes and flow cytometric B lymphocyte population analysis [3–5]. Genetic and genome-wide association analyses have revealed novel susceptibility loci significantly associated with CVID (nucleotide polymorphisms and copy number variants), and identified variants shared among CVID patients as well as other variants unique to individual patients [6–18]. At this point in time, CVID is considered a complex disease that can be caused by a variety of underlying defects.
The index equine CVID case was published in 2002, and over 30 cases have been diagnosed by our laboratory since then [19–21]. The common elements of presentation in these equine CVID patients consisted of late-onset recurrent bacterial infections, severe hypogammaglobulinemia, marked B cell lymphopenia, and failure to mount humoral immune responses to protein vaccination [19–21]. Equine CVID patients sustain production of T lymphocyte, monocyte, phagocyte, eosinophil, basophil, and platelet populations, as well as functional phagocytosis, oxidative burst, and T cell proliferation capacity. Immunohistochemical analyses of lymphoid tissues collected from equine CVID patients reveal the presence of large amounts of T lymphocytes, with absence of germinal centers and few scattered B lymphocytes [19, 21, 22]. Our previous studies indicated that mRNA expression of E2A, PAX5, and CD19 was significantly reduced in the bone marrow of equine CVID patients when compared to healthy horses, supported by the lack of PAX5/BSAP and CD19 protein expression in the bone marrow of affected horses [22].
B lymphocytes are continuously generated over an individual’s lifetime from hematopoietic stem cells (HSCs) in the bone marrow. B lymphocyte differentiation has been studied intensively in humans and mice, and it follows a tightly-regulated and ordered progression, with the essential transcription factors functioning in a complex network of auto-regulation, cross-regulation, and positive and negative feedback loops [23–25]. Differentiation progresses from HSCs to multipotent progenitors (MPPs), to lymphoid-primed multipotent progenitors (LMPPs), and then common lymphoid progenitors (CLPs); after this point, B lineage specification and commitment take place [26].
The prominent role of epigenetic mechanisms in regulation of B lymphopoiesis has become appreciated recently. B lymphocyte transcription factors E2A, EBF1, and PAX5 interact with chromatin remodeling factors to promote H3 acetylation marks on activated genes, and increase methylation on H3K4 to poise target genes for expression at later stages of differentiation [23, 27–29]. E2A also remodels chromatin of the immunoglobulin locus to enable recombination, along with IKAROS [30, 31]. EBF1 also facilitates DNA demethylation and initiates nucleosome remodeling of target genes in association with the SWI/SNF complex [32, 33]. Progressive histone remodeling and DNA demethylation during B lymphopoiesis has been demonstrated for PAX5, CD19, and CD79A genes [27, 32, 34]. PAX5 represses a set of non-B lineage genes by associating with histone deacetylases and chromatin remodeling factors [35, 36]. Demethylation near B lymphocyte transcription factor binding sites becomes prevalent as multipotent progenitors commit to the B lineage [37].
The goal of this study was to apply transcriptome and epigenome analyses to gain a comprehensive and unbiased profile of gene expression and regulation in the bone marrow of equine CVID patients. This approach facilitates assessment of gene expression over the spectrum of B lymphocyte differentiation, bone marrow stromal cell gene expression, non-coding RNAs, and expressed genes of viral origin. We hypothesize that equine CVID patients studied here share a common gene expression profile of the critical B lymphocyte genes in the bone marrow involved in the etiology of this form of CVID, and epigenetic mechanisms may explain the non-familial and late-onset characteristics of this condition.
2. MATERIALS AND METHODS
2.1. Tissue samples
These experiments were approved by the Cornell University Center for Animal Resources and Education and Institutional Animal Care and Use Committee for the use of vertebrates in research. Bone marrow from equine CVID patients (n = 7) were collected during necropsy through the Cornell University Veterinary Pathology service or forwarded overnight on dry ice to our laboratory by attending veterinarians, and stored at −80°C. The equine CVID patients sampled for this study were clinical cases from the United States (California, Florida, Illinois, Kentucky, Maryland, Pennsylvania) and differed in sex (3 males, 4 females), breed (4 Thoroughbred, 3 Quarter Horse), age (2–19 years), and none were related. The common elements of CVID presentation in these 7 horses consisted of recurrent bacterial infections, hypogammaglobulinemia, and B cell lymphopenia measured by flow cytometry. The clinical and immunological parameters of most equine CVID patients included in this study have been described previously, including horses of other breeds than the ones used in this study [20]. Healthy control adult Thoroughbred horse bone marrow control samples (n = 3) were collected within 1 hour of euthanasia, snap frozen in liquid nitrogen, and stored at −80°C until use; these were archived from research investigations performed by us and other investigators over the years at Cornell University College of Veterinary Medicine.
2.2. RNA isolation and RNA-Seq library preparation and sequencing
RNA was isolated from snap-frozen tissues following homogenization by QIAshredder (Qiagen, Valencia, CA) as directed by the RNeasy kit (Qiagen) including on-column digestion of contaminating genomic DNA. RNA was quantified with a Nanodrop (Thermo Fisher Scientific, Inc., Waltham, MA). RNA purity and integrity was assessed using a BioAnalyzer 2100 (Agilent, Santa Clara, CA), and all RNA samples had RNA integrity number values ≥ 8.0. RNA-Seq library preparations were performed according to Illumina’s standard mRNA-seq kit protocol, and sequences of about 100 bases in length were obtained on an Illumina HiSeq2000 by the Cornell University Institute of Biotechnology Genomics Facility, Ithaca, NY. The RNA-Seq dataset is available in GenBank as BioProject PRJNA266428.
2.3. RNA-Seq alignment, gene expression analysis, and statistical tests
Quality of the raw sequence reads was assessed with FastQC (Version 0.9.0, Babraham Bioinformatics), and low quality bases were gradually trimmed from the 3′ end until length of 40 nucleotides with seqtk’s trimfq tool (https://github.com/lh3/seqtk). Trimmed reads were aligned to the Equine reference genome sequence, EquCab2.0, with TopHat version 2.0.8 [38] using Ensembl gene model 75 as the annotation reference for alignment and all downstream analysis. Cufflinks version 2.1.1 software was used to quantify genes and transcripts according to the gene model, and conduct reference-guided transcript assembly [39]. The read counts per gene were extracted from cufflinks outputs using in-house perl script and used for count-based analysis with edgeR package [40]. Genes with very low expression were filtered out unless the count per million reads ≥ 0.1 in at least 3 samples, leaving 15,508 out of 26,991 genes. The gene expression pattern among samples was inspected by multidimensional scaling (MDS) in edgeR. Differential expression analysis was conducted with 2-group test between the control group and each of the 2 CVID groups by fitting a negative binomial generalized log-linear model (GLM) to the read counts for each gene. False discovery rate of 0.05 was chosen as the threshold for significant differences in expression after adjusting p-values for multiple tests with the Benjamini and Hochberg method.
Single nucleotide polymorphism (SNP) variants were called from RNA-Seq data with Novoalign (Novocraft Technologies, Selangor, Malaysia) and GATK [41].
Gene ontology was analyzed with the highest stringency parameters of the Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resources 6.7 [42, 43].
Equine endogenous retroviruses were identified from the horse genome sequence in silico and assessed by RNA-Seq data previously [44–46].
Complete equine herpes virus (EHV) strain sequences were obtained from GenBank as follows: EHV1 NC_001491; EHV2 NC_001650; EHV4 NC_001844; EHV8 NC_017826.1; and EHV9 NC_011644.1. For EHV strains without published genome sequences, all available gene sequences were used: EHV3 AF081188, AF514778, and AF514779; EHV5 AF050671.1, AF141886.1, AF495531.1, DQ471427.1, DQ471428.1, DQ471429.1, DQ471430.1, DQ471431.1, DQ471432.1, DQ471433.1, DQ471434.1, DQ471435.1, DQ504440.1, EF182710.1, EF182711.1, EF182712.1, EF515178.1, GQ154073.1, GQ154074.1, GQ325592.1, GQ325593.1, GQ325594.1, GQ325595.1, GQ325596.1, GQ325597.1, GQ325598.1, GQ325599.1, GU065283.1, GU065284.1, GU065285.1, HM234087.1, HM234088.1, HM234089.1, HM234090.1, JN982959.1, JN982960.1, JN982961.1, JX125459.1, L01473.1; and EHV7 EU165547.
2.4. Reduced representation bisulfite sequencing and analysis
Genomic DNA was isolated from equine CVID patients (n = 2) and healthy control horse (n = 1) core bone marrow samples with Qiagen DNeasy Blood and Tissue Kit and unmethylated lambda DNA was obtained (Promega, Madison, WI). Reduced representation bisulfite sequence (RRBS) libraries were prepared by the Cornell Epigenetics Core Facility, Weill Cornell Medical College, New York, NY per Illumina protocol. Libraries were sequenced on the Hi-Seq 2000 at Cornell Institute of Biotechnology, Ithaca, NY. After removal of adapter and primer sequences used in RRBS library construction, sequence reads went through an adaptive quality trimming of low quality trailing bases from the 3′ end. Such adaptive quality trimming (also adaptor trimming) was performed with cutadapt (http://code.google.com/p/cutadapt/). For bisulfite mapping, reads were converted into a C-to-T and a G-to-A version and then aligned to equivalently converted versions of the reference genome, and the methylation state of positions involving cytosines was inferred by comparing the read sequence with the corresponding genomic sequence. Sequence reads that produce a unique best alignment from the four alignment processes against the bisulfite genomes were then compared to the normal genomic sequence, and the methylation state of all cytosine positions in the read was inferred using Bismark (v0.6.0) [47]. The CpGs with read depth ≥ 5 were kept as informative CpGs. To score CpG island (CGI) methylation, we required that the methylation level was determined for ≥10% of their total CpGs and a CGI must have ≥ 5 informative CpGs. Then CGIs with an average methylation level ≥75% and ≤25% were called methylated and unmethylated, respectively. The horse CGI list was created by Wu et al. [48] using the model-based method. The RRBS sequence dataset is available in GenBank as BioProject PRJNA266432.
2.5. Amplification and cloning of bisulfite-treated genomic DNA and analysis
Genomic DNA was isolated from equine CVID patients (n = 7) and healthy control horse (n = 6) frozen bone marrow core samples as directed by the DNeasy Blood & Tissue Kit (Qiagen). Bisulfite treatment of genomic DNA was performed as directed by the MethylEasy Xceed kit (Genetic Signatures, Randwick, Australia). Primers to amplify bisulfite-treated genomic DNA were designed with MethPrimer [49]. The PAX5 enhancer region was amplified with a nested PCR strategy entailing first round primers 5′ TTTTTGGTAAAGTAGAGGATTTGAG 3′ and 5′ AAATAAAATAAAAAAACCTTCAATAAC 3′, followed by amplification with nested primers 5′ TTGAGGTTAGGTGATTAATTTTAGG 3′ and 5′ AATAAAATAAAAAAACCTTCAATAAC 3′, which generated a 182 base pair product and encompassed 6 CpG sites. The CD19 promoter region was amplified with primers 5′ GGGGAATAGAAAGTGATTTAATAGA 3′ and 5′ AACCTAATAAACACTAAACCATAAATATCT 3′, which generated a 218 base pair product and encompassed 5 CpG sites. Amplification of 20 ng bisulfite-treated genomic DNA was performed with TaKaRa Ex Taq DNA polymerase (Clontech, Mountain View, CA) with the following cycling program: 98°C for 3 minutes; 40 cycles of 98°C for 10 seconds, 50°C for 30 seconds, 72°C for 30 seconds; and a final extension of 72°C for 10 minutes. Amplicons were excised from the agarose gel and purified with the GeneJET Gel Extraction and DNA Cleanup Micro Kit (Thermo Fisher Scientific, Inc.). Purified amplicons were then cloned with the CloneJET PCR Cloning Kit (Thermo Fisher Scientific, Inc.) and transformed into NEB 5-alpha Competent E. coli (New England BioLabs, Inc., Ipswich, MA). Single colonies were expanded in Luria broth with ampicillin and plasmid DNA was isolated with the GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific, Inc.). One microgram of plasmid DNA was submitted for sequencing at the Cornell University Institute of Biotechnology, Ithaca, NY. Initial sequence analysis was performed with Geneious software version 6.1.6 (Biomatters, Ltd., Auckland, New Zealand, available from http://www.geneious.com/) [50]. Analysis of bisulfite conversion efficiency, methylation level, and identification of identical bisulfite sequences was performed with QUMA Quantification tool for Methylation Analysis [51]. Statistical comparison of methylation levels between healthy control and equine CVID groups was performed with Fisher’s exact test. To control for multiple measurements from each horse and test for effects of disease status, CpG site, and the interaction of status*site on methylation, a GEE logistic model with an exchangeable working correlation structure. Statistical analyses were performed in SPSS v.22 (IBM SPSS Inc., IBM Corporation, Somers, NY).
3. RESULTS and DISCUSSION
Transcriptome sequencing (RNA-Seq) was undertaken to obtain an unbiased and comprehensive insight to differential gene expression in the bone marrow of equine CVID patients. Core bone marrow samples were utilized rather than isolated cells to investigate comprehensive expression of B cell genes, genes indicative of multiple hematopoietic lineages, and bone marrow stromal cell genes without the need for individual gene assays.
3.1. RNA-Seq gene quantification and validation
RNA-Seq libraries were generated from bone marrow RNA isolated from equine CVID patients (n = 7) and healthy control horses (n = 3). Approximately 19 million sequence reads were obtained from each library, which were then mapped to the EquCab2.0 genome, resulting in quantification of 15,508 known Ensembl (v75) genes. RNA-Seq transcript quantification was validated with quantitative RT-PCR of 12 genes (supplemental figure 1). Overall, the fold changes in gene expression between healthy control and CVID horse bone marrow were similar with RNA-Seq and qRT-PCR methods, with Spearman correlation r > 0.79 and p ≤ 0.003.
3.2. Functional annotation of differentially expressed genes in CVID horse bone marrow
The bone marrow transcriptome profiles of equine CVID patients and healthy control horses were studied with multidimensional scaling and three distinct groups became apparent: one group comprised of healthy control samples and two CVID subgroups (Figure 1). CVID group 1 consisted of patients 1–4 and CVID group 2 consisted of patients 5–7. This segregation coincided with patient breed: patients in group 1 are Thoroughbreds and patients in group 2 are Quarter Horses. We used this segregation strategically in order to subtract genes that would have been differentially expressed due to breed or polymorphisms, and only considered differences between healthy control and CVID samples, rather than differences between CVID groups. This result supported RNA-Seq as a tool for classification of CVID patients, providing sensitive and unbiased data.
Figure 1. Multidimensional scaling plot of bone marrow transcriptomes.

Dimension 1 (x-axis) and dimension 2 (y-axis) separate samples based on expression of 15,508 genes and create 3 groups for statistical analysis: healthy controls (C4 and C5 overlap; and C8), CVID group 1 (patients #1–4), and CVID group 2 (patients #5–7).
Gene expression analysis of bone marrow transcriptomes revealed that 103 genes were differentially expressed between healthy controls and all CVID patients. To appreciate the biological importance of the 103 differentially expressed genes, the lists of significantly down-regulated or up-regulated genes were analyzed with the Database for Annotation, Visualization and Integrated Discovery (DAVID). ‘Immunoglobulin’ was the most over-represented gene term of the 48 down-regulated genes (Table 1). In the list of 55 up-regulated genes, the terms ‘chemokine activity’ and ‘complement activation’ were over-represented (Table 1). This analysis indicated that the dominant signatures of the CVID horse bone marrow transcriptome were loss of Ig gene expression and increased expression of genes involved in inflammatory responses.
Table 1.
Differentially expressed genes (DEGs) between healthy and CVID-affected bone marrow.
| Group comparison | Number of DEG | Over-represented gene term | Fold enrichment | # genes | FDR corrected p-value |
|---|---|---|---|---|---|
| Healthy vs. all CVID | 48 down-regulated | Immunoglobulin C1-set | 337.5 | 7 | 2.88 × 10−9 |
| Genes: IGHD, IGHE, IGHG1, IGHG2, IGHG3, IGHG4, IGKC | |||||
| 55 up-regulated | Chemokine activity | 41.3 | 6 | 3.08 × 10−4 | |
| Genes: CCL4, CCL5, CCL19, CXCL2, CXCL9, IL8 | |||||
| Complement activation | 38.3 | 5 | 1.10 × 10−2 | ||
| Genes: C1R, C1S, C4A, CFH, SERPING1 | |||||
| Healthy vs. CVID group 1 | 71 down-regulated | Blood coagulation | 17.2 | 7 | 4.19 × 10−3 |
| (CVID group 2 not DE) | Genes: CD36, GP6, ITGA2, ITGB3, MMRN1, P2RY12, TREML1 | ||||
| 68 up-regulated | Ectodermal dysplasia | 40.8 | 3 | 2.77 | |
| Genes: KRT6A, KRT14, KRT16 | raw p-value 2.36 × 10 −3 | ||||
| Healthy vs. CVID group 2 | 918 down-regulated | Mitosis | 5.1 | 51 | 1.19 × 10−18 |
| (CVID group 1 not DE) | Genes: ANAPC7, ANLN, ASPM, BUB1B, CCNB1, CCNF, CDC25B, CDCA8, CDK2, CENPE, CENPF, CIT, CKAP5, DLGAP5, ERCC6L, ESPL1, FAM83D, FBXO5, HAUS3, HAUS6, INCENP, KATNB1, KIF2C, KIF15, KIF18A, KIF20B, KIF23, KNTC1, NCAPD2, NCAPD3, NCAPG2, NCAPH, NDC80, NEK2, NUP43, NUSAP1, PBK, PBRM1, PDS5B, RAD21, SGOL1, SMC1A, SMC3, SMC4, SPAG5, STAG2, TERF1, TPX2, TUBB, VCPIP1, WEE1 | ||||
| Kinesin motor region | 6.8 | 12 | 1.34 × 10−3 | ||
| Genes: CENPE, KIF2C, KIF3A, KIF4A, KIF14, KIF15, KIF18A, KIF18B, KIF20A, KIF23, KIF24, KIF27 | |||||
| Heme biosynthetic process | 10.4 | 8 | 8.80 × 10−3 | ||
| Genes: ALAD, ALAS2, CPOX, FECH, FXN, HMBS, PPOX, UROS | |||||
| 1,083 up-regulated | Laminin EGF-like domain | 7.5 | 10 | 6.19 × 10−3 | |
| Genes: HSPG2, LAMA1, LAMA2, LAMA4, LAMB1, LAMB2, LAMC3, NTN1, NTN4, STAB1 | |||||
Further, 139 additional genes were differentially expressed in the bone marrow of CVID group 1 versus healthy controls and 2,001 other genes were differentially expressed in bone marrow of CVID group 2 versus healthy controls (Figure 2). To gain insight to the differences between the subgroups of CVID patients, we then applied the same enrichment analysis to these groups of genes (Table 1). In the 139 genes that were only differentially expressed in the bone marrow of CVID group 1, 71 were down-regulated, with enrichment of genes involved in blood coagulation. Of the 68 up-regulated genes in the bone marrow of CVID group 1, no enrichment terms were significant at the FDR-corrected level, but when considering raw p-values, enrichment of genes involved in ectodermal dysplasia was indicated based on the predominance of keratin gene expression. Recently, a role for keratins in cell differentiation, growth, and adhesion of non-epithelial cell lineages has been described, which is applicable to the bone marrow context [52].
Figure 2. Differential gene expression profiles between healthy control horse bone marrow and equine CVID patient bone marrow.
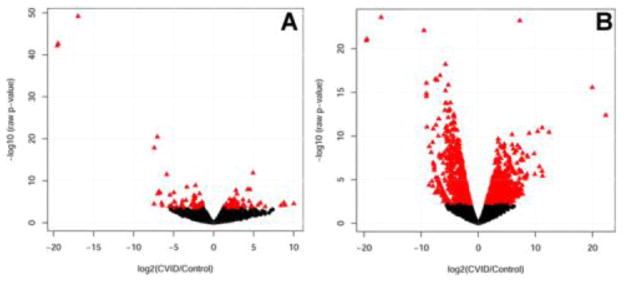
Gene expression is plotted by log2 fold change on the x-axis and −log10 FDR-adjusted p-value on the y-axis. Genes with significant differences in expression level compared to healthy control horses (FDR-adjusted p-value > 0.05) are represented by triangles in panel A) for CVID group 1; and panel B) for CVID group 2. Genes with equivalent expression are shown in filled black circles.
In the 2,001 genes that were only differentially expressed in the bone marrow of CVID group 2, 918 were down-regulated, with enrichment of genes involved mitosis, kinesin motors, and heme biosynthetic process. In the set of 1,083 up-regulated genes, over-represented biological terms included laminin EGF-like domain. Together, these genes regulate cell adhesion, migration, and differentiation. The expression differences of these genes may be due to both CVID and breed differences.
3.3. Differentiation of hematopoietic lineages in CVID horse bone marrow
Next, we utilized the bone marrow transcriptome dataset to investigate the mRNA expression of transcription factors, surface markers, and cytokines indicative of HSCs, bone marrow stromal cells, and 9 mature lineages that differentiate from HSCs (Figure 3A). Expression levels of a set of genes known to be expressed in the undifferentiated HSC stage were compared plotted for equine CVID patients and healthy control horses (BCL11A, CREBBP, GATA2, IRF8, MYB, RUNX1, TAL1, CD10/MME, CD34, SCA1/ATXN1, FLT3, KIT, and FLT3LG) [64]. Down-regulation was observed for BCL11A, TAL1, FLT3, and KIT in one of the CVID groups (FDR ≤ 0.028, Figure 3B). Loss of these genes may impair many aspects of hematopoiesis (long-term HSCs, CLPs, early B cells, T and NK lineages, erythroid differentiation, and myeloid progenitors) [53–60]. Because these genes were only down-regulated in one sub-group of CVID patients and the effect of deficiency of these genes are not restricted to B lymphopoiesis, they were not considered further.
Figure 3. Expression of hematopoietic stem cell lineage differentiation genes in the bone marrow of healthy control horses and equine CVID patients.

Gene expression is presented in counts per million reads (CPM) and plotted on the y-axis log2 scale for the genes listed on the x-axis. Transcription factor, cell surface, and cytokine genes are categorized by lineage-specific expression: panel A) schematic of hematopoietic lineage differentiation; B) hematopoietic stem cell; C) bone marrow stroma; D) megakaryocyte; E) erythrocyte; F) monocyte; G) neutrophil, eosinophil, and basophil; H) T lymphocyte and natural killer (NK) cell; I) B lymphocyte. Significant differences in expression were determined by p-values corrected for false discovery rate (FDR). ‘*’ denotes FDR < 0.05, ‘**’ denotes FDR < 0.01, ‘***’ denotes FDR< 0.001.
KITLG, IL6, IL7, and IL11 are cytokines expressed in the bone marrow milieu, which we have attributed to the bone marrow stroma compartment (Figure 3C) [61, 62]. Up-regulated expression of IL6 and IL11 was only detected in the bone marrow of CVID group 2 patients (FDR < 0.001).
Megakaryocyte differentiation is derived from the megakaryocyte and erythroid progenitor (MEP), a precursor of the common myeloid progenitor (CMP, Figure 3A) [63]. Megakaryocyte markers (NF-E2, THPO, ITGB3/CD61, and CD36) were either equivalent between healthy control horses and CVID samples or decreased in only one group (Figure 3D) [64].
Megakaryocyte and erythroid progenitors also propagate the erythroid lineage. Erythroid transcription factors FLI1, FOG/ZNF408, and erythropoietin (EPO) were expressed at equivalent levels among all equine CVID patients and healthy control horses (Figure 3E) [65]. However, expression of hemogen (HEMGN/EDAG), beta and epsilon globin chains (HBB, HBE1), and mature lineage markers erythropoietin receptor (EPOR), glycophorin A (CD235A/GYPA), and transferrin receptor (TFRC) were down-regulated in CVID group 2 horses compared to healthy control horses and CVID group 1 (FDR ≤ 0.003, Figure 3E) [65]. Accordingly, clinical anemia, perhaps inflammatory in origin due to recurrent bacterial infections, was documented in the CVID group 2 patient (#5) with the most drastic down-regulation of these genes.
The granulocyte and macrophage progenitor (GMP) can differentiate into monocytes, neutrophils, basophils, and eosinophils [63]. Monocyte markers PU.1/SPI1, MAFB, CSF1R, ANPEP, and M-CSF/CSF1 were expressed at comparable levels (Figure 3F) [66, 67]. The mature monocyte lineage marker CD14 was significantly up-regulated in the bone marrow of all equine CVID patients (FDR ≤ 0.033). CD14 binds and can be up-regulated by lipopolysaccharide and other bacterial wall constituents [68]. These CVID patients experienced bacterial infections, and because most other myeloid markers were not differentially expressed, we presume that the increase in CD14 expression was due to bacterial exposure rather than increased myelopoiesis.
Expression of the granulocyte-inducing cytokine, G-CSF/CSF3, only differed in one CVID sub-group (FDR < 0.001, Figure 3G) [69]. Gene expression of the mature neutrophil marker CD177 was increased approximately 100 times in the bone marrow of all equine CVID patients, although significance levels were different for CVID groups (CVID group 1 FDR > 0.05, raw p-value = 0.007, CVID group 2 FDR = 0.022) [70]. Correspondingly, neutrophilia was documented in most of these CVID patients, likely in response to bacterial infections. Basophil and eosinophil marker expression did not differ between healthy control and CVID bone marrow transcriptomes (IL5, CCR3, EPX) [71–73].
Lymphoid lineages T, natural killer (NK), and B cells differentiate from the CLP [63]. Gene expression of T lymphocyte markers did not differ in the bone marrow of equine CVID patients (NOTCH1, GATA3, CD3D, CD3E, CD3G, CD4, CD7, CD8A, CD8B, CD40LG, and IL7R; Figure 3H) [74–76]. In the NK lineage, gene expression of markers IL15, NCAM1/CD56, and NCR1/NKp46 were assessed, and only IL15 was increased in CVID group 2 bone marrow (FDR = 0.031) [77, 78]. Gene expression of CD2, a surface marker of T, NK, and B cells, was significantly up-regulated in the bone marrow of group 2 CVID patients (FDR = 0.012) [79, 80].
In the B lymphocyte lineage, expression of PAX5, the transcription factor responsible for B lymphocyte commitment, was significantly down-regulated as no PAX5 mRNA expression was detected in six of the seven CVID horse bone marrow samples (FDR < 0.024, Figure 3I). PAX5 activates CD19 gene expression; CD19 levels were variable in the bone marrow of equine CVID patients but were not significantly decreased after FDR correction [81]. However, MS4A1/CD20 gene expression was down-regulated in all CVID horse bone marrow samples (FDR ≤ 0.031). CD22 is a marker of mature and memory B cells, and CD22 gene expression was comparable between healthy control and CVID horse bone marrow [82].
Overall, gene expression of all hematopoietic lineages was detected in all CVID horses with significant increases in the CD14 myeloid marker and the CD177 neutrophil marker. Of the genes described above, the only down-regulated genes shared by all CVID horse bone marrow samples were the B cell markers PAX5 and CD20 (FDR ≤ 0.031). Within CVID group 2, a clear gene signature of anemia was present based on down-regulation of TAL1, HBB, HBE1, HEMGN, EPOR, GYPA, and TFRC genes. These data suggest that hematopoietic development is normal or responsive to the infection challenges of these patients, with production of myeloid and lymphoid cells, supporting the presence of an adequate bone marrow environment; in addition, the only obvious cell line deficiency in the central or peripheral compartment was B lymphocytes.
3.4. Stages of B lymphocyte differentiation in CVID horse bone marrow
Given the pronounced peripheral B lymphopenia and loss of B cell-specific gene and protein expression in the bone marrow of the equine CVID patients, we further analyzed gene expression specific to B lymphopoiesis [21, 22]. For this purpose, genes were assigned to discrete B lymphocyte differentiation stages based on the literature, but it is important to note that genes are expressed often over multiple stages and may be sustained until plasma cell differentiation, or expression levels may change between differentiation stages. In the bone marrow transcriptome profiles, many genes important for B cell lineage differentiation had low expression in the bone marrow of healthy control horses (Figure 4, less than 20 counts per million). Because genes with low expression are lower in statistical power and often have increased variance, we considered the p-value to identify significant differences in gene expression if the difference was at least 2-fold, in addition to the FDR corrected p-value.
Figure 4. Expression of B lymphocyte-specific genes during differentiation from hematopoietic stem cells in the bone marrow of healthy control horses and equine CVID patients.

Gene expression is presented in counts per million reads (CPM) and plotted on the y-axis log scale for the genes listed on the x-axis. Genes are categorized by B lymphocyte differentiation stage. Significant differences in expression were determined by p-values corrected for false discovery rate (FDR). ‘*’ denotes FDR < 0.05, ‘**’ denotes FDR < 0.01, ‘***’ denotes FDR< 0.001.
In the undifferentiated HSC and MPP stages, gene expression of crucial B lymphocyte markers IKAROS, IRF8, PU.1, RUNX1, CD24, CD34, and KIT was comparable between healthy control and CVID horse bone marrow samples (Figure 4) [53, 83–85]. However, differential gene expression was detected for BCL11A in CVID horse bone marrow (CVID group 1 p = 0.029, CVID group 2 FDR < 0.001). BCL11A loss impacts B lymphopoiesis and production of T and NK lineages; defects in T and NK lineages are not observed in equine CVID patients [56]. E2A/TCF3 expression was decreased in CVID group 2 (FDR = 0.026), although a significant reduction in E2A/TCF3 gene expression was detected in all CVID horse bone marrow samples by qRT-PCR (p < 0.05) [22]. E2A regulates cell cycle progression and primes lymphoid lineage-associated genes in HSCs, maintains appropriate numbers of MPPs and LMPPs, represses myeloid lineage genes, and activates expression of B lineage-specific genes such as EBF1, FOXO1, and BCL11A [23, 86, 87]. CD43/SPN (FDR = 0.003), FLT3 (FDR = 0.028) and KIT (FDR < 0.001) were down-regulated in one CVID group.
Expression of genes assigned to the CLP and early B stages was comparable between healthy control and CVID groups.
In the pro-B cell stage, PAX5 was significantly down-regulated in all CVID horse bone marrow samples (FDR < 0.024). PAX5 commits a cell to the B lineage by repressing at least 110 genes of T lymphocyte or myeloid lineages, activating 170 additional genes specific for the B lymphocyte lineage, importantly CD19, and providing positive feedback for EBF1 [88–90]. CD19 gene expression was decreased in CVID group 1 bone marrow (p < 0.005). Expression of PAX5 and CD19 genes were found to be significantly decreased in the same CVID horse bone marrow samples with qRT-PCR (p < 0.03) [22]. Expression of RAG2 was absent in all CVID group 2 horse bone marrow samples (p < 0.008) and absent in 3 of 4 CVID group 1 samples, although not statistically significant (p = 0.060). Decreased expression or loss of multiple genes representative of the pro-B cell stage in CVID horse bone marrow samples by both RNA-Seq and qRT-PCR suggests a block in B lymphocyte differentiation at the pro-B cell stage.
Expression levels of genes indicative of the subsequent pre-B cell stage (CD20/MS4A1, IGHM, and Ig light chain constant region IGKC, IGLC1, and IGLC7) were reduced in CVID horse bone marrow (FDR < 0.031, Figure 4) [91, 92]. Similarly, expression of genes specific to later B cell differentiation stages in CVID horse bone marrow was also significantly decreased (DPEP2, IGJ) or absent (IGHD) [93, 94]. This loss of gene expression substantiated the blockade of differentiation at the pro-B cell stage shared among all CVID patients. Notably, equine CVID patient 1 still maintained the highest gene expression levels for IGHM, IGKC, IGLC1, IGLC7, and IGJ.
Genes specific to plasma cells (PRDM1/BLIMP1, CD138/SDC1) remained at expression levels equivalent to healthy control horses, and contribute to expression level of genes such as CD79A, which were initiated during B lymphopoiesis and sustained throughout maturation and plasma cell differentiation (Figure 4) [95, 96]. Presumably expression of these genes represents pre-existing, long-lived plasma cells.
In summary, PAX5 is the most severely down-regulated gene identified in the B lymphocyte differentiation pathway, and is consistent with a developmental block entering the pro-B cell stage. In mouse models, loss of PAX5 expression promotes cells to differentiate into other lymphoid or myeloid lineages, and deletion of PAX5 from mature B lymphocytes causes de-differentiation into the CLP stage then progression to functional T lymphocytes [81, 97]. It does not seem feasible that equine CVID patients could lack PAX5 expression from birth (analogous to a knock-out mouse). Equine CVID patients have a median age of 10.5 years at the time of recurrent bacterial infections and diagnosis, with a range of 2 to 23 years. Given the natural conditions of equine management, it is unlikely for a horse to be healthy for 10 or 23 years (not experiencing recurrent infections) without any B lymphocytes. We hypothesize that aberrant epigenetic silencing of PAX5 occurs in a B lymphocyte progenitor cell at some postnatal time (perhaps variable among patients), B lymphopoiesis in the bone marrow is eventually lost, and B lymphocytes in the periphery become depleted in CVID patients, as B lymphocytes must be continuously produced. An epigenetic mechanism could explain the late-onset and non-familial manifestation of CVID.
The loss of PAX5 expression in CVID horse bone marrow was puzzling given that E2A and EBF1 genes were still expressed albeit in low levels. E2A can be subjected to post-translation regulation via binding of inhibitor of DNA binding (ID) proteins, KITLG, or TAL1 [98–100]. E2A deficient mice have a complete block in B lymphopoiesis prior to EBF1 expression and some T lymphocyte impairments [101–103]. MPPs deficient in E2A also fail to fully up-regulate FLT3, this may explain the more pronounced decrease in FLT3 expression found in CVID group 2 bone marrow [86]. Dosage of E2A is also important, as haploinsufficiency impairs differentiation [104]. Together, this suggests the E2A function is impaired, yet expression of downstream genes FOXO1 and EBF1 was maintained.
The presence of comparable EBF1 expression levels was also perplexing, as it is dependent upon E2A for expression and should activate PAX5 expression. EBF1 is capable of rescuing B lymphopoiesis in E2A-deficient progenitors by activating PAX5 gene expression, yet PAX5 was not detected in most CVID horse bone marrow samples [23, 105]. EBF1 is also regulated at the post-transcriptional level, which may be the case in CVID [106].
More recent and refined experiments revealed that 12–20% of CLPs express PAX5, presumably due to lineage priming [27]. Collectively, considering the lack of PAX5 expression and decreased E2A expression, it seems likely that the initial defect in CVID occurs in an early progenitor cell, and becomes fully manifested in the pro-B cell stage with the loss of PAX5.
3.5. Expression of genes documented to bear causal human CVID mutations
Genetic mutations have been described in 9 genes that may cause CVID in affected human patients, although family members of CVID patients are usually unaffected; these account for <10% of human CVID cases [6–17]. Most causal mutations have been identified in genes encoding B lymphocyte surface proteins and B lymphocyte receptor signaling proteins (CD19, CD20, CD21, CD27, CD81, BAFF-R, ICOS, PLCG2, LRBA). We investigated the expression of these genes in equine CVID bone marrow transcriptome data. Down-regulation of CD19 (p < 0.005 in CVID group 1) and CD20 (FDR < 0.031) genes were discussed in section 3.4. CD81, which is expressed by both B and T lymphocytes, was not different between healthy control and CVID horse bone marrow samples. ICOS gene expression was increased in CVID group 1 horse bone marrow samples at p ≤ 0.010, and TACI was down-regulated 14 fold in CVID group 1 bone marrow transcriptomes (FDR < 0.008). Only expression patterns of CD19 and CD20 are consistent with human CVID cohorts; however, down-regulation of B cell genes preceding CD19 and CD20 expression (i.e., PAX5) precludes the assignment of CD19 and CD20 as a causal role of disease status in equine CVID patients.
3.6. Sequence variation analysis
An advantage of RNA-Seq is the gathering of sequence content in addition to transcript quantification. Comparison of the sequences obtained herein with the reference genome revealed 9,552 single nucleotide polymorphisms (SNPs) predicted to have a functional effect. Ten of those identified were exclusive to equine CVID patients, including: non-synonymous coding change in PILRA; frameshifts in ADAMTS4, EFNB1, and RELB; splice site donors in LAMP1, pleckstrin, and ZNF219; splice site acceptors in C16orf62 homolog, CCL19, and WWP2 (supplemental Table 1). We recognize that the sample size in this dataset precludes powerful statistical analysis of SNPs contained in the transcriptomes but the expression levels of these SNPs were examined to determine whether any might warrant further investigation. The gene expression level of C16orf62 homolog, EFNB1, LAMP1, PILRA, pleckstrin, WWP2, and ZNF219 in CVID horse bone marrow was comparable to that of healthy control horses. CCL19 was up-regulated in all CVID horse bone marrow samples, and ADAMTS4 and RELB were up-regulated only in CVID group 2 samples (FDR ≤ 0.003). Further analysis of genetic variants associated with equine CVID is being investigated with genome-wide association studies in our laboratory.
3.7. Virus expression analysis
We next examined whether viral gene expression was different in CVID horse bone marrow samples. Endogenous retrovirus (ERV) mRNA expression was determined by comparing nearly 1,000 reported equine endogenous retroviruses with the bone marrow transcriptome dataset [44–46]. Of the 128 ERVs expressed in the bone marrow, only two ERVs exhibited significant differential expression between equine CVID patients and healthy control horses: EqERV3 (genome position chromosome 1:27393304 to 27404726, p = 0.001) and POL_gamma_21 (genome position chromosome 23:7188674 to 7189847, p < 0.001) (supplemental Table 2). Expression of EqERV3 was detected in healthy control horses and 3 of 7 CVID patients (#5–7); of those, only 1 equine CVID patient (#5) exhibited significant up-regulation. ERV POL_gamma_21 was expressed by healthy control horses and 5 of 7 CVID patients (#1–5), but significantly down-regulated only in group 2 CVID patients. Thus, we concluded that ERV were unlikely to contribute to CVID disease.
The RNA-Seq approach also facilitated analysis of equine herpesvirus gene expression. Publicly available sequences for equine herpesvirus strains 1, 2, 3, 4, 5, 7, 8, and 9 were compared to the RNA-Seq dataset obtained in this study. No EHV sequences were detected in healthy control horses, or in 6 of the 7 CVID patients. For 1 equine CVID patient (#3), < 5 reads mapped to the EHV reference sequences. These findings suggest that the equine herpes viruses noted above are unlikely to contribute to the loss of B cell production in equine CVID patients.
3.8. Reduced representation bisulfite sequencing
Given the non-familial and late-onset aspects of CVID together with the loss of PAX5 gene expression despite the presence of upstream transcription factors, we hypothesized that the loss of B lymphopoiesis was due to aberrant methylation of essential genes. To obtain a genome-wide assessment of methylation status, reduced representation bisulfite (RRBS) libraries were prepared from bone marrow genomic DNA of healthy control (n = 1) and CVID (n = 2) horses, as well as commercial unmethylated lambda DNA. Inclusion of unmethylated lambda DNA allowed confirmation that the bisulfite conversion of DNA was efficient (99.4%). Approximately 55 – 69 million sequence reads per sample were obtained and aligned to the equine genome sequence (EquCab2.0), with an alignment rate of 38.4–41.5% (Table 2). These rates are within the range of 19.4 – 44.6% reported for human RRBS alignments to the human genome sequence [107]. Over 2 million unique CpG sites were sequenced for each sample and 24.2% of these were methylated in healthy control horse bone marrow, whereas 28.0 – 31.0%of CpG sites were methylated in CVID horse bone marrow (Table 2). CpG islands were generally 100% unmethylated (70.6% in healthy control bone marrow) or 100% methylated in bone marrow genomic DNA (22.2%); 7.2% of CpG islands harbored partial methylation between 25% and 75%. By principal component analysis of CpG methylation, the healthy control horse bone marrow was clearly distinct from the CVID horse bone marrow (Figure 5). In total, 1,021 unique promoter or intragenic CpG islands were identified by RRBS. Of these, 185 CGIs gained methylation and 245 CGIs lost methylation in the bone marrow of both CVID patients. To appreciate the relevance of these findings to CVID, the lists of differentially expressed genes identified by RNA-Seq and differentially methylated genes identified by RRBS were merged, which revealed that the PAX5 gene was both down-regulated and gained methylation in CVID horse bone marrow (supplemental Table 3).
Table 2.
RRBS library mapping to reference genome and CpG content
| Sample | Total reads | Aligned reads | Alignment rate | # unique CpGs | Mean CpG coverage | C methylated in CpG context |
|---|---|---|---|---|---|---|
| Control | 59,563,167 | 22,886,821 | 38.4% | 2,238,235 | 27.0x | 24.2% |
| CVID 3 | 55,756,378 | 23,157,707 | 41.5% | 2,321,898 | 26.7x | 28.0% |
| CVID 4 | 68,852,020 | 27,149,164 | 39.4% | 2,630,177 | 27.7x | 31.0% |
Figure 5. Principal Component Analysis of CpG methylation between healthy control horse and equine CVID patient bone marrow genomic DNA.

Principal component analysis plot of genome-wide CpG methylation from healthy control horse and equine CVID patient bone marrow genomic DNA. The two first principal components are plotted and the proportion of variance explained by each component is scaled on the axes. The plot shows a clear demarcation of CpG methylation profiles between healthy and equine CVID bone marrow.
3.9. Methylation of PAX5 regulatory regions
In order to validate differential methylation of the PAX5 gene in CVID horse bone marrow, we amplified the PAX5 enhancer region from bisulfite-treated bone marrow genomic DNA from healthy control (n = 6) and CVID (n = 7) horse samples. The PAX5 enhancer was selected because methylation does not have an apparent role in regulating the PAX5 promoter, whereas the PAX5 enhancer becomes demethylated in multipotent hematopoietic progenitors [27]. Transcription factors PU.1, IRF4, IRF8, and NF-kB have been demonstrated to bind to the enhancer, and putative motifs for E2A (E-box) and RUNX1 binding are also present in the horse PAX5 enhancer sequence [27]. Cloning and sequencing ≥ 10 unique clones revealed that, compared to healthy control horse bone marrow genomic DNA, CVID horse bone marrow genomic DNA harbors greater methylation over 6 CpG sites (28.3 – 33.3% vs. 31.7 – 65.0%, respectively, p = 0.000 by Fisher’s exact test, Figure 6). A GEE logistic model with an exchangeable working correlation structure was performed to test for the effects of status, site and the interaction of status*site on methylation, while controlling for multiple measurements on each horse which were not independent of one another. The GEE model results confirmed that CVID status had a significant effect on methylation levels (p ≤ 0.001), and further revealed that CpG site also had a significant effect on methylation levels (p ≤ 0.001). The site analysis found that the first CpG in this region was marked by significantly more methylation than the last CpG site. This finding may have regulatory implications because the first CpG is found within the composite ETS/ISRE-consensus element (EICE) that fuses the ETS-binding motif (5′-GGAA-3′) with the IRF4-binding motif (5′-AANNGAAA-3′), and thus methylation may be regulating transcription factor binding. No binding sites are predicted for the last CpG site, perhaps indicating that there is less need for regulation by methylation at this site. No significant interaction was found between CVID status and CpG site (p = 0.855).
Figure 6. Methylation of PAX5 enhancer regulatory region in healthy control horses and equine CVID patients.
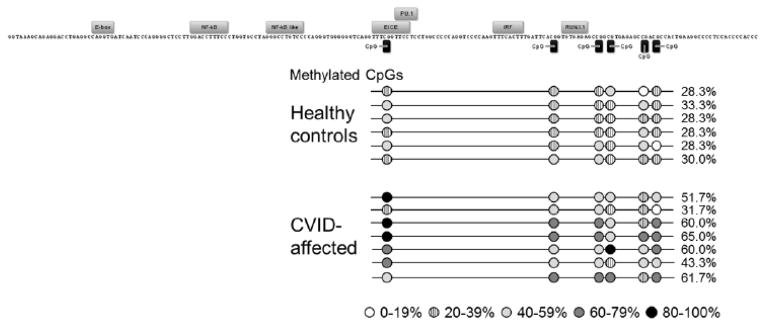
Methylation status was determined by amplification of bisulfite-treated bone marrow genomic DNA and sequencing of 10 unique clones per horse. Transcription factor binding sites are labeled above motif sequence and CpG sites are marked by black rectangles. Average methylation of each CpG is shown by circles shaded according to level of methylation. DNA from healthy horses (n=6) had 29.4% total methylation and CVID-affected horses (n=7) had 53.3% total methylation in this region, a significant increase (p = 0.000).
Thus, both increased methylation and decreased mRNA expression of the PAX5 gene was observed in the bone marrow of CVID horses. Increased methylation of the PAX5 enhancer suggests that the causal defect may lie in the multipotent hematopoietic progenitor stage because demethylation of this region should occur at that stage. This agrees with the RNA-Seq data in that the cause of equine CVID may initiate in an early stage of B lymphopoiesis and manifest as loss of PAX5 expression in the pro-B cell stage (Figure 8).
Figure 8. Overview of B lymphocyte differentiation block in equine CVID patients based on transcriptome and epigenome data analyses.
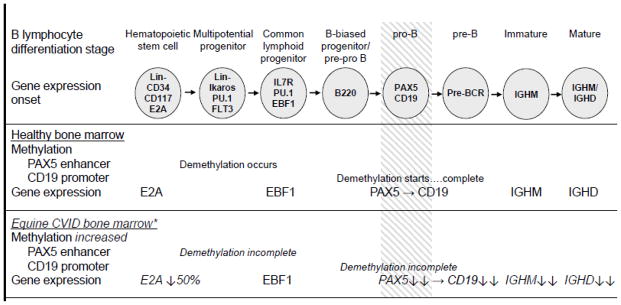
Stages of B lymphocyte differentiation are shown at the top. Changes in methylation and gene expression shown for healthy bone marrow yielding successful B lymphopoiesis (top panel) and equine CVID patient bone marrow with diminished B lymphopoiesis (bottom panel). The block defined in equine CVID B lymphopoiesis is indicated by shading. * Data shown are statistically different from control samples.
3.10. CD19 promoter methylation assessment
Next, we measured the methylation of CD19, a transcriptional target of PAX5, in the same healthy control and CVID horse bone marrow samples. Demethylation of the CD19 promoter initiates at the pre-pro B cell stage, between the CLP and pro-B cell stages, and is complete at the pre-B cell stage [34]. Amplification, cloning, and sequencing of 10 unique clones from each sample revealed that healthy control bone marrow samples carried 36.0 – 54.0% methylation over 5 CpG sites, in contrast to 52.0 – 92.0% methylation found in CVID horse bone marrow (p = 0.000, Figure 7). Increased methylation of the CD19 promoter further substantiates the placement of the B differentiation block in equine CVID.
Figure 7. Methylation of CD19 promoter region in healthy control horses and equine CVID patients.

Methylation status was determined by amplification of bisulfite-treated bone marrow genomic DNA and sequencing of 10 unique clones per horse. Transcription factor binding sites are labeled above motif sequence and CpG sites are marked by black rectangles. Average methylation of each CpG is shown by circles shaded according to level of methylation. DNA from 6 healthy horses had 44.3% methylation in this region and CVID-affected horses (n=7) had 74.0% total methylation, a significant increase (p = 0.000).
3.11. Correspondence between equine CVID and human CVID
Like human CVID patients, equine CVID patients present with late-onset hypogammaglobulinemia and recurrent bacterial infections (commonly, pneumonia, sinusitis, hepatitis, meningitis) that are treated with antimicrobials; however, the periodic administration of IgG to horses is impractical due to costs, and these patients are submitted to euthanasia when clinical management is not successful. We established an archive of equine CVID patient tissues which facilitated the bone marrow analyses presented herein; this is an advantage of equine CVID as it would be difficult to generate the same data from human CVID patients. Accordingly, only two descriptions of bone marrow biopsies from human CVID patients have been published [108, 109].
Although equine CVID patients present an apparently homogeneous manifestation of disease (impaired B lymphocyte development in the bone marrow), about 15% of human CVID patients present marked B cell lymphopenia that suggests impaired B cell development, whereas the majority of patients show heterogeneous phenotype. Enumeration of cells sorted into B cell differentiation stages from a heterogeneous group of human CVID bone marrow aspirates revealed a subset of patients (9 of 25) with a significant decrease in pre-B and immature B cells in the bone marrow and a corresponding decrease in the number of circulating CD19+ cells; no other hematopoietic abnormalities were identified [108]. Recently, similar findings were obtained in a smaller human CVID cohort (n=12, reduced number of pre-B-II and immature B cells in bone marrow correlated with fewer circulating transitional B cells; hematopoiesis otherwise normal) [109]. Not all of the human CVID donors in these studies exhibited B lymphopenia, suggesting that disturbances in later stages of B cell differentiation may not always be reflected in the periphery, or importantly, identification of reduced populations may vary with the B cell flow cytometric labeling strategy (ie, CD19 alone vs transitional phenotype). These findings of underlying bone marrow disturbances in human CVID patients, with or without B lymphopenia, substantiates the usefulness of the equine CVID model for gaining insight into B cell differentiation blockages.
Nevertheless, the natural and homogeneous presentation of CVID in horses allowed the identification of an epigenetic mechanism with gene expression implications that could be applied to any gene or form of CVID in human patients. The exact dysfunctional epigenetic mechanism and key genes may be different between equine and human patients and most likely among human patients; but the identification of an underlying mechanism that leads to a late-onset impairment of cell development or dysfunction provides novel tools to move forward into the spectra of dysfunctional gene regulation of the many forms of CVID. Ultimately, we anticipate that understanding the molecular events that initiate and maintain epigenetic gene silencing could lead to the development of therapeutic strategies that reverse gene silencing in CVID.
4. CONCLUSION
Analysis of transcriptome and epigenome profiles revealed a loss of the B cell signature PAX5 gene expression, with increased methylation of the PAX5 enhancer in the bone marrow of equine CVID patients. Differences in gene expression were measured between patients, perhaps due to a progressive nature of CVID, supported by the clinical findings and history, and molecular analyses. Also, we used breed segregation strategically in order to subtract genes that would have been differentially expressed due to breed or polymorphisms. B lymphopenia due to incomplete lymphopoiesis was shared among all patients, as well as the molecular signatures of PAX5 repression. Herein, we utilized core bone marrow samples to assess concomitantly hematopoietic environment and hematopoiesis of multiple lineages, and identified a single cell deficiency. These findings lead us to further explore the implications of E2A down-regulation, a B cell signature transcription factor upstream of PAX5, including isolating and enumerating early progenitor populations (HSC, LMPP, and CLP). Integrating gene expression with epigenetic status may be a key insight to understanding the onset of CVID and investigating novel therapeutic strategies.
Supplementary Material
Highlights.
Transcriptome profiles revealed pro-B cell differentiation block in equine CVID
1,021 unique CpG islands were differentially methylated in CVID horse bone marrow
PAX5 had lower expression and increased methylation in CVID horse bone marrow
Acknowledgments
L.S. was partially supported by the NYSTEM-funded Cornell Mammalian Cell Reprogramming Core. RNA-Seq and RRBS sequence analysis was aided by the Cornell University Biotechnology Resource Center Bioinformatics Facility. The authors thank Dr. Jonathan Flax for helpful discussion and Mr. Brendan Kraft for technical assistance. This study was supported by National Institutes of Health New Director’s Innovator Award DP2OD007216. The funding agency had no role in study design, collection or interpretation of data, writing of the manuscript, or in publication decisions.
Abbreviations
- bHLH
transcription factor basic helix-loop-helix motif
- CGI
CpG island
- CLP
common lymphoid progenitor
- CMP
common myeloid progenitor
- CpG
cytosine-phosphate-guanine dinucleotide motif
- CPM
count per million reads
- CVID
Common variable immunodeficiency
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- EHV
equine herpes virus
- EICE
composite ETS/ISRE-consensus element
- ERV
endogenous retrovirus
- FDR
false discovery rate
- GEE
generalized estimating equations
- GMP
granulocyte and macrophage progenitor
- HSC
hematopoietic stem cell
- ID
inhibitor of DNA binding proteins
- Ig
immunoglobulin
- LMPP
lymphoid-primed multipotent progenitor
- MDS
multidimensional scaling
- MEP
megakaryocyte and erythroid progenitor
- MPP
multipotent progenitor
- NK
natural killer cells
- RNA-Seq
RNA sequencing
- RRBS
reduced representation bisulfite sequence
- SNP
single nucleotide polymorphism
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sanford JP, Favour CB, Tribeman MS. Absence of serum gamma globulins in an adult. N Engl J Med. 1954;250:1027–1029. doi: 10.1056/NEJM195406172502403. [DOI] [PubMed] [Google Scholar]
- 2.Cunningham-Rundles C. The many faces of common variable immunodeficiency. Hematology Am Soc Hematol Educ Program. 2012;2012:301–305. doi: 10.1182/asheducation-2012.1.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, Vlkova M, Hernandez M, Detkova D, Bos PR, Poerksen G, von Bernuth H, Baumann U, Goldacker S, Gutenberger S, Schlesier M, Bergeron-van der Cruyssen F, Le Garff M, Debre P, Jacobs R, Jones J, Bateman E, Litzman J, van Hagen PM, Plebani A, Schmidt RE, Thon V, Quinti I, Espanol T, Webster AD, Chapel H, Vihinen M, Oksenhendler E, Peter HH, Warnatz K. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 4.Mouillot G, Carmagnat M, Gerard L, Garnier JL, Fieschi C, Vince N, Karlin L, Viallard JF, Jaussaud R, Boileau J, Donadieu J, Gardembas M, Schleinitz N, Suarez F, Hachulla E, Delavigne K, Morisset M, Jacquot S, Just N, Galicier L, Charron D, Debre P, Oksenhendler E, Rabian C DEFI Study Group, B-cell and T-cell phenotypes in CVID patients correlate with the clinical phenotype of the disease. J Clin Immunol. 2010;30:746–755. doi: 10.1007/s10875-010-9424-3. [DOI] [PubMed] [Google Scholar]
- 5.Pandolfi F, Milito C, Conti V, Pagliari D, Frosali S, Cianci R, Quinti I. Common variable immunodeficiency - new insight into the pathogenesis and the quest for a workable classification. J Biol Regul Homeost Agents. 2013;27:285–289. [PubMed] [Google Scholar]
- 6.Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Drager R, Eibel H, Fischer B, Schaffer AA, Mages HW, Kroczek RA, Peter HH. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4:261–268. doi: 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
- 7.van Zelm MC, Reisli I, van der Burg M, Castano D, van Noesel CJ, van Tol MJ, Woellner C, Grimbacher B, Patino PJ, van Dongen JJ, Franco JL. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901–1912. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 8.Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Bohm J, Kienzler AK, Pan-Hammarstrom Q, Hammarstrom L, Rakhmanov M, Schlesier M, Grimbacher B, Peter HH, Eibel H. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A. 2009;106:13945–13950. doi: 10.1073/pnas.0903543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Zelm MC, Smet J, Adams B, Mascart F, Schandene L, Janssen F, Ferster A, Kuo CC, Levy S, van Dongen JJ, van der Burg M. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010;120:1265–1274. doi: 10.1172/JCI39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IA, Dolman KM, Beaumont T, Tedder TF, van Noesel CJ, Eldering E, van Lier RA. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest. 2010;120:214–222. doi: 10.1172/JCI40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiel J, Kimmig L, Salzer U, Grudzien M, Lebrecht D, Hagena T, Draeger R, Voelxen N, Bergbreiter A, Jennings S, Gutenberger S, Aichem A, Illges H, Hannan JP, Kienzler AK, Rizzi M, Eibel H, Peter HH, Warnatz K, Grimbacher B, Rump JA, Schlesier M. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J Allergy Clin Immunol. 2012;129:801–810.e6. doi: 10.1016/j.jaci.2011.09.027. [DOI] [PubMed] [Google Scholar]
- 12.Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, Subramanian N, Bunney TD, Baxendale RW, Martins MS, Romberg N, Komarow H, Aksentijevich I, Kim HS, Ho J, Cruse G, Jung MY, Gilfillan AM, Metcalfe DD, Nelson C, O’Brien M, Wisch L, Stone K, Douek DC, Gandhi C, Wanderer AA, Lee H, Nelson SF, Shianna KV, Cirulli ET, Goldstein DB, Long EO, Moir S, Meffre E, Holland SM, Kastner DL, Katan M, Hoffman HM, Milner JD. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. 2012;366:330–338. doi: 10.1056/NEJMoa1102140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, Herholz P, Trujillo-Vargas CM, Phadwal K, Simon AK, Moutschen M, Etzioni A, Mory A, Srugo I, Melamed D, Hultenby K, Liu C, Baronio M, Vitali M, Philippet P, Dideberg V, Aghamohammadi A, Rezaei N, Enright V, Du L, Salzer U, Eibel H, Pfeifer D, Veelken H, Stauss H, Lougaris V, Plebani A, Gertz EM, Schaffer AA, Hammarstrom L, Grimbacher B. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90:986–1001. doi: 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Montfrans JM, Hoepelman AI, Otto S, van Gijn M, van de Corput L, de Weger RA, Monaco-Shawver L, Banerjee PP, Sanders EA, Jol-van der Zijde CM, Betts MR, Orange JS, Bloem AC, Tesselaar K. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol. 2012;129:787–793.e6. doi: 10.1016/j.jaci.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pan-Hammarstrom Q, Salzer U, Du L, Bjorkander J, Cunningham-Rundles C, Nelson DL, Bacchelli C, Gaspar HB, Offer S, Behrens TW, Grimbacher B, Hammarstrom L. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. 2007;39:429–430. doi: 10.1038/ng0407-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sekine H, Ferreira RC, Pan-Hammarstrom Q, Graham RR, Ziemba B, de Vries SS, Liu J, Hippen K, Koeuth T, Ortmann W, Iwahori A, Elliott MK, Offer S, Skon C, Du L, Novitzke J, Lee AT, Zhao N, Tompkins JD, Altshuler D, Gregersen PK, Cunningham-Rundles C, Harris RS, Her C, Nelson DL, Hammarstrom L, Gilkeson GS, Behrens TW. Role for Msh5 in the regulation of Ig class switch recombination. Proc Natl Acad Sci U S A. 2007;104:7193–7198. doi: 10.1073/pnas.0700815104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keller MD, Jyonouchi S. Chipping away at a mountain: genomic studies in common variable immunodeficiency. Autoimmun Rev. 2013;12:687–689. doi: 10.1016/j.autrev.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orange JS, Glessner JT, Resnick E, Sullivan KE, Lucas M, Ferry B, Kim CE, Hou C, Wang F, Chiavacci R, Kugathasan S, Sleasman JW, Baldassano R, Perez EE, Chapel H, Cunningham-Rundles C, Hakonarson H. Genome-wide association identifies diverse causes of common variable immunodeficiency. J Allergy Clin Immunol. 2011;127:1360–7.e6. doi: 10.1016/j.jaci.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flaminio MJ, LaCombe V, Kohn CW, Antczak DF. Common variable immunodeficiency in a horse. J Am Vet Med Assoc. 2002;221:1296–302. 1267. doi: 10.2460/javma.2002.221.1296. [DOI] [PubMed] [Google Scholar]
- 20.Pellegrini-Masini A, Bentz AI, Johns IC, Parsons CS, Beech J, Whitlock RH, Flaminio MJ. Common variable immunodeficiency in three horses with presumptive bacterial meningitis. J Am Vet Med Assoc. 2005;227:114–22. 87. doi: 10.2460/javma.2005.227.114. [DOI] [PubMed] [Google Scholar]
- 21.Flaminio MJ, Tallmadge RL, Salles-Gomes CO, Matychak MB. Common variable immunodeficiency in horses is characterized by B cell depletion in primary and secondary lymphoid tissues. J Clin Immunol. 2009;29:107–116. doi: 10.1007/s10875-008-9221-4. [DOI] [PubMed] [Google Scholar]
- 22.Tallmadge RL, Such KA, Miller KC, Matychak MB, Felippe MJ. Expression of essential B cell development genes in horses with common variable immunodeficiency. Mol Immunol. 2012;51:169–176. doi: 10.1016/j.molimm.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, Ideker T, Glass CK, Murre C. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol. 2010;11:635–643. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mansson R, Welinder E, Ahsberg J, Lin YC, Benner C, Glass CK, Lucas JS, Sigvardsson M, Murre C. Positive intergenic feedback circuitry, involving EBF1 and FOXO1, orchestrates B-cell fate. Proc Natl Acad Sci U S A. 2012;109:21028–21033. doi: 10.1073/pnas.1211427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Q, Iida R, Yokota T, Kincade PW. Early events in lymphopoiesis: an update. Curr Opin Hematol. 2013;20:265–272. doi: 10.1097/MOH.0b013e3283612628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grosschedl R. Establishment and Maintenance of B Cell Identity. Cold Spring Harb Symp Quant Biol. 2013;78:23–30. doi: 10.1101/sqb.2013.78.020057. [DOI] [PubMed] [Google Scholar]
- 27.Decker T, Pasca di Magliano M, McManus S, Sun Q, Bonifer C, Tagoh H, Busslinger M. Stepwise activation of enhancer and promoter regions of the B cell commitment gene Pax5 in early lymphopoiesis. Immunity. 2009;30:508–520. doi: 10.1016/j.immuni.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 28.Treiber T, Mandel EM, Pott S, Gyory I, Firner S, Liu ET, Grosschedl R. Early B cell factor 1 regulates B cell gene networks by activation, repression and transcription-independent poising of chromatin. Immunity. 2010;32:714–725. doi: 10.1016/j.immuni.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 29.McManus S, Ebert A, Salvagiotto G, Medvedovic J, Sun Q, Tamir I, Jaritz M, Tagoh H, Busslinger M. The transcription factor Pax5 regulates its target genes by recruiting chromatin-modifying proteins in committed B cells. EMBO J. 2011;30:2388–2404. doi: 10.1038/emboj.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynaud D, Demarco IA, Reddy KL, Schjerven H, Bertolino E, Chen Z, Smale ST, Winandy S, Singh H. Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nat Immunol. 2008;9:927–936. doi: 10.1038/ni.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakamoto S, Wakae K, Anzai Y, Murai K, Tamaki N, Miyazaki M, Miyazaki K, Romanow WJ, Ikawa T, Kitamura D, Yanagihara I, Minato N, Murre C, Agata Y. E2A and CBP/p300 act in synergy to promote chromatin accessibility of the immunoglobulin kappa locus. J Immunol. 2012;188:5547–5560. doi: 10.4049/jimmunol.1002346. [DOI] [PubMed] [Google Scholar]
- 32.Maier H, Ostraat R, Gao H, Fields S, Shinton SA, Medina KL, Ikawa T, Murre C, Singh H, Hardy RR, Hagman J. Early B cell factor cooperates with Runx1 and mediates epigenetic changes associated with mb-1 transcription. Nat Immunol. 2004;5:1069–1077. doi: 10.1038/ni1119. [DOI] [PubMed] [Google Scholar]
- 33.Gao H, Lukin K, Ramirez J, Fields S, Lopez D, Hagman J. Opposing effects of SWI/SNF and Mi-2/NuRD chromatin remodeling complexes on epigenetic reprogramming by EBF and Pax5. Proc Natl Acad Sci U S A. 2009;106:11258–11263. doi: 10.1073/pnas.0809485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walter K, Bonifer C, Tagoh H. Stem cell-specific epigenetic priming and B cell-specific transcriptional activation at the mouse Cd19 locus. Blood. 2008;112:1673–1682. doi: 10.1182/blood-2008-02-142786. [DOI] [PubMed] [Google Scholar]
- 35.Eberhard D, Jimenez G, Heavey B, Busslinger M. Transcriptional repression by Pax5 (BSAP) through interaction with corepressors of the Groucho family. EMBO J. 2000;19:2292–2303. doi: 10.1093/emboj/19.10.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linderson Y, Eberhard D, Malin S, Johansson A, Busslinger M, Pettersson S. Corecruitment of the Grg4 repressor by PU.1 is critical for Pax5-mediated repression of B-cell-specific genes. EMBO Rep. 2004;5:291–296. doi: 10.1038/sj.embor.7400089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee ST, Xiao Y, Muench MO, Xiao J, Fomin ME, Wiencke JK, Zheng S, Dou X, de Smith A, Chokkalingam A, Buffler P, Ma X, Wiemels JL. A global DNA methylation and gene expression analysis of early human B-cell development reveals a demethylation signature and transcription factor network. Nucleic Acids Res. 2012;40:11339–11351. doi: 10.1093/nar/gks957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 43.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Kuyl AC. Characterization of a full-length endogenous beta-retrovirus, EqERV-beta1, in the genome of the horse (Equus caballus) Viruses. 2011;3:620–628. doi: 10.3390/v3060620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Etxebarria K, Jugo BM. Detection and characterization of endogenous retroviruses in the horse genome by in silico analysis. Virology. 2012;434:59–67. doi: 10.1016/j.virol.2012.08.047. [DOI] [PubMed] [Google Scholar]
- 46.Brown K, Moreton J, Malla S, Aboobaker AA, Emes RD, Tarlinton RE. Characterisation of retroviruses in the horse genome and their transcriptional activity via transcriptome sequencing. Virology. 2012;433:55–63. doi: 10.1016/j.virol.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 47.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu H, Caffo B, Jaffee HA, Irizarry RA, Feinberg AP. Redefining CpG islands using hidden Markov models. Biostatistics. 2010;11:499–514. doi: 10.1093/biostatistics/kxq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 50.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res. 2008;36:W170–5. doi: 10.1093/nar/gkn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vijayaraj P, Kroeger C, Reuter U, Hartmann D, Magin TM. Keratins regulate yolk sac hematopoiesis and vasculogenesis through reduced BMP-4 signaling. Eur J Cell Biol. 2010;89:299–306. doi: 10.1016/j.ejcb.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 53.Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, Wunsche P, Zeisberger P, Brocks D, Gu L, Herrmann C, Haas S, Essers MA, Brors B, Eils R, Huber W, Milsom MD, Plass C, Krijgsveld J, Trumpp A. Identification of Regulatory Networks in HSCs and Their Immediate Progeny via Integrated Proteome, Transcriptome, and DNA Methylome Analysis. Cell Stem Cell. 2014 doi: 10.1016/j.stem.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 54.Thoren LA, Liuba K, Bryder D, Nygren JM, Jensen CT, Qian H, Antonchuk J, Jacobsen SE. Kit regulates maintenance of quiescent hematopoietic stem cells. J Immunol. 2008;180:2045–2053. doi: 10.4049/jimmunol.180.4.2045. [DOI] [PubMed] [Google Scholar]
- 55.Yu Y, Wang J, Khaled W, Burke S, Li P, Chen X, Yang W, Jenkins NA, Copeland NG, Zhang S, Liu P. Bcl11a is essential for lymphoid development and negatively regulates p53. J Exp Med. 2012;209:2467–2483. doi: 10.1084/jem.20121846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu P, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T, Jenkins NA, Copeland NG. Bcl11a is essential for normal lymphoid development. Nat Immunol. 2003;4:525–532. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- 57.Porcher C, Swat W, Rockwell K, Fujiwara Y, Alt FW, Orkin SH. The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell. 1996;86:47–57. doi: 10.1016/s0092-8674(00)80076-8. [DOI] [PubMed] [Google Scholar]
- 58.Aplan PD, Nakahara K, Orkin SH, Kirsch IR. The SCL gene product: a positive regulator of erythroid differentiation. EMBO J. 1992;11:4073–4081. doi: 10.1002/j.1460-2075.1992.tb05500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beaudin AE, Boyer SW, Forsberg EC. Flk2/Flt3 promotes both myeloid and lymphoid development by expanding non-self-renewing multipotent hematopoietic progenitor cells. Exp Hematol. 2014;42:218–229.e4. doi: 10.1016/j.exphem.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu X, Satpathy AT, Kc W, Liu P, Murphy TL, Murphy KM. Bcl11a controls Flt3 expression in early hematopoietic progenitors and is required for pDC development in vivo. PLoS One. 2013;8:e64800. doi: 10.1371/journal.pone.0064800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balduino A, Mello-Coelho V, Wang Z, Taichman RS, Krebsbach PH, Weeraratna AT, Becker KG, de Mello W, Taub DD, Borojevic R. Molecular signature and in vivo behavior of bone marrow endosteal and subendosteal stromal cell populations and their relevance to hematopoiesis. Exp Cell Res. 2012;318:2427–2437. doi: 10.1016/j.yexcr.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsuda-Hashii Y, Takai K, Ohta H, Fujisaki H, Tokimasa S, Osugi Y, Ozono K, Matsumoto K, Nakamura T, Hara J. Hepatocyte growth factor plays roles in the induction and autocrine maintenance of bone marrow stromal cell IL-11, SDF-1 alpha, and stem cell factor. Exp Hematol. 2004;32:955–961. doi: 10.1016/j.exphem.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 63.Rieger MA, Schroeder T. Hematopoiesis. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a008250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang Y, Bluteau D, Debili N, Vainchenker W. From hematopoietic stem cells to platelets. J Thromb Haemost 5 Suppl. 2007;1:318–327. doi: 10.1111/j.1538-7836.2007.02472.x. [DOI] [PubMed] [Google Scholar]
- 65.Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood. 2011;118:6258–6268. doi: 10.1182/blood-2011-07-356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huber R, Pietsch D, Gunther J, Welz B, Vogt N, Brand K. Regulation of monocyte differentiation by specific signaling modules and associated transcription factor networks. Cell Mol Life Sci. 2014;71:63–92. doi: 10.1007/s00018-013-1322-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Subramani J, Ghosh M, Rahman MM, Caromile LA, Gerber C, Rezaul K, Han DK, Shapiro LH. Tyrosine phosphorylation of CD13 regulates inflammatory cell-cell adhesion and monocyte trafficking. J Immunol. 2013;191:3905–3912. doi: 10.4049/jimmunol.1301348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Landmann R, Knopf HP, Link S, Sansano S, Schumann R, Zimmerli W. Human monocyte CD14 is upregulated by lipopolysaccharide. Infect Immun. 1996;64:1762–1769. doi: 10.1128/iai.64.5.1762-1769.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84:1737–1746. [PubMed] [Google Scholar]
- 70.Bayat B, Werth S, Sachs UJ, Newman DK, Newman PJ, Santoso S. Neutrophil transmigration mediated by the neutrophil-specific antigen CD177 is influenced by the endothelial S536N dimorphism of platelet endothelial cell adhesion molecule-1. J Immunol. 2010;184:3889–3896. doi: 10.4049/jimmunol.0903136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Clutterbuck EJ, Sanderson CJ. Human eosinophil hematopoiesis studied in vitro by means of murine eosinophil differentiation factor (IL5): production of functionally active eosinophils from normal human bone marrow. Blood. 1988;71:646–651. [PubMed] [Google Scholar]
- 72.Hausmann OV, Gentinetta T, Fux M, Ducrest S, Pichler WJ, Dahinden CA. Robust expression of CCR3 as a single basophil selection marker in flow cytometry. Allergy. 2011;66:85–91. doi: 10.1111/j.1398-9995.2010.02431.x. [DOI] [PubMed] [Google Scholar]
- 73.Curran CS. Human eosinophil adhesion and receptor expression. Methods Mol Biol. 2014;1178:129–141. doi: 10.1007/978-1-4939-1016-8_12. [DOI] [PubMed] [Google Scholar]
- 74.von Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med. 1995;181:1519–1526. doi: 10.1084/jem.181.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hozumi K, Negishi N, Tsuchiya I, Abe N, Hirano K, Suzuki D, Yamamoto M, Engel JD, Habu S. Notch signaling is necessary for GATA3 function in the initiation of T cell development. Eur J Immunol. 2008;38:977–985. doi: 10.1002/eji.200737688. [DOI] [PubMed] [Google Scholar]
- 76.Lane P, Traunecker A, Hubele S, Inui S, Lanzavecchia A, Gray D. Activated human T cells express a ligand for the human B cell-associated antigen CD40 which participates in T cell-dependent activation of B lymphocytes. Eur J Immunol. 1992;22:2573–2578. doi: 10.1002/eji.1830221016. [DOI] [PubMed] [Google Scholar]
- 77.Schotte R, Dontje W, Nagasawa M, Yasuda Y, Bakker AQ, Spits H, Blom B. Synergy between IL-15 and Id2 promotes the expansion of human NK progenitor cells which can be counteracted by the E protein HEB required to drive T cell development. J Immunol. 2010;184:6670–6679. doi: 10.4049/jimmunol.0901508. [DOI] [PubMed] [Google Scholar]
- 78.Zamai L, Galeotti L, Del Zotto G, Canonico B, Mirandola P, Papa S. Identification of a NCR+/NKG2D+/LFA-1(low)/CD94(−) immature human NK cell subset. Cytometry A. 2009;75:893–901. doi: 10.1002/cyto.a.20789. [DOI] [PubMed] [Google Scholar]
- 79.Bierer BE, Sleckman BP, Ratnofsky SE, Burakoff SJ. The biologic roles of CD2, CD4, and CD8 in T-cell activation. Annu Rev Immunol. 1989;7:579–599. doi: 10.1146/annurev.iy.07.040189.003051. [DOI] [PubMed] [Google Scholar]
- 80.Punnonen J, de Vries JE. Characterization of a novel CD2+ human thymic B cell subset. J Immunol. 1993;151:100–110. [PubMed] [Google Scholar]
- 81.Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401:556–562. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 82.Dorken B, Moldenhauer G, Pezzutto A, Schwartz R, Feller A, Kiesel S, Nadler LM. HD39 (B3), a B lineage-restricted antigen whose cell surface expression is limited to resting and activated human B lymphocytes. J Immunol. 1986;136:4470–4479. [PubMed] [Google Scholar]
- 83.Merkenschlager M. Ikaros in immune receptor signaling, lymphocyte differentiation, and function. FEBS Lett. 2010;584:4910–4914. doi: 10.1016/j.febslet.2010.09.042. [DOI] [PubMed] [Google Scholar]
- 84.Wang H, Lee CH, Qi C, Tailor P, Feng J, Abbasi S, Atsumi T, Morse HC., 3rd IRF8 regulates B-cell lineage specification, commitment and differentiation. Blood. 2008;112:4028–4038. doi: 10.1182/blood-2008-01-129049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hunte BE, Capone M, Zlotnik A, Rennick D, Moore TA. Acquisition of CD24 expression by Lin-CD43+B220(low)ckit(hi) cells coincides with commitment to the B cell lineage. Eur J Immunol. 1998;28:3850–3856. doi: 10.1002/(SICI)1521-4141(199811)28:11<3850::AID-IMMU3850>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 86.Yang Q, Kardava L, St Leger A, Martincic K, Varnum-Finney B, Bernstein ID, Milcarek C, Borghesi L. E47 controls the developmental integrity and cell cycle quiescence of multipotential hematopoietic progenitors. J Immunol. 2008;181:5885–5894. doi: 10.4049/jimmunol.181.9.5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dias S, Mansson R, Gurbuxani S, Sigvardsson M, Kee BL. E2A proteins promote development of lymphoid-primed multipotent progenitors. Immunity. 2008;29:217–227. doi: 10.1016/j.immuni.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schebesta A, McManus S, Salvagiotto G, Delogu A, Busslinger GA, Busslinger M. Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity. 2007;27:49–63. doi: 10.1016/j.immuni.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 89.Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, Busslinger M. Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity. 2006;24:269–281. doi: 10.1016/j.immuni.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 90.Roessler S, Gyory I, Imhof S, Spivakov M, Williams RR, Busslinger M, Fisher AG, Grosschedl R. Distinct promoters mediate the regulation of Ebf1 gene expression by interleukin-7 and Pax5. Mol Cell Biol. 2007;27:579–594. doi: 10.1128/MCB.01192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Uchida J, Lee Y, Hasegawa M, Liang Y, Bradney A, Oliver JA, Bowen K, Steeber DA, Haas KM, Poe JC, Tedder TF. Mouse CD20 expression and function. Int Immunol. 2004;16:119–129. doi: 10.1093/intimm/dxh009. [DOI] [PubMed] [Google Scholar]
- 92.van Zelm MC, van der Burg M, de Ridder D, Barendregt BH, de Haas EF, Reinders MJ, Lankester AC, Revesz T, Staal FJ, van Dongen JJ. Ig gene rearrangement steps are initiated in early human precursor B cell subsets and correlate with specific transcription factor expression. J Immunol. 2005;175:5912–5922. doi: 10.4049/jimmunol.175.9.5912. [DOI] [PubMed] [Google Scholar]
- 93.Hystad ME, Myklebust JH, Bo TH, Sivertsen EA, Rian E, Forfang L, Munthe E, Rosenwald A, Chiorazzi M, Jonassen I, Staudt LM, Smeland EB. Characterization of early stages of human B cell development by gene expression profiling. J Immunol. 2007;179:3662–3671. doi: 10.4049/jimmunol.179.6.3662. [DOI] [PubMed] [Google Scholar]
- 94.Pioli PD, Debnath I, Weis JJ, Weis JH. Zfp318 Regulates IgD Expression by Abrogating Transcription Termination within the Ighm/Ighd Locus. J Immunol. 2014;193:2546–2553. doi: 10.4049/jimmunol.1401275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol. 2011;23:341–349. doi: 10.1016/j.smim.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 96.Robillard N, Wuilleme S, Moreau P, Bene MC. Immunophenotype of normal and myelomatous plasma-cell subsets. Front Immunol. 2014;5:137. doi: 10.3389/fimmu.2014.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cobaleda C, Jochum W, Busslinger M. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature. 2007;449:473–477. doi: 10.1038/nature06159. [DOI] [PubMed] [Google Scholar]
- 98.Kee BL. E and ID proteins branch out. Nat Rev Immunol. 2009;9:175–184. doi: 10.1038/nri2507. [DOI] [PubMed] [Google Scholar]
- 99.Riley RL, Knowles J, King AM. Levels of E2A protein expression in B cell precursors are stage-dependent and inhibited by stem cell factor (c-kit ligand) Exp Hematol. 2002;30:1412–1418. doi: 10.1016/s0301-472x(02)00953-0. [DOI] [PubMed] [Google Scholar]
- 100.O’Neil J, Shank J, Cusson N, Murre C, Kelliher M. TAL1/SCL induces leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell. 2004;5:587–596. doi: 10.1016/j.ccr.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 101.Zhuang Y, Soriano P, Weintraub H. The helix-loop-helix gene E2A is required for B cell formation. Cell. 1994;79:875–884. doi: 10.1016/0092-8674(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 102.Bain G, Maandag EC, Izon DJ, Amsen D, Kruisbeek AM, Weintraub BC, Krop I, Schlissel MS, Feeney AJ, van Roon M. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. 1994;79:885–892. doi: 10.1016/0092-8674(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 103.Bain G, Robanus Maandag EC, te Riele HP, Feeney AJ, Sheehy A, Schlissel M, Shinton SA, Hardy RR, Murre C. Both E12 and E47 allow commitment to the B cell lineage. Immunity. 1997;6:145–154. doi: 10.1016/s1074-7613(00)80421-5. [DOI] [PubMed] [Google Scholar]
- 104.Herblot S, Aplan PD, Hoang T. Gradient of E2A activity in B-cell development. Mol Cell Biol. 2002;22:886–900. doi: 10.1128/MCB.22.3.886-900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Seet CS, Brumbaugh RL, Kee BL. Early B cell factor promotes B lymphopoiesis with reduced interleukin 7 responsiveness in the absence of E2A. J Exp Med. 2004;199:1689–1700. doi: 10.1084/jem.20032202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mansson R, Zandi S, Welinder E, Tsapogas P, Sakaguchi N, Bryder D, Sigvardsson M. Single-cell analysis of the common lymphoid progenitor compartment reveals functional and molecular heterogeneity. Blood. 2010;115:2601–2609. doi: 10.1182/blood-2009-08-236398. [DOI] [PubMed] [Google Scholar]
- 107.Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, Jager N, Gnirke A, Stunnenberg HG, Meissner A. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 2010;28:1106–1114. doi: 10.1038/nbt.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ochtrop ML, Goldacker S, May AM, Rizzi M, Draeger R, Hauschke D, Stehfest C, Warnatz K, Goebel H, Technau-Ihling K, Werner M, Salzer U, Eibel H, Schlesier M, Peter HH. T and B lymphocyte abnormalities in bone marrow biopsies of common variable immunodeficiency. Blood. 2011;118:309–318. doi: 10.1182/blood-2010-11-321695. [DOI] [PubMed] [Google Scholar]
- 109.Anzilotti C, Kienzler AK, Lopez-Granados E, Gooding S, Davies B, Pandit H, Lucas M, Price A, Littlewood T, van der Burg M, Patel SY, Chapel H. Key stages of bone marrow B-cell maturation are defective in patients with common variable immunodeficiency disorders. J Allergy Clin Immunol. 2015 doi: 10.1016/j.jaci.2014.12.1943. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.